
New Antineoplastic Naphthohydroquinones Attached to Labdane and Rearranged Diterpene Skeletons

 ,
,  , and
, and 
Abstract
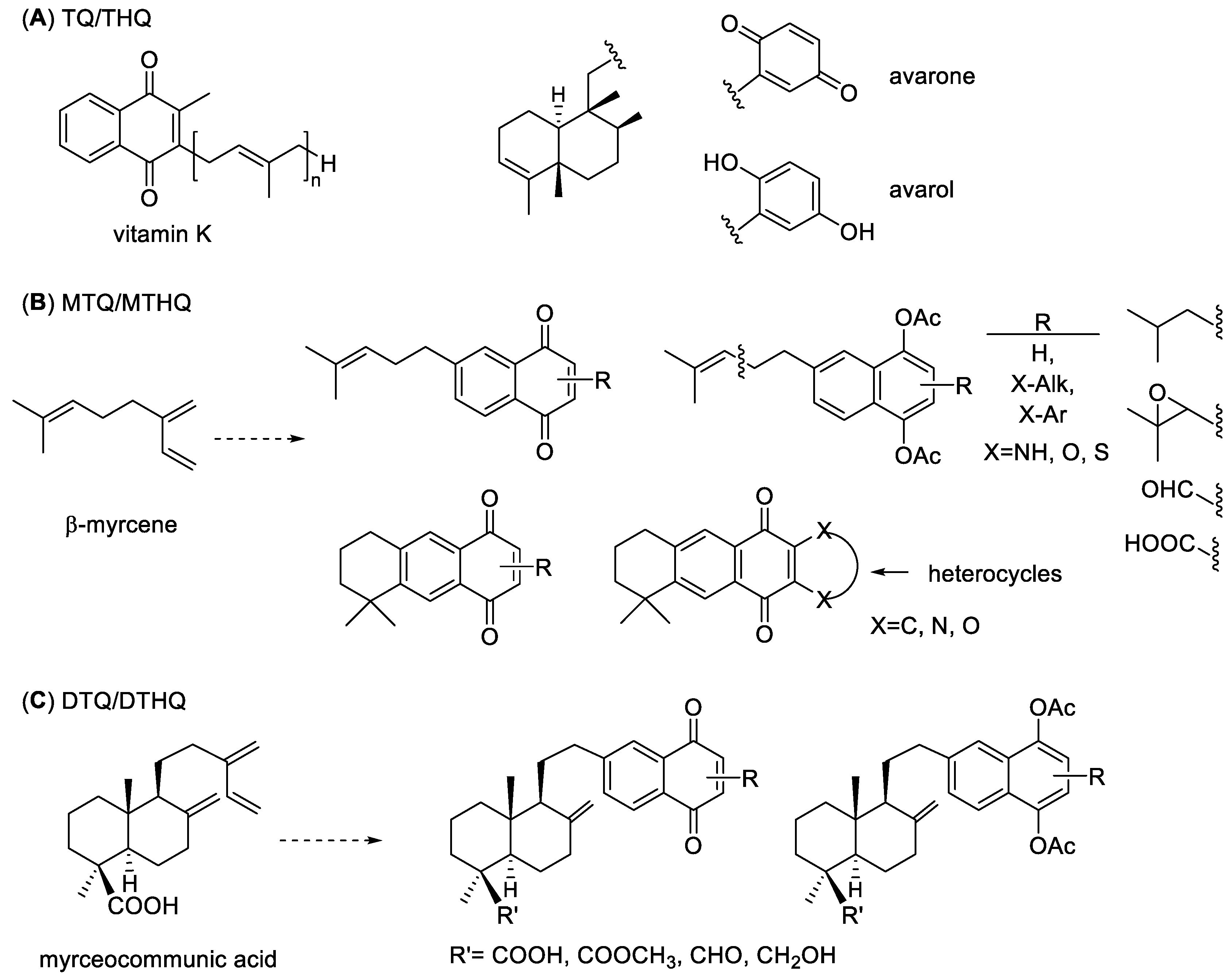
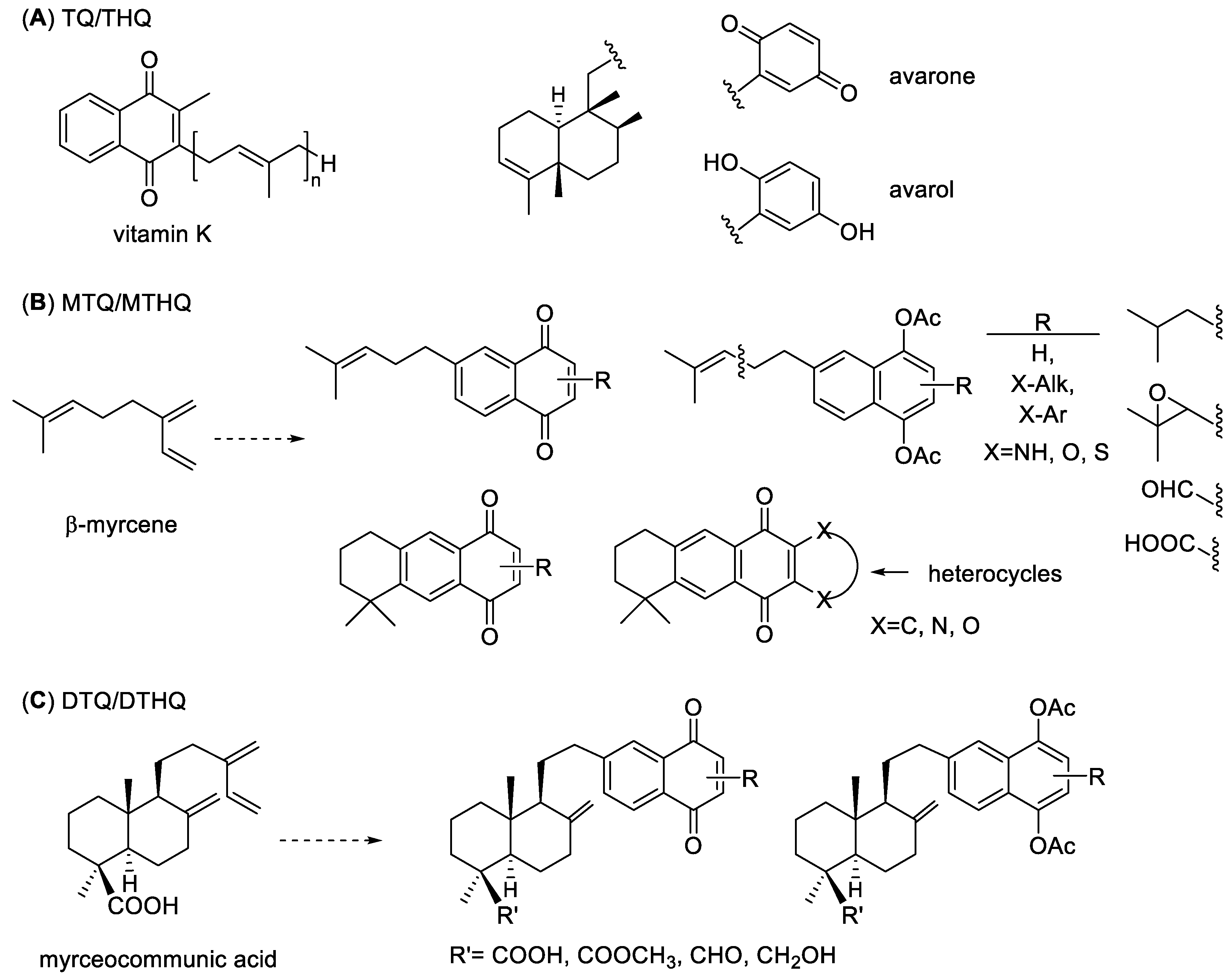
:1. Introduction
2. Results and Discussion
2.1. Chemistry
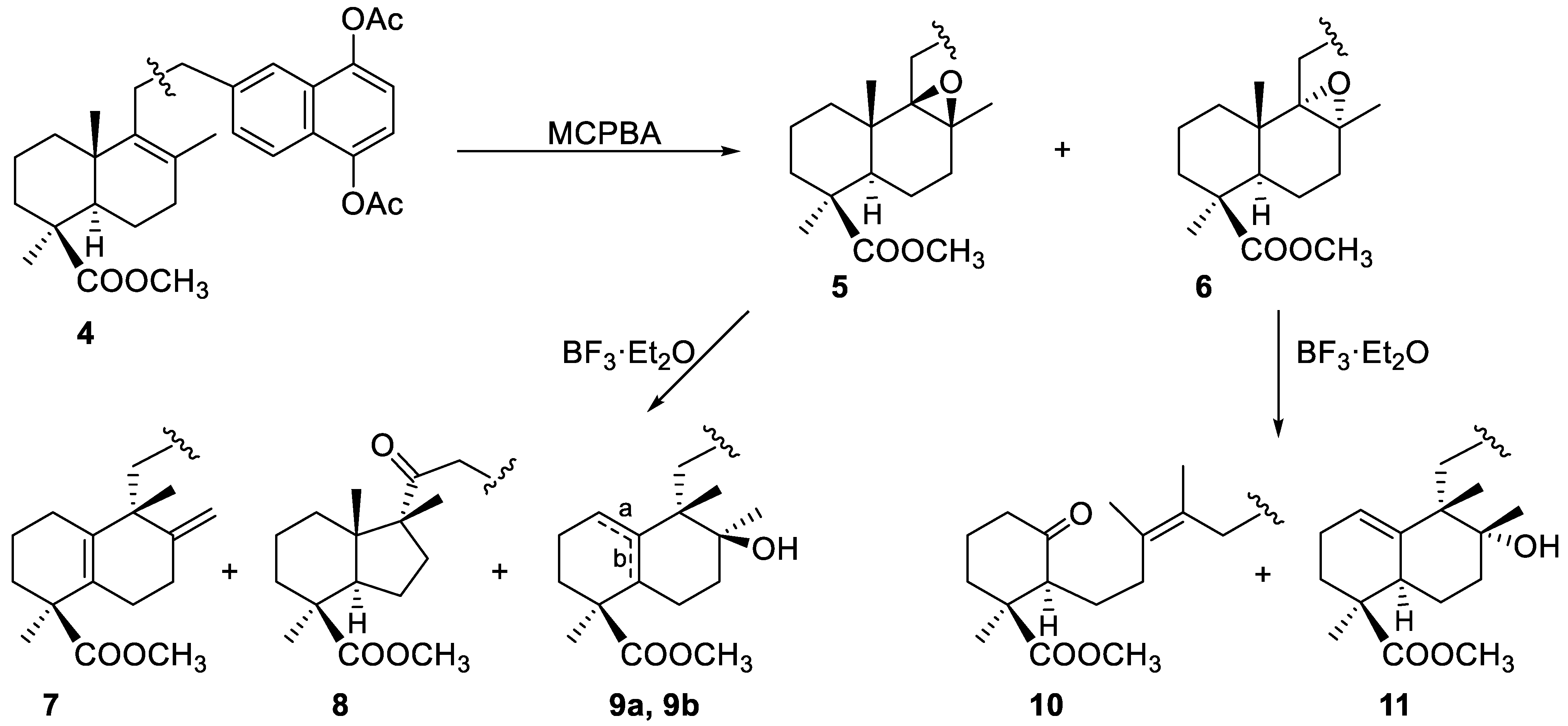
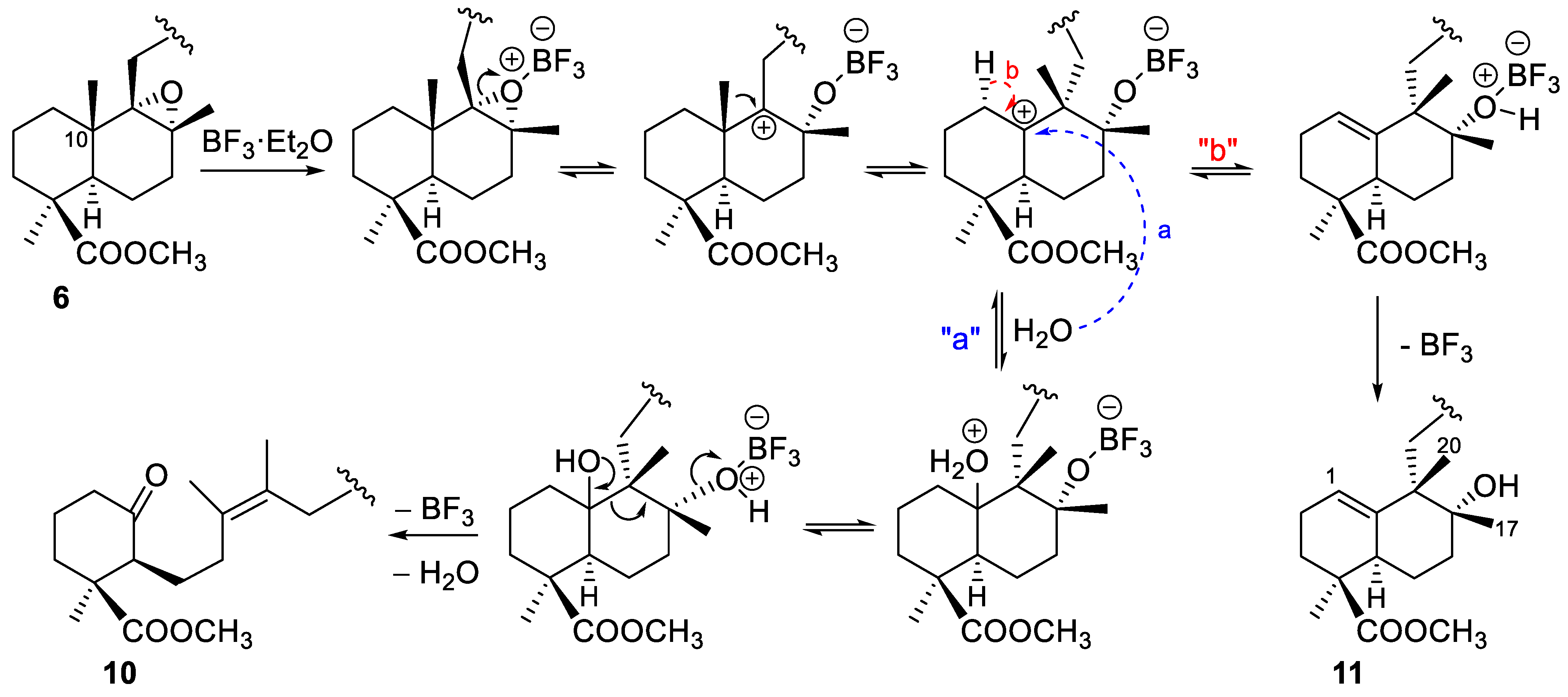
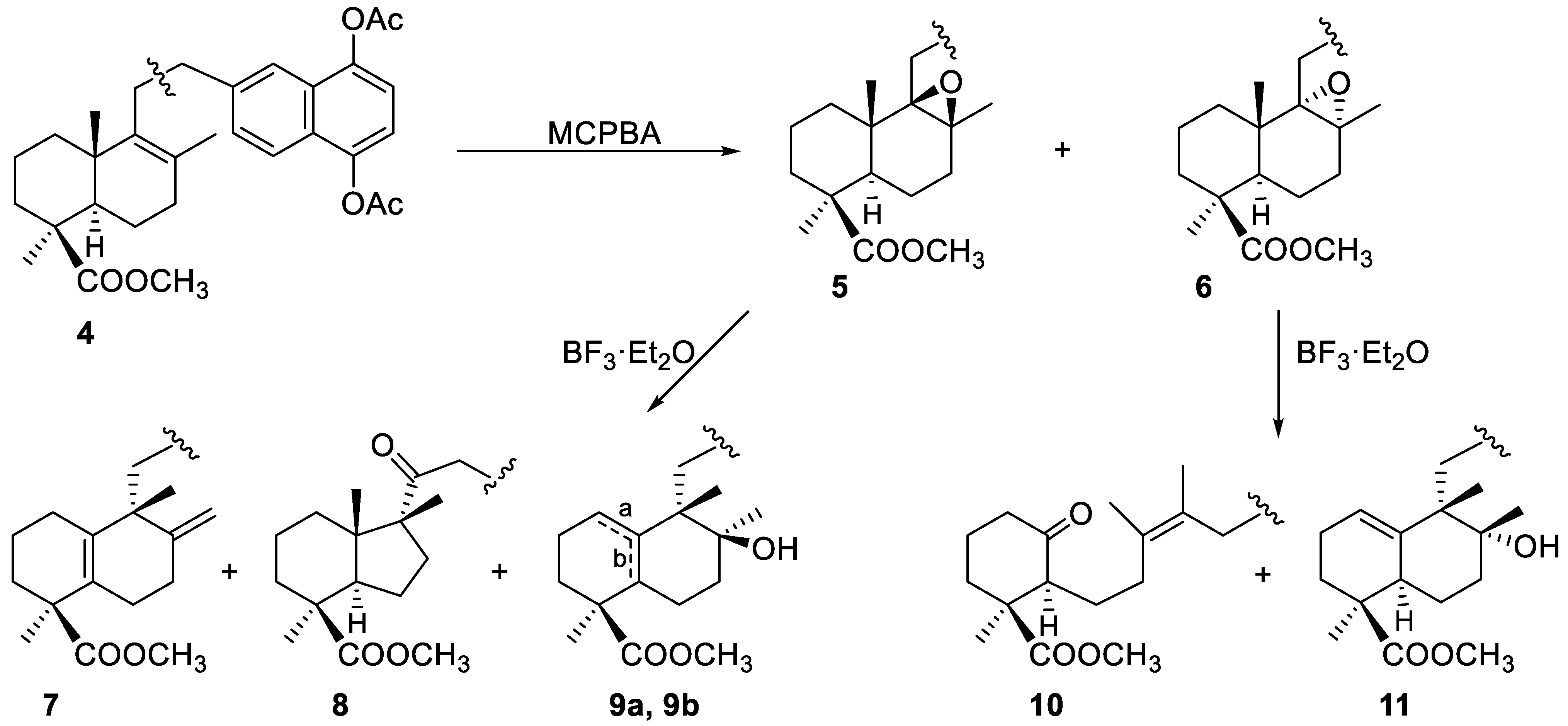
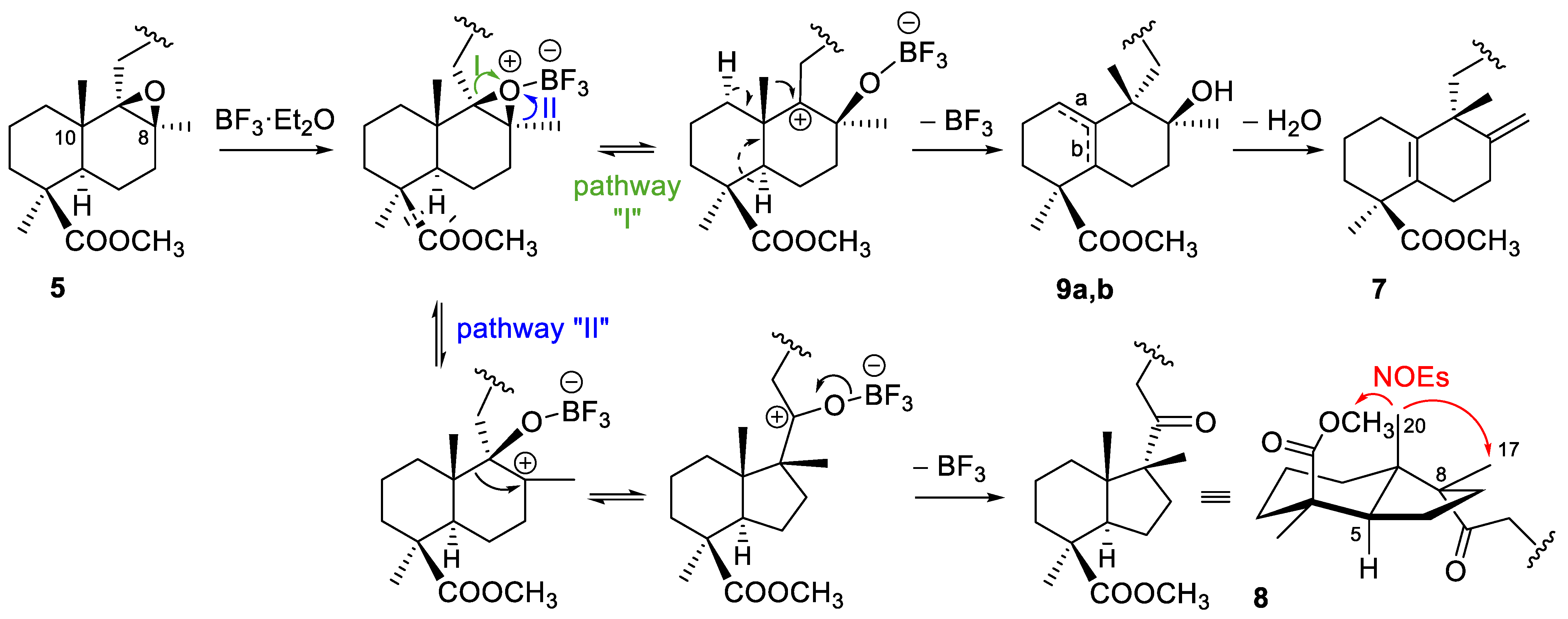
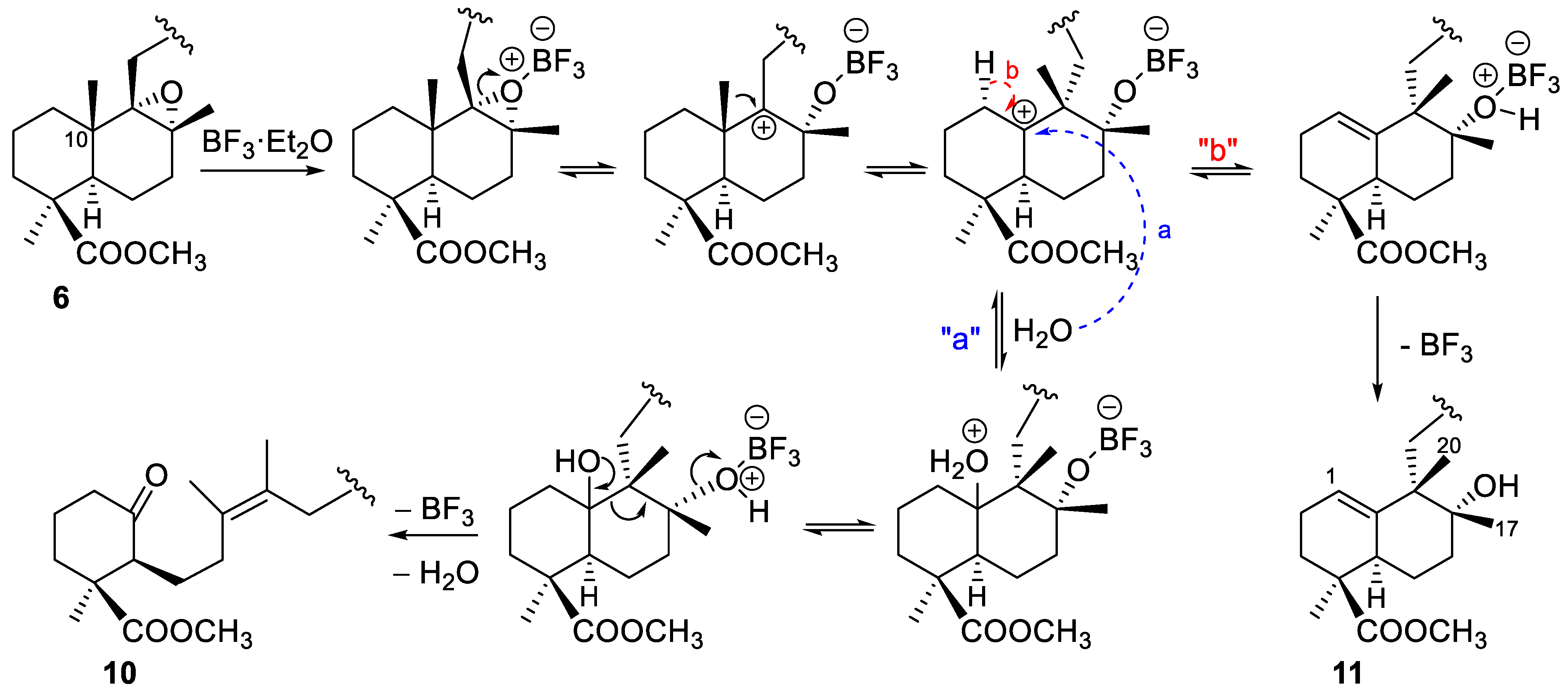
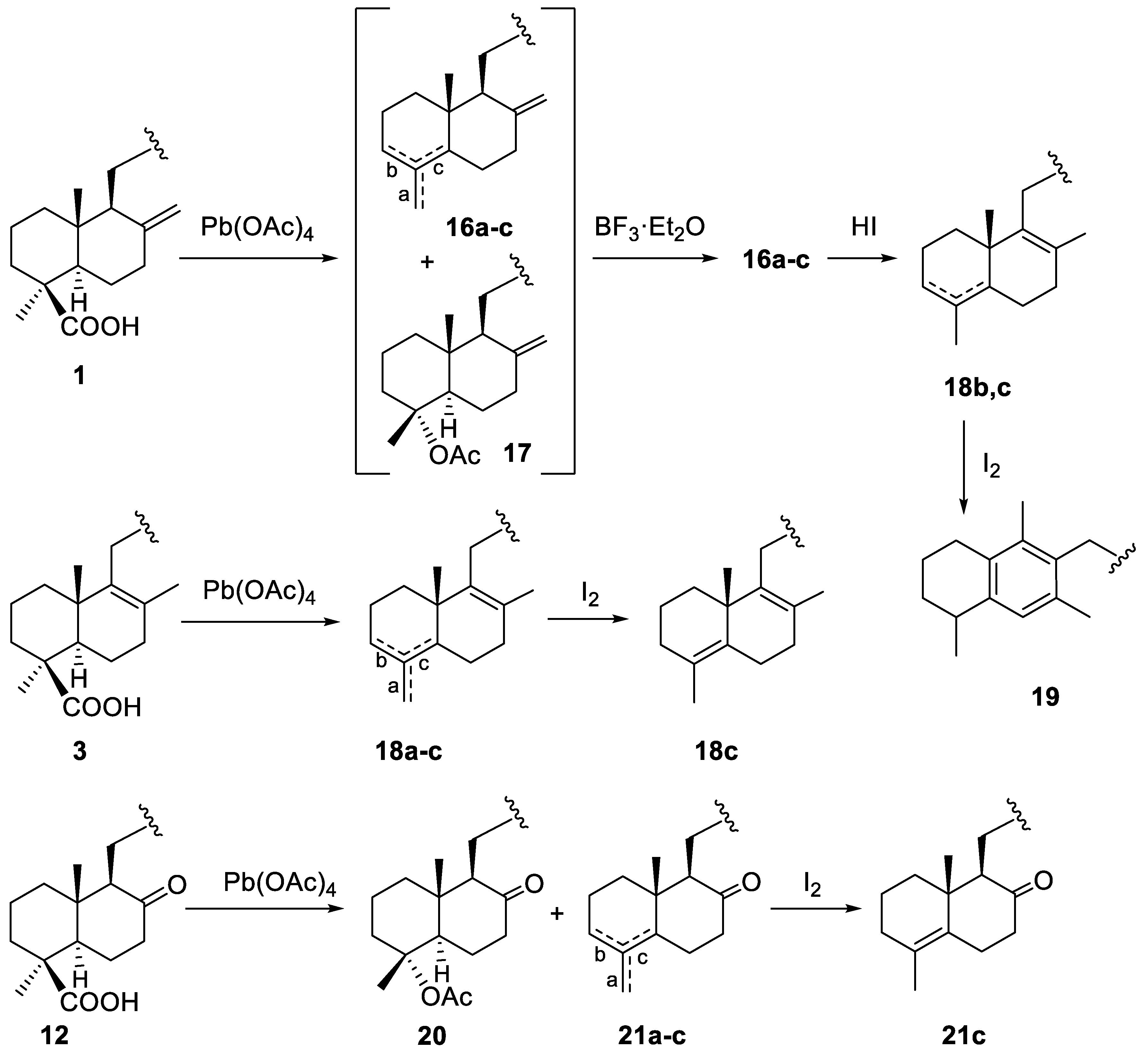
2.1.1. Epoxidation and Rearrangement Reactions
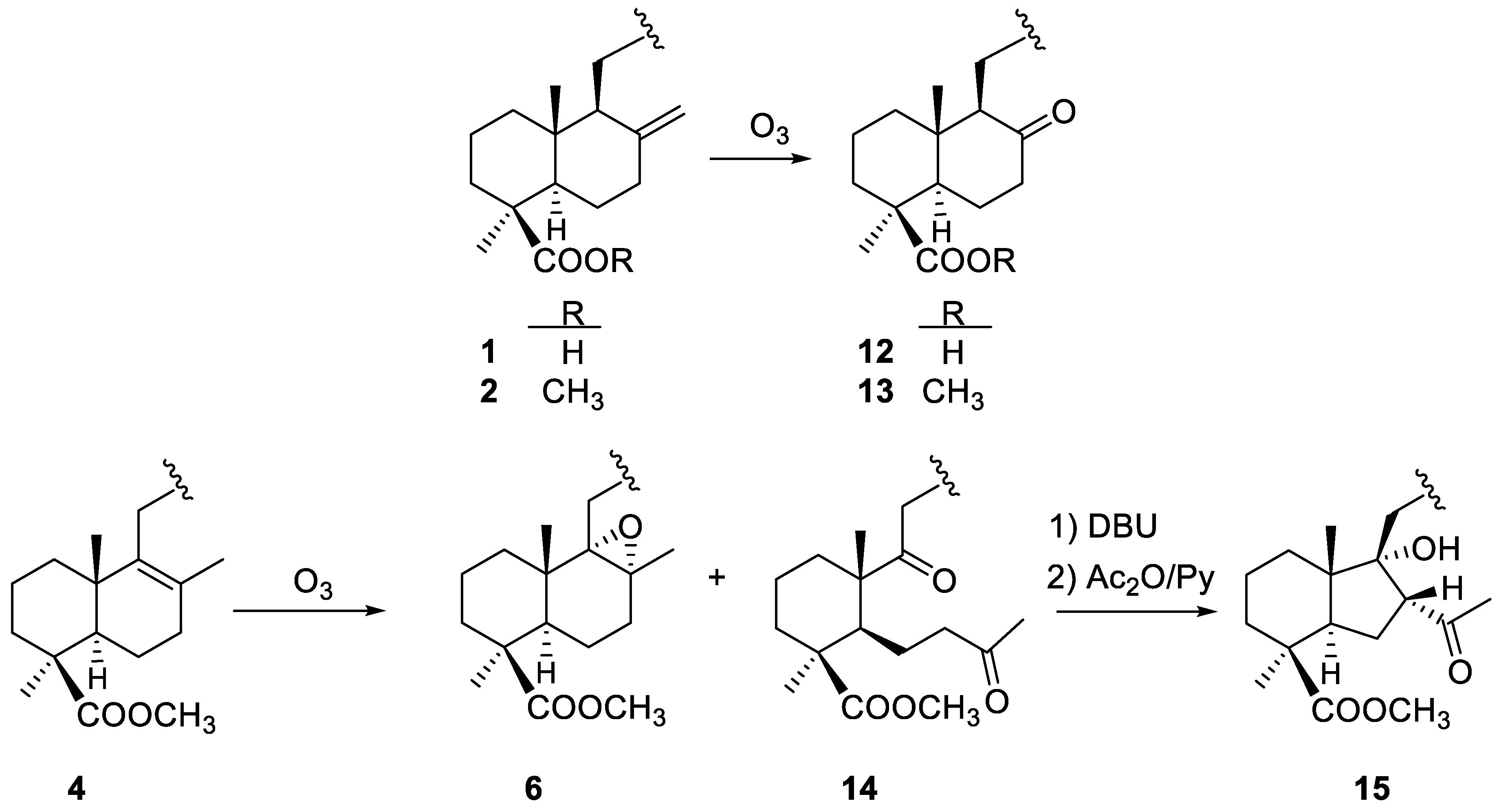
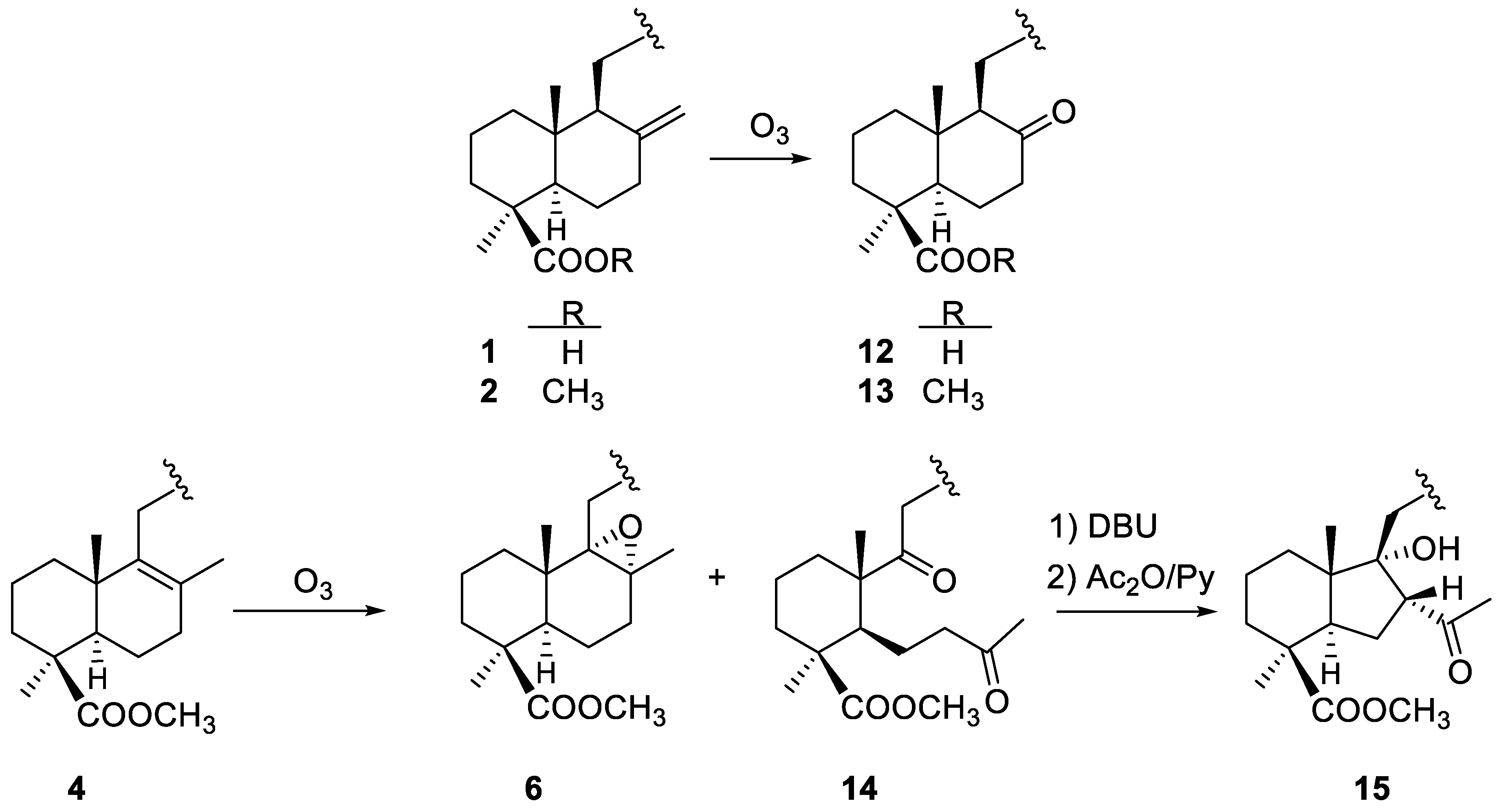
2.1.2. Ozonolysis
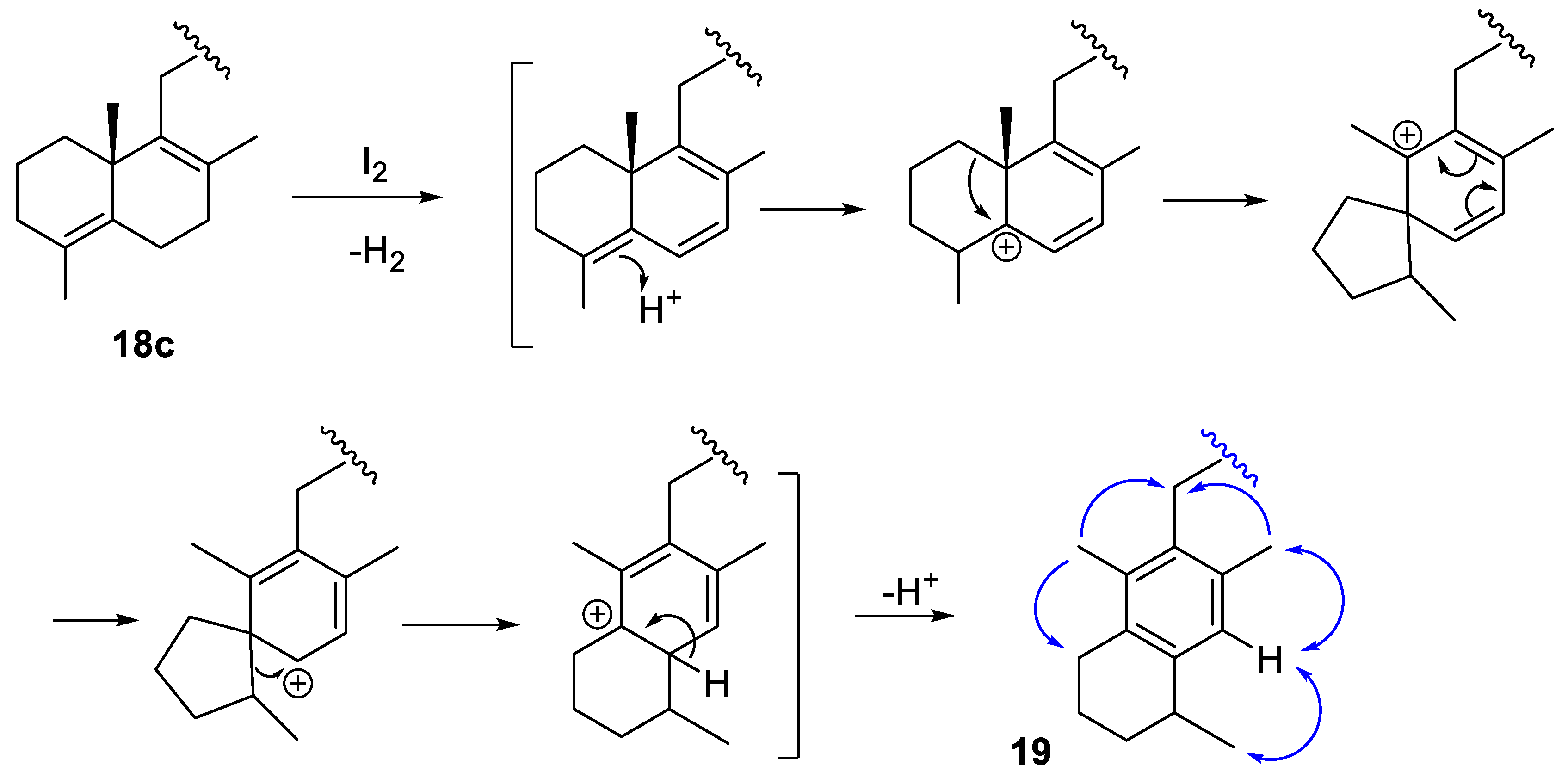
2.1.3. Decarboxylation Reactions
2.2. Biological Evaluation
3. Materials and Methods
3.1. Chemistry
3.2. Biological Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Menna, M.; Imperatore, C.; D’Aniello, F.; Aiello, A. Meroterpenes from marine invertebrates: Structures, occurrence and ecological implications. Mar. Drugs 2013, 11, 1602–1643. [Google Scholar] [CrossRef] [Green Version]
- García, P.A.; Hernández, A.P.; San Feliciano, A.; Castro, M.A. Bioactive prenyl- and terpenyl-quinones/hydroquinones of marine origin. Mar. Drugs 2018, 16, 292. [Google Scholar]
- Sunassee, S.N.; Davies-Coleman, M.T. Cytotoxic and antioxidant marine prenylated quinones and hydroquinones. Nat. Prod. Rep. 2012, 29, 513–535. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Gordaliza, M.; Castro, A.; Mahiques, M.M.; San Feliciano, A.; Garcia-Gravalos, M.D. Further antineoplastic terpenylquinones and terpenylhydroquinones. Bioorg. Med. Chem. 1998, 6, 31–41. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Reinoso, P.; Miguel del Corral, J.M.; Castro, M.A.; Gordaliza, M.; Gupta, M.P.; Solis, P.; San Feliciano, A. Cytotoxic-antineoplastic activity of hydroquinone derivatives. Eur. J. Med. Chem. 2002, 37, 177–182. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Ojeda, C.; Escobar, J.; Gallardo, C.; Miguel del Corral, J.M.; Castro, A.; Cuevas, C.; San Feliciano, A. Synthesis, characterisation and cytotoxicity of chloro derivatives of prenylnaphthohydroquinone. Bioorg. Med. Chem. 2005, 13, 3841–3846. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Castro, M.A.; Gordaliza, M.; Martin, M.L.; Gualberto, S.A.; Gamito, A.M.; Cuevas, C.; San Feliciano, A. Synthesis and cytotoxicity of new aminoterpenylquinones. Bioorg. Med. Chem. 2005, 13, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Gordaliza, M.; Garcia, P.A.; Gamito, A.M.; Gualberto, S.A.; Batista, R.; San Feliciano, A. A novel synthetic route to cytotoxic 1,4-anthraquinones from 1,4-benzoquinones. Synthesis 2005, 19, 3202–3208. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Ojeda, C.; Miguel del Corral, J.M.; Castro, M.A.; Mollinedo, F.; San Feliciano, A. Synthesis and evaluation as antitumor agents of 1,4-naphthohydroquinone derivatives conjugated with amino acids and purines. Arch. Pharm. 2013, 346, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Castro, M.A.; Gamito, A.M.; Tangarife-Castano, V.; Roa-Linares, V.; Miguel del Corral, J.M.; Mesa-Arango, A.C.; Betancur-Galvis, L.; Francesch, A.M.; San Feliciano, A. New 1,4-anthracenedione derivatives with fused heterocyclic rings: Synthesis and biological evaluation. RSC Adv. 2015, 5, 1244–1261. [Google Scholar] [CrossRef] [Green Version]
- Miguel del Corral, J.M.; Gordaliza, M.; Castro, M.A.; Mahiques, M.M.; Chamorro, P.; Molinari, A.; Garcia-Gravalos, M.D.; Broughton, H.B.; San Feliciano, A. New selective cytotoxic diterpenylquinones and diterpenylhydroquinones. J. Med. Chem. 2001, 44, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Miguel del Corral, J.M.; Castro, M.A.; Rodriguez, M.L.; Chamorro, P.; Cuevas, C.; San Feliciano, A. New cytotoxic diterpenylnaphthohydroquinone derivatives obtained from a natural diterpenoid. Bioorg. Med. Chem. 2007, 15, 5760–5774. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Miguel del Corral, J.M.; Rodriguez, M.L.; San Feliciano, A. An easy route to pentacyclic terpenylquinones. Tetrahedron Lett. 2012, 53, 519–521. [Google Scholar] [CrossRef]
- Hernandez, A.P.; Diez, P.; Garcia, P.A.; Miguel del Corral, J.M.; Perez-Andres, M.; Diez, D.; San Feliciano, A.; Fuentes, M.; Castro, M.A. New hybrids derived from podophyllic aldehyde and diterpenylhydroquinones with selectivity toward osteosarcoma cells. ACS Med. Chem. Lett. 2018, 9, 328–333. [Google Scholar] [CrossRef]
- Castro, M.A.; Gamito, A.M.; Tangarife-Castatno, V.; Zapata, B.; Miguel del Corral, J.M.; Mesa-Arango, A.C.; Betancur-Galvis, L.; San Feliciano, A. Synthesis and antifungal activity of terpenyl-1,4-naphthoquinone and 1,4-anthracenedione derivatives. Eur. J. Med. Chem. 2013, 67, 19–27. [Google Scholar] [CrossRef]
- Roa-Linares, V.C.; Miranda-Brand, Y.; Tangarife-Castatno, V.; Ochoa, R.; Garcia, P.A.; Castro, M.A.; Betancur-Galvis, L.; San Feliciano, A. Anti-herpetic, anti-Dengue and antineoplastic activities of simple and heterocycle-fused derivatives of terpenyl-1,4-naphthoquinone and 1,4-anthraquinone. Molecules 2019, 24, 1279. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pertejo, Y.; Escudero-Martínez, J.M.; Reguera, R.M.; Balaña-Fouce, R.; Garcia, P.A.; Jambrina, P.G.; San Feliciano, A.; Castro, M.A. Antileishmanial activity of terpenylquinones on Leishmania infantum and their effects on Leishmania topoisomerase IB. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 70–79. [Google Scholar] [CrossRef]
- El Haib, A.; Benharref, A.; Parrès-Maynadié, S.; Manoury, E.; Daran, J.C.; Urrutigoïty, M.; Gouygou, M. Molecular rearrangemente of epoxide derived from sesquitepenes by Lewis acid catalysis. Tetrahedron Assymmetry 2010, 21, 1272–1277. [Google Scholar] [CrossRef]
- Geroge, J.H.; McArdle, M.; Baldwin, J.E.; Adlington, R.M. Biomimetic rearrangements of simplified labdane diterpenoids. Tetrahedron 2010, 66, 6321–6330. [Google Scholar] [CrossRef]
- Mohanraj, S.; Herz, W. Photosensitized oxygenation of labda-8(17),12-diene, labda-8(17), 13-diene, and the biformenes. Synthesis of pumiloxide. J. Org. Chem. 1981, 46, 1362–1366. [Google Scholar] [CrossRef]
- Smith, M.B.; March, J. March’s Advanced Organic Chemistry: Reactions, Mechanism, and Structure, 6th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Bailey, P.S.; Hwang, H.H.; Chiang, C.-Y. Mechanisms of epoxidation during ozonation of carbon-carbon double bonds. J. Org. Chem. 1985, 50, 231–234. [Google Scholar] [CrossRef]
- Hagiwara, H.; Takeuchi, F.; Nozawa, M.; Hoshi, T.; Suzuki, T. The first total synthesis and determination of the absolute configuration of chapecoderin A, B and C. Tetrahedron 2004, 60, 1983–1989. [Google Scholar] [CrossRef]
- Yang, Z.; Kitano, Y.; Chiba, K.; Shibata, N.; Kurokawa, H.; Doi, Y.; Arakawa, Y.; Tada, M. Synthesis of variously oxidized abietane diterpenes and their antibacterial activities against MRSA and VRE. Bioorg. Med. Chem. 2001, 9, 347–356. [Google Scholar] [CrossRef]
- Fernández-Mateos, A.; Ferrero-Barrueco, O.; De Pascual-Teresa, J.; Rubio-González, R. Synthesis of (-)4,8β-dimethyl testolactone from (+)O-15-methyl isoagathate. Tetrahedron 1991, 47, 4375–4382. [Google Scholar] [CrossRef]
- Marcos, I.S.; Basabe, P.; Laderas, M.; Díez, D.; Jorge, A.; Rodilla, J.M.; Moro, R.F.; Lithgow, A.M.; Barata, I.G.; Urones, J.G. Side-chain migration reactions and ring B aromatization in labdanes: Scope and limitations. Synthesis of isogregenedane type tetrahydronaphthalenic diterpenes. Tetrahedron 2003, 59, 2333–2343. [Google Scholar] [CrossRef]
- National Cancer Institute. Common Cancer Types. Available online: https://www.cancer.gov/types/common-cancers (accessed on 8 January 2021).
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Waren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | A-549 | HT-29 | MB-231 |
|---|---|---|---|
| 1 | 0.24 | 0.24 | nt |
| 2 | 0.12 | 0.50 | nt |
| 3 | 0.20 | 0.20 | nt |
| 4 | 0.24 | 0.24 | nt |
| 5 | 0.96 | 0.96 | 3.25 |
| 6 | 0.24 | 0.24 | 1.21 |
| 7 | 5.00 | 5.00 | nt |
| 8 | 0.19 | 0.19 | nt |
| 9a | 3.25 | 4.21 | 3.25 |
| 10 | 4.80 | 4.80 | 1.26 |
| 11 | 3.64 | 3.64 | 2.68 |
| 12 | 10.10 | 10.10 | 4.25 |
| 13 | 4.90 | 4.90 | nt |
| 14 | 3.01 | >5.41 | nt |
| 15 | 2.97 | 2.97 | 1.54 |
| 16a–c | 2.02 | 3.82 | 2.07 |
| 16a | 2.24 | 4.25 | 2.06 |
| 17 | 0.83 | 0.86 | 1.06 |
| 18a | 0.22 | 0.22 | nt |
| 18c | 0.22 | 0.22 | nt |
| 19 | 1.42 | 2.47 | 1.78 |
| 20 | 0.86 | 0.86 | 1.06 |
| 21a | 3.12 | 2.90 | 2.45 |
| 21c | 5.00 | 5.00 | nt |
| Doxorubicin | 0.31 | 0.26 | 0.16 |
| H | 3 * | 5 ** | 6 ** | 7 * | 8 ** | 9a ** | 10 * | 11 ** | 12 * | 13 * |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5.52 m | 5.61 m | ||||||||
| 14 | 7.42 dd (8.8, 1.8) | 7.37 dd (8.6, 1.6) | 7.35 dd (8.6, 1.3) | 7.32 dd (8.8, 1.8) | 7.38 d (8.6) | 7.39 dd (8.8, 1.6) | 7.42 dd (8.8, 1. 3) | 7.39 dd (8.6, 1.6) | 7.38 dd (8.8, 1.5) | 7.34 d (8.8) |
| 15 | 7.77 d (8.8) | 7.77 d (8.6) | 7.77 d (8.6) | 7.75 d (8.8) | 7.77 d (8.6) | 7.78 d (8.8) | 7.78 d (8.8) | 7.80 d (8.6) | 7.78 d (8.8) | 7.75 d (8.8) |
| 16 | 7.59 bs | 7.57 bs | 7.57 bs | 7.55 bs | 7.63 s | 7.59 bs | 7.64 bs | 7.59 bs | 7.51 bs | 7.50 bs |
| 17 | 1.70 s | 1.32 s | 1.38 s | 5.10 bs 4.95 bs | 1.17 s | 1.13 s | 1.62 s | 1.11 s | - | - |
| 18 | 1.29 s | 1.17 s | 1.16 s | 1.17 s | 1.15 s | 1.37 s | 1.28 s | 1.40 s | 1.27 s | 1.16 s |
| 20 | 0.91 s | 0.86 s | 0.93 s | 0.86 s | 0.65 s | 1.15 s | 1.69 s | 1.19 s | 0.63 s | 0.50 s |
| 2′ | 7.21 d (8.2) | 7.21 d (8.2) | 7.22 d (8.2) | 7.21 d (8.2) | 7.21 d (8.2) | 7.16 d (8.4) | 7.21 d (8.2) | 7.23 d (8.2) | 7.21 d (8.0) | 7.16 d (8.0) |
| 3′ | 7.16 d (8.2) | 7.16 d (8.2) | 7.16 d (8.2) | 7.16 d (8.2) | 7.17 d (8.2) | 7.21 d (8.4) | 7.15 d (8.2) | 7.18 d (8.2) | 7.16 d (8.0) | 7.11 d (8.0) |
| 2 x OAc | 2.44 s, 2.46 s | 2.44 s, 2.46 s | 2.45 s, 2.47 s | 2.44 s, 2.46 s | 2.44 s, 2.48 s | 2.45 s, 2.46 s | 2.45 s, 2.50 s | 2.45 s, 2.47 s | 2.45 s, 2.46 s | 2.36 s, 2.38 s |
| 19-OCH3 | - | 3.63 s | 3.63 s | 3.63 s | 3.63 s | 3.70 s | 3.61 s | 3.72 s | - | 3.54 s |
| C | 3 * | 5 ** | 6 ** | 7 * | 8 ** | 9a ** | 10 * | 11 ** | 12 * | 13 * |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 37.7 | 34.2 | 37.4 | 40.8 | 35.1 | 120.5 | 39.3 | 122.4 | 39.1 | 38.9 |
| 2 | 19.5 | 18.8 | 18.6 | 19.3 | 20.3 | 22.3 | 21.9 | 22.4 # | 19.5 | 19.4 |
| 3 | 37.0 | 37.4 | 37.4 | 35.3 | 36.8 | 24.7 | 33.8 | 24.3 | 37.4 | 37.5 |
| 4 | 43.7 | 43.5 | 43.9 | 47.9 # | 44.0 | 44.4 | 50.5 | 44.4 | 44.1 | 43.9 |
| 5 | 53.4 | 45.6 | 54.7 | 134.6 | 52.9 | 41.4 | 58.0 | 41.4 | 54.6 | 54.3 |
| 6 | 29.6 | 18.9 | 20.1 | 26.4 | 23.1 | 27.5 | 25.0 | 27.4 # | 25.5 | 25.4 |
| 7 | 30.3 | 30.4 | 33.3 | 34.2 | 33.2 | 37.3 | 33.3 | 35.4 | 43.0 | 42.8 |
| 8 | 127.5 | 63.1 | 64.8 | 151.9 | 60.6 | 75.9 | 128.0 | 75.0 | 211.9 | 211.4 |
| 9 | 138.5 | 70.9 | 70.9 | 37.2 # | 216.1 | 48.5 | 128.7 | 49.0 | 61.5 | 61.3 |
| 10 | 39.8 | 39.1 | 38.9 | 128.6 | 47.0 | 140.9 | 210.0 | 140.8 | 43.6 | 43.2 |
| 11 | 20.6 | 29.0 | 33.3 | 23.9 | 43.2 | 35.7 | 36.2 | 38.0 | 23.1 | 23.0 |
| 12 | 34.2 | 33.2 | 35.8 | 31.1 | 30.4 | 30.9 | 35.4 | 31.4 | 34.8 | 34.6 |
| 13 | 141.9 | 141.6 | 141.5 | 142.4 | 140.5 | 142.3 | 141.5 | 141.8 | 141.2 | 140.9 |
| 14 | 128.2 | 128.2 | 128.0 | 128.4 | 128.4 | 128.5 | 128.6 | 128.2 | 128.4 | 128.2 |
| 15 | 121.6 | 121.7 | 121.8 | 121.6 | 121.8 | 121.7 | 121.5 | 121.9 | 121.7 | 121.5 |
| 16 | 119.4 | 119.6 | 119.5 | 119.9 | 120.4 | 119.8 | 119.9 | 119.8 | 120.1 | 119.8 |
| 17 | 19.8 | 22.2 | 21.0 | 106.4 | 20.1 | 24.0 # | 18.3 | 17.2 | - | - |
| 18 | 28.5 | 28.7 | 28.3 | 26.2 | 28.3 | 22.3 | 24.3 | 22.4 | 28.8 | 28.4 |
| 19 | 184.4 | 178.0 | 177.8 | 177.7 | 178.1 | 177.7 | 175.6 | 177.5 | 182.9 | 176.7 |
| 20 | 17.9 | 15.5 | 14.5 | 23.4 | 14.8 | 16.8 # | 18.6 | 22.7 | 13.3 | 12.9 |
| 1′ | 143.8 | 143.9 | 143.9 | 143.8 | 143.9 | 143.8 | 144.0 | 143.9 | 143.9 | 143.7 |
| 2′ | 117.6 | 116.7 | 116.8 | 116.6 | 116.9 | 116.7 | 116.6 | 116.8 | 116.8 | 117.5 |
| 3′ | 116.6 | 117.7 | 117.8 | 117.7 | 117.8 | 117.7 | 117.6 | 117.8 | 117.7 | 116.6 |
| 4′ | 144.2 | 144.3 | 144.3 | 144.3 | 144.3 | 144.3 | 144.3 | 144.3 | 144.3 | 144.1 |
| 5′ | 126.1 | 126.1 | 126.1 | 126.0 | 126.2 | 125.2 | 126.1 | 126.1 | 126.2 | 125.9 |
| 6′ | 127.7 | 127.8 | 127.8 | 127.8 | 127.7 | 127.8 | 127.8 | 127.8 | 127.7 | 127.5 |
| OAc (2xCO) | 169.4 | 169.3 | 169.4 | 169.3 | 169.3 169.2 | 169.4 | 169.5 169.3 | 169.3 | 169.3 | 169.0 |
| OAc (2xCH3) | 20.9 | 20.9 21.0 | 21.0 | 21.0 | 21.0 | 21.0 | 21.0 | 20.9 | 20.9 | 20.7 |
| 19-OCH3 | - | 51.1 | 51.2 | 51.9 | 51.1 | 51.5 | 51.7 | 51.5 | - | 51.0 |
| H | 14 ** | 15 ** | 16a * | 17 * | 18a * | 18c * | 19 ** | 20 * | 21a ** | 21c ** |
|---|---|---|---|---|---|---|---|---|---|---|
| 6 | 6.94 bs | |||||||||
| 14 | 7.39 dd (8.8, 1.3) | 7.36 dd (8.6, 1.4) | 7.39 dd (8.8, 1.8) | 7.39 dd (8.8, 1.8) | 7.44 dd (8.8, 1.5) | 7.45 d (8.4) | 7.47 d (8.6) | 7.39 dd (8.4, 1.5) | 7.41 dd (8.8, 1.8) | 7.41 dd (8.8, 1.8) |
| 15 | 7.78 d (8.8) | 7.78 d (8.6) | 7.78 d (8.8) | 7.79 d (8.8) | 7.79 d (8.8) | 7.80 d (8.4) | 7.82 d (8.6) | 7.78 d (8.4) | 7.79 d (8.8) | 7.78 d (8.8) |
| 16 | 7.63 bs | 7.60 bs | 7.57 bs | 7.56 bs | 7.61 d (1.5) | 7.62 bs | 7.65 bs | 7.35 bs | 7.56 bs | 7.56 bs |
| 17 | 2.04 s | 2.22 s | 5.02 bs 4.72 bs | 4.99 bs 4.71 bs | 1.70 s | 1.75 s | 2.35 s | - | - | - |
| 18 | 1.12 s | 1.20 s | 4.72 bs 4.41 bs | 1.40 bs | 4.79 d (1.5) 4.52 d (1.5) | 1.66 s | 1.30 d (7.0) | 1.43 s | 4.80 d (1.5) 4.49 d (1.5) | 1.64 s |
| 20 | 1.00 s | 0.70 s | 0.53 s | 0.69 s | 0.84 s | 1.19 s | 2.23 s | 0.73 s | 0.56 s | 0.86 s |
| 2′ | 7.21 d (8.3) | 7.20 d (8.3) | 7.21 d (8.3) | 7.22 d (8.4) | 7.22 d (8.2) | 7.23 d (8.2) | 7.22 d (8.2) | 7.21 d (8.4) | 7.21 d (8.0) | 7.20 d (8.0) |
| 3′ | 7.16 d (8.3) | 7.15 d (8.3) | 7.16 d (8.3) | 7.16 d (8.4) | 7.17 d (8.2) | 7.17 d (8.2) | 7.19 d (8.2) | 7.16 d (8.4) | 7.16 d (8.0) | 7.15 d (8.0) |
| 2 x OAc | 2.47 s, 2.44 s | 2.47 s, 2.44 s | 2.47 s, 2.46 s | 2.47 s, 2.46 s | 2.47 s, 2.45 s | 2.48 s, 2.46 s | 2.47 s, 2.46 s | 2.47 s, 2.46 s | 2.47 s, 2.46 s | 2.44 s, 2.42 s |
| 19-OCH3 | 3.64 s | 3.68 s | - | - | - | - | - | - | - | - |
| Others | 5.43 bs (OH) | 1.94 s (4-OAc) | 1.94 s (4-OAc) |
| C | 14 ** | 15 ** | 16a * | 17 * | 18a * | 18c * | 19 ** | 20 * | 21a ** | 21c ** |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34.5 | 30.4 | 38.5 | 37.7 | 36.9 | 36.9 | 27.8 | 37.7 | 38.7 | 37.1 |
| 2 | 18.8 | 19.9 | 23.9 | 19.9 | 21.3 | 18.8 | 20.4 | 19.6 | 23.2 | 19.3 |
| 3 | 34.5 | 37.2 | 36.7 | 37.4 | 36.9 | 35.2 | 30.7 | 37.1 | 36.5 | 34.9 |
| 4 | 46.1 | 43.9 | 150.5# | 85.9 | 151.0 | 135.2 # | 32.7 | 85.4 | 148.8 | 132.5# |
| 5 | 48.0 | 51.8 | 53.7 | 55.7 | 48.7 | 142.1 # | 139.9 | 52.4 | 49.9 | 127.2# |
| 6 | 23.3 | 27.9 | 27.7 | 22.7 | 30.7 | 30.8 | 127.9 | 23.3 | 26.5 | 26.3 |
| 7 | 46.1 | 53.2 | 37.5 | 37.7 | 36.3 | 33.9 | 132.9 # | 42.1 | 42.0 | 42.1 |
| 8 | 208.5 | 216.1 | 148.1 # | 147.7 | 126.9 # | 123.0 # | 135.2 # | 211.4 | 212.0 | 212.5 |
| 9 | 214.7 | 84.3 | 51.3 | 53.9 | 138.4 # | 138.2 # | 134.2 | 62.5 | 60.6 | 59.6 |
| 10 | 52.6 | 49.2 | 41.8 | 40.4 | 40.7 | 39.5 | 133.2 | 43.2 | 44.6 | 42.8 |
| 11 | 39.9 | 38.8 | 25.9 | 23.8 | 23.4 | 23.6 | 31.9 | 23.3 | 24.0 | 24.0 |
| 12 | 30.5 | 31.3 | 34.9 | 34.8 | 32.5 | 32.1 | 36.0 | 34.9 | 35.1 | 32.6 |
| 13 | 140.5 | 141.6 | 142.0 | 141.9 | 142.0 | 142.1 | 141.3 | 141.2 | 141.4 | 141.3 |
| 14 | 128.3 | 128.3 | 128.5 | 128.4 | 128.3 | 128.3 | 128.3 | 128.3 | 128.5 | 128.4 |
| 15 | 122.0 | 121.7 | 121.6 | 121.7 | 121.7 | 121.7 | 121.8 | 121.8 | 121.7 | 121.6 |
| 16 | 120.4 | 119.9 | 119.9 | 119.9 | 119.5 | 119.5 | 119.7 | 120.1 | 120.1 | 119.9 |
| 17 | 29.8 | 31.5 | 106.2 & | 107.1 | 19.8 | 19.9 | 20.0 | - | - | - |
| 18 | 27.0 | 28.2 | 107.4 & | 19.4 | 105.9 | 25.6 | 23.0 | 19.4 | 107.4 | 19.7 |
| 19 | 177.7 | 177.8 | - | - | - | - | - | - | - | - |
| 20 | 18.8 | 14.9 | 12.3 | 14.3 | 18.8 | 19.0 | 15.0 | 14.6 | 13.1 | 12.7 |
| 1′ | 144.0 | 143.9 | 144.0 | 144.0 | 143.9 | 143.9 | 143.9 | 143.9 | 144.0 | 143.8 |
| 2′ | 117.1 | 116.7 | 116.7 | 116.7 | 117.7 | 117.7 | 116.8 | 116.8 | 116.8 | 116.6 |
| 3′ | 118.0 | 117.7 | 117.7 | 117.7 | 116.7 | 116.7 | 117.7 | 117.7 | 117.7 | 117.6 |
| 4′ | 144.4 | 144.3 | 144.4 | 144.4 | 144.4 | 144.3 | 144.3 | 144.3 | 144.3 | 144.2 |
| 5′ | 126.3 | 126.1 | 126.2 | 126.2 | 126.2 | 126.1 | 126.2 | 126.2 | 126.2 | 126.0 |
| 6′ | 127.8 | 127.8 | 127.8 | 127.9 | 127.9 | 127.9 | 127.9 | 127.7 | 127.8 | 127.6 |
| OAc (2xCO) | 169.5 | 169.3 169.2 | 169.4 | 169.2 | 169.4 | 169.4 | 169.3 | 169.4 | 169.4 | 169.2 |
| OAc (2xCH3) | 21.1 | 20.9 21.0 | 21.0 | 20.9 | 21.0 | 20.9 | 20.9 | 21.0 | 21.0 | 20.8 |
| 19-OCH3 | 51.6 | 51.3 | ||||||||
| 4-OAc | 22.7 170.2 | 22.7 170.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández, Á.P.; Chamorro, P.; Rodríguez, M.L.; Miguel del Corral, J.M.; García, P.A.; Francesch, A.; San Feliciano, A.; Castro, M.Á. New Antineoplastic Naphthohydroquinones Attached to Labdane and Rearranged Diterpene Skeletons. Molecules 2021, 26, 474. https://doi.org/10.3390/molecules26020474
Hernández ÁP, Chamorro P, Rodríguez ML, Miguel del Corral JM, García PA, Francesch A, San Feliciano A, Castro MÁ. New Antineoplastic Naphthohydroquinones Attached to Labdane and Rearranged Diterpene Skeletons. Molecules. 2021; 26(2):474. https://doi.org/10.3390/molecules26020474
Chicago/Turabian StyleHernández, Ángela P., Pablo Chamorro, Mª Lucena Rodríguez, José M. Miguel del Corral, Pablo A. García, Andrés Francesch, Arturo San Feliciano, and Mª Ángeles Castro. 2021. "New Antineoplastic Naphthohydroquinones Attached to Labdane and Rearranged Diterpene Skeletons" Molecules 26, no. 2: 474. https://doi.org/10.3390/molecules26020474
APA StyleHernández, Á. P., Chamorro, P., Rodríguez, M. L., Miguel del Corral, J. M., García, P. A., Francesch, A., San Feliciano, A., & Castro, M. Á. (2021). New Antineoplastic Naphthohydroquinones Attached to Labdane and Rearranged Diterpene Skeletons. Molecules, 26(2), 474. https://doi.org/10.3390/molecules26020474