
Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS




Abstract
:
1. Introduction
2. Results and Discussion
2.1. LC-MS
2.2. Method Validation
2.2.1. Quantitation of the Ratio of CSA and CSB in Reference Sample
2.2.2. Standard Curves
2.2.3. Accuracy and Precision
2.2.4. Stability
2.2.5. Robustness
2.2.6. Limit of Detection and Limit of Quantitation
2.2.7. Carry-Over
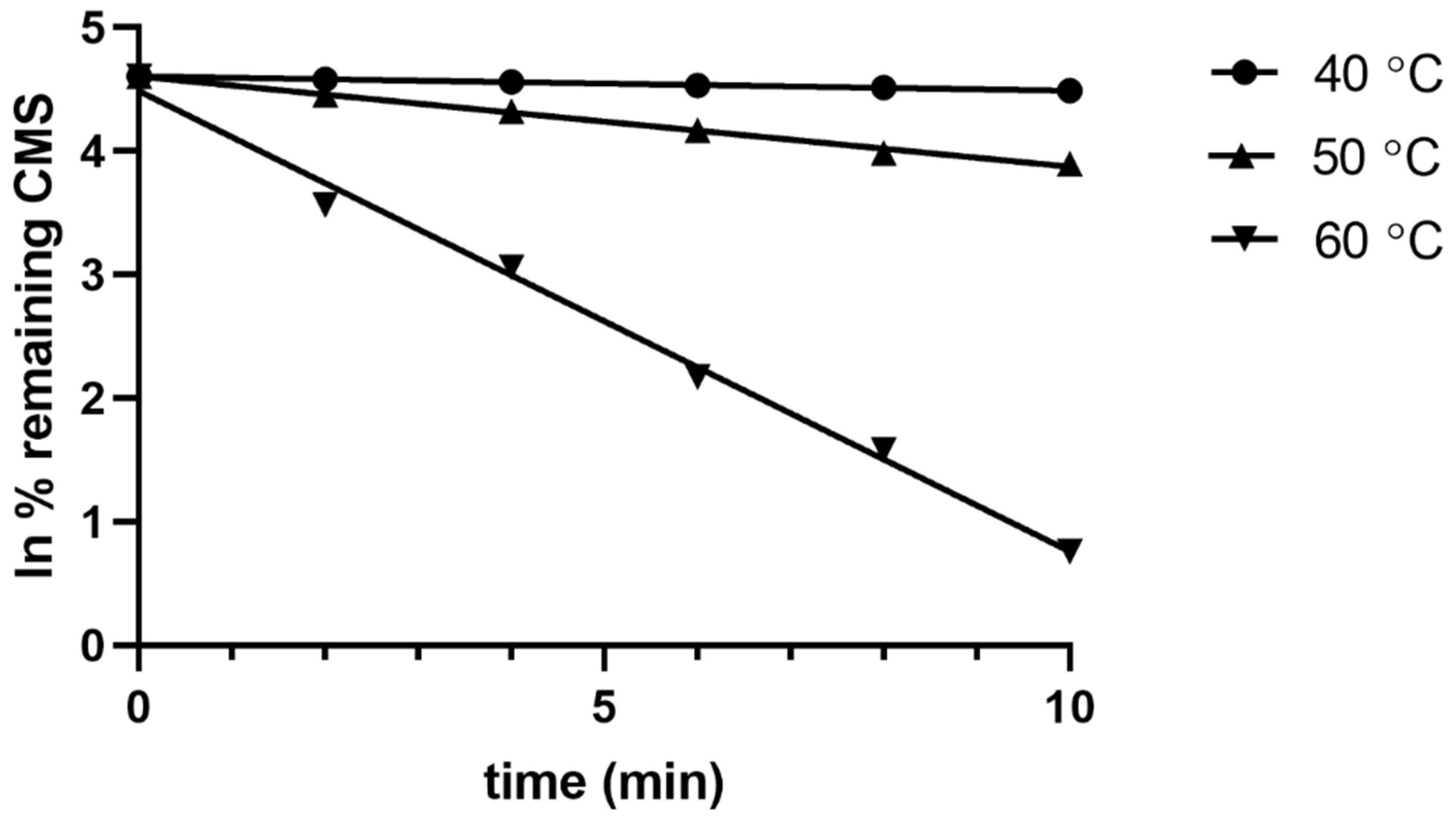
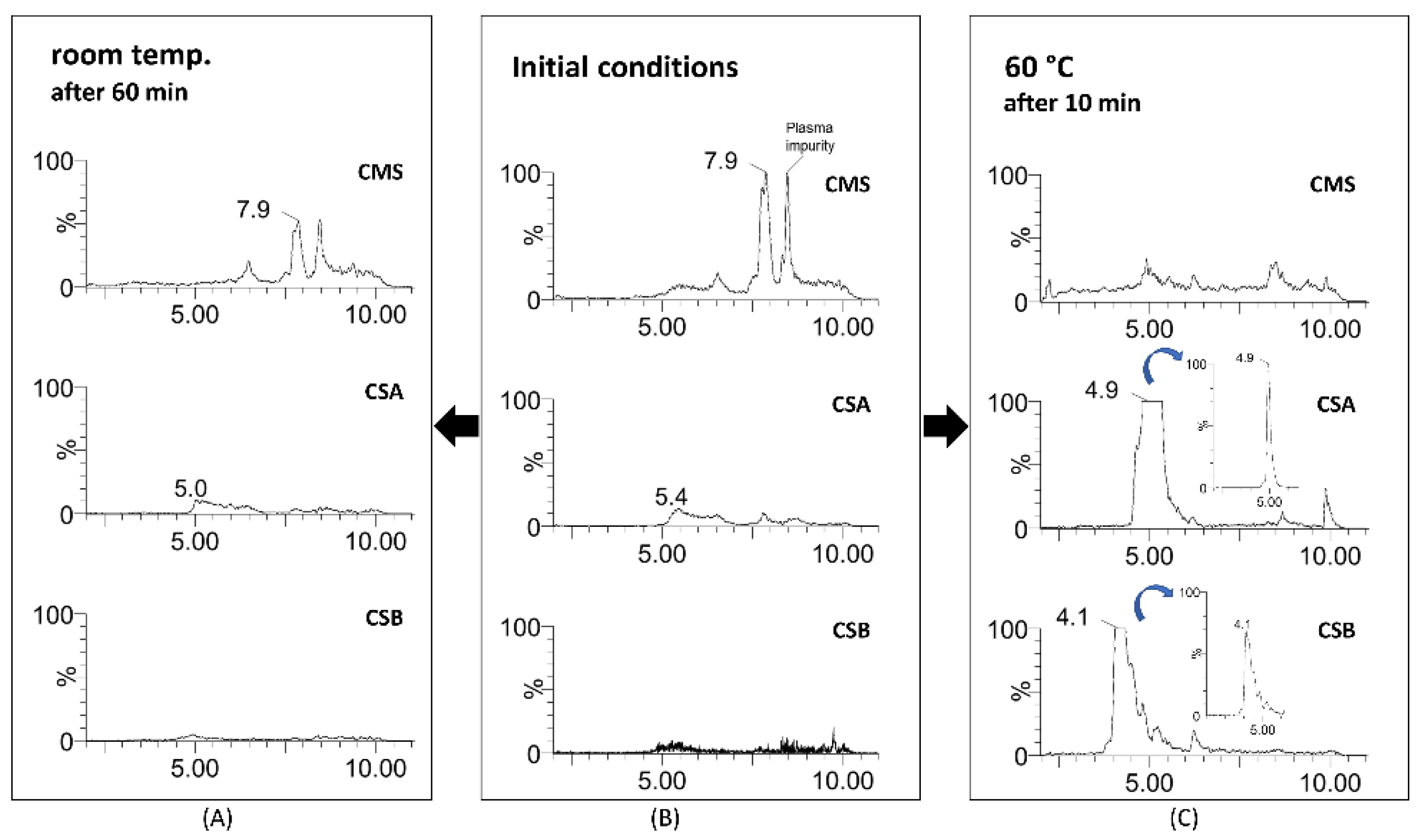
2.3. Acidic Hydrolysis of CMS
2.4. Application in Plasma Samples
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Liquid Chromatography
3.3. Mass Spectrometry
3.4. Preparation of Standard Solutions
3.5. Assay Validation
3.6. Acidic Hydrolysis of CMS
3.7. Application to Plasma Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kumar, K.H.; Gill, J.S.; Arora, S.; Khanna, S.P. Prevalence of multidrug-resistant, extensively drug-resistant, and pandrug-resistant Pseudomonas aeruginosa from a tertiary level Intensive Care Unit. J. Glob. Infect. Dis. 2016, 8, 155–159. [Google Scholar] [CrossRef]
- El-Sayed Ahmed, M.A.E.-G.; Zhong, L.-L.; Shen, C.; Yang, Y.; Doi, Y.; Tian, G.-B. Colistin and its role in the Era of antibiotic resistance: An extended review (2000–2019). Emerg. Microbes Infect. 2020, 9, 868–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Fujikawa, K. Studies on the Chemical Structure of Colistin. J. Biochem. 1964, 56, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Govaerts, C.; Rozenski, J.; Orwa, J.; Van Schepdael, A.; Hoogmartens, J. Mass spectrometric fragmentation of cyclic peptides belonging to the polymyxin and colistin antibiotics studied by ion trap and quadrupole/orthogonal-acceleration time-of-flight technology. Rapid Commun. Mass Spectrom. 2002, 16, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Brunel, J.-M.; Dubus, J.-C.; Reynaud-Gaubert, M.; Rolain, J.-M. Colistin: An update on the antibiotic of the 21st century. Expert Rev. Anti-Infect. Ther. 2012, 10, 917–934. [Google Scholar] [CrossRef] [PubMed]
- Berlana, D.; Llop, J.M.; Fort, E.; Badia, M.B.; Jódar, R. Use of colistin in the treatment of multiple-drug-resistant gram-negative infections. Am. J. Health-Syst. Pharm. 2005, 62, 39–47. [Google Scholar] [CrossRef]
- Dagla, I.; Tsarbopoulos, A.; Gikas, E. Design of Experiments guided Multivariate Calibration for the quantitation of injectable Colistimethate sodium by Ultra Performance Liquid Chromatography—High Resolution Mass Spectrometry. Talanta 2020, 220, 121406. [Google Scholar] [CrossRef]
- He, H.; Li, J.-C.; Nation, R.L.; Jacob, J.; Chen, G.; Lee, H.J.; Tsuji, B.T.; Thompson, P.E.; Roberts, K.; Velkov, T.; et al. Pharmacokinetics of four different brands of colistimethate and formed colistin in rats. J. Antimicrob. Chemother. 2013, 68, 2311–2317. [Google Scholar] [CrossRef] [Green Version]
- Sivanesan, S.; Roberts, K.; Wang, J.; Chea, S.-E.; Thompson, P.E.; Li, J.; Nation, R.L.; Velkov, T. Pharmacokinetics of the Individual Major Components of Polymyxin B and Colistin in Rats. J. Nat. Prod. 2017, 80, 225–229. [Google Scholar] [CrossRef]
- Dagla, I.; Karkoula, E.; Baira, E.; Tsarbopoulos, A.; Gikas, E. Analytical methodologies used for the determination of colistin in biological fluids. Is it still a challenge? J. Pharm. Biomed. Anal. 2019, 164, 777–788. [Google Scholar] [CrossRef]
- Zabidi, M.S.; Abu Bakar, R.; Musa, N.; Yusuf, W.N.W. Analytical methodologies for measuring colistin levels in pharmacokinetic studies. J. Liq. Chromatogr. Relat. Technol. 2020, 43, 671–686. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Coulthard, K.; Johnson, D.W. A simple method for the assay of colistin in human plasma, using pre-column derivatization with 9-fluorenylmethyl chloroformate in solid-phase extraction cartridges and re-versed-phase high-performance liquid chromatography. J. Chromatogr. B 2001, 761, 167–175. [Google Scholar] [CrossRef]
- Reed, M.D.; Stern, R.C.; Ms, M.A.O.; Blumer, J.L. The Pharmacokinetics of Colistin in Patients with Cystic Fibrosis. J. Clin. Pharmacol. 2001, 41, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Li, J.; Nation, R.L.; Nicolazzo, J.A. Brain Penetration of Colistin in Mice Assessed by a Novel High-Performance Liquid Chromatographic Technique. Antimicrob. Agents Chemother. 2009, 53, 4247–4251. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Wu, X.-J.; Fan, Y.-X.; Guo, B.; Zhang, J. Development and validation of a UHPLC–MS/MS assay for colistin methanesulphonate (CMS) and colistin in human plasma and urine using weak-cation exchange solid-phase extraction. J. Pharm. Biomed. Anal. 2016, 124, 303–308. [Google Scholar] [CrossRef]
- Bihan, K.; Lu, Q.; Enjalbert, M.; Apparuit, M.; Langeron, O.; Rouby, J.-J.; Funck-Brentano, C.; Zahr, N. Determination of Colistin and Colistimethate Levels in Human Plasma and Urine by High-Performance Liquid Chromatography–Tandem Mass Spec-trometry. Ther. Drug Monit. 2016, 38, 796–803. Available online: https://insights.ovid.com/pubmed?pmid=27684296 (accessed on 13 July 2017). [CrossRef]
- Dotsikas, Y.; Markopoulou, C.K.; Koundourellis, J.E.; Loukas, Y.L. Validation of a novel LC-MS/MS method for the quantitation of colistin A and B in human plasma. J. Sep. Sci. 2010, 34, 37–45. [Google Scholar] [CrossRef]
- Gikas, E.; Bazoti, F.N.; Katsimardou, M.; Anagnostopoulos, D.; Papanikolaou, K.; Inglezos, I.; Skoutelis, A.; Daikos, G.L.; Tsarbopoulos, A. Determination of colistin A and colistin B in human plasma by UPLC–ESI high resolution tandem MS: Application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2013, 83, 228–236. [Google Scholar] [CrossRef]
- Li, J.; Milne, R.W.; Nation, R.L.; Turnidge, J.D.; Coulthard, K.; Valentine, J. Simple Method for Assaying Colistin Methanesulfonate in Plasma and Urine Using High-Performance Liquid Chromatography. Antimicrob. Agents Chemother. 2002, 46, 3304–3307. [Google Scholar] [CrossRef] [Green Version]
- Jansson, B.; Karvanen, M.; Cars, O.; Plachouras, D.; Friberg, L.E. Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J. Pharm. Biomed. Anal. 2009, 49, 760–767. [Google Scholar] [CrossRef]
- Gobin, P.; Lemaître, F.; Marchand, S.; Couet, W.; Olivier, J.-C. Assay of Colistin and Colistin Methanesulfonate in Plasma and Urine by Liquid Chromatography-Tandem Mass Spectrometry. Antimicrob. Agents Chemother. 2010, 54, 1941–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, T.; Tissot, F.; Gardiol, C.; Corti, N.; Wehrli, S.; Guidi, M.; Wagner, A.; Buclin, T.; Couet, W.; Marchetti, O.; et al. High-throughput hydrophilic interaction chromatography coupled to tandem mass spectrometry for the optimized quantification of the anti-Gram-negatives antibiotic colistin A/B and its pro-drug colistimethate. J. Chromatogr. A 2014, 1369, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Marchand, S.; Lamarche, I.; Gobin, P.; Couet, W. Dose-ranging pharmacokinetics of colistin methanesulphonate (CMS) and colistin in rats following single intravenous CMS doses. J. Antimicrob. Chemother. 2010, 65, 1753–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, M.; Gregoire, N.; Megarbane, B.; Gobin, P.; Balayn, D.; Marchand, S.; Mimoz, O.; Couet, W. Population Pharmacokinetics of Colistin Methanesulfonate and Colistin in Critically Ill Patients with Acute Renal Failure Requiring Intermittent Hemodialysis. Antimicrob. Agents Chemother. 2016, 60, 1788–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corcione, S.; Baietto, L.; Malvasio, V.; Stella, M.; Di Perri, G.; D’Avolio, A.; De Rosa, F.G. Pharmacokinetics of colistin methanesulfonate (CMS) in burn patients. J. Antimicrob. Chemother. 2016, 72, 319–321. [Google Scholar] [CrossRef] [Green Version]
- EMA. European Medicines Agency Completes Review of Polymyxin-Based Medicines; European Medicines Agency: London, UK, 2015; Volume 44, pp. 1–4. Available online: http://www.ema.europa.eu/docs/en_GB/document_library/Referrals_document/Polymyxin_31/WC500176333.pdf (accessed on 16 December 2014).
- Tran, T.B.; Velkov, T.; Nation, R.L.; Forrest, A.; Tsuji, B.T.; Bergen, P.J.; Li, J. Pharmacokinetics/pharmacodynamics of colistin and polymyxin B: Are we there yet? Int. J. Antimicrob. Agents 2016, 48, 592–597. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.F.; Karaiskos, I.; Plachouras, D.; Karvanen, M.; Pontikis, K.; Jansson, B.; Papadomichelakis, E.; Antoniadou, A.; Giamarellou, H.; Armaganidis, A.; et al. Application of a Loading Dose of Colistin Methanesulfonate in Critically Ill Patients: Population Pharmacokinetics, Protein Binding, and Prediction of Bacterial Kill. Antimicrob. Agents Chemother. 2012, 56, 4241–4249. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Authors | Acid | Concentration (M) | Reaction Time (min) | Temp (°C) |
|---|---|---|---|---|
| Li et al. [19] | sulfuric | 0.08–0.1 | 10 | room |
| Jansson et al. [20] | sulfuric | 1 | 15–20 | room |
| Gobin et al. [21] | sulfuric | 0.5 | 60 | room |
| Gikas et al. [18] | sulfuric | 0.5 | 60 | room |
| Mercier et al. [22] | sulfuric | 1 | 30 | room |
| Zhao et al. [15] | sulfuric | 0.5 | 15 | room |
| Bihan et al. [16] | sulfuric | 1 | 60 | room |
| Analyte (μg mL−1) | Accuracy * (n = 3, %Error) | Repeatability * (n = 3, %RSD) | Intermediate Precision (%RSD) | Autosampler Stability (n = 3, %RSD) | Benchtop Stability (n = 3, %RSD) | Robustness (n = 3, %RSD) |
|---|---|---|---|---|---|---|
| CSA | ||||||
| 1.81 | <6.15 | <3.71 | 1.86 | 2.78 | 3.45 | <4.45 |
| 2.41 | <1.41 | <2.55 | 1.28 | |||
| 3.61 | <11.05 | <5.61 | 9.07 | 1.15 | 3.12 | <3.12 |
| 4.82 | <4.71 | <1.73 | 4.23 | |||
| 6.02 | <2.26 | <5.14 | 3.44 | |||
| 6.62 | <3.21 | <5.42 | 3.58 | 1.96 | 2.95 | <2.14 |
| CSB | ||||||
| 4.19 | <1.71 | <5.71 | 5.16 | 2.53 | 3.56 | <4.27 |
| 5.59 | <1.68 | <3.80 | 2.48 | |||
| 8.39 | <2.02 | <4.39 | 5.59 | 1.84 | 3.78 | <2.95 |
| 11.18 | <5.33 | <2.55 | 2.13 | |||
| 13.98 | <5.07 | <3.82 | 2.93 | |||
| 15.38 | <4.61 | <4.85 | 3.71 | 1.69 | 3.17 | <1.97 |
| CMS | ||||||
| 2 | <11.01 | <5.41 | 5.96 | |||
| 6 | <13.21 | <5.71 | 5.74 | 2.35 | 7.54 | <4.12 |
| 8 | <7.91 | <4.27 | 5.06 | |||
| 12 | <4.79 | <4.54 | 5.98 | 1.87 | 5.58 | <4.05 |
| 16 | <4.71 | <3.84 | 2.30 | |||
| 20 | <1.53 | <1.85 | 1.29 | |||
| 22 | <0.40 | <0.53 | 0.37 | 2.56 | 5.87 | <3.94 |
| LOD (μg mL−1) | LOQ (μg mL−1) | |||||
| CSA | 0.29 | 0.88 | ||||
| CSB | 1.38 | 4.18 | ||||
| CMS | 1.92 | 5.83 |
| Temp (°C) | kt (min−1) | t1/2 (0.693/K) (min) |
|---|---|---|
| 20 * | 0.0003 | 2310.00 |
| 40 ** | 0.0146 | 47.47 |
| 50 ** | 0.0709 | 9.78 |
| 60 ** | 0.4485 | 1.55 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagla, I.; Tsarbopoulos, A.; Gikas, E. Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS. Molecules 2021, 26, 447. https://doi.org/10.3390/molecules26020447
Dagla I, Tsarbopoulos A, Gikas E. Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS. Molecules. 2021; 26(2):447. https://doi.org/10.3390/molecules26020447
Chicago/Turabian StyleDagla, Ioanna, Anthony Tsarbopoulos, and Evagelos Gikas. 2021. "Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS" Molecules 26, no. 2: 447. https://doi.org/10.3390/molecules26020447
APA StyleDagla, I., Tsarbopoulos, A., & Gikas, E. (2021). Colistimethate Acidic Hydrolysis Revisited: Arrhenius Equation Modeling Using UPLC-QToF MS. Molecules, 26(2), 447. https://doi.org/10.3390/molecules26020447