
Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine


Abstract
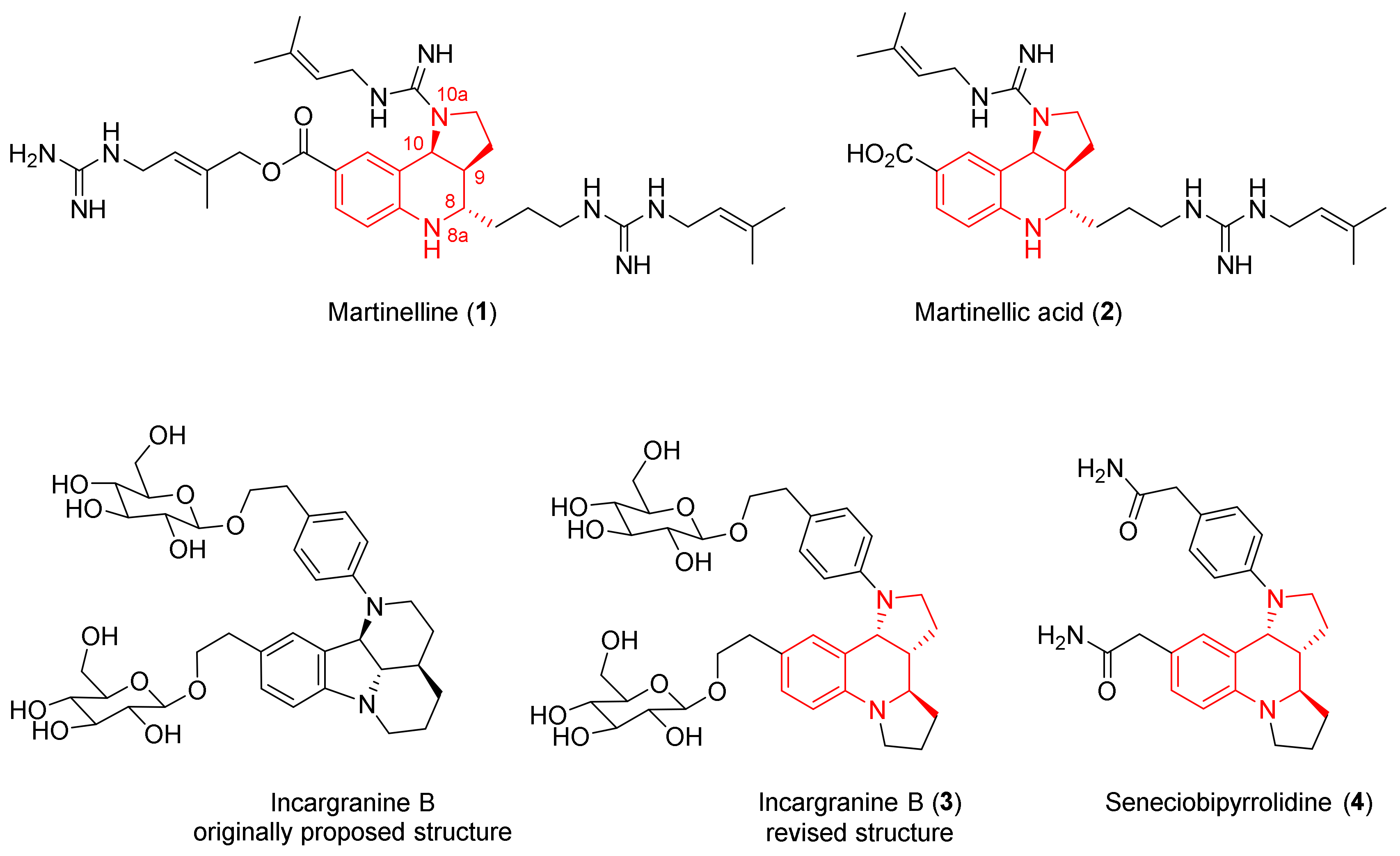
:1. Introduction
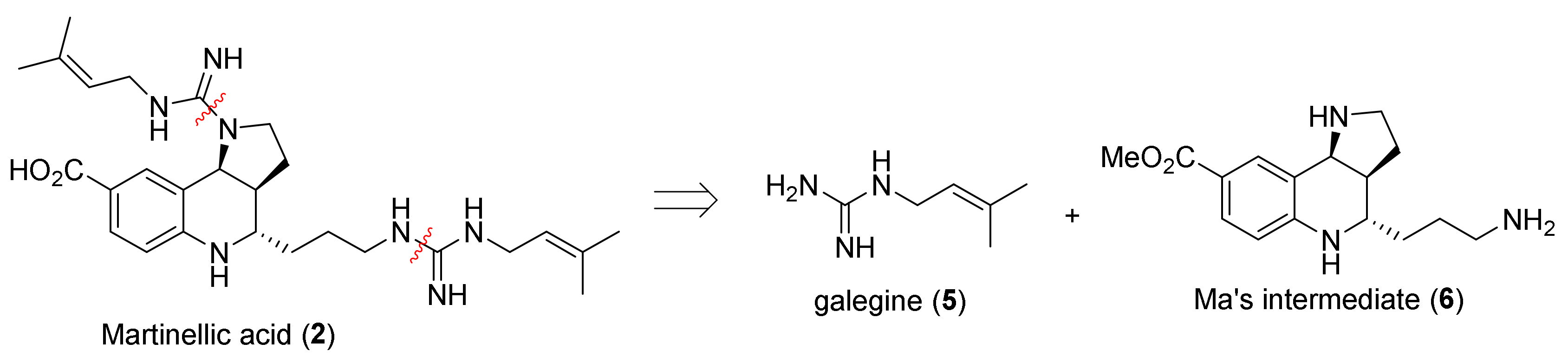
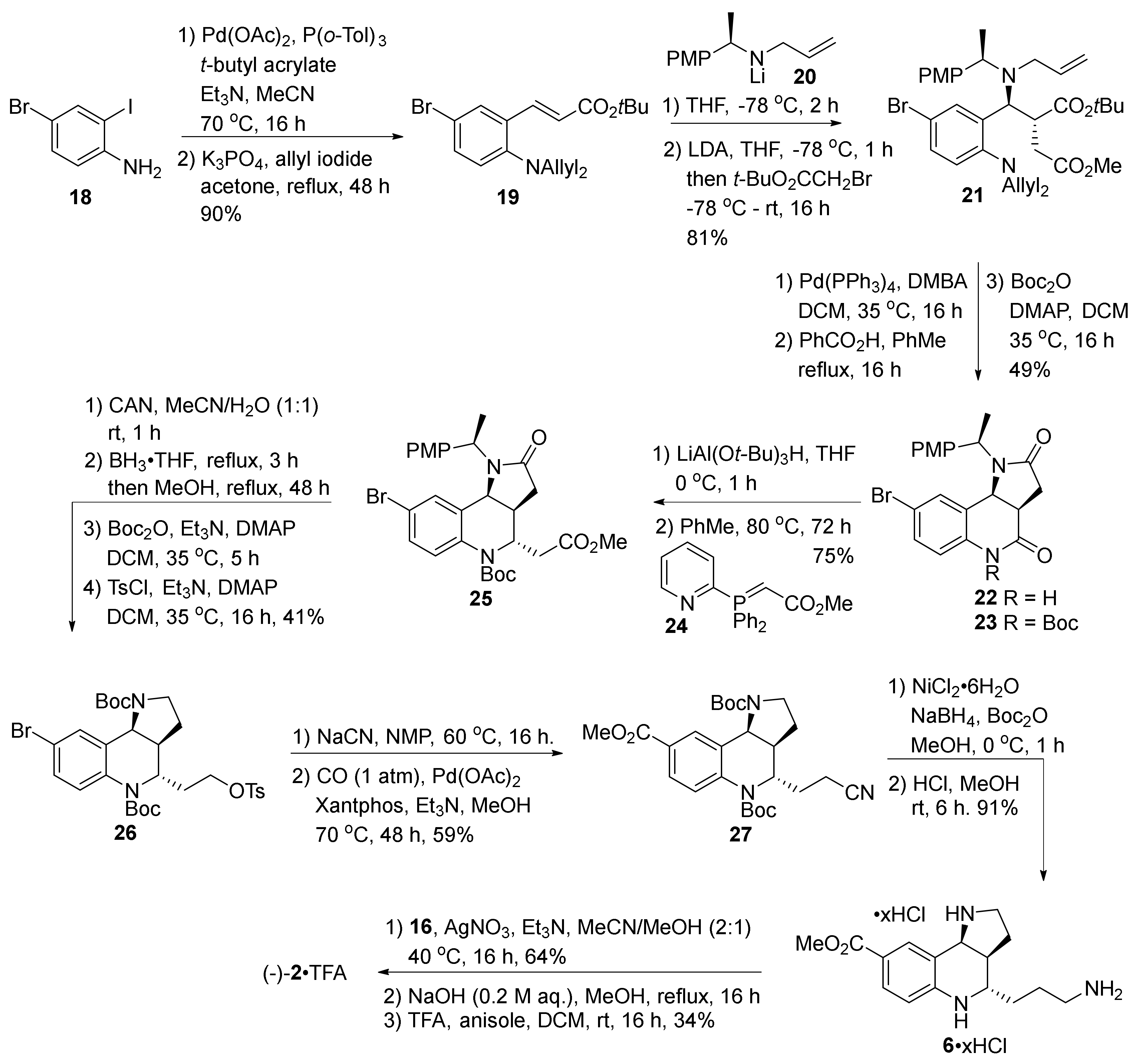
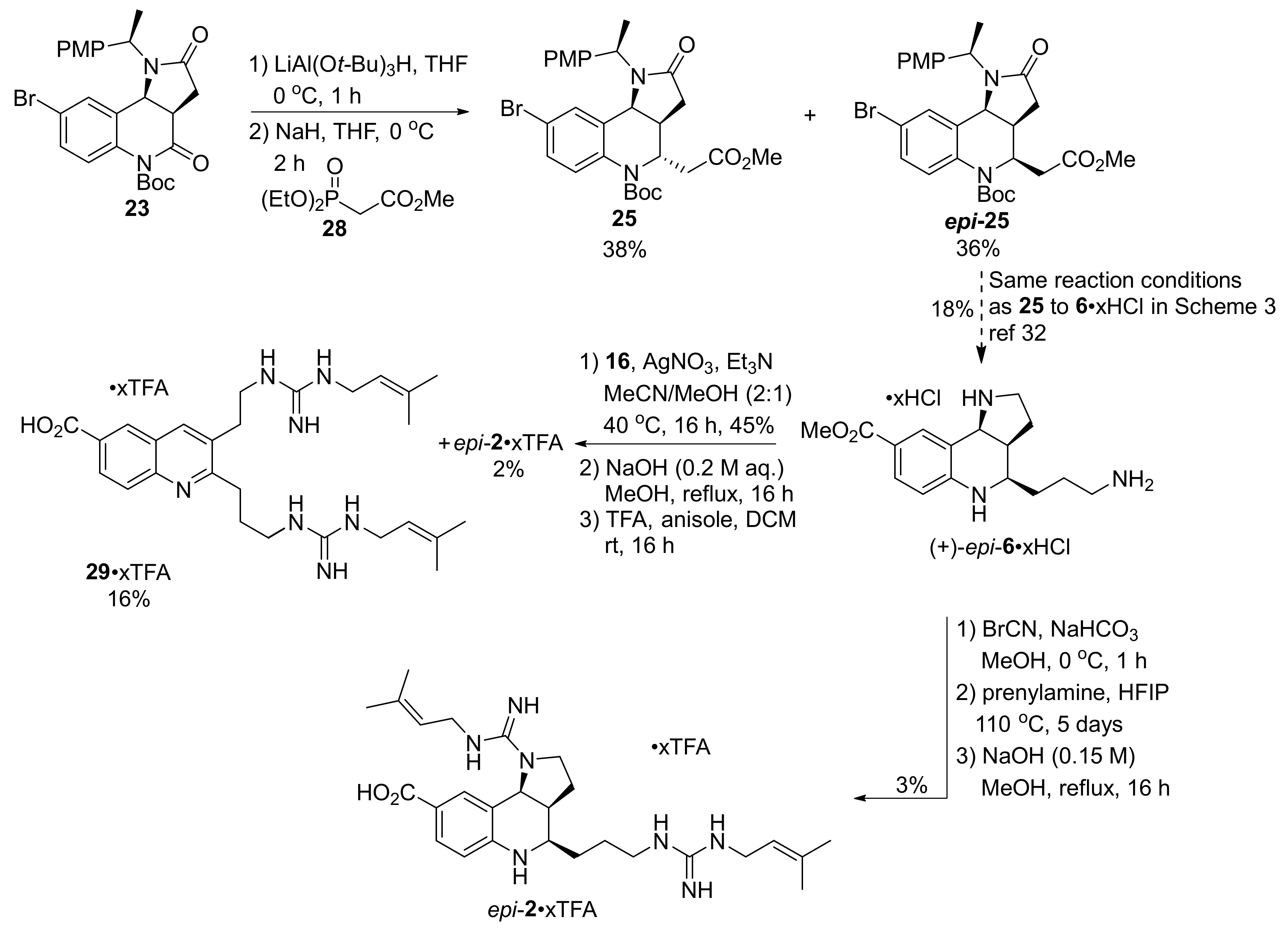
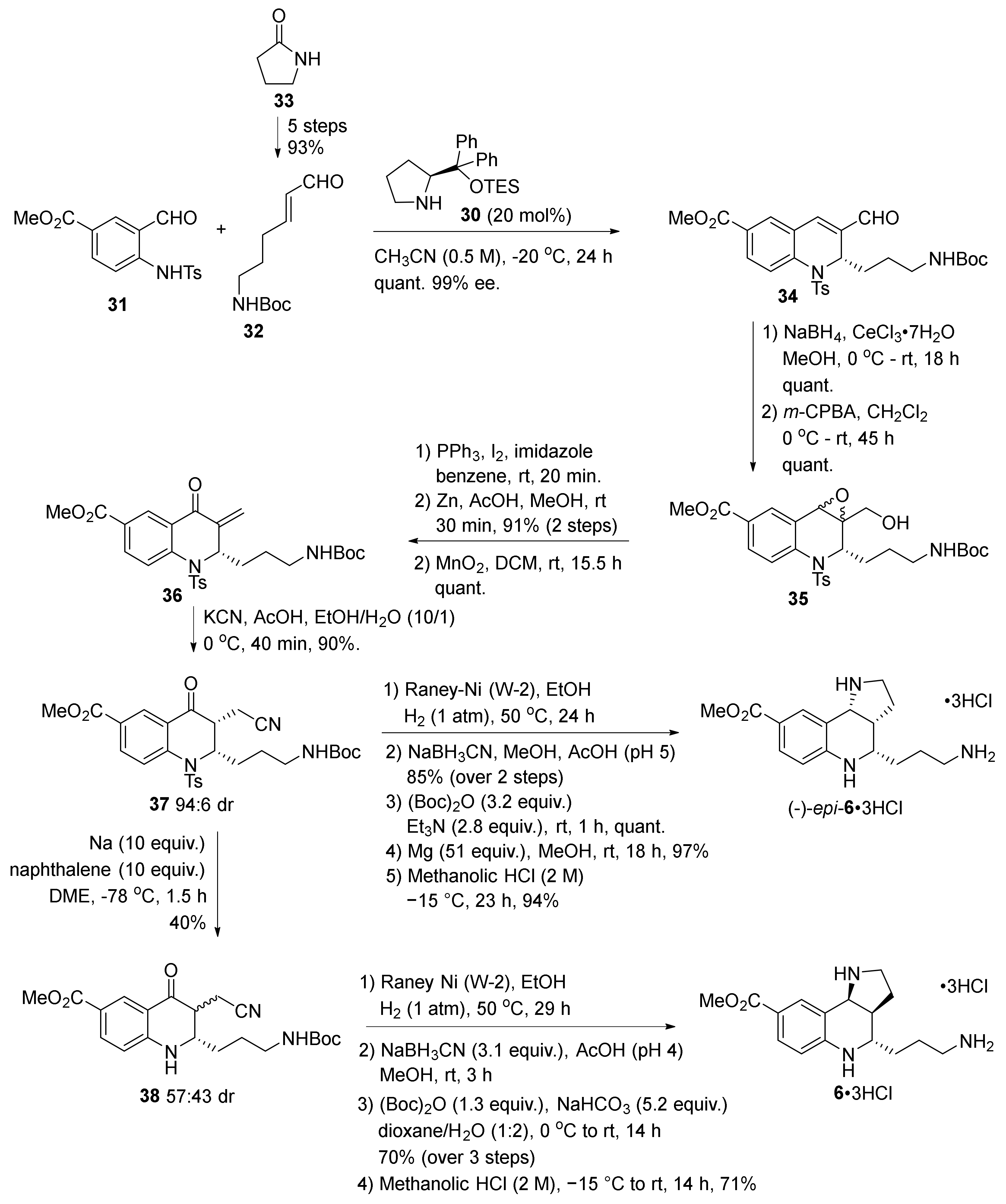
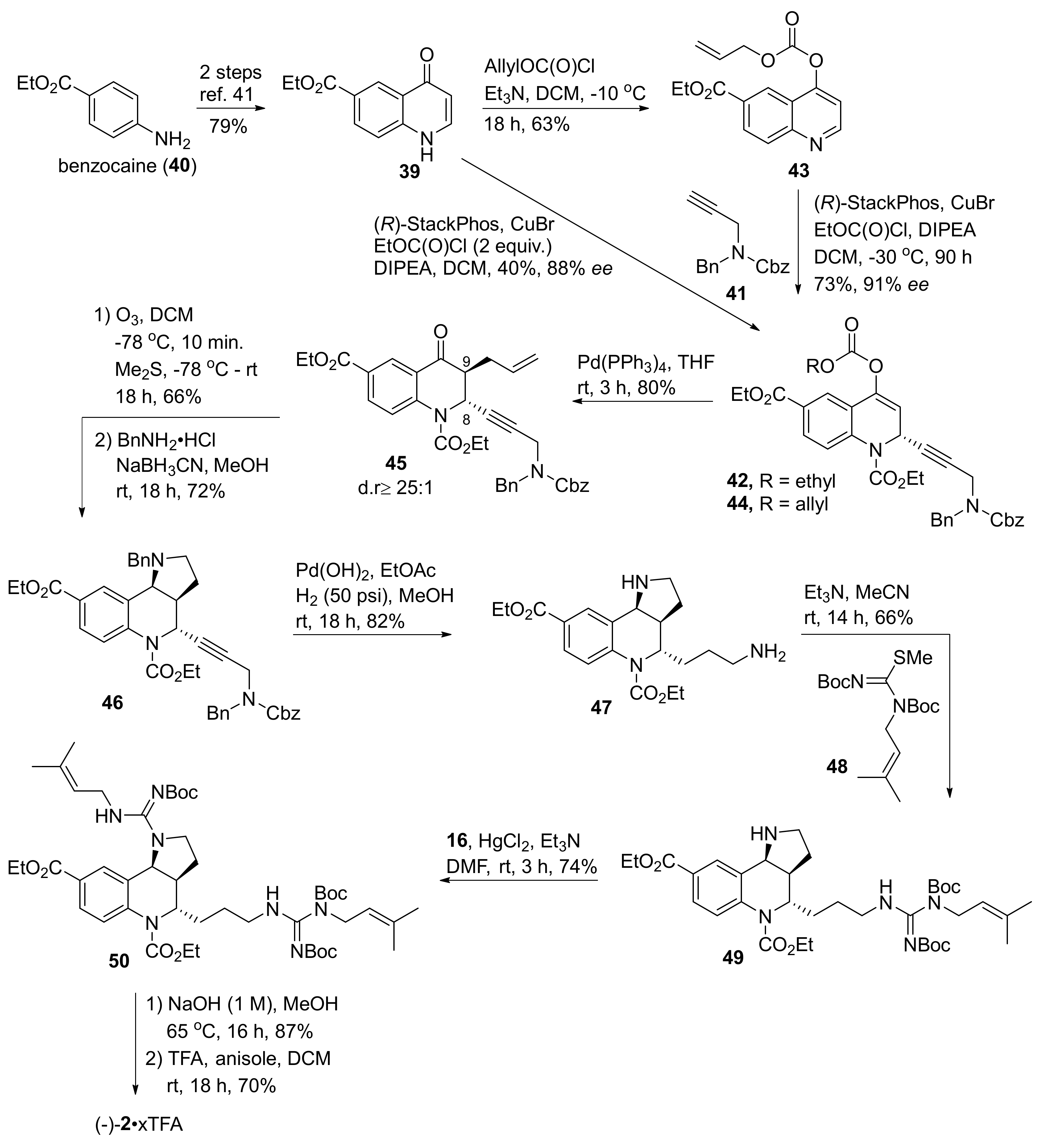
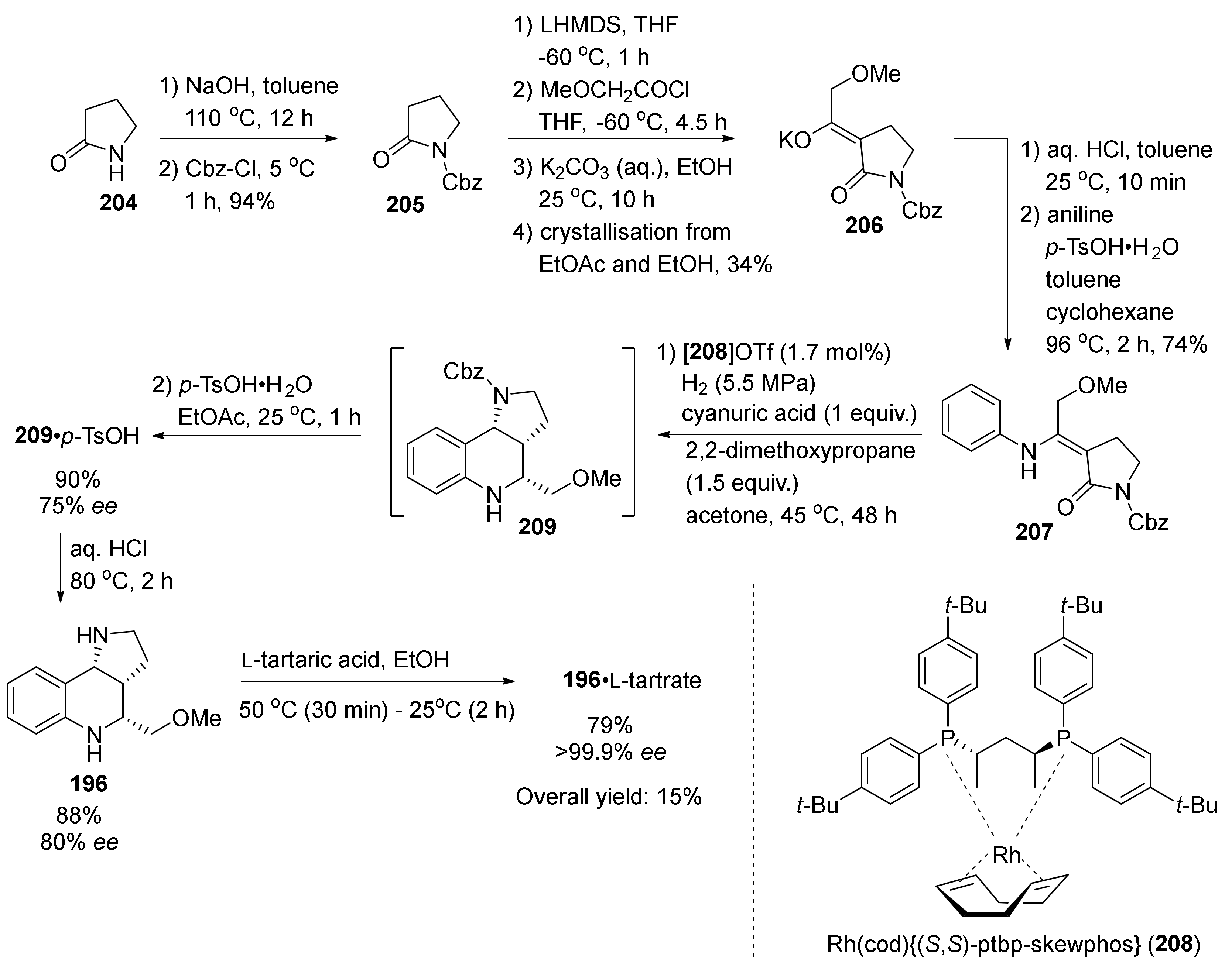
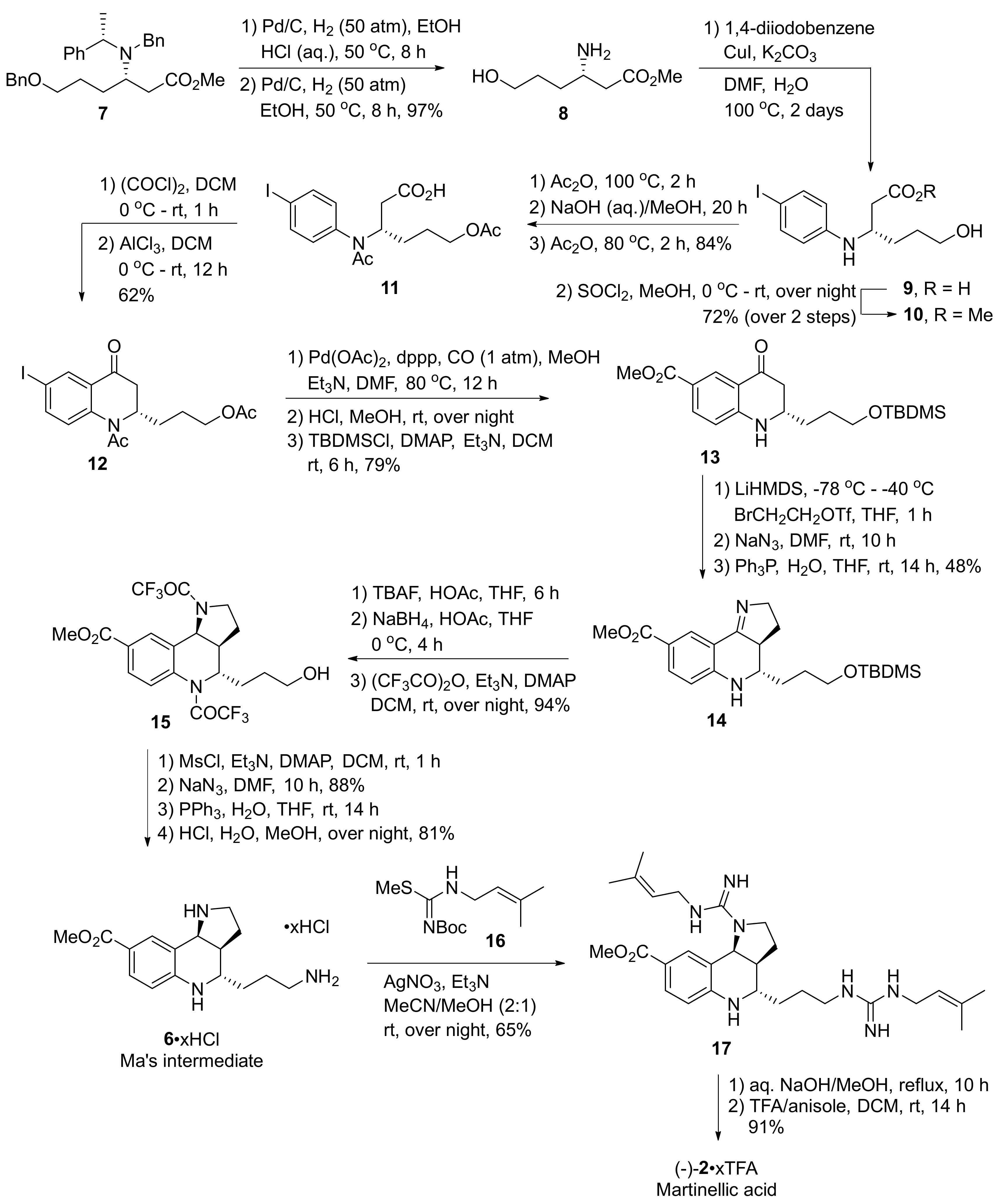
2. Total and Formal Synthesis of Martinellic Acid (2)
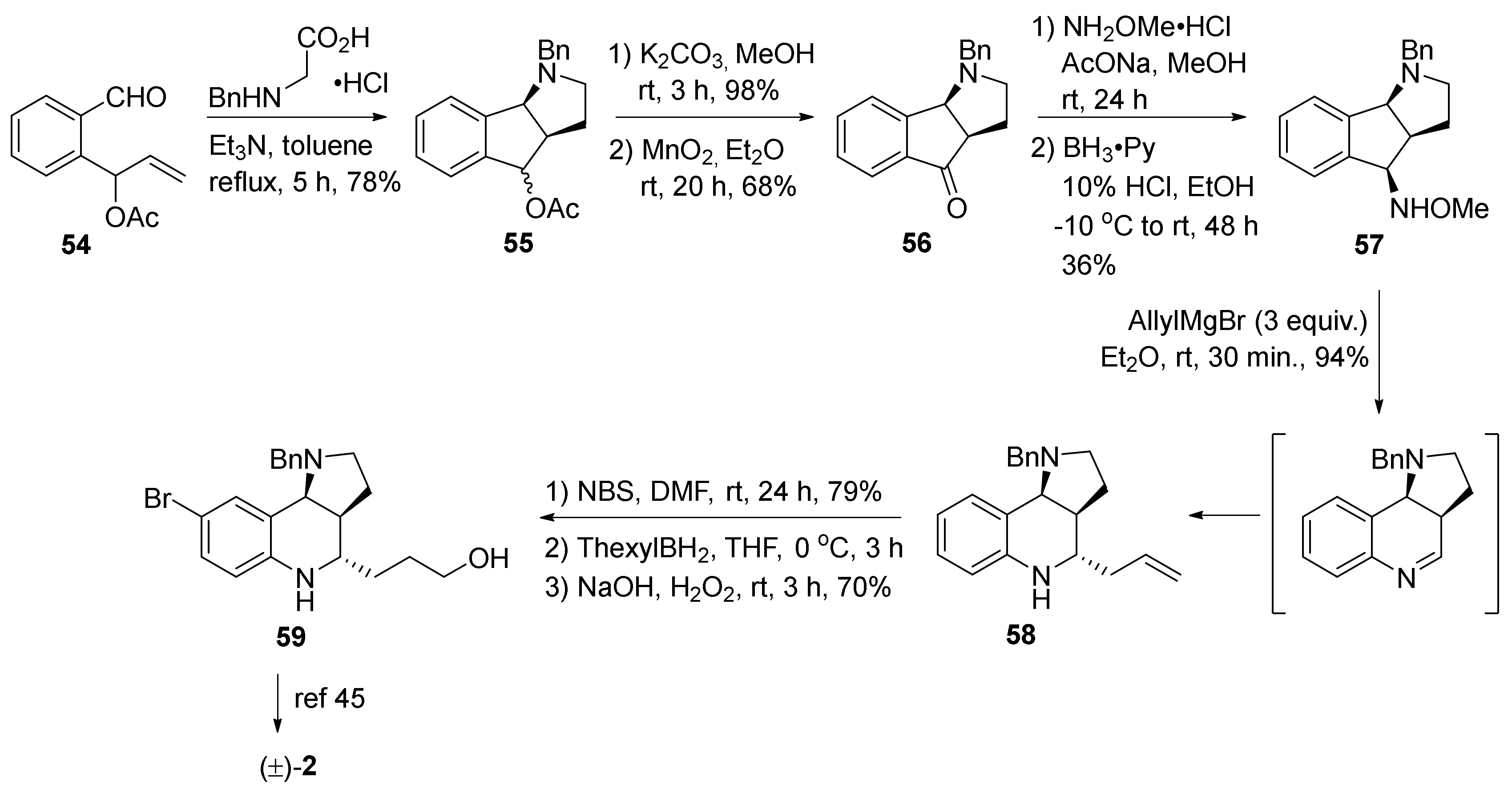
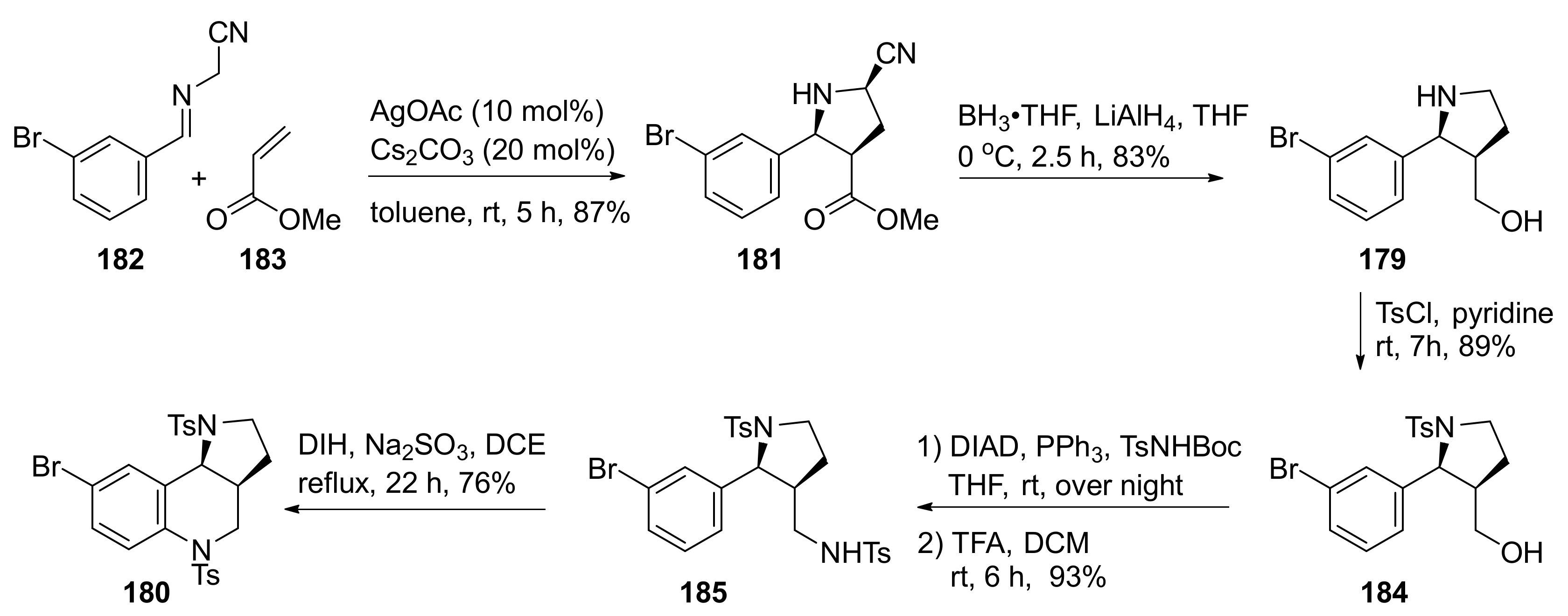
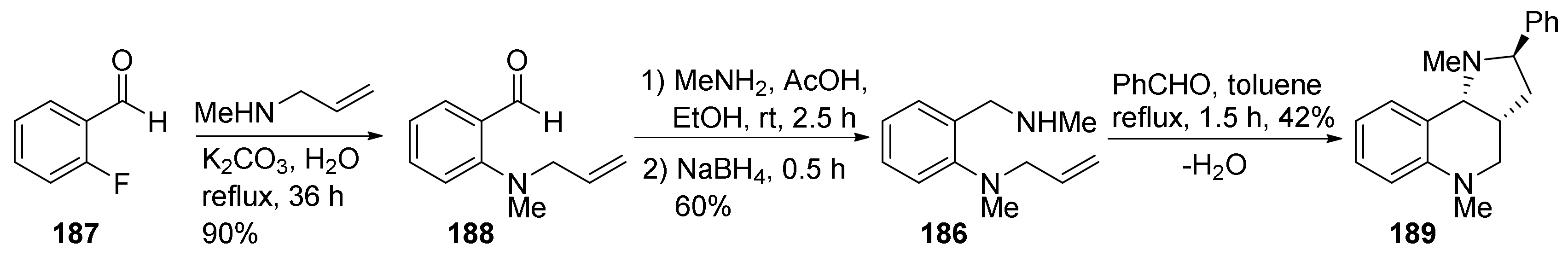
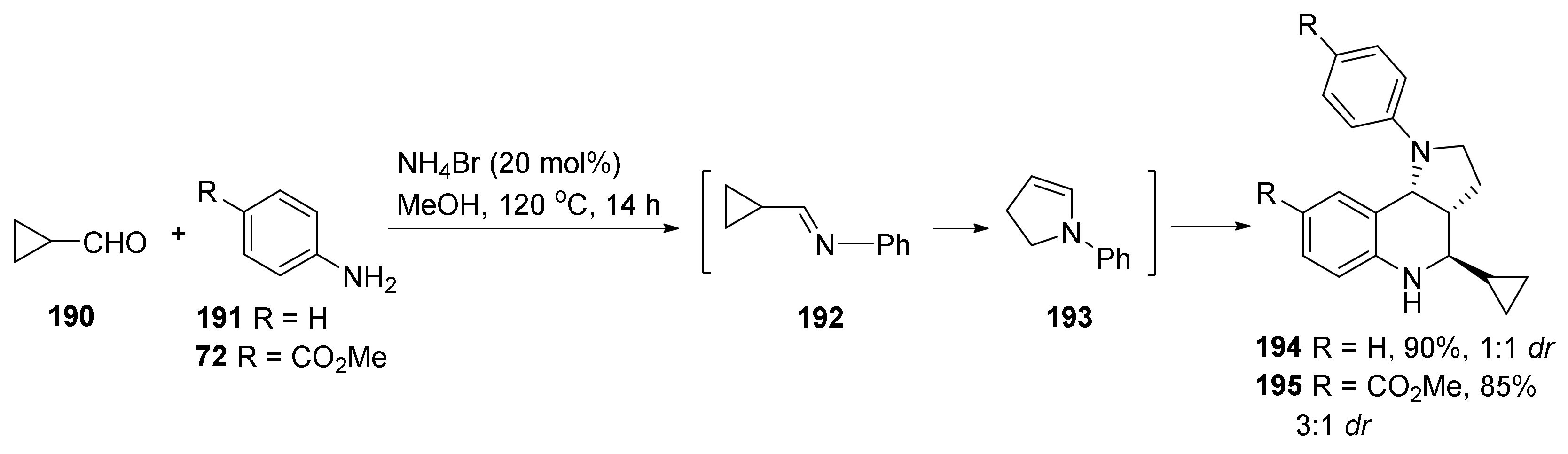
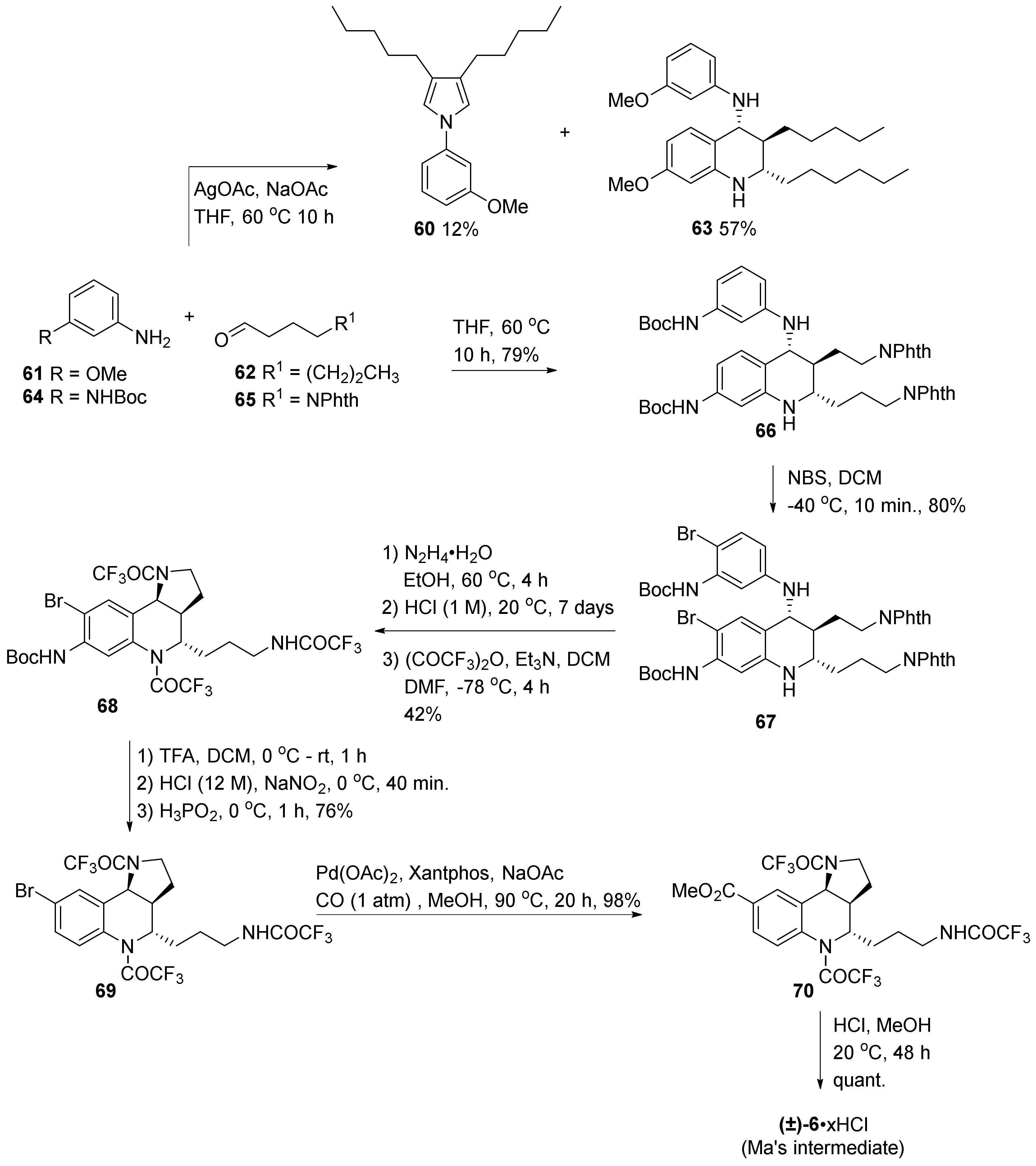
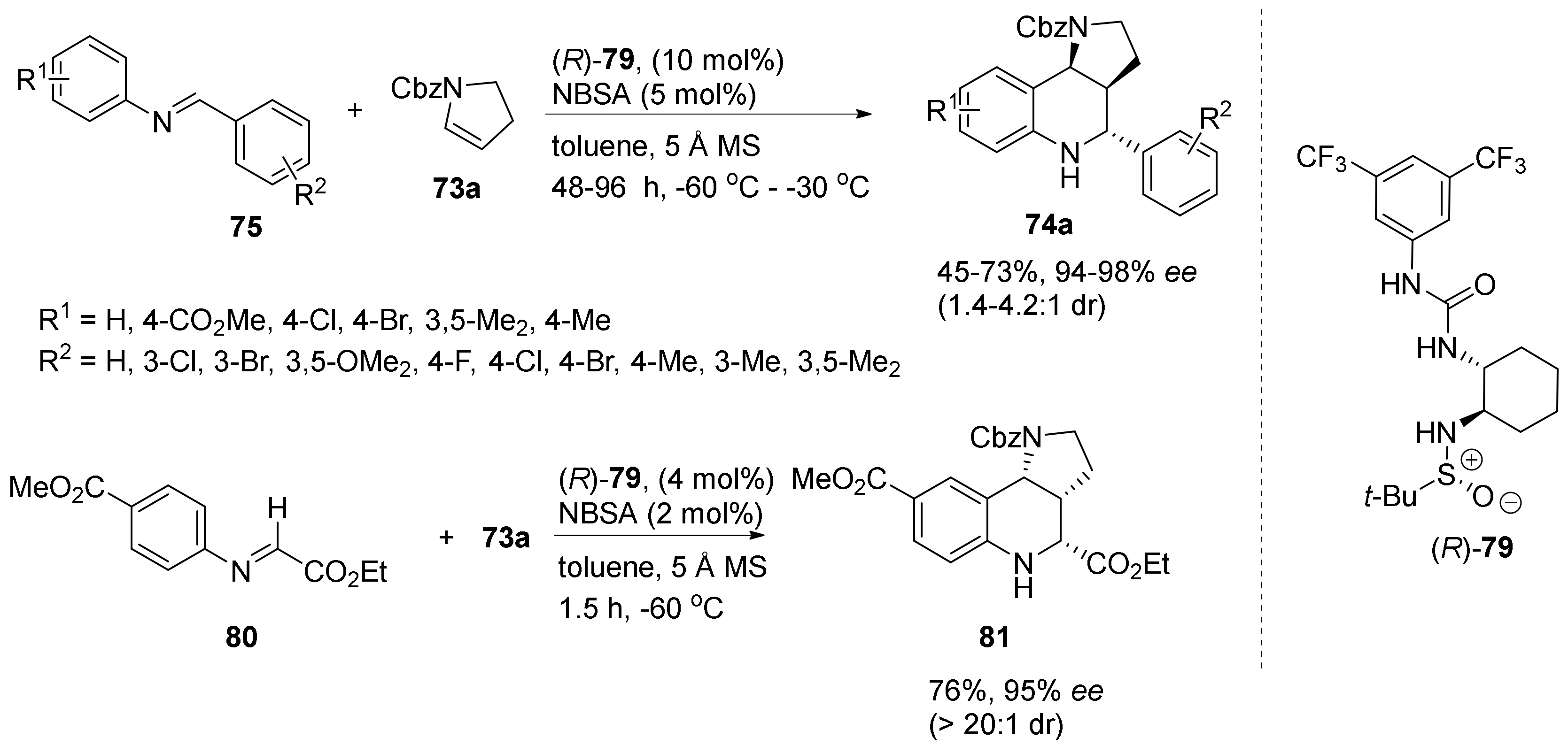
3. Tricyclic Core Scaffold Synthesis
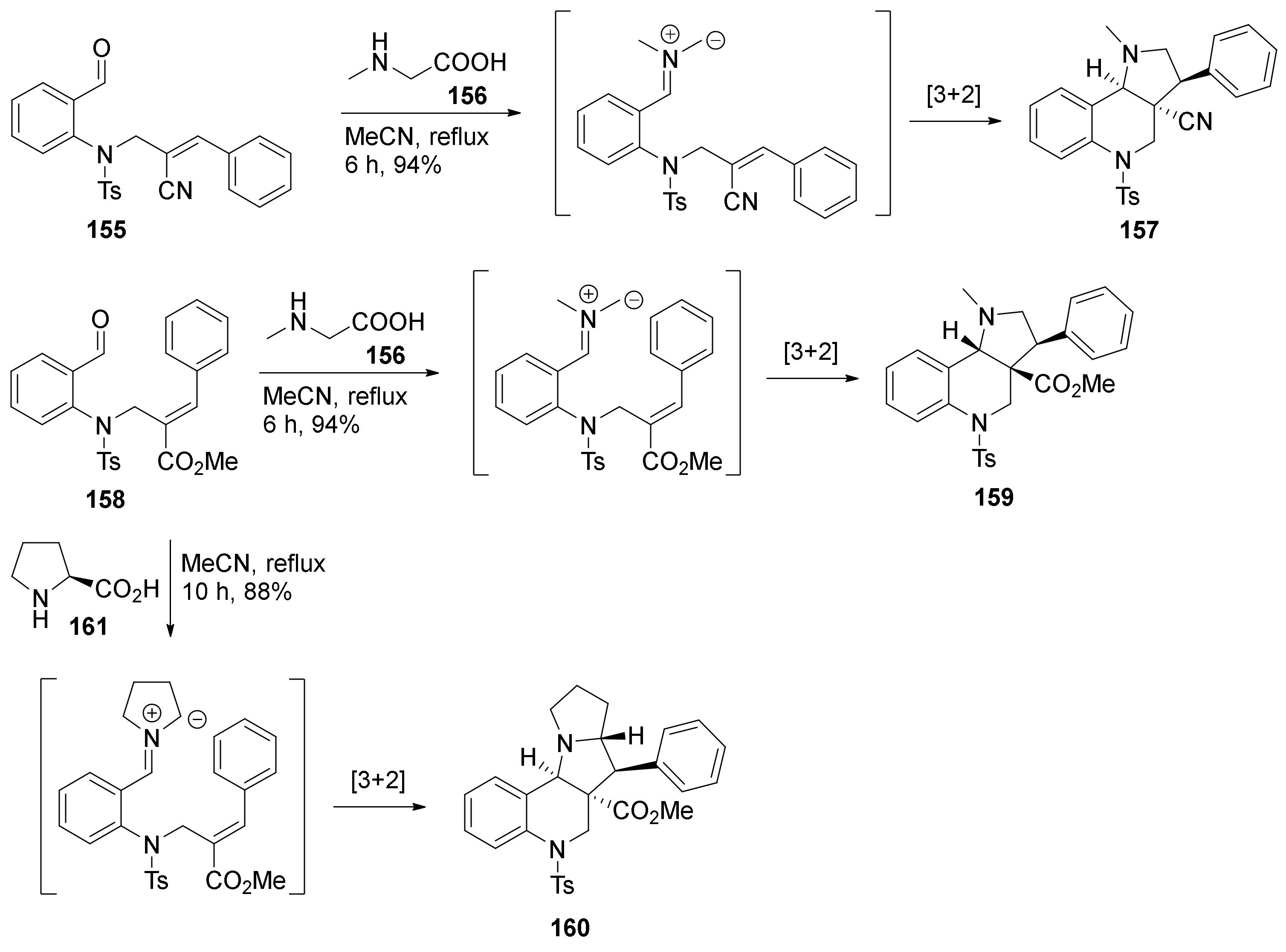
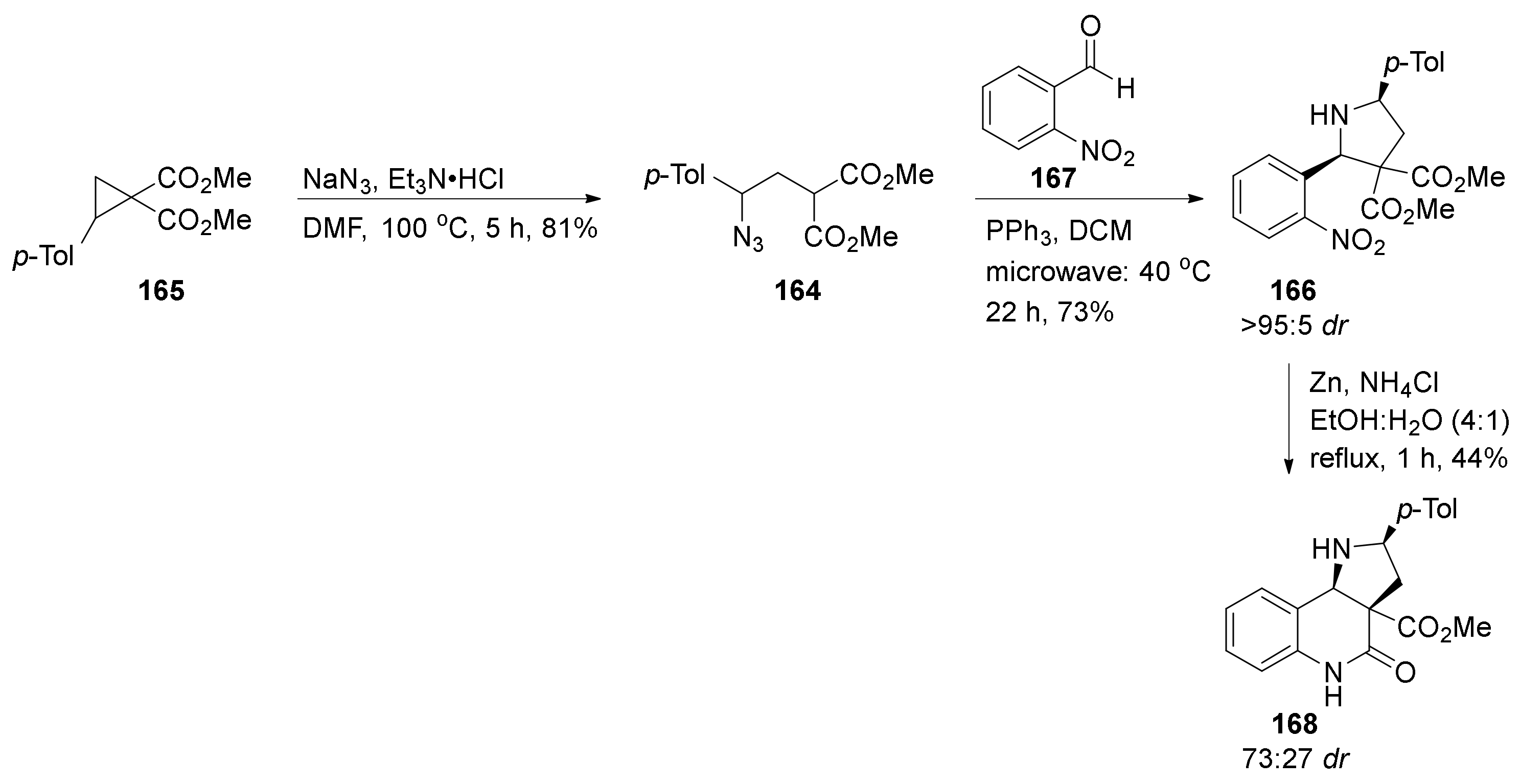
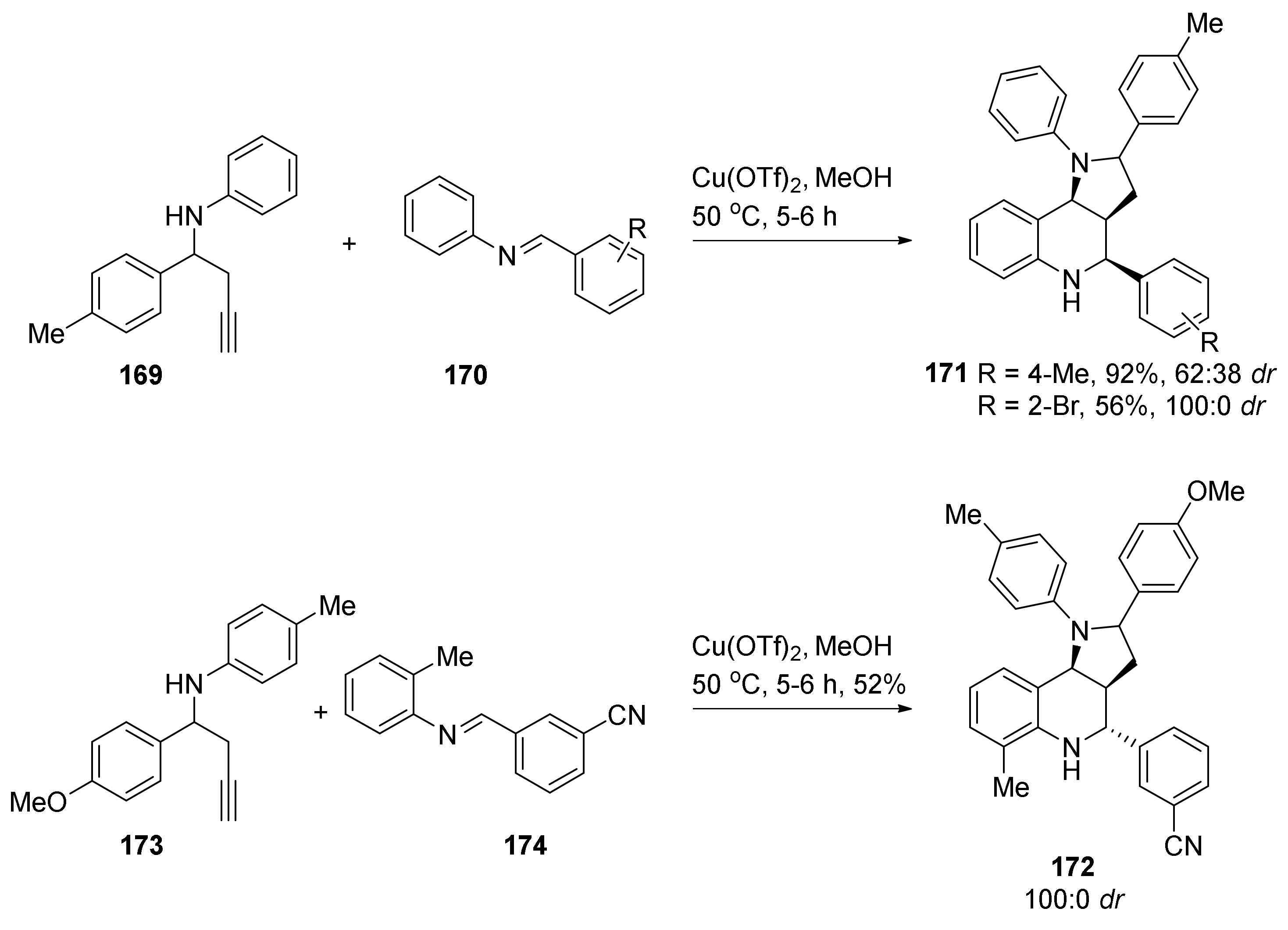
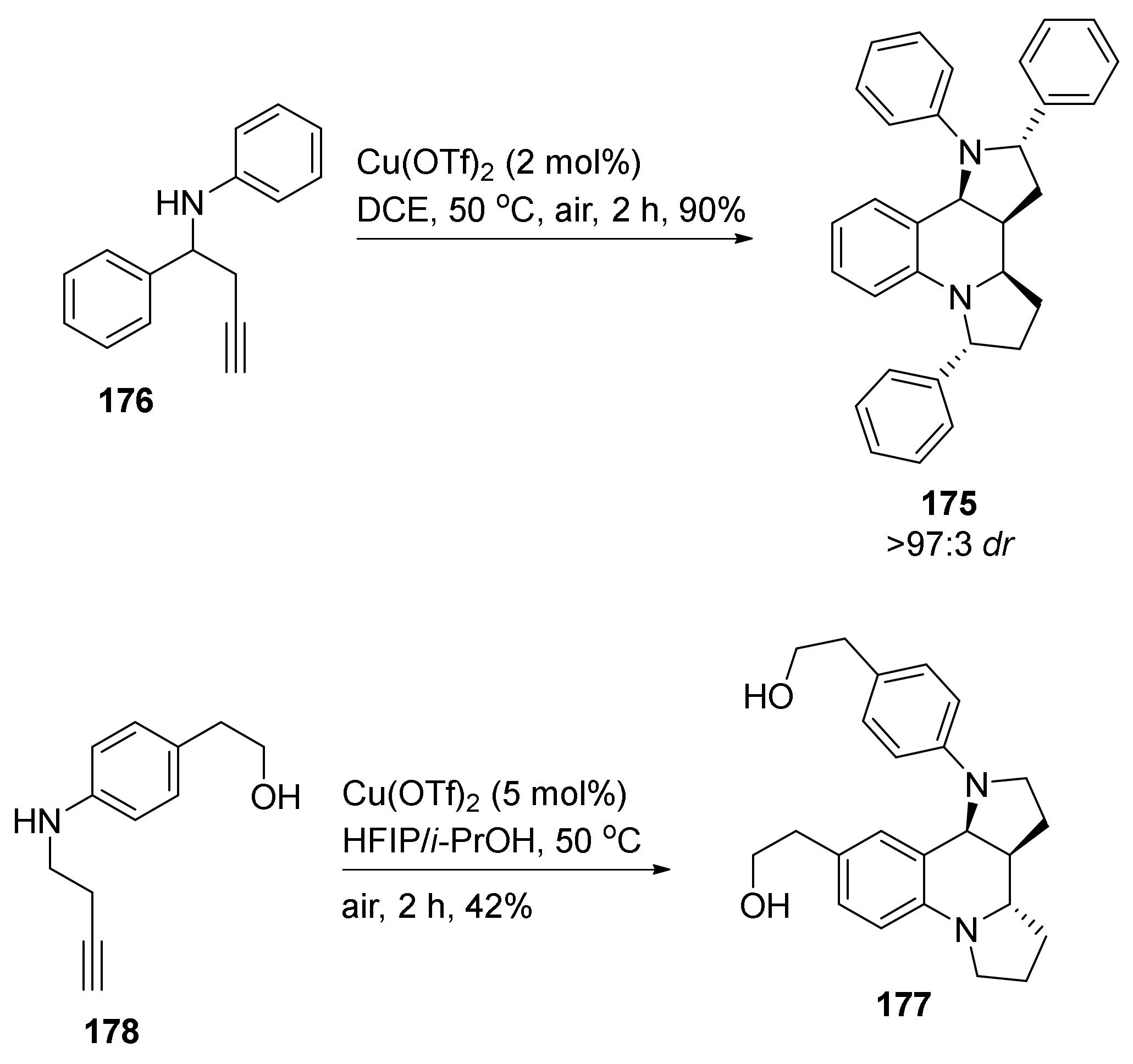
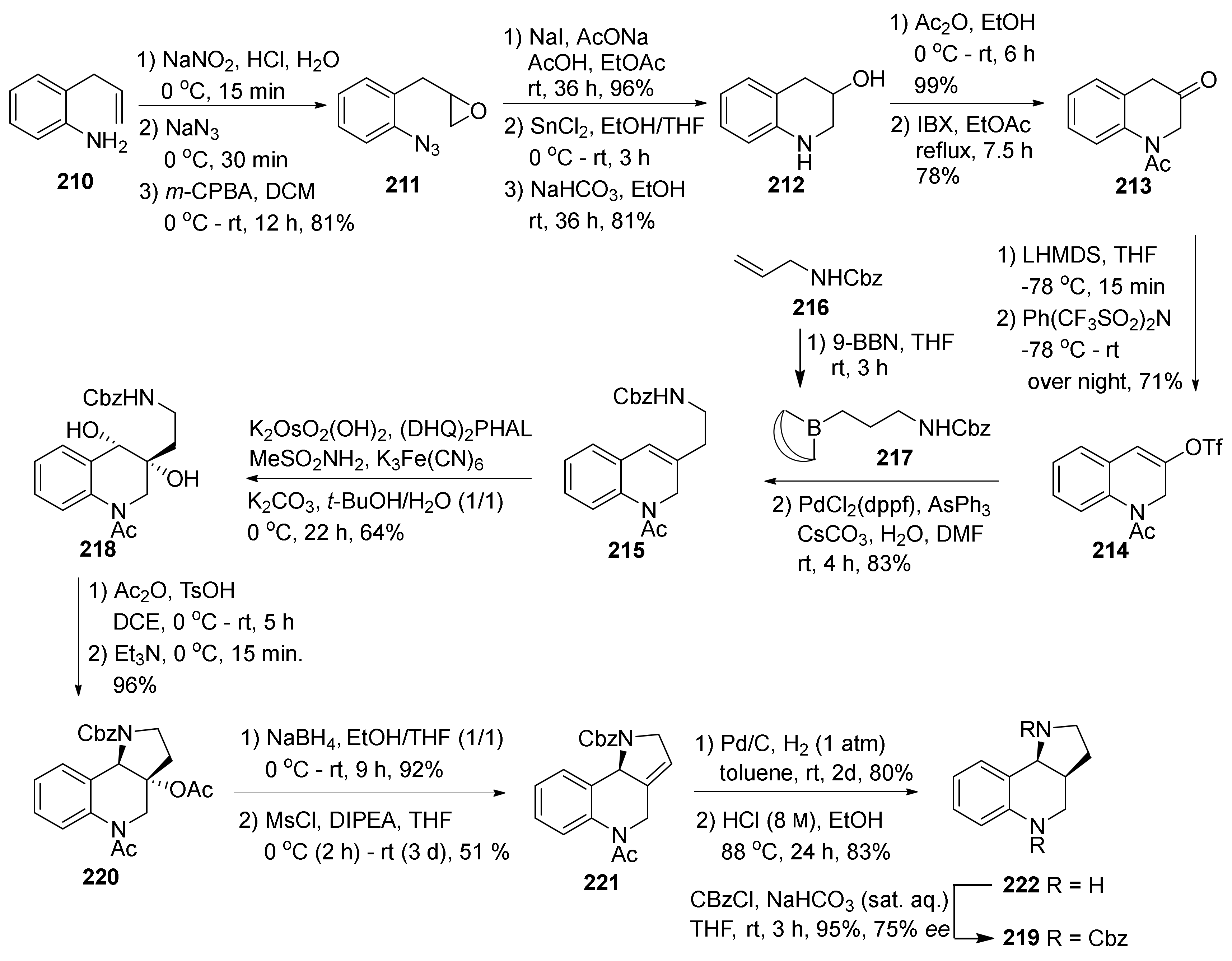
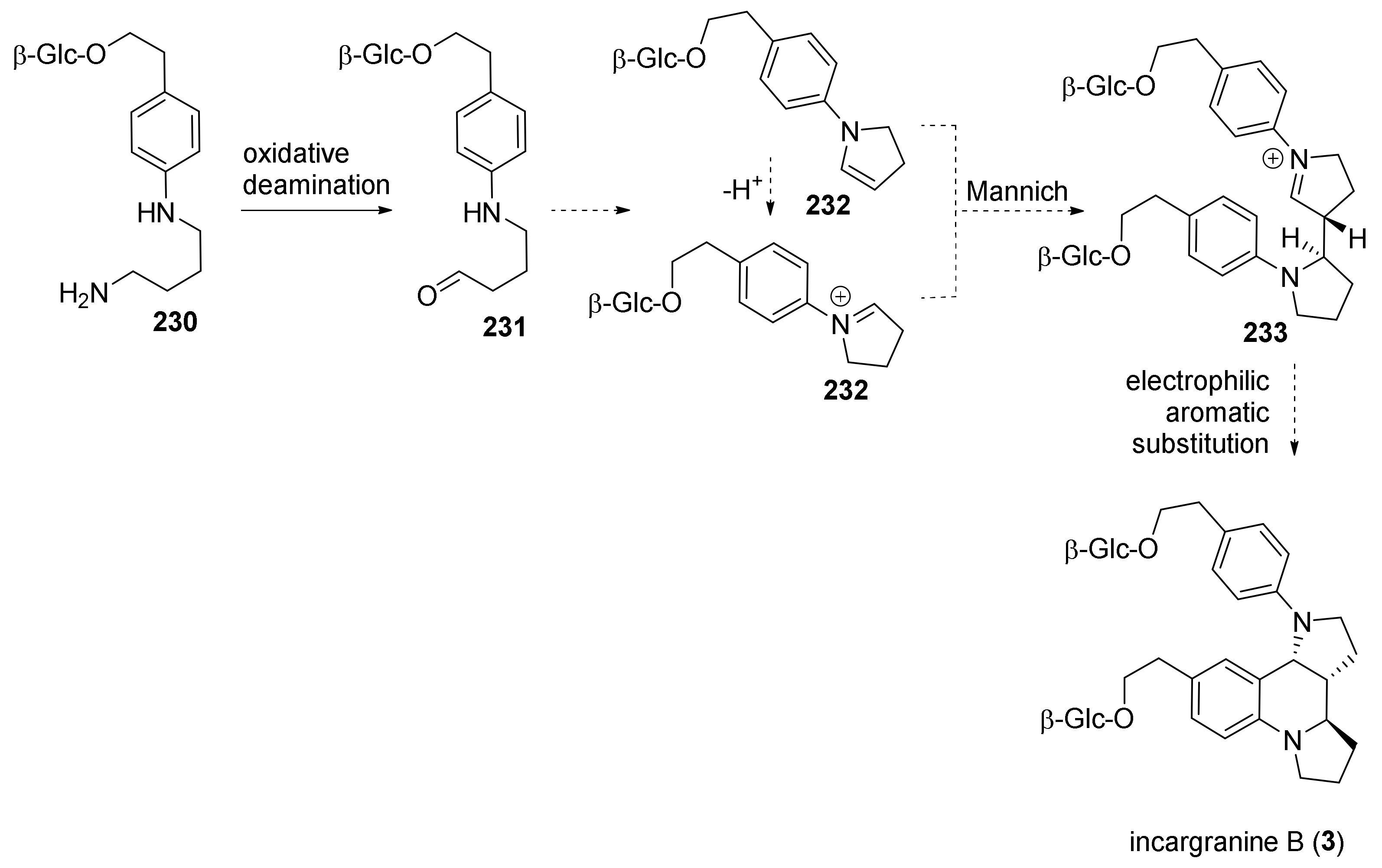
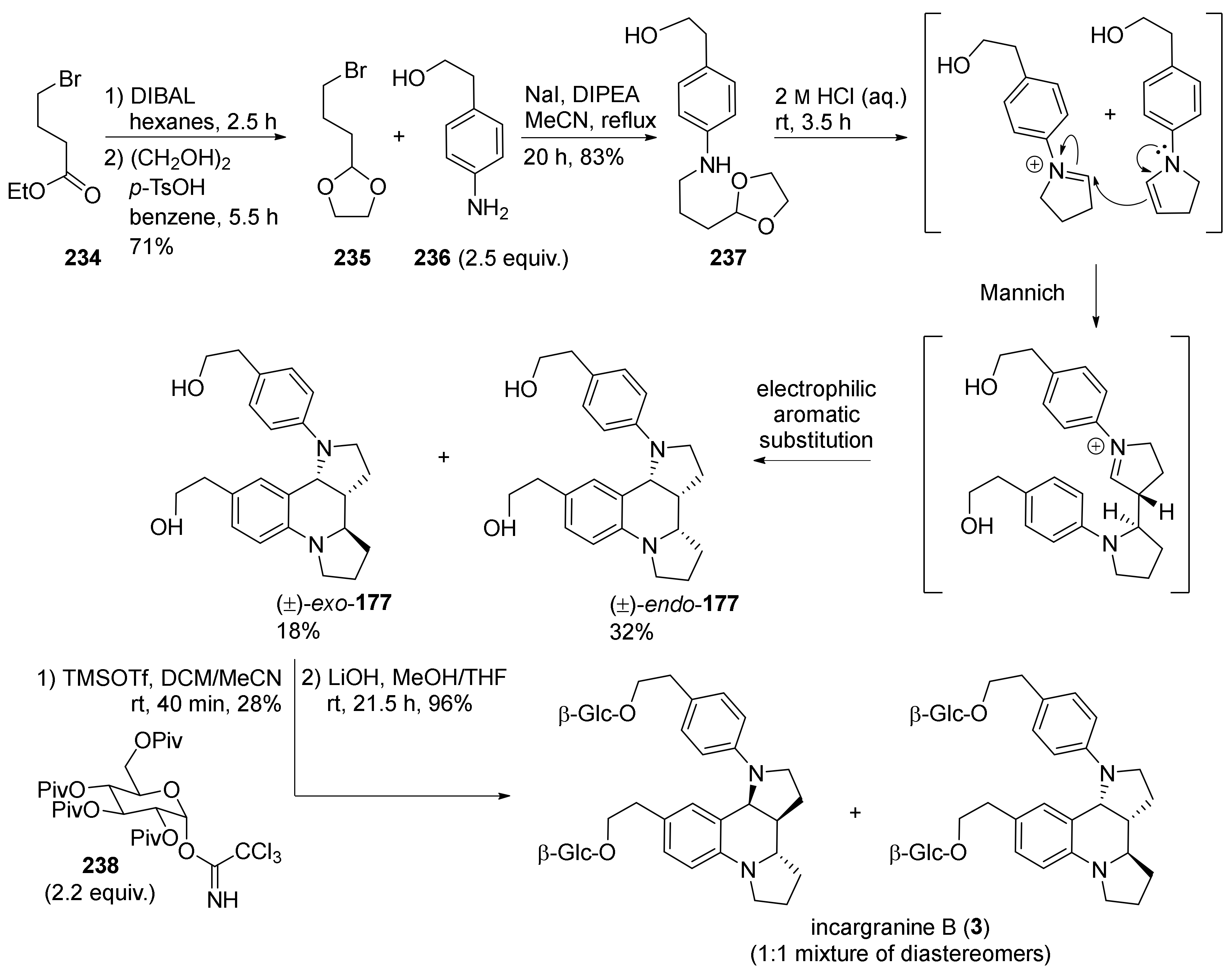
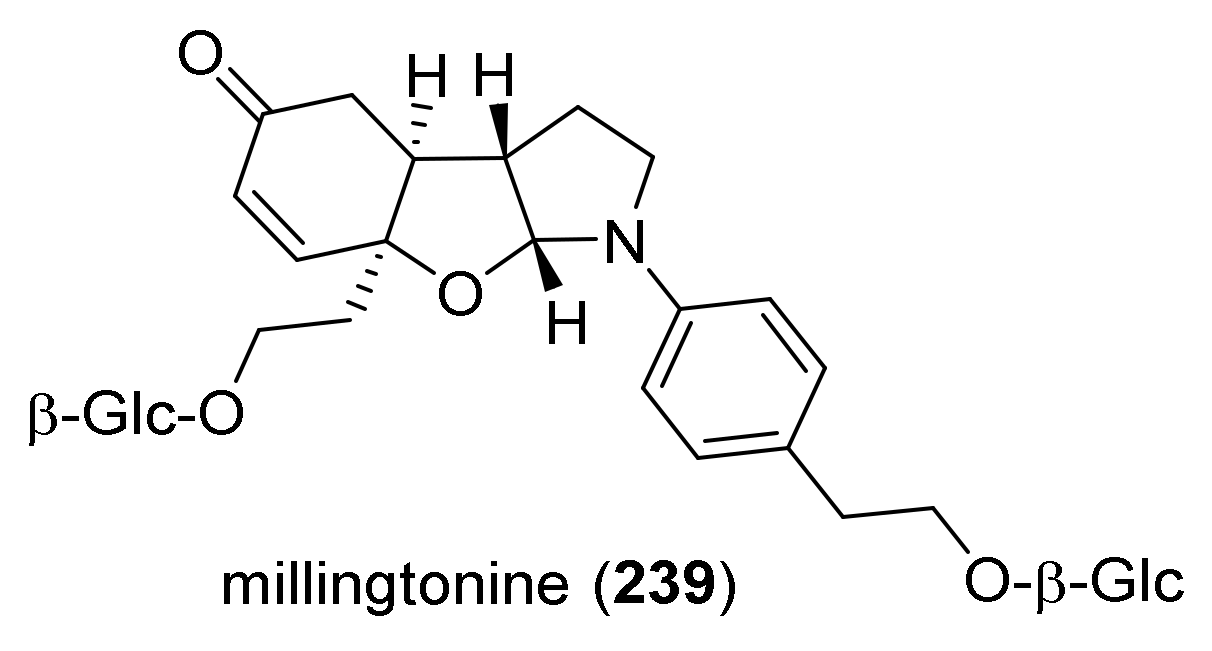
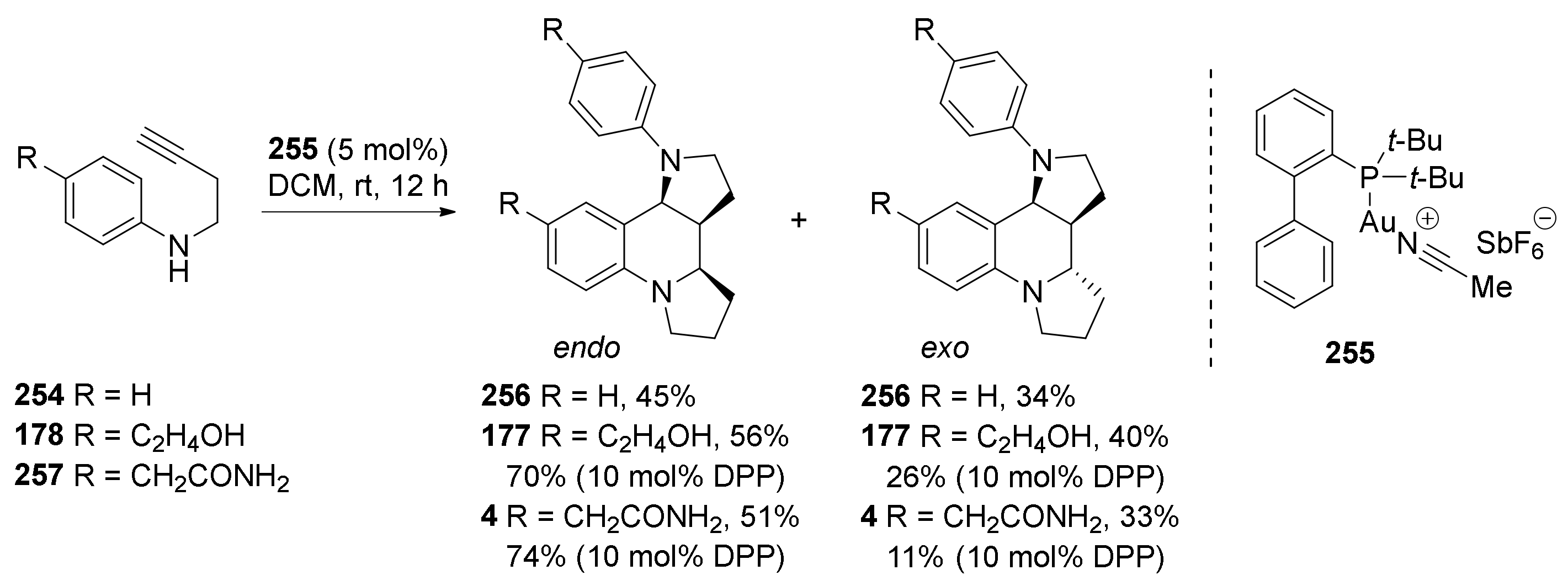
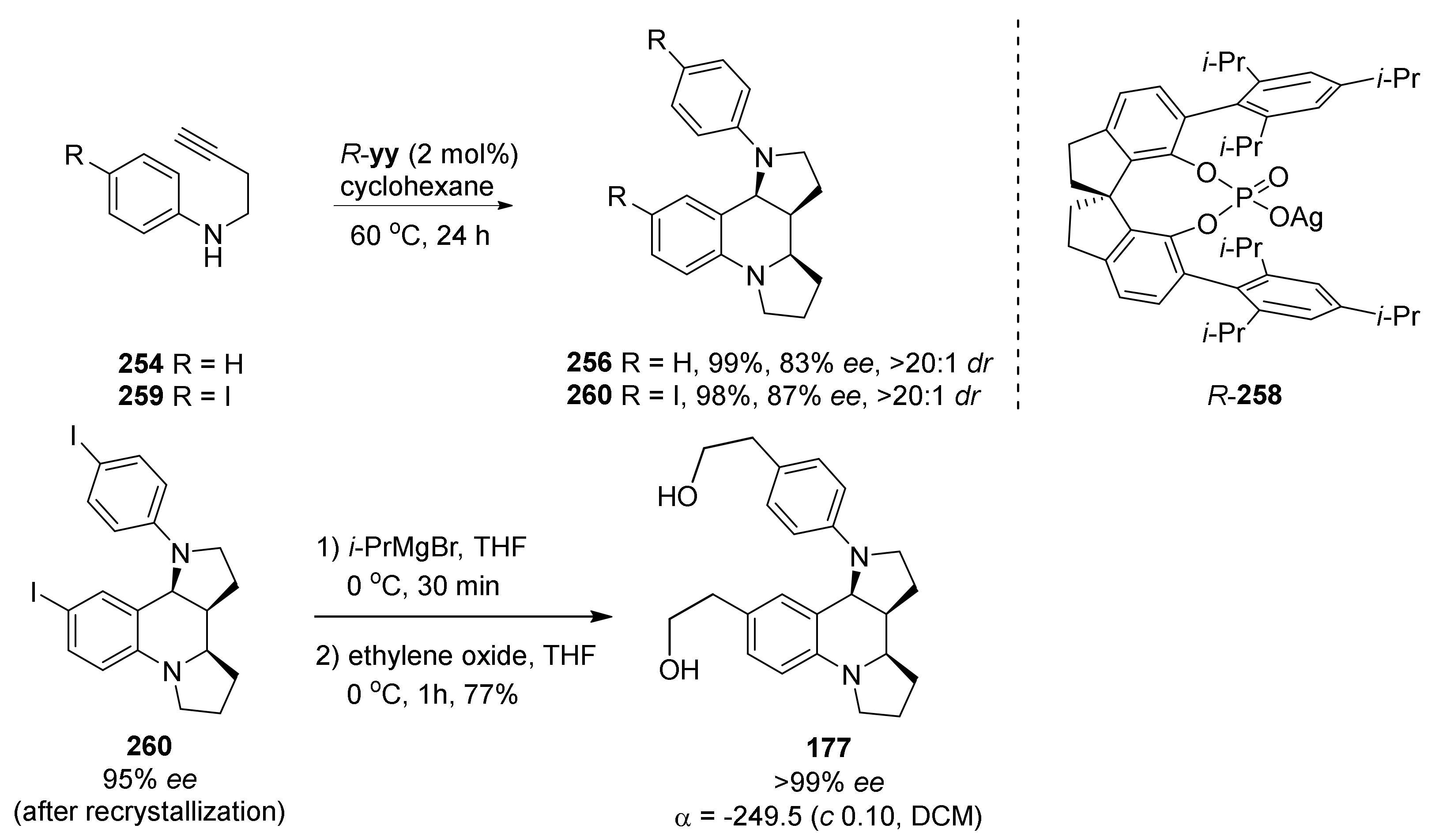
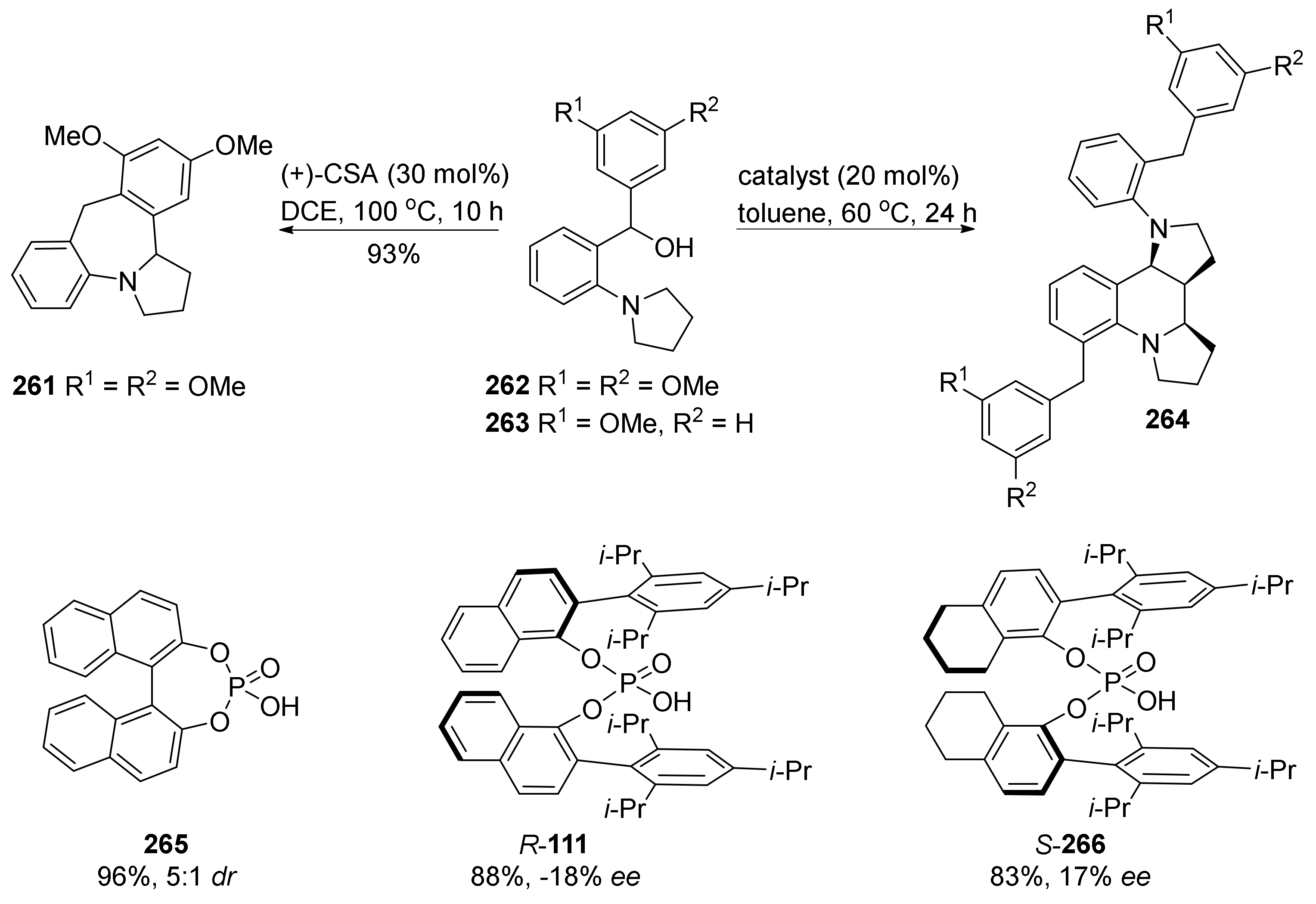
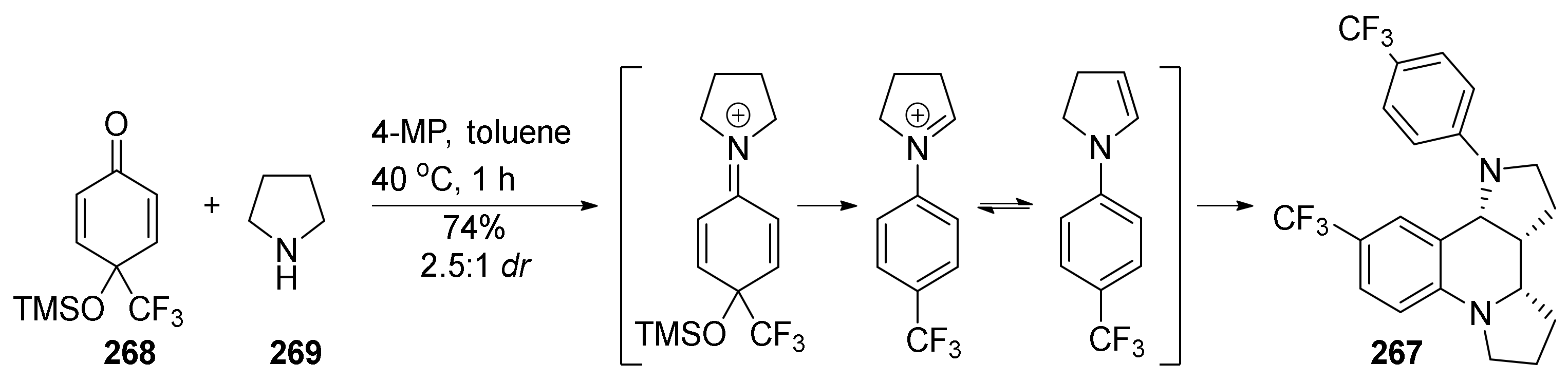
4. Synthesis of Dipyrroloquinolines Towards Natural Products Seneciobipyrrolidine (3) and Incargranine B (4)
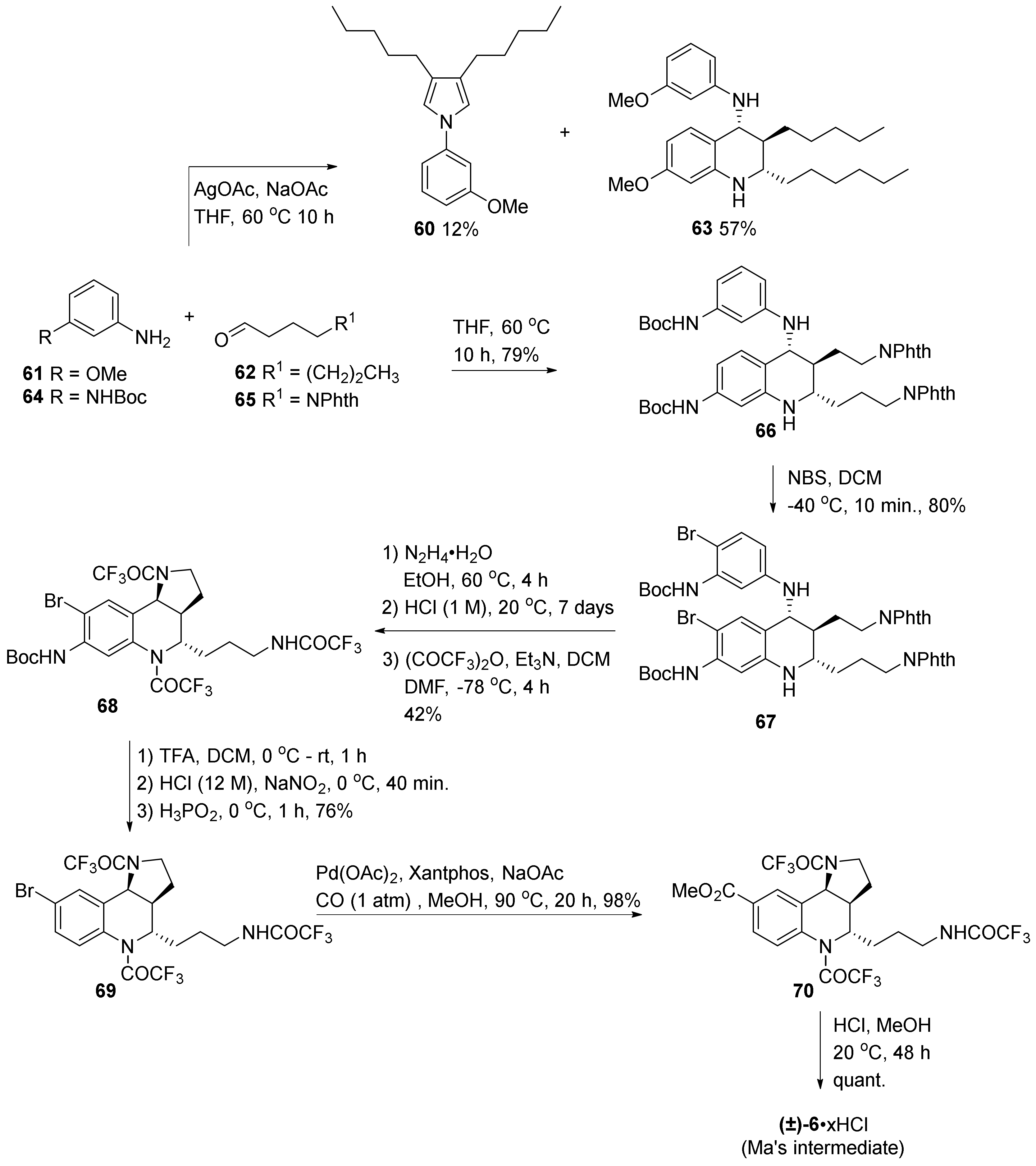
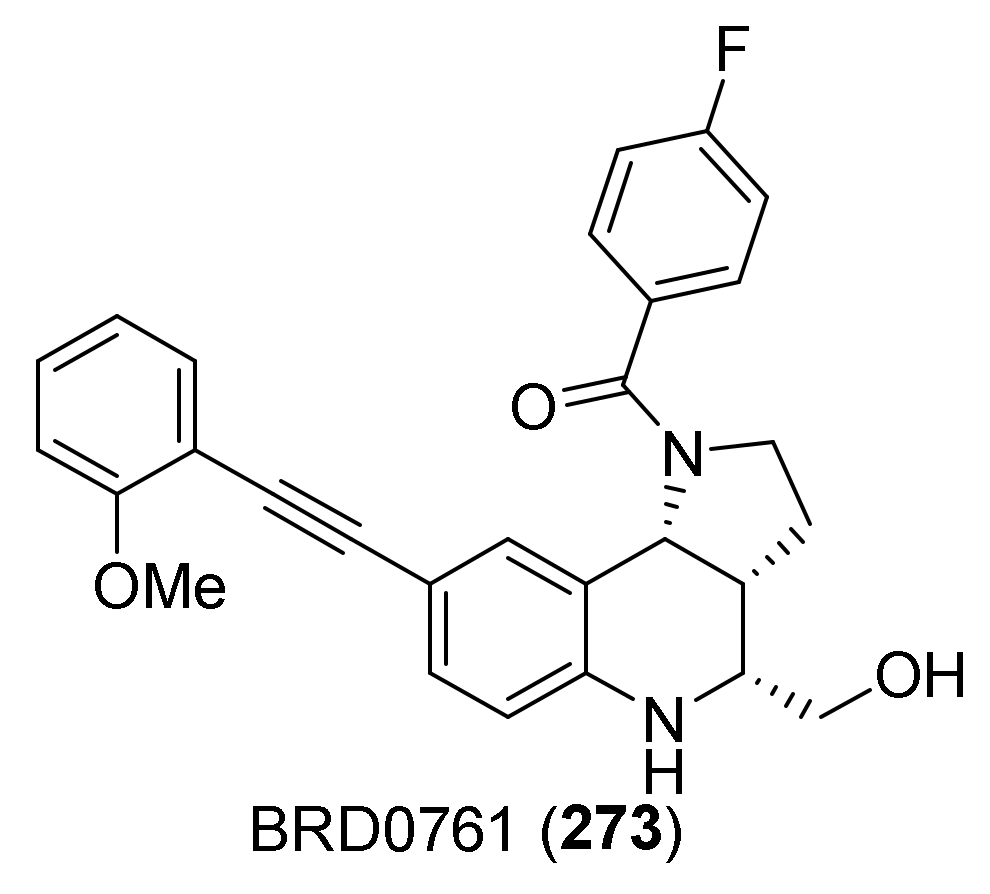
5. Elaborating Biological Properties
6. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guo, L.-D.; Chen, Y.; Xu, J. Total Synthesis of Daphniphyllum Alkaloids: From Bicycles to Diversified Caged Structures. Acc. Chem. Res. 2020, 53, 7390–7394. [Google Scholar] [CrossRef]
- Baudoin, O. Multiple Catalytic C–H Bond Functionalization for Natural Product Synthesis. Angew. Chem. Int. Ed. 2020, 59, 17798–17809. [Google Scholar] [CrossRef]
- Frontier, A.J.; Hernandez, J.J. New Twists in Nazarov Cyclization Chemistry. Acc. Chem. Res. 2020, 53, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; He, H. Advances in N-Heterocyclic Carbene Catalysis for Natural Product Synthesis. Eur. J. Org. Chem. 2020, 2020, 5917–5925. [Google Scholar] [CrossRef]
- Mandal, S.; Thirupathi, B. Strategies for the construction of γ-spirocyclic butenolides in natural product synthesis. Org. Biomol. Chem. 2020, 18, 5287–5314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhou, Y.-J.; Liang, X.-W. Total synthesis of natural products using photocycloaddition reactions of arenes. Org. Biomol. Chem. 2020, 18, 5558–5566. [Google Scholar] [CrossRef] [PubMed]
- Kalita, S.J.; Cheng, F.; Huang, Y.Y. Recent advances of applying boron-reagents in asymmetric total syntheses of natural products and bio-active molecules. Adv. Synth. Catal. 2020, 362, 2778–2800. [Google Scholar] [CrossRef]
- Fernandes, R.A.; Kumar, P.; Choudhary, P. Advances in catalytic and protecting-group-free total synthesis of natural products: A recent update. Chem. Commun. 2020, 56, 8569–8590. [Google Scholar] [CrossRef]
- Hu, Y.-J.; Li, L.-X.; Han, J.-C.; Min, L.; Li, C.-C. Recent Advances in the Total Synthesis of Natural Products Containing Eight-Membered Carbocycles (2009–2019). Chem. Rev. 2020, 120, 5910–5953. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J.; Snader, K.M. Natural Products in Drug Discovery and Development. J. Nat. Prod. 1997, 60, 52–60. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. The influence of natural products upon drug discovery. Nat. Prod. Rep. 2000, 17, 215–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural product scaffolds as leads to drugs. Future Med. Chem. 2009, 1, 1415–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Last 25 Years. J. Nat. Prod. 2007, 70, 461–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the 30 Years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Monteleone, S.; Fuchs, J.E.; Liedl, K.R. Molecular connectivity predefines polypharmacology: Aliphatic rings, chirality, and sp3 centers enhance target selectivity. Front. Pharmacol. 2017, 8, 552. [Google Scholar] [CrossRef] [Green Version]
- Witherup, K.M.; Ransom, R.W.; Graham, A.C.; Bernard, A.M.; Salvatore, M.J.; Lumma, W.C.; Anderson, P.S.; Pitzenberger, S.M.; Varga, S.L. Martinelline and martinellic acid, novel G-protein linked receptor antagonists from the tropical plant Martinella iquitosensis (Bignoniaceae). J. Am. Chem. Soc. 1995, 117, 6682–6685. [Google Scholar] [CrossRef]
- Elvis-Offiah, U.B.; Bafor, E.E.; Eze, G.I.; Igbinumwen, O.; Viegelmann, C.; Edrada-Ebel, R. In vivo investigation of female reproductive functions and parameters in nonpregnant mice models and mass spectrometric analysis of the methanol leaf extract of Emilia Coccinea (Sims) G Dons. Physiol. Rep. 2016, 4, e13047. [Google Scholar] [CrossRef]
- Zulfiker, A.H.M.; Sohrabi, M.; Qi, J.; Matthews, B.; Wei, M.Q.; Grice, I.D. Multi-constituent identification in Australian cane toad skin extracts using high-performance liquid chromatography high-resolution tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 129, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Abhimannue, A.P.; Mohan, M.C.; Kumar, B.P. Inhibition of Tumor Necrosis Factor-α and Interleukin-1β Production in Lipopolysaccharide-Stimulated Monocytes by Methanolic Extract of Elephantopus scaber Linn and Identification of Bioactive Components. Appl. Biochem. Biotechnol. 2016, 179, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Gentry, A.H.; Cook, K. Martinella (Bignoniaceae): A widely used eye medicine of South America. J. Ethnopharmacol. 1984, 11, 337–343. [Google Scholar] [CrossRef]
- Ogunlesi, M.; Okiei, W.; Ademoye, M. Medicinal Plants Used in Treating Eye Infections in Nigeria. In A Textbook of Medicinal Plants from Nigeria; Odugbemi, T., Ed.; University of Lago Press: Lago, Nigeria, 2008; p. 305. [Google Scholar]
- Shen, Y.H.; Su, Y.Q.; Tian, J.M.; Lin, S.; Li, H.L.; Tang, J.; Zhang, W.D. A Unique Indolo-[1,7]naphthyridine Alkaloid from Incarvillea mairei var. grandiflora (Wehrh.) Grierson. Helv. Chim. Acta 2010, 93, 2393–2396. [Google Scholar] [CrossRef]
- Brown, P.D.; Willis, A.C.; Sherburn, M.S.; Lawrence, A.L. Total synthesis and structural revision of the alkaloid incargranine B. Angew. Chem. Int. Ed. 2013, 52, 13273–13275. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Chou, G.; Wang, Z. Three New Alkaloids from Senecio scandens. Chem. Nat. Compd. 2014, 50, 329–332. [Google Scholar] [CrossRef]
- Wang, D.; Huang, L.; Chen, S. Senecio scandens Buch.-Ham.: A review on its ethnopharmacology, phytochemistry, pharmacology, and toxicity. J. Ethnopharmacol. 2013, 149, 1–23. [Google Scholar] [CrossRef]
- Ma, D.; Xia, C.; Jiang, J.; Zhang, J.; Tang, W. Aromatic nucleophilic substitution or CuI-catalyzed coupling route to martinellic acid. J. Org. Chem. 2003, 68, 442–451. [Google Scholar] [CrossRef]
- Ikeda, S.; Shibuya, M.; Iwabuchi, Y. Asymmetric total synthesis of martinelline and martinellic acid. Chem. Commun. 2007, 504–506. [Google Scholar] [CrossRef]
- Shirai, A.; Miyata, O.; Tohnai, N.; Miyata, M.; Procter, D.J.; Sucunza, D.; Naito, T. Total Synthesis of (−)-Martinellic Acid via Radical Addition–Cyclization–Elimination Reaction. J. Org. Chem. 2008, 73, 4464–4475. [Google Scholar] [CrossRef]
- Davies, S.G.; Fletcher, A.M.; Lee, J.A.; Lorkin, T.J.; Roberts, P.M.; Thomson, J.E. A diastereodivergent strategy for the asymmetric syntheses of (−)-martinellic acid and (−)-4-epi-martinellic acid. Tetrahedron 2013, 69, 9779–9803. [Google Scholar] [CrossRef]
- Nyerges, M. Construction of pyrrolo [3,2-c] quinolines: Recent advances in the synthesis of the martinelline alkaloids. Heterocycles 2004, 63, 1685–1712. [Google Scholar] [CrossRef]
- Lovely, C.J.; Bararinarayana, V. Synthetic studies toward the Martinella alkaloids. Curr. Org. Chem. 2008, 12, 1431–1453. [Google Scholar] [CrossRef]
- Eichholzer, J.V.; Lewis, I.A.; Macleod, J.K.; Oelrichs, P.B.; Vallely, P.J. Galegine and a new dihydroxyalkylacetamide from Verbesina enceloiodes. Phytochemistry 1982, 21, 97–99. [Google Scholar] [CrossRef]
- Reuter, G. Arginin als Vorstufe von Galegin in Galega officinalis L. Zur Biochemie und Physiologie von Galegin in Galega officinalis L., III. Mitt. Arch. Pharm. 1963, 296, 516–522. [Google Scholar] [CrossRef]
- Ma, D.; Xia, C.; Jiang, J.; Zhang, J. First total synthesis of martinellic acid, a naturally occurring bradykinin receptor antagonist. Org. Lett. 2001, 3, 2189–2191. [Google Scholar] [CrossRef]
- Powell, D.A.; Batey, R.A. Total synthesis of the alkaloids martinelline and martinellic acid via a hetero Diels–Alder multicomponent coupling reaction. Org. Lett. 2002, 4, 2913–2916. [Google Scholar] [CrossRef]
- Xia, C.; Heng, L.; Ma, D. Total synthesis of (±)-martinelline. Tetrahedron Lett. 2002, 43, 9405–9409. [Google Scholar] [CrossRef]
- Davies, S.G.; Fletcher, A.M.; Lee, J.A.; Lorkin, T.J.; Roberts, P.M.; Thomson, J.E. Asymmetric synthesis of (−)-martinellic acid. Org. Lett. 2013, 15, 2050–2053. [Google Scholar] [CrossRef]
- Rong, Z.; Li, Q.; Lin, W.; Jia, Y. Reagent-free synthesis of 2,3,4-polysubstituted tetrahydroquinolines: Application to the formal synthesis of (±)-martinellic acid and martinelline. Tetrahedron Lett. 2013, 54, 4432–4434. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, Y.; Arai, H.; Makino, K.; Hamada, Y. Enantioselective synthesis of martinelline chiral core and its diastereomer using asymmetric tandem Michael–aldol reaction. Tetrahedron 2008, 64, 11568–11579. [Google Scholar] [CrossRef]
- He, Y.; Mahmud, H.; Moningka, R.; Lovely, C.J.; Dias, H.R. Cyclization reactions of N-acryloyl-2-aminobenzaldehyde derivatives: Formal total synthesis of martinellic acid. Tetrahedron 2006, 62, 8755–8769. [Google Scholar] [CrossRef]
- Hadden, M.; Nieuwenhuyzen, M.; Osborne, D.; Stevenson, P.J.; Thompson, N.; Walker, A.D. Synthesis of the heterocyclic core of martinelline and martinellic acid. Tetrahedron 2006, 62, 3977–3984. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; O’Hare, S.M. Total synthesis of (±)-martinellic acid. Org. Lett. 2001, 3, 4217–4220. [Google Scholar] [CrossRef] [PubMed]
- Hadden, M.; Nieuwenhuyzen, M.; Osborne, D.; Stevenson, P.J.; Thompson, N. Synthesis of the heterocyclic core of the alkaloids martinelline and martinellic acid. Tetrahedron Lett. 2001, 42, 6417–6419. [Google Scholar] [CrossRef]
- Batey, R.A.; Powell, D.A. Multi-component coupling reactions: Synthesis of a guanidine containing analog of the hexahydropyrrolo [3,2-c] quinoline alkaloid martinelline. Chem. Commun. 2001, 22, 2362–2363. [Google Scholar] [CrossRef]
- Davies, S.G.; Ichihara, O.; Walters, I. Asymmetric-synthesis of syn-a-alkyl-b-amino acids. J. Chem. Soc. Perkin Trans. 1994, 9, 1141–1147. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Xie, H.; Zu, L.; Jiang, W.; Duesler, E.N.; Wang, W. Chiral Diphenylprolinol TES Ether Promoted Conjugate Addition–Aldol-Dehydration Reactions between α, β-Unsaturated Aldehydes and 2-N-Protected Amino Benzaldehydes. Org. Lett. 2007, 9, 965–968. [Google Scholar] [CrossRef]
- Hara, O.; Sugimoto, K.; Makino, K.; Hamada, Y. New synthesis of a pyrroloquinoline skeleton, the martinelline core, using a tandem Michael-aldol strategy. Synlett 2004, 2004, 1625–1627. [Google Scholar]
- Pappoppula, M.; Aponick, A. Enantioselective Total Synthesis of (−)-Martinellic Acid. Angew. Chem. Int. Ed. 2015, 54, 15827–15830. [Google Scholar] [CrossRef]
- Lackey, K.E. Compounds and Methods of Treatment. U.S. Patent 20080234267A1, 25 September 2008. [Google Scholar]
- Behenna, D.C.; Stoltz, B.M. The enantioselective Tsuji allylation. J. Am. Chem. Soc. 2004, 126, 15044–15045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, M.; Kawai, S.; Hayashi, M.; Naito, T.; Miyata, O. Efficient entry into 2-substituted tetrahydroquinoline systems through alkylative ring expansion: Stereoselective formal synthesis of (±)-martinellic acid. J. Org. Chem. 2010, 75, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Batey, R.; Simoncic, P.; Smyj, R.; Lough, A. A three-component coupling protocol for the synthesis of substituted hexahydropyrrolo [3,2-c] quinolines. Chem. Commun. 1999, 7, 651–652. [Google Scholar] [CrossRef]
- Kouznetsov, V.V. Recent synthetic developments in a powerful imino Diels-Alder reaction (Povarov reaction): Application to the synthesis of N-polyheterocycles and related alkaloids. Tetrahedron 2009, 65, 2721–2750. [Google Scholar] [CrossRef]
- Fochi, M.; Caruana, L.; Bernardi, L. Catalytic asymmetric aza-Diels–Alder reactions: The Povarov cycloaddition reaction. Synthesis 2014, 46, 135–157. [Google Scholar] [CrossRef]
- Hadden, M.; Stevenson, P.J. Regioselective synthesis of pyrroloquinolines—Approaches to Martinelline. Tetrahedron Lett. 1999, 40, 1215–1218. [Google Scholar] [CrossRef]
- He, L.; Liu, H.-B.; Zhao, L.; Wang, D.-X.; Wang, M.-X. Lewis acid-catalyzed reaction between tertiary enamides and imines of salicylaldehydes: Expedient synthesis of novel 4-chromanamine derivatives. Tetrahedron 2015, 71, 523–531. [Google Scholar] [CrossRef]
- Yu, J.; Jiang, H.-J.; Zhou, Y.; Luo, S.-W.; Gong, L.-Z. Sodium Salts of Anionic Chiral Cobalt(III) Complexes as Catalysts of the Enantioselective Povarov Reaction. Angew. Chem. Int. Ed. 2015, 54, 11209–11213. [Google Scholar] [CrossRef]
- Liu, X.; Toy, P.H. Halogen Bond-Catalyzed Povarov Reactions. Adv. Synth. Catal. 2020, 362, 3437–3441. [Google Scholar] [CrossRef]
- Reyes-Gutiérrez, P.E.; Amatov, T.T.; Švec, P.; Císařová, I.; Šaman, D.; Pohl, R.; Teplý, F.; Pospíšil, L. Helquats as Promoters of the Povarov Reaction: Synthesis of 1,2,3,4-Tetrahydroquinoline Scaffolds Catalyzed by Helicene-Viologen Hybrids. ChemPlusChem 2020, 85, 2212–2218. [Google Scholar] [CrossRef]
- Škopić, M.K.; Götte, K.; Gramse, C.; Dieter, M.; Pospich, S.; Raunser, S.; Weberskirch, R.; Brunschweiger, A. Micellar Brønsted Acid Mediated Synthesis of DNA-Tagged Heterocycles. J. Am. Chem. Soc. 2019, 141, 10546–10555. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric Cooperative Catalysis of Strong Brønsted Acid–Promoted Reactions Using Chiral Ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard, B.; O’Shea, M.W.; Donckele, E.; Kesavan, S.; Akella, L.B.; Xu, H.; Jacobsen, E.N.; Marcaurelle, L.A. Application of a catalytic asymmetric Povarov reaction using chiral ureas to the synthesis of a tetrahydroquinoline library. ACS Comb. Sci. 2012, 14, 621–630. [Google Scholar] [CrossRef]
- Kinzel, T.; Zhang, Y.; Buchwald, S.L. A new palladium precatalyst allows for the fast Suzuki–Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J. Am. Chem. Soc. 2010, 132, 14073–14075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, B.-L.; Ding, C.-H.; Yang, X.-F.; Wan, X.-L.; Hou, X.-L. Kinetic resolution of 2, 3-dihydro-2-substituted 4-quinolones by palladium-catalyzed asymmetric allylic alkylation. J. Am. Chem. Soc. 2009, 131, 18250–18251. [Google Scholar] [CrossRef]
- Gigant, N.; Gillaizeau, I. Construction of nitrogen-fused tetrahydroquinolines via a domino reaction. Org. Lett. 2012, 14, 4622–4625. [Google Scholar] [CrossRef]
- Song, Z.; Zhao, Y.-M.; Zhai, H. One-Step Construction of Tetrahydro-5 H-indolo [3,2-c] quinolines from Benzyl Azides and Indoles via a Cascade Reaction Sequence. Org. Lett. 2011, 13, 6331–6333. [Google Scholar] [CrossRef]
- Desai, P.; Schildknegt, K.; Agrios, K.A.; Mossman, C.; Milligan, G.L.; Aube, J. Reactions of alkyl azides and ketones as mediated by Lewis acids: Schmidt and Mannich reactions using azide precursors. J. Am. Chem. Soc. 2000, 122, 7226–7232. [Google Scholar] [CrossRef]
- Dagousset, G.; Retailleau, P.; Masson, G.; Zhu, J. Chiral phosphoric acid-catalyzed enantioselective three-component Povarov reaction using cyclic enethioureas as dienophiles: Stereocontrolled access to enantioenriched hexahydropyrroloquinolines. Chem. Eur. J. 2012, 18, 5869–5873. [Google Scholar] [CrossRef]
- Roy, S.; Reiser, O. A Catalytic Multicomponent Approach for the Stereoselective Synthesis of cis-4,5-Disubstituted Pyrrolidinones and Tetrahydro-3H-pyrrolo[3,2-c]quinolines. Angew. Chem. Int. Ed. 2012, 51, 4722–4725. [Google Scholar] [CrossRef]
- Wood, J.; Bagi, C.M.; Akuche, C.; Bacchiocchi, A.; Baryza, J.; Blue, M.-L.; Brennan, C.; Campbell, A.-M.; Choi, S.; Cook, J.H.; et al. 4,5-Disubstituted cis-pyrrolidinones as inhibitors of type II 17β-hydroxysteroid dehydrogenase. Part 3. Identification of lead candidate. Bioorg. Med. Chem. Lett. 2006, 16, 4965–4968. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hirao, H.; Li, Y.; Zhou, J. Palladium-Catalyzed Asymmetric Intermolecular Cyclization. Angew. Chem. Int. Ed. 2013, 52, 8676–8680. [Google Scholar] [CrossRef] [PubMed]
- Comesse, S.; Sanselme, M.; Daïch, A. New and expeditious tandem sequence aza-Michael/intramolecular nucleophilic substitution route to substituted γ-lactams: Synthesis of the tricyclic core of (±)-Martinellines. J. Org. Chem. 2008, 73, 5566–5569. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, R.; Lawson, A.M.; Daïch, A.; Comesse, S. Synthesis of highly functionalized pyrrolidines as tunable templates for the direct access to (±)-coerulescine and the tricyclic core of martinellines. Org. Biomol. Chem. 2013, 11, 1818–1821. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, T.; Bao, W. FeCl3-Catalyzed Cascade Cyclization in One Pot: Synthesis of Ring-Fused Tetrahydroquinoline Derivatives from Arylamines and N-Substituted Lactams. J. Org. Chem. 2013, 78, 8155–8160. [Google Scholar] [CrossRef]
- Yang, M.; Su, B.; Wang, Y.; Chen, K.; Jiang, X.; Zhang, Y.-F.; Zhang, X.-S.; Chen, G.; Cheng, Y.; Cao, Z.; et al. Silver-catalysed direct amination of unactivated C–H bonds of functionalized molecules. Nat. Commun. 2014, 5, 4707. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Zhang, H.-R.; Zhu, S.-Y.; Zhu, P.; Hui, X.-P. Highly stereoselective synthesis of functionalized pyrrolo [3, 2-c] quinolines via N-heterocyclic carbene catalyzed cascade sequence. Org. Lett. 2014, 16, 5048–5051. [Google Scholar] [CrossRef]
- Wang, J.; Li, Y.; Sun, J.; Wang, H.; Jin, Z.; Chi, Y.R. Carbene-Catalyzed Enantioselective Addition of Benzylic Carbon to Unsaturated Acyl Azolium for Rapid Synthesis of Pyrrolo[3,2-c]quinolines. ACS Catal. 2018, 8, 9859–9864. [Google Scholar] [CrossRef]
- Boomhoff, M.; Yadav, A.K.; Appun, J.; Schneider, C. Modular, flexible, and stereoselective synthesis of pyrroloquinolines: Rapid assembly of complex heterocyclic scaffolds. Org. Lett. 2014, 16, 6236–6239. [Google Scholar] [CrossRef]
- Appun, J.; Stolz, F.; Naumov, S.; Abel, B.; Schneider, C. Modular Synthesis of Dipyrroloquinolines: A Combined Synthetic and Mechanistic Study. J. Org. Chem. 2018, 83, 1737–1744. [Google Scholar] [CrossRef]
- Bakthadoss, M.; Kannan, D.; Srinivasan, J.; Vinayagam, V. Highly regio-and diastereo-selective synthesis of novel tri-and tetra-cyclic perhydroquinoline architectures via an intramolecular [3+2] cycloaddition reaction. Org. Biomol. Chem. 2015, 13, 2870–2874. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.-A.; Li, J.; Xia, P.-J.; Zhou, Z.-F.; Deng, Z.-X.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Diastereoselective Intramolecular [3+2]-Annulation of Donor–Acceptor Cyclopropane with Imine-Assembling Hexahydropyrrolo [3,2-c] quinolinone Scaffolds. J. Org. Chem. 2016, 81, 11185–11194. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, A.S.; Ivanova, O.A.; Chagarovskiy, A.O.; Stebunov, N.S.; Orlov, N.V.; Shumsky, A.N.; Budynina, E.M.; Rybakov, V.B.; Trushkov, I.V. Domino Staudinger/aza-Wittig/Mannich Reaction: An Approach to Diversity of Di- and Tetrahydropyrrole Scaffolds. Chem. Eur. J. 2016, 22, 17967–17971. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, K.L.; Villemson, E.V.; Budynina, E.M.; Ivanova, O.A.; Trushkov, I.V.; Melnikov, M.Y. Ring Opening of Donor–Acceptor Cyclopropanes with the Azide Ion: A Tool for Construction of N-Heterocycles. Chem. Eur. J. 2015, 21, 4975–4987. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, C.; Huang, K.; Liu, L.; Chang, W.; Li, J. Copper-catalyzed cascade reaction via intramolecular hydroamination cyclization of homopropargylic amines and intermolecular Povarov reaction with imines. Org. Lett. 2016, 18, 2367–2370. [Google Scholar] [CrossRef]
- Liu, L.; Wang, C.; Liu, Q.; Kong, Y.; Chang, W.; Li, J. Copper (II) Trifluoromethanesulfonate Catalyzed Hydroamination Cyclization–Dimerization Cascade Reaction of Homopropargylic Amines for the Construction of Complex Fused Nitrogen-Containing Tetracycles. Eur. J. Org. Chem. 2016, 2016, 3684–3690. [Google Scholar] [CrossRef]
- Li, J.; Lin, N.; Yu, L.; Zhang, Y. Synthesis of β-prolinols via [3+2] cycloaddition and one-pot programmed reduction: Valuable building blocks for polyheterocycles. Tetrahedron Lett. 2016, 57, 5777–5780. [Google Scholar] [CrossRef]
- Xie, H.; Gong, B.; Zhong, X.; Cui, H.; Xiang, J. Intramolecular Cycloaddition of Azomethine Ylides Activated by Aromatic Rings: Scope and Limitations. Chem. HeterocycIic Comp. 2016, 52, 484–492. [Google Scholar] [CrossRef]
- Cai, J.; Li, F.; Deng, G.-J.; Ji, X.; Huang, H. The cyclopropylimine rearrangement/Povarov reaction cascade for the assembly of pyrrolo [3, 2-c] quinoline derivatives. Green Chem. 2016, 18, 3503–3506. [Google Scholar] [CrossRef]
- Kondo, Y.; Nishikimi, Y. Process for Production of Optically Active Hexahydropyrroloquinoline and Intermediate for the Process. U.S. Patents WO 2010038434, 8 April 2010. [Google Scholar]
- Tatsuta, K.; Kondo, Y. Process for Preparation of Hexahydropyrroloquinoline. U.S. Patents JP 2011256110A, 30 September 2011. [Google Scholar]
- Yamada, M.; Usutani, H.; Ito, T.; Yamano, M. Construction of a (3aR, 4R, 9bR)-Hexahydropyrrolo- quinoline by Stereoselective Hydrogen-Mediated Domino Cyclization. Org. Process Res. Dev. 2019, 23, 535–547. [Google Scholar] [CrossRef]
- Lindbäck, E.; Sydnes, M.O. Catalytic Enantioselective Synthesis of the Partially Reduced Tricyclic Pyrrolo [3,2-c] quinoline Core Structure of the Martinella Alkaloids. ChemistrySelect 2016, 1, 1837–1840. [Google Scholar] [CrossRef]
- Kolb, H.C.; Andersson, P.G.; Sharpless, K.B. Toward an understanding of the high enantioselectivity in the osmium-catalyzed asymmetric dihydroxylation (AD). 1. Kinetics. J. Am. Chem. Soc. 1994, 116, 1278–1291. [Google Scholar] [CrossRef]
- Reddy, J.S.; Rao, B.V. A short, efficient, and stereoselective total synthesis of a pyrrolidine alkaloid: (−)-codonopsinine. J. Org. Chem. 2007, 72, 2224–2227. [Google Scholar] [CrossRef] [PubMed]
- Haarr, M.B.; Sydnes, M.O. Synthetic approach towards the martinella alkaloids. Unpublished work. 2020. [Google Scholar]
- Wittig, G.; Sommer, H. Zum Verhalten ungesättigter Ammoniumsalze gegenüber Protonenacceptoren. Justus Liebigs Ann. Chem. 1955, 594, 1–14. [Google Scholar] [CrossRef]
- Swan, G.A.; Wilcock, J.D. Reduction of some N-alkyl- and N-aryl-pyrrolidin-2-ones and-piperidin-2-ones by lithium aluminium hydride. J. Chem. Soc. Perkin Trans. 1974, 885–891. [Google Scholar] [CrossRef]
- Kerr, G.H.; Meth-Cohn, O.; Mullock, E.B.; Suschitzky, H. Reactions of NN-dialkylanilines with diethyl azodicarboxylate and with ozone. J. Chem. Soc. Perkin Trans. 1974, 1614–1619. [Google Scholar] [CrossRef]
- Khandelwal, G.D.; Swan, G.A.; Roy, R.B. Dehydrogenation of some aromatic tertiary amines by gamma radiation and by peroxides. J. Chem. Soc. Perkin Trans. 1974, 891–896. [Google Scholar] [CrossRef]
- Rao, G.A.; Periasamy, M. Cycloaddition of enamine and iminium ion intermediates formed in the reaction of N-arylpyrrolidines with T-HYDRO. Synlett 2015, 26, 2231–2236. [Google Scholar] [CrossRef]
- Minakata, S.; Ohshima, Y.; Takemiya, A.; Ryu, I.; Komatsu, M.; Ohshiro, Y. Catalytic oxidation of amines utilizing binuclear copper (II) complex of 7-azaindole. Chem. Lett. 1997, 26, 311–312. [Google Scholar] [CrossRef]
- Buswell, M.; Fleming, I. The reaction of phenyldimethylsilyllithium with N-phenylpyrrolidone. Chem. Commun. 2003, 202–203. [Google Scholar] [CrossRef] [PubMed]
- Min, C.; Sanchawala, A.; Seidel, D. Dual C–H Functionalization of N-Aryl Amines: Synthesis of Polycyclic Amines via an Oxidative Povarov Approach. Org. Lett. 2014, 16, 2756–2759. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yao, X.; Zhang, L.; Ni, P.; Cheng, R.; Ye, J. Direct Arylation of α-Amino C(sp3)-H Bonds by Convergent Paired Electrolysis. Angew. Chem. Int. Ed. 2019, 58, 16548–16552. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Yang, Q.; Zhang, L.; Luo, S. Photoredox Mediated Acceptorless Dehydrogenative Coupling of Saturated N-Heterocycles. ACS Catal. 2019, 9, 3589–3594. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Chang, W.; Li, J. The Divergent Cascade Reactions of Arylalkynols with Homopropargylic Amines or Electron-Deficient Olefins: Access to the Spiro-Isobenzofuran-b-pyrrolo- quinolines or Bridged-Isobenzofuran Polycycles. J. Org. Chem. 2018, 83, 7799–7813. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.-Q.; Xu, J.-T.; Feng, Z.-T.; Liang, H.; Wang, Z.-Y.; Qin, Y.; Xu, P.-F. Dual C(sp3)–H Bond Functionalization of N-Heterocycles through Sequential Visible-Light Photocatalyzed Dehydrogenation/ [2+2] Cycloaddition Reactions. Angew. Chem. Int. Ed. 2018, 57, 5110–5114. [Google Scholar] [CrossRef]
- Kong, Y.; Liu, Y.; Wang, B.; Li, S.; Liu, L.; Chang, W.; Li, J. The Catalyst-Controlled Divergent Cascade Reactions of Homo-Propargylic Amines and Nitrones: Synthesis of Pyrrolo-Isoxazolidines and γ-Lactams. Adv. Synth. Catal. 2018, 360, 1240–1252. [Google Scholar] [CrossRef]
- Wang, F.; He, Y.; Tian, M.; Zhang, X.; Fan, X. Synthesis of α-Formylated N-Heterocycles and Their 1,1-Diacetates from Inactivated Cyclic Amines Involving an Oxidative Ring Contraction. Org. Lett. 2018, 20, 864–867. [Google Scholar] [CrossRef]
- Snider, B.B.; Ahn, Y.; Foxman, B.M. Synthesis of the tricyclic triamine core of martinelline and martinellic acid. Tetrahedron Lett. 1999, 40, 3339–3342. [Google Scholar] [CrossRef]
- Fustero, S.; Bello, P.; Miró, J.; Sánchez-Roselló, M.; Maestro, M.A.; Gonzalez, J.; del Pozo, C. Gold catalyzed stereoselective tandem hydroamination–formal aza-Diels–Alder reaction of propargylic amino esters. Chem. Commun. 2013, 49, 1336–1338. [Google Scholar] [CrossRef]
- Ma, C.-L.; Li, X.-H.; Yu, X.-L.; Zhu, X.-L.; Hu, Y.-Z.; Dong, X.-W.; Tan, B.; Liu, X.-Y. Gold-catalyzed tandem synthesis of bioactive spiro-dipyrroloquinolines and its application in the one-step synthesis of incargranine B aglycone and seneciobipyrrolidine (I). Org. Chem. Front. 2016, 3, 324–329. [Google Scholar] [CrossRef]
- Yu, X.-L.; Kuang, L.; Chen, S.; Zhu, X.-L.; Li, Z.-L.; Tan, B.; Liu, X.-Y. Counteranion-controlled unprecedented diastereo-and enantioselective tandem formal Povarov reaction for construction of bioactive octahydro-dipyrroloquinolines. ACS Catal. 2016, 6, 6182–6190. [Google Scholar] [CrossRef]
- Li, S.-S.; Zhou, L.; Wang, L.; Zhao, H.; Yu, L.; Xiao, J. Organocatalytic C (sp3)–H functionalization via carbocation-initiated cascade [1,5]-hydride transfer/cyclization: Synthesis of dihydrodibenzo [b,e] azepines. Org. Lett. 2018, 20, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wang, M.; Pan, L.; Li, Y.; Liu, Q. Csp 3–H bond functionalization of amines via tunable iminium ions: Divergent synthesis of trifluoromethylated arylamines. Chem. Commun. 2018, 54, 8721–8724. [Google Scholar] [CrossRef] [PubMed]
- Kajino, M.; Hird, N.W.; Tarui, N.; Banno, H.; Kawano, Y.; Inatomi, N. Preparation of Fused Quinoline Derivatives as NK2 Receptor Antagonists for Functional Gastrointestinal Diseases. U.S. Patent WO 2005105802, 10 November 2005. [Google Scholar]
- Tanaka, T.; Matsumoto-Okano, S.; Inatomi, N.; Fujioka, Y.; Kamiguchi, H.; Yamaguchi, M.; Imanishi, A.; Kawamoto, M.; Miura, K.; Nishikawa, Y. Establishment and validation of a rabbit model for in vivo pharmacodynamic screening of tachykinin NK2 antagonists. J. Pharmacol. Sci. 2012, 118, 487–495. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Tanaka, A.; Nakamura, A.; Matsushita, K.; Imanishi, A.; Matsumoto-Okano, S.; Inatomi, N.; Miura, K.; Toyoda, M.; Mizojiri, G. Effects of TAK-480, a Novel Tachykinin NK2–Receptor Antagonist, on Visceral Hypersensitivity in Rabbits and Ricinoleic Acid–Induced Defecation in Guinea Pigs. J. Pharmacol. Sci. 2012, 120, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, T.; Konishi, T.; Yamano, M. Asymmetric catalytic hydrogenation for large scale preparation of optically active 2-(N-benzoylamino) cyclohexanecarboxylic acid derivatives. Catal. Sci. Technol. 2012, 2, 2146–2152. [Google Scholar] [CrossRef]
- Furuya, K.; Yamamoto, N.; Ohyabu, Y.; Makino, A.; Morikyu, T.; Ishige, H.; Kuzutani, K.; Endo, Y. The Novel Non-steroidal Selective Androgen Receptor Modulator S-101479 Has Additive Effects with Bisphosphonate, Selective Estrogen Receptor Modulator, and Parathyroid Hormone on the Bones of Osteoporotic Female Rats. Biol. Pharm. Bull. 2012, 35, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Furuya, K.; Yamamoto, N.; Ohyabu, Y.; Morikyu, T.; Ishige, H.; Albers, M.; Endo, Y. Mechanism of the tissue-specific action of the selective androgen receptor modulator S-101479. Biol. Pharm. Bull. 2013, 36, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Duvall, J.R.; Bedard, L.; Naylor-Olsen, A.M.; Manson, A.L.; Bittker, J.A.; Sun, W.; Fitzgerald, M.E.; He, Z.; Lee, M.D., IV; Marie, J.-C.; et al. Identification of highly specific diversity-oriented synthesis-derived inhibitors of Clostridium difficile. ACS Infect. Dis. 2017, 3, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Prosser, G.A.; Rodenburg, A.; Khoury, H.; de Chiara, C.; Howell, S.; Snijders, A.P.; de Carvalho, L.P.S. Glutamate racemase is the primary target of β-chloro-D-alanine in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6091–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, B.; Basarab, G.; Comita-Prevoir, J.; Gowravaram, M.; Hill, P.; Kiely, A.; Loch, J.; MacPherson, L.; Morningstar, M.; Mullen, G. Potent and selective inhibitors of Helicobacter pylori glutamate racemase (MurI): Pyridodiazepine amines. Bioorg. Med. Chem. Lett. 2009, 19, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, T.; Fisher, S.L.; Kern, G.; Folmer, R.H.; Xue, Y.; Newton, D.T.; Keating, T.A.; Alm, R.A.; de Jonge, B.L. Exploitation of structural and regulatory diversity in glutamate racemases. Nature 2007, 447, 817–822. [Google Scholar] [CrossRef]
- Park, S.W.; Casalena, D.E.; Wilson, D.J.; Dai, R.; Nag, P.P.; Liu, F.; Boyce, J.P.; Bittker, J.A.; Schreiber, S.L.; Finzel, B.C. Target-based identification of whole-cell active inhibitors of biotin biosynthesis in Mycobacterium tuberculosis. Chem. Biol. 2015, 22, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M.; Wu, P.; Heler, R.; Mok, B.; Lim, D.; Siriwardena, S.U.; Paul, B. A high-throughput platform to identify small-molecule inhibitors of CRISPR-Cas9. Cell 2019, 177, 1067–1079. [Google Scholar] [CrossRef]
- Choudhary, A.; Fox, K.; Subramanian, H.; Franco, E. Compositions and Methods for Regulating Proteins and Nucleic Acids Activities. U.S. Patent 20200239879A1, 30 July 2020. [Google Scholar]
- Choudhary, A.; Maji, B.; Gangopadhyay, S.A.; Lee, M.; Shi, M. Inhibitors of RNA-Guided Nuclease Target Binding and Uses Thereof. U.S. Patent WO 2020068304, 2 April 2020. [Google Scholar]



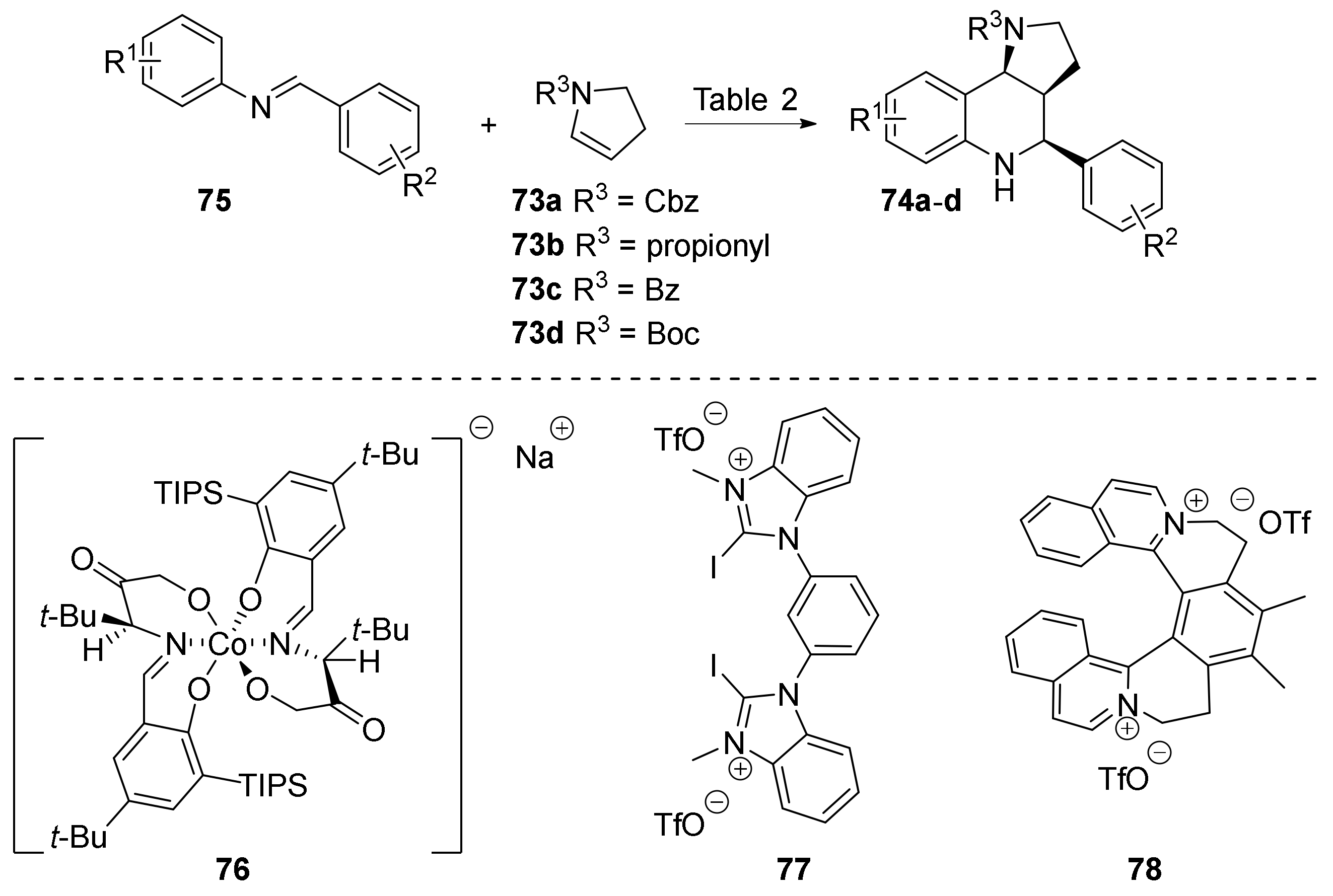
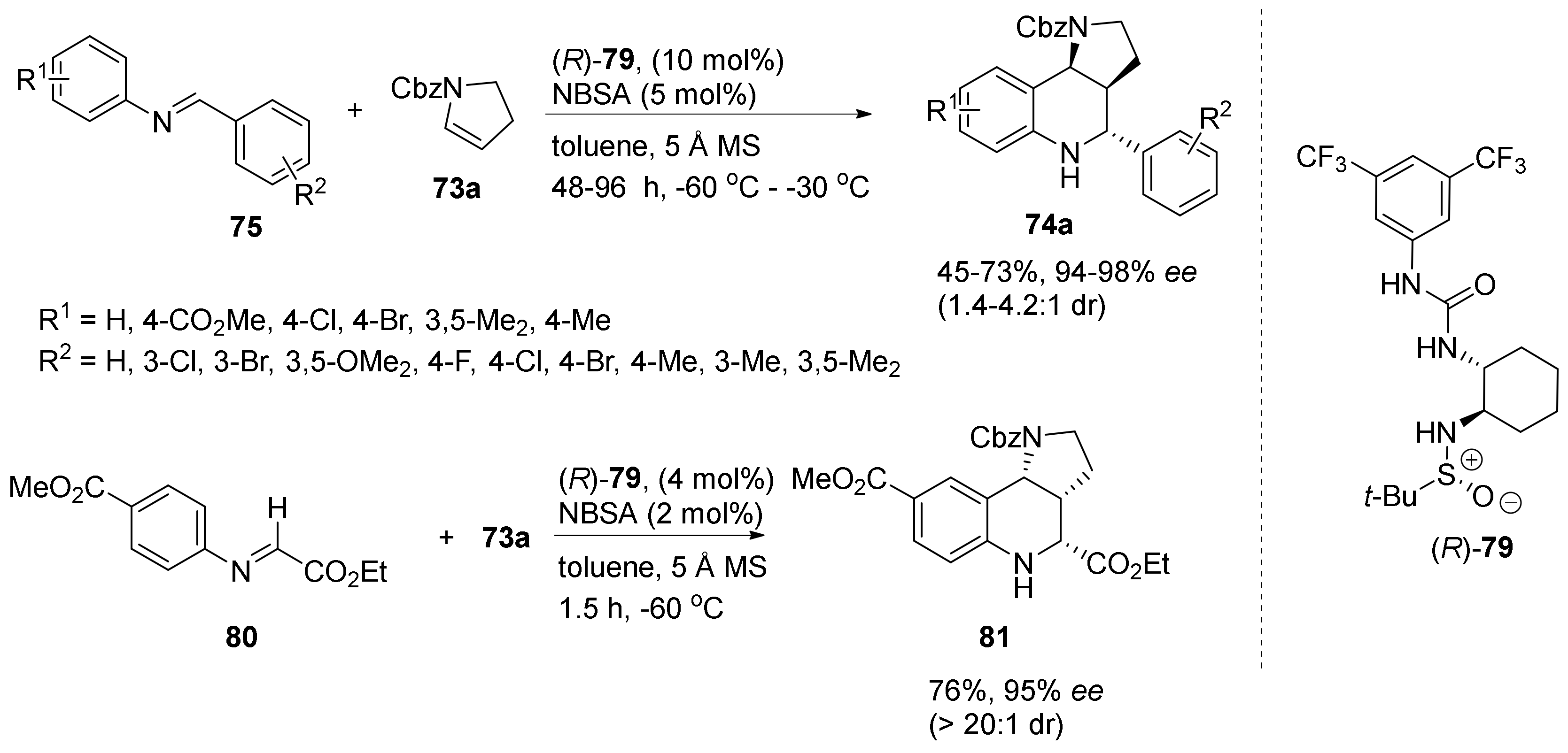
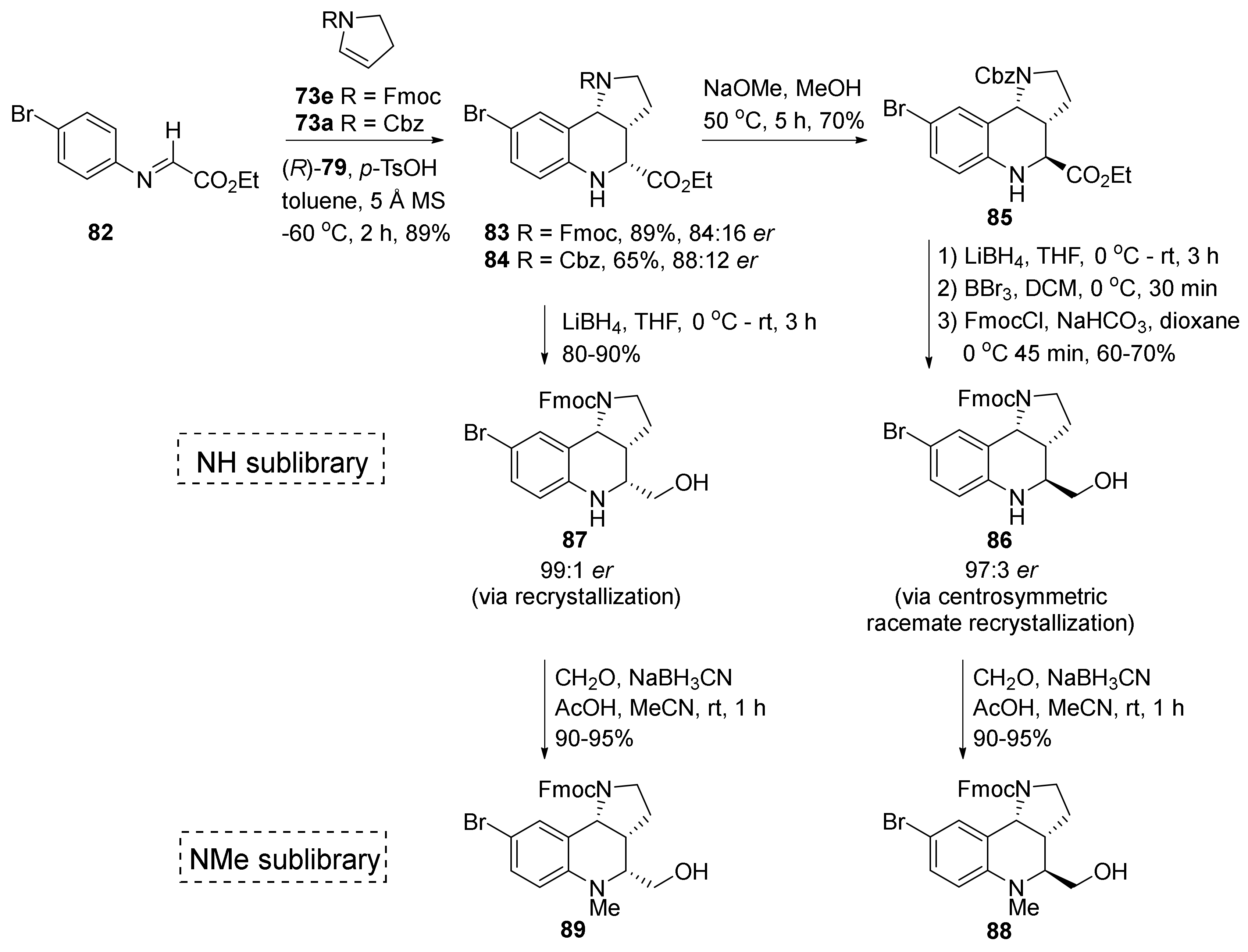
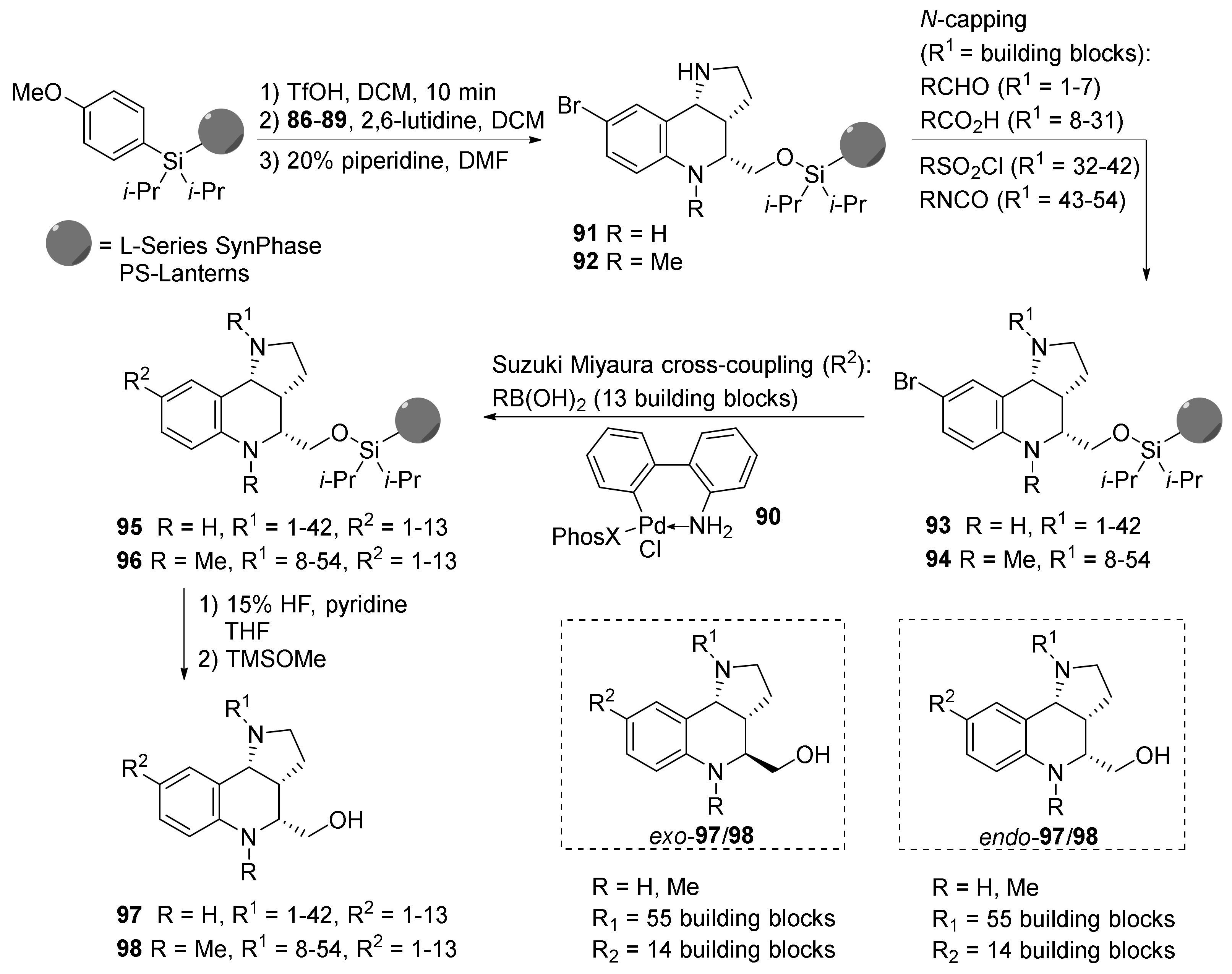
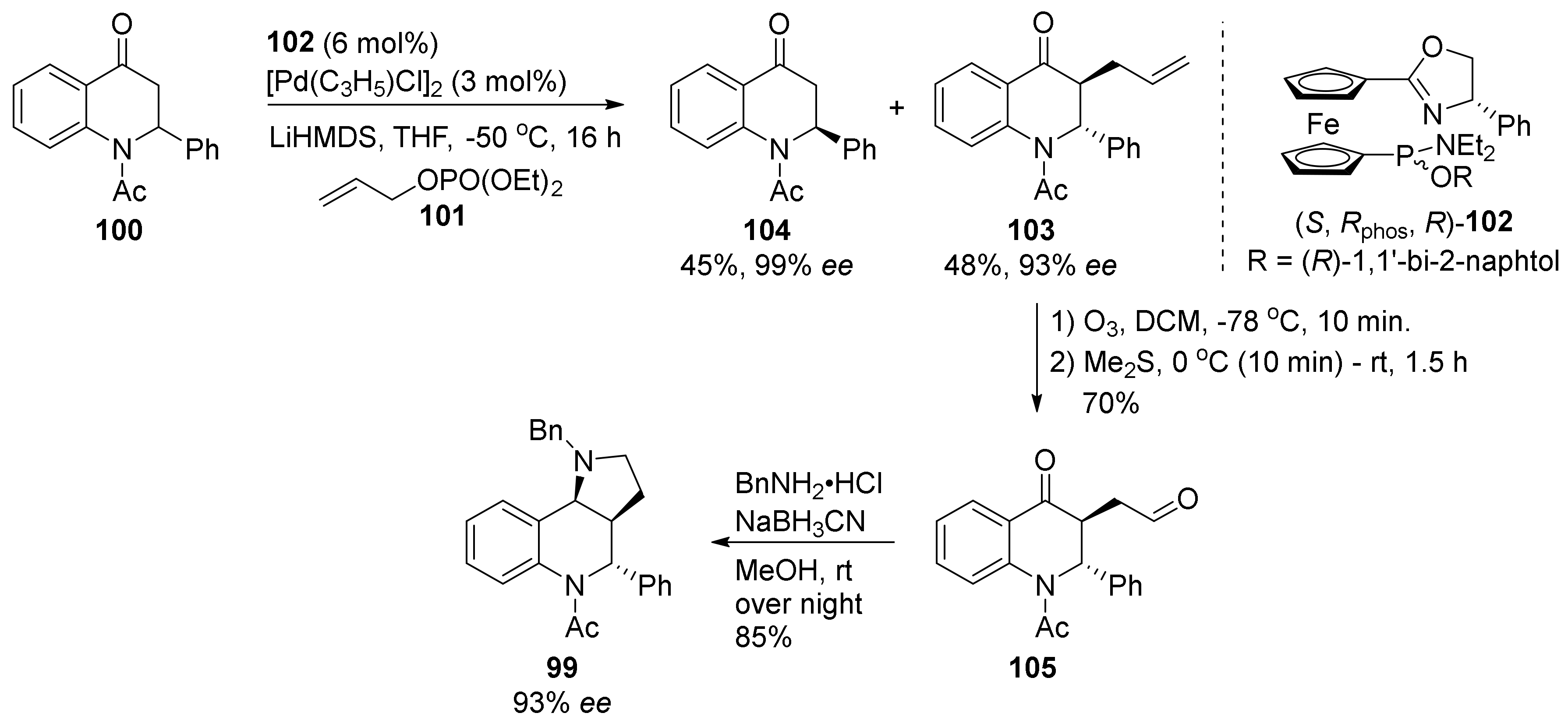
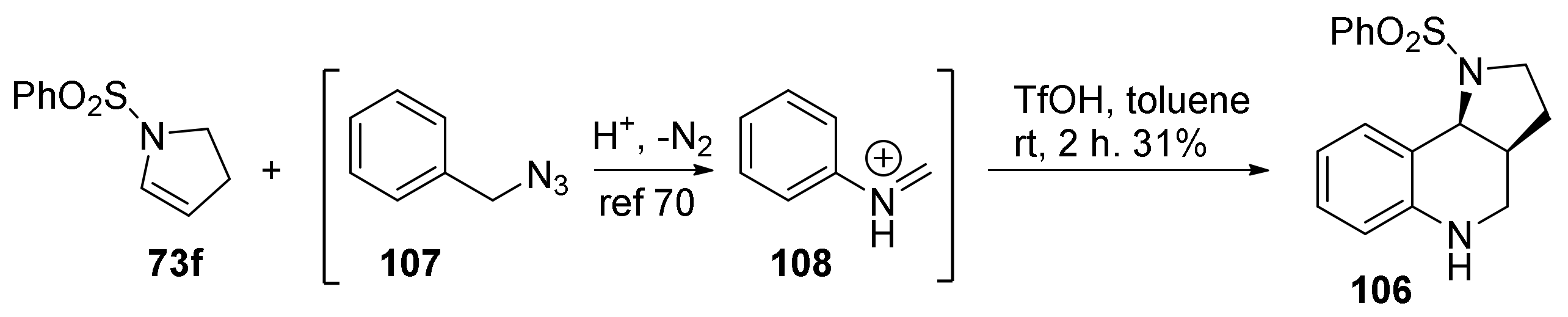
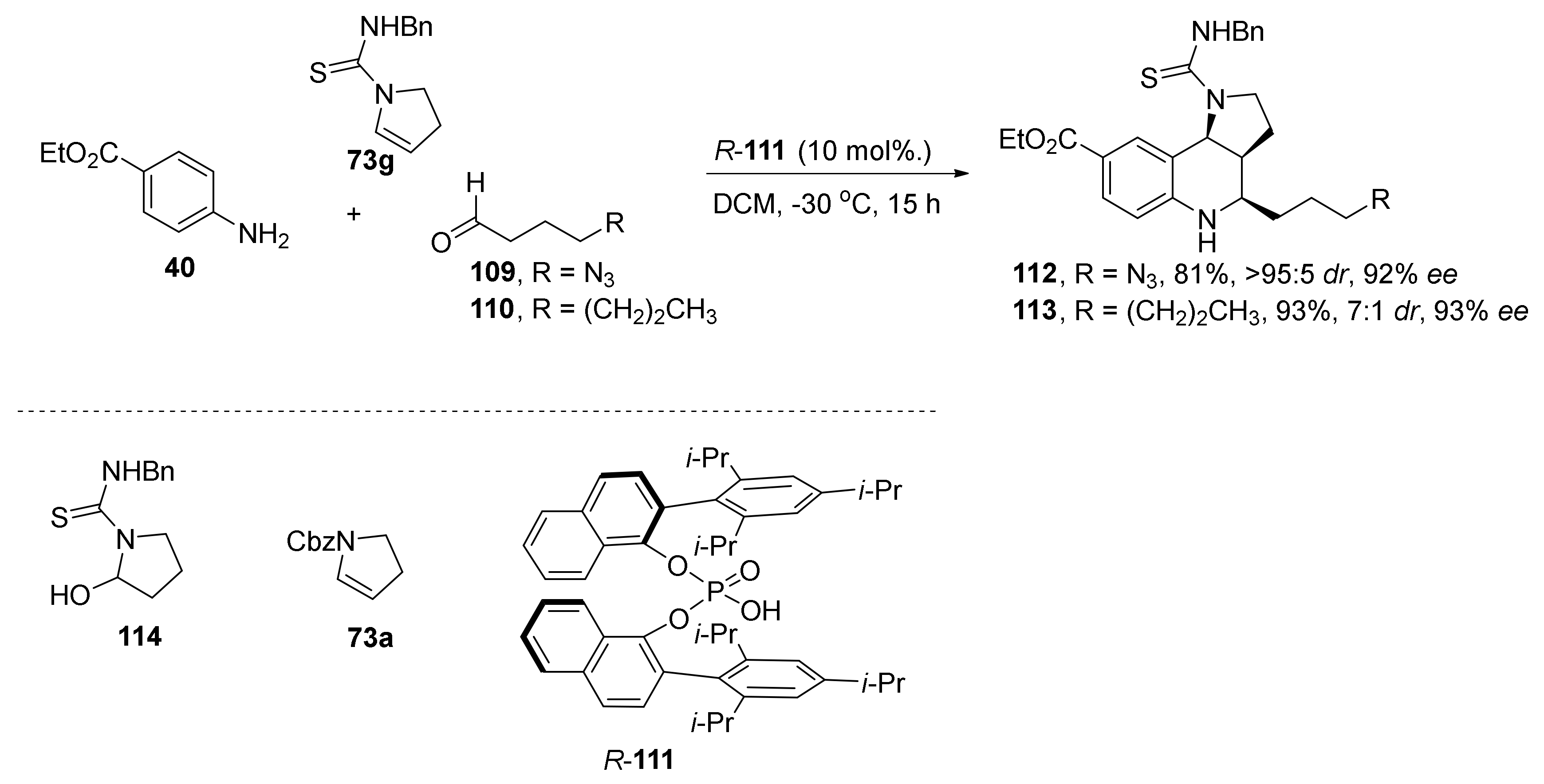
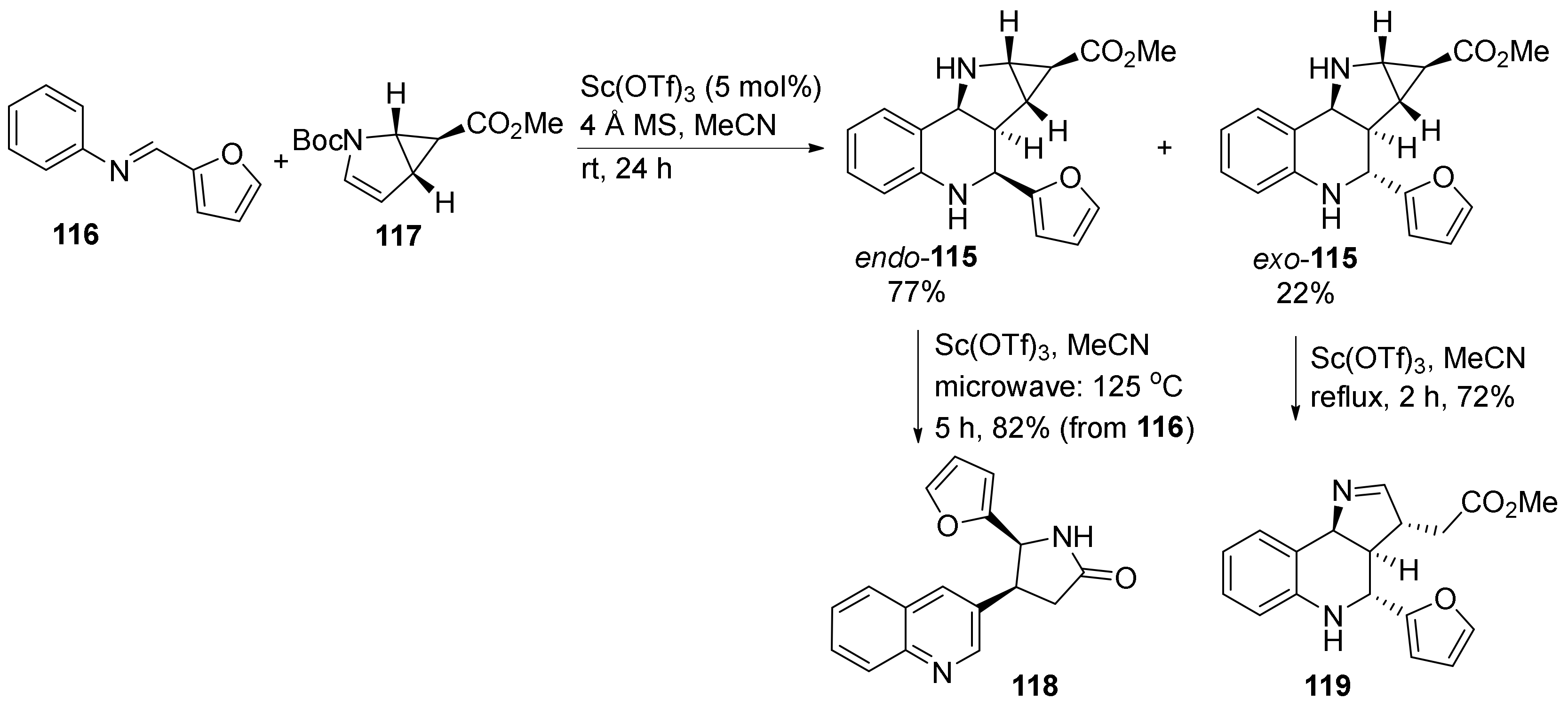






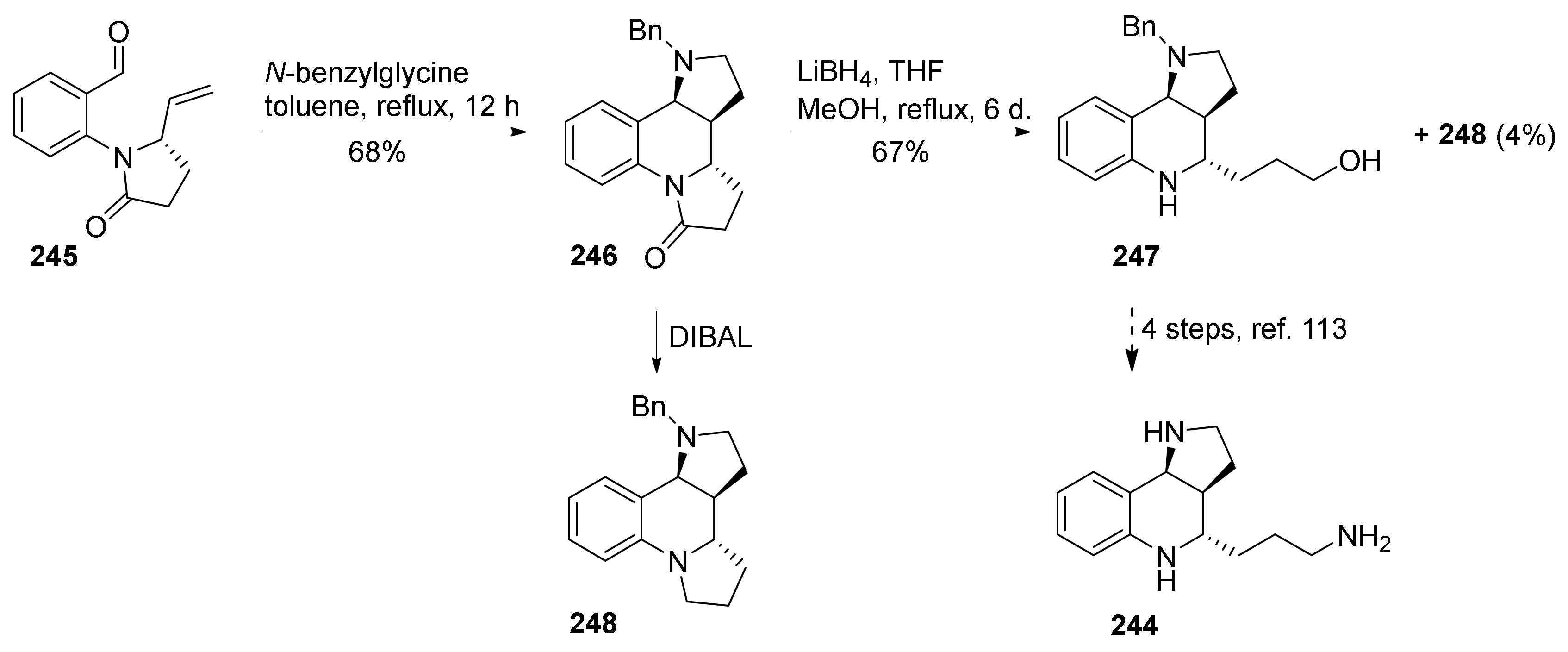
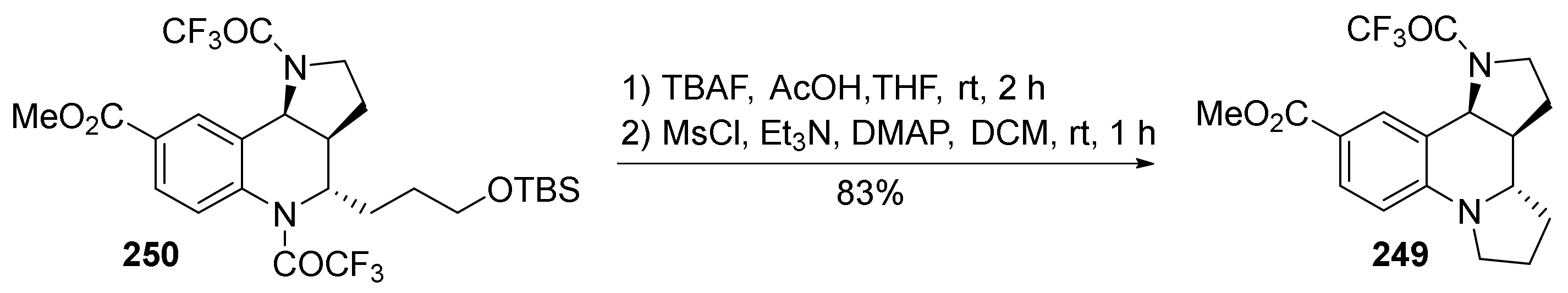
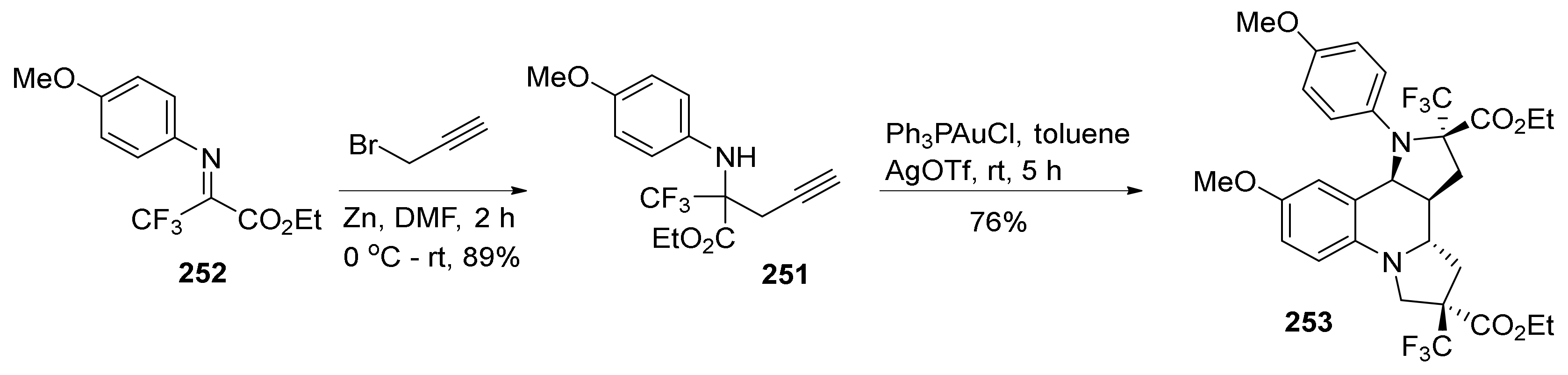
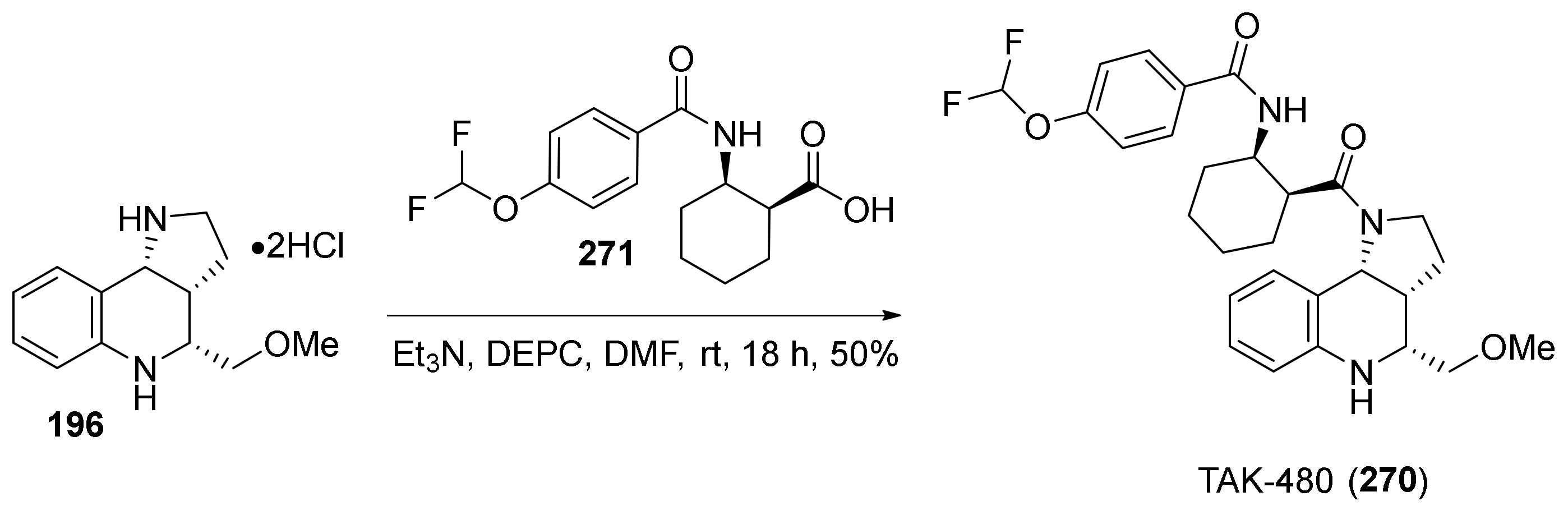

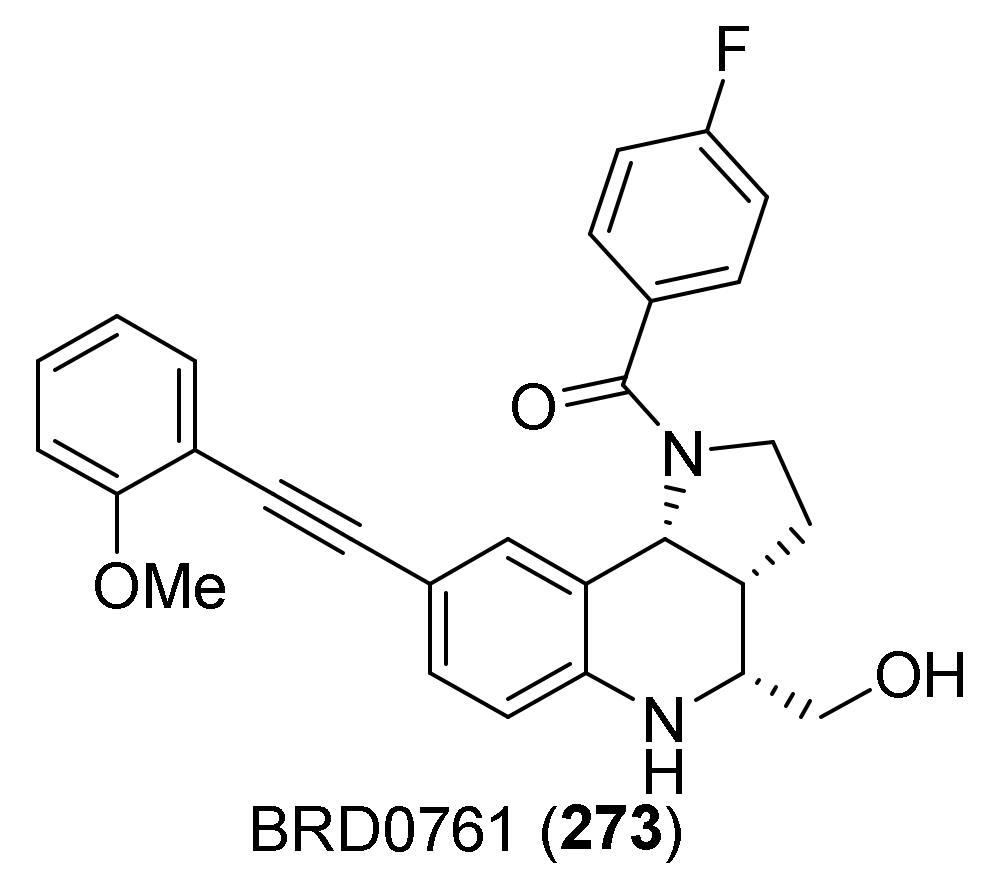

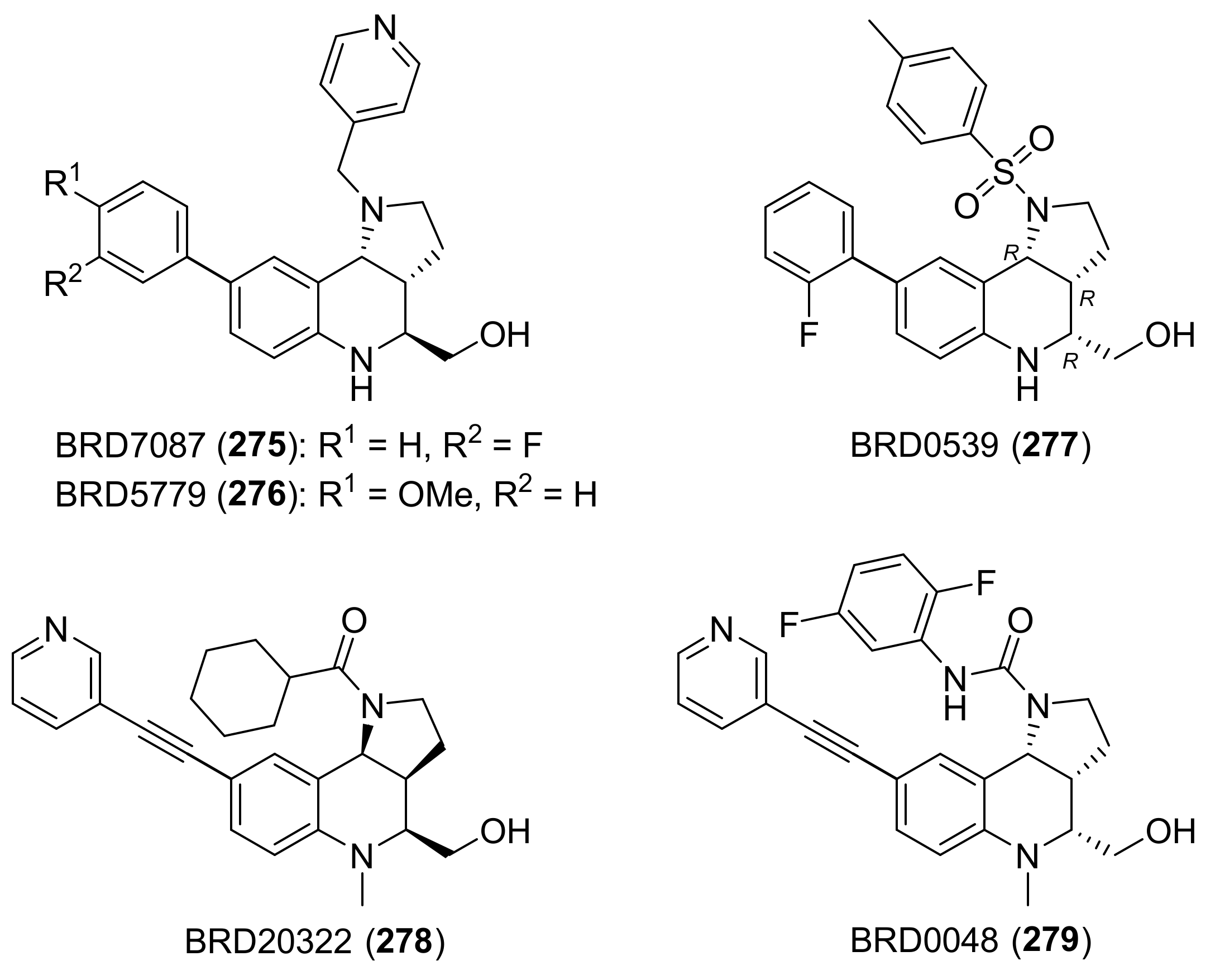

{kind=link}
{kind=link}
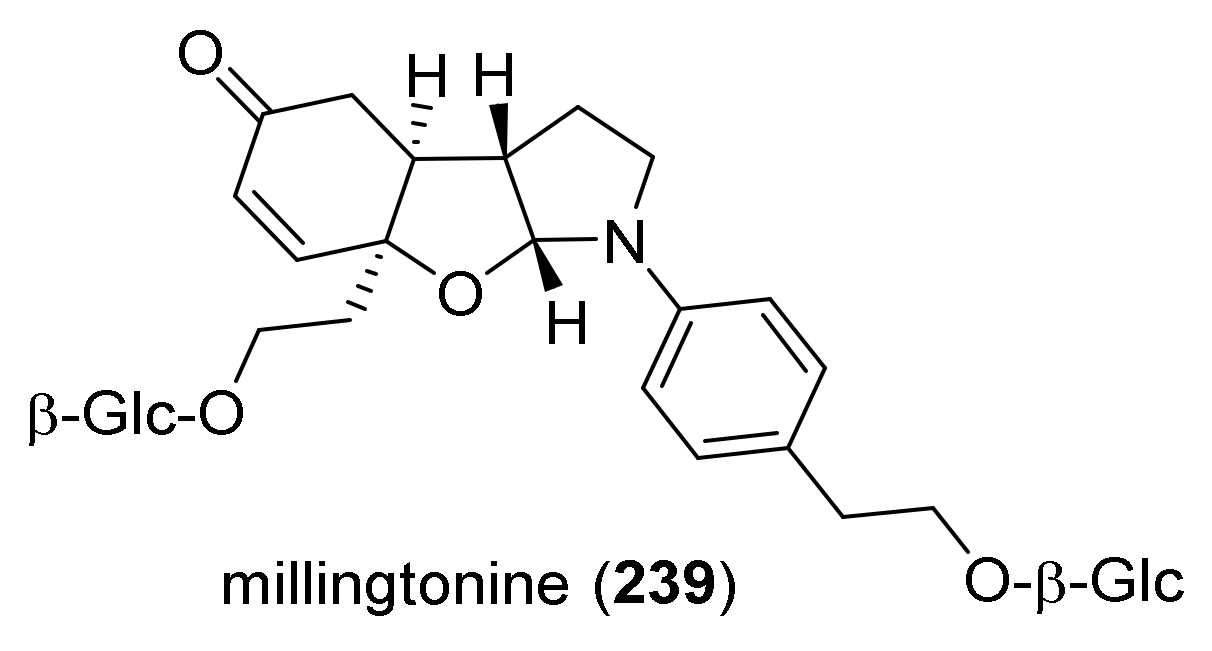{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
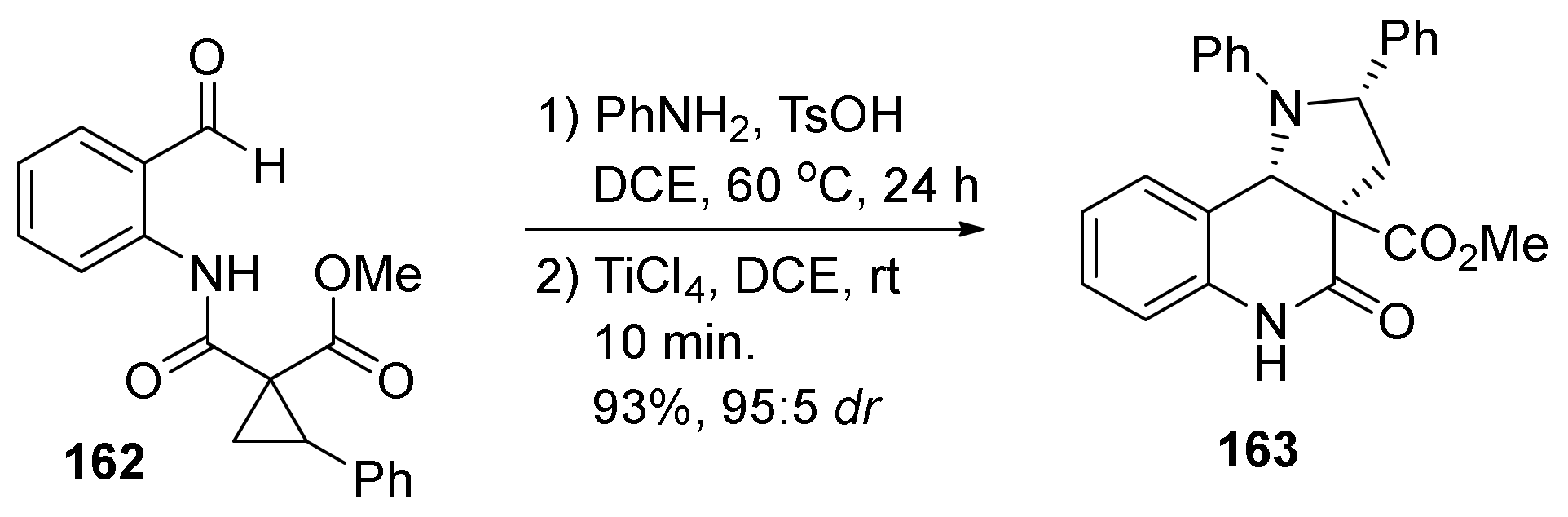{kind=link}
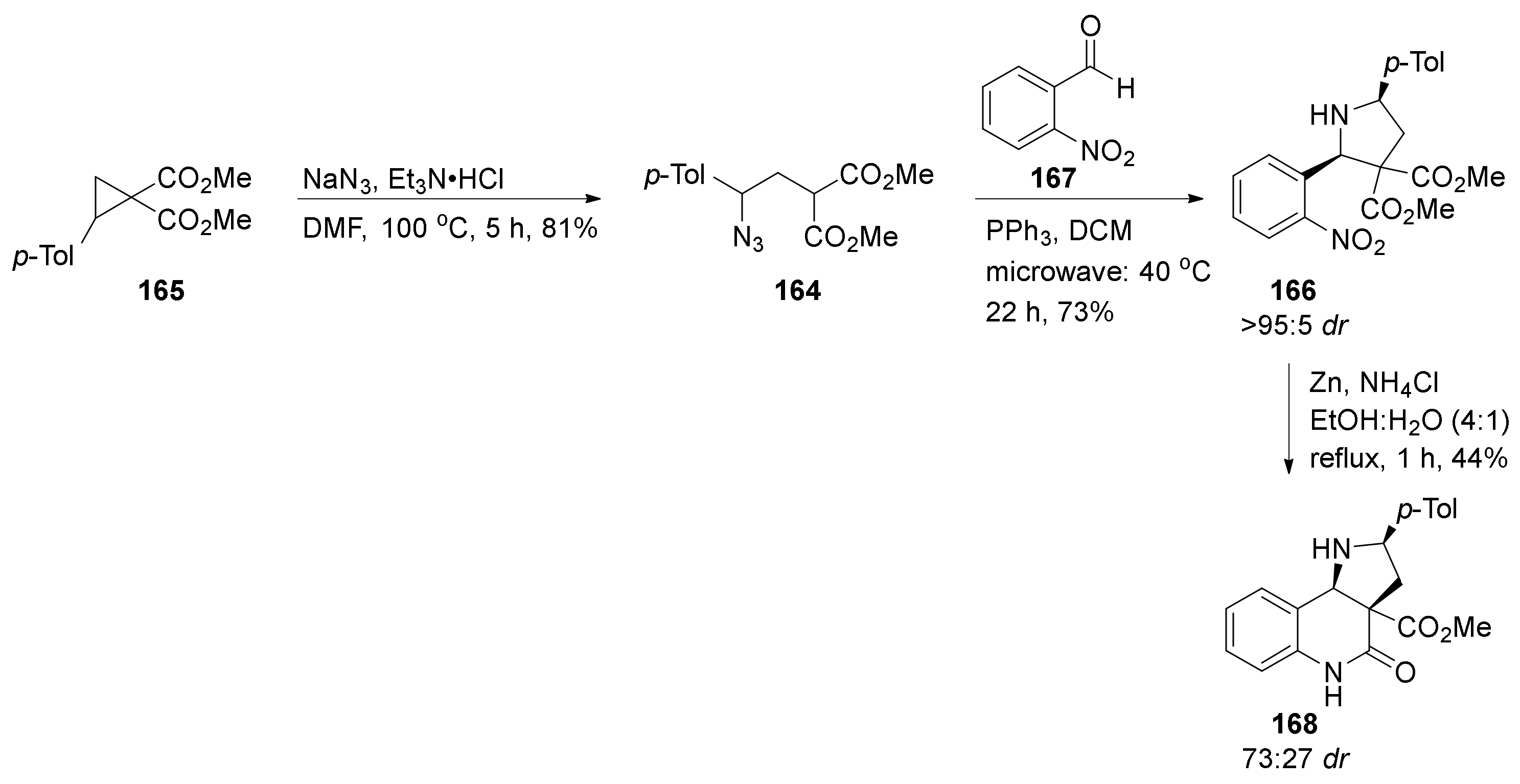{kind=link}
{kind=link}
{kind=link}
{kind=link}
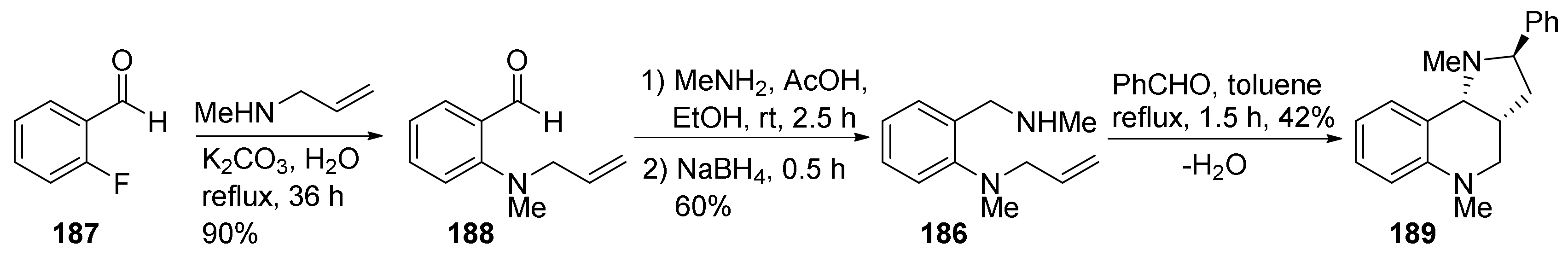{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
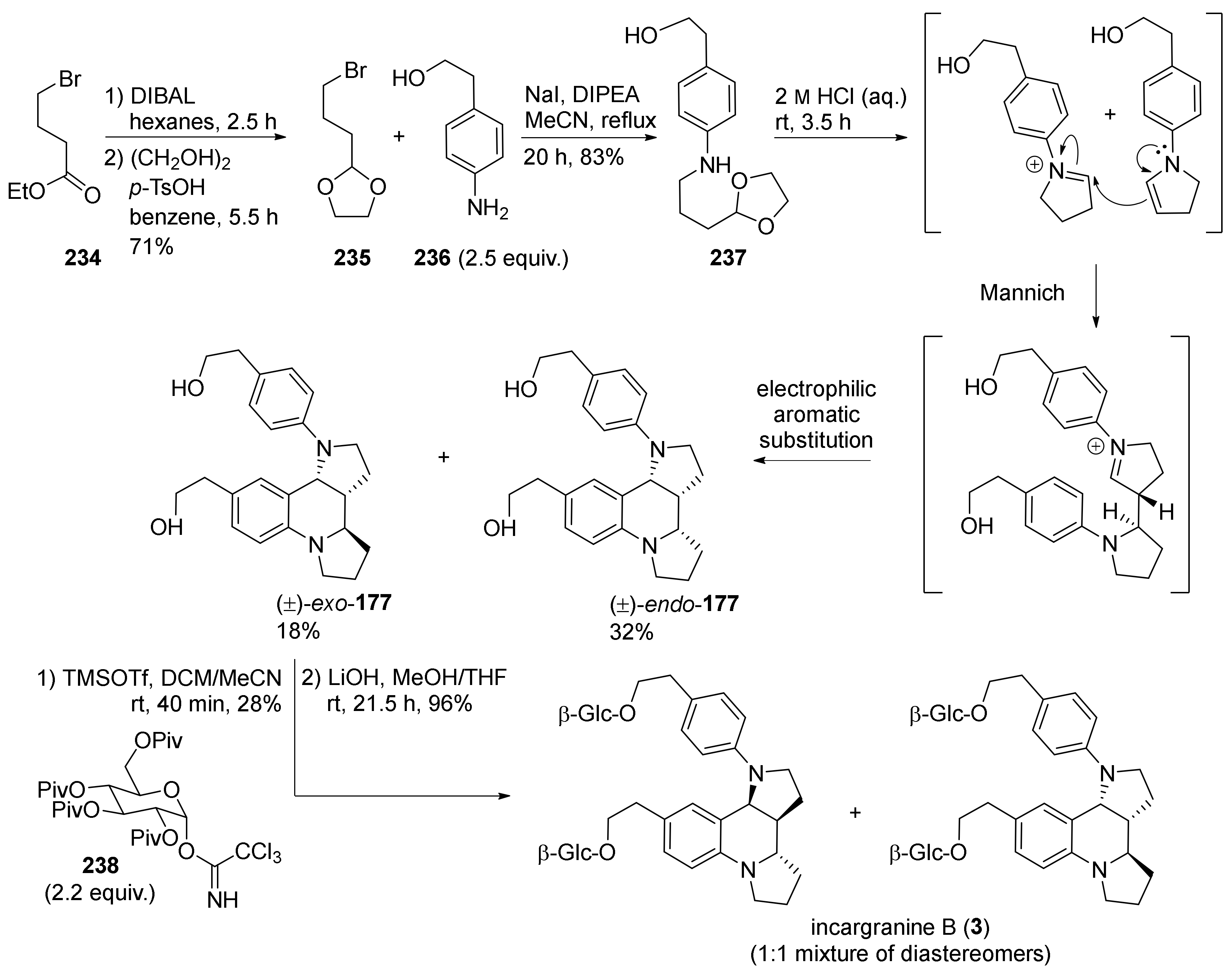{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Optical Rotation [αD] | Concentration (mg/ 10 cm3) | Reference |
|---|---|---|---|
| 1 (isolated) | +9.4 | 0.02 | [19] |
| (−)-1 | −108.0 | 0.09 | [30] |
| (+)-1 | +98.6 | 0.02 | [30] |
| 2 (isolated) | −8.5 | 0.01 | [19] |
| (−)-2 | −122.7 | 0.37 | [29] |
| (−)-2 | −118 | 0.3 | [32] |
| (−)-2 | −164.3 | 0.14 | [30] |
| (+)-2 | +165.5 | 0.11 | [30] |
| (−)-2 | −164.8 | 0.33 | [31] |
| 3 (isolated) | −12 | 0.275 | [25] |
| (±)-3 | −16.7 | 0.275 | [26] |
| 4 (isolated) | −72.9 | 0.10 | [27] |
| Entry | Reaction Conditions | R1 | R2 | Enamide | Yield | Endo:Exo | Reference |
|---|---|---|---|---|---|---|---|
| 1 | InCl3 (2 equiv.), MeCN, rt, 30 min | H | H | 73b | 41% | 1:1 | [58] |
| 2 | H | 2-NO2 | 73b | 50% | 2:1 | ||
| 3 | Zn(OTf)2 (10 mol%), DCM, rt | H | 2-OH | 73c | 42% | >20:1 | [59] |
| 4 | 76 (10 mol%), 5 Å MS, n-hexane −40 °C, 72 h | H | H | 73a | 94% | >20:1 | [60] |
| 5 | 77 (10 mol%), MeCN, rt, 0.5–1 h | H | H | 73a | 95% | 43:57 | [61] |
| 6 | 78 (10 mol%), MeCN, rt, 27 h | 4-OMe | H | 73d | 86% | 53:47 | [62] |
| 7 | Micellar-SO3H, H2O, 25 °C, 18 h | H | 4-O-DNA | 73d | >90% | NR | [63] |
| Entry | Starting Material | Conditions | Yield a | Endo:Exo | Reference |
|---|---|---|---|---|---|
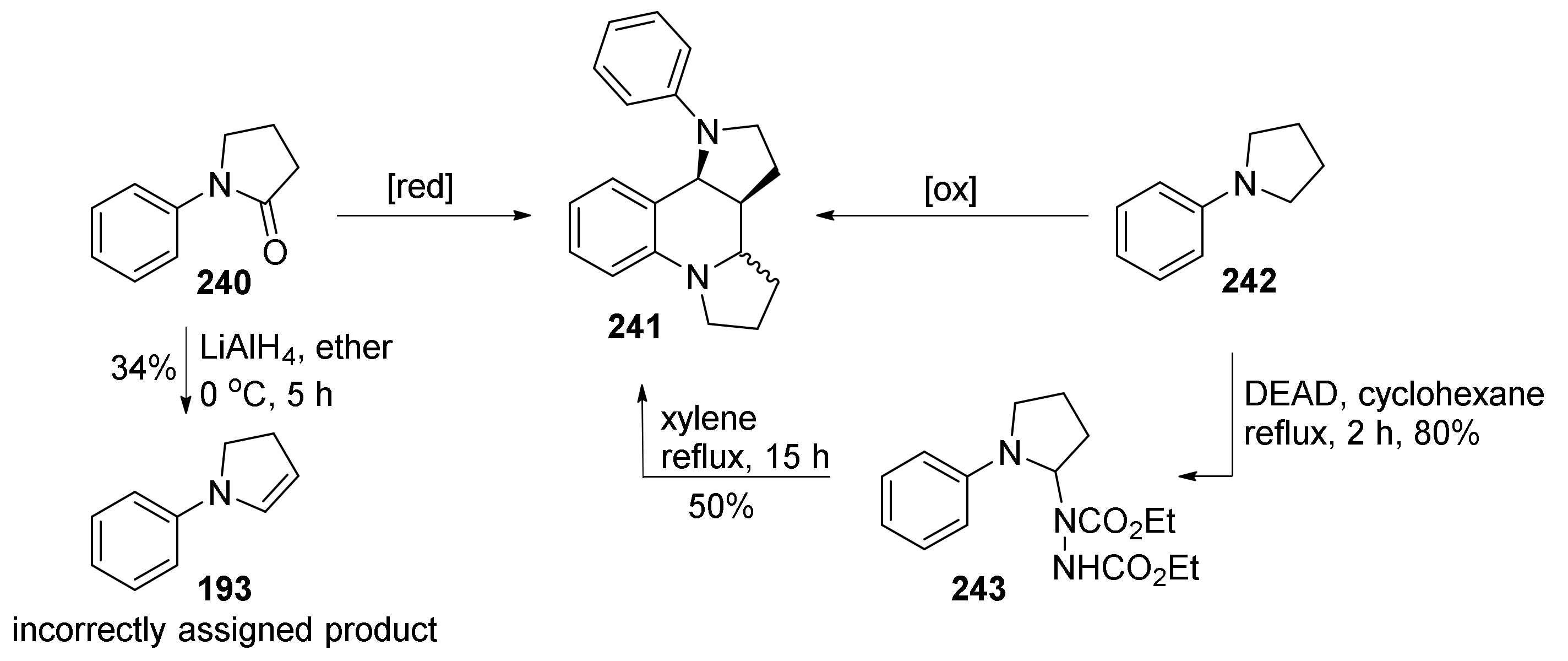
| 1 | 240 | LiAlH4, ether, rt, 3.5 h | 27% | 38:36 | [100] |
| 2 | 242 | O3, n-hexane, 0 °C | 41% | NR | [101] |
| 3 | 242 | 1) DEAD, cyclohexane, reflux, 2 h, 80% 2) xylene, reflux, 15 h | 50% | 28:22 | [101] |
| 4 | 242 | γ-irradiation, 17 d | 9% | ~1:1 | [102] |
| 5 | 242 | di-t-butylperoxide, 140 °C, 44 h | NR | ~1:1 | [102] |
| 6 | 242 | Dibenzoyl peroxide, MeCN, 0 °C, 9 h | 28% | 1:0 | [102] |
| 7 | 242 | t-BuOOH, NaOAc•3H2O, cyclohexane 70 °C, 24 h | 72% | 1:0 | [103] |
| 8 | 242 | Cu(OAc)2, O2, Et3N | 26% | 11:15 | [104] |
| 9 | 240 | PhMe2SiLi, −78 to −20 °C | 47% | 31:16 | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haarr, M.B.; Sydnes, M.O. Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules 2021, 26, 341. https://doi.org/10.3390/molecules26020341
Haarr MB, Sydnes MO. Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules. 2021; 26(2):341. https://doi.org/10.3390/molecules26020341
Chicago/Turabian StyleHaarr, Marianne B., and Magne O. Sydnes. 2021. "Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine" Molecules 26, no. 2: 341. https://doi.org/10.3390/molecules26020341
APA StyleHaarr, M. B., & Sydnes, M. O. (2021). Synthesis of the Hexahydropyrrolo-[3,2-c]-quinoline Core Structure and Strategies for Further Elaboration to Martinelline, Martinellic Acid, Incargranine B, and Seneciobipyrrolidine. Molecules, 26(2), 341. https://doi.org/10.3390/molecules26020341