
Expanding the Scope of the Cleavable N-(Methoxy)oxazolidine Linker for the Synthesis of Oligonucleotide Conjugates


Abstract
:1. Introduction
2. Results
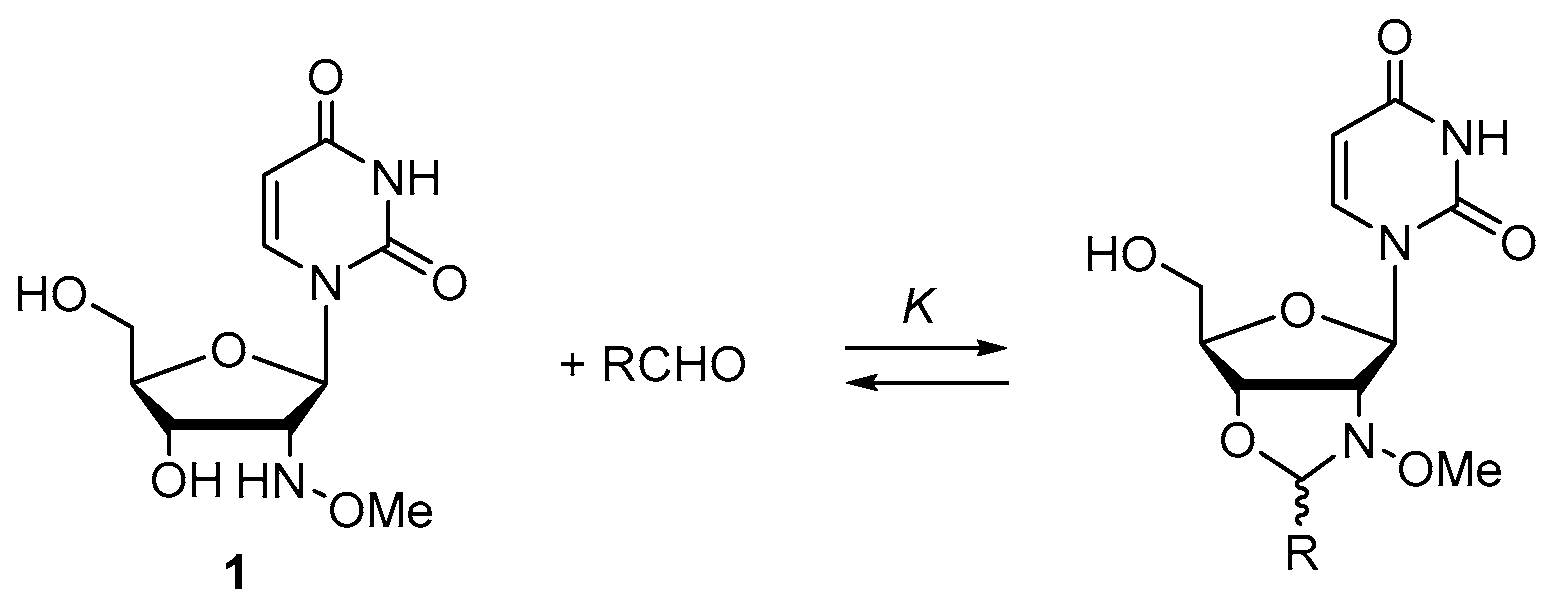
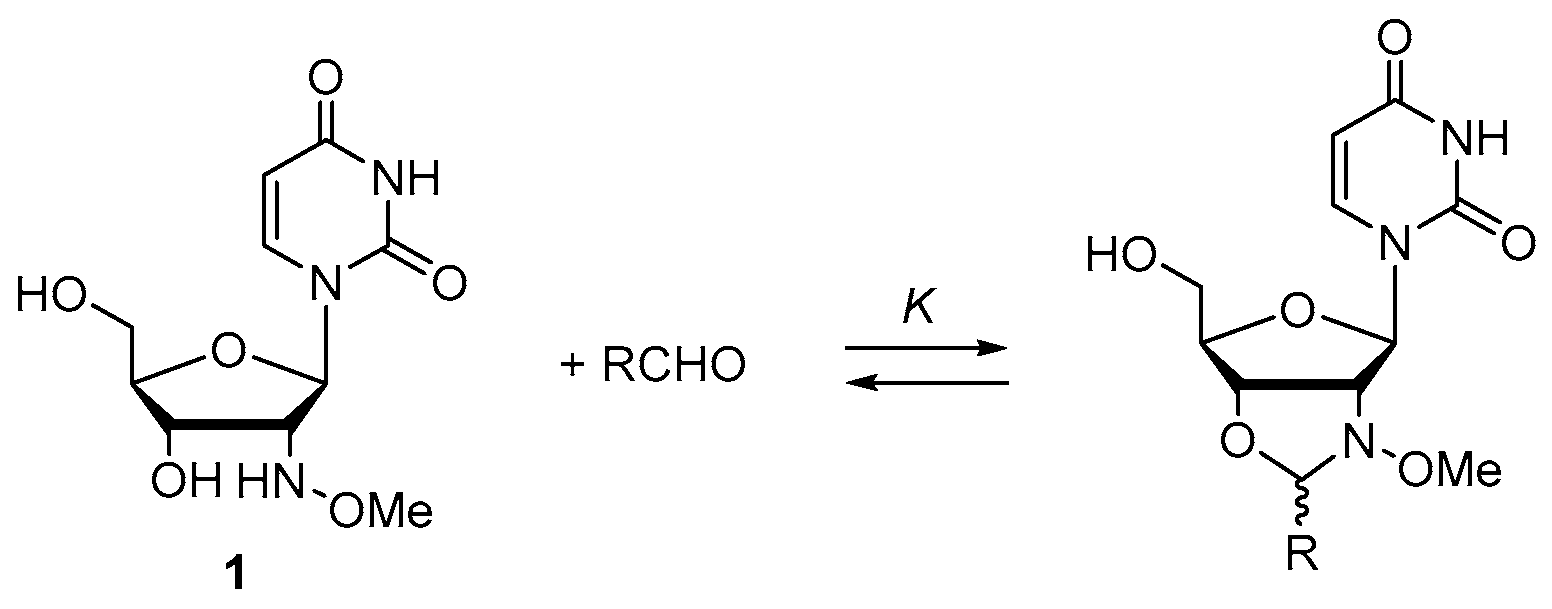
2.1. Small Molecular Model Study
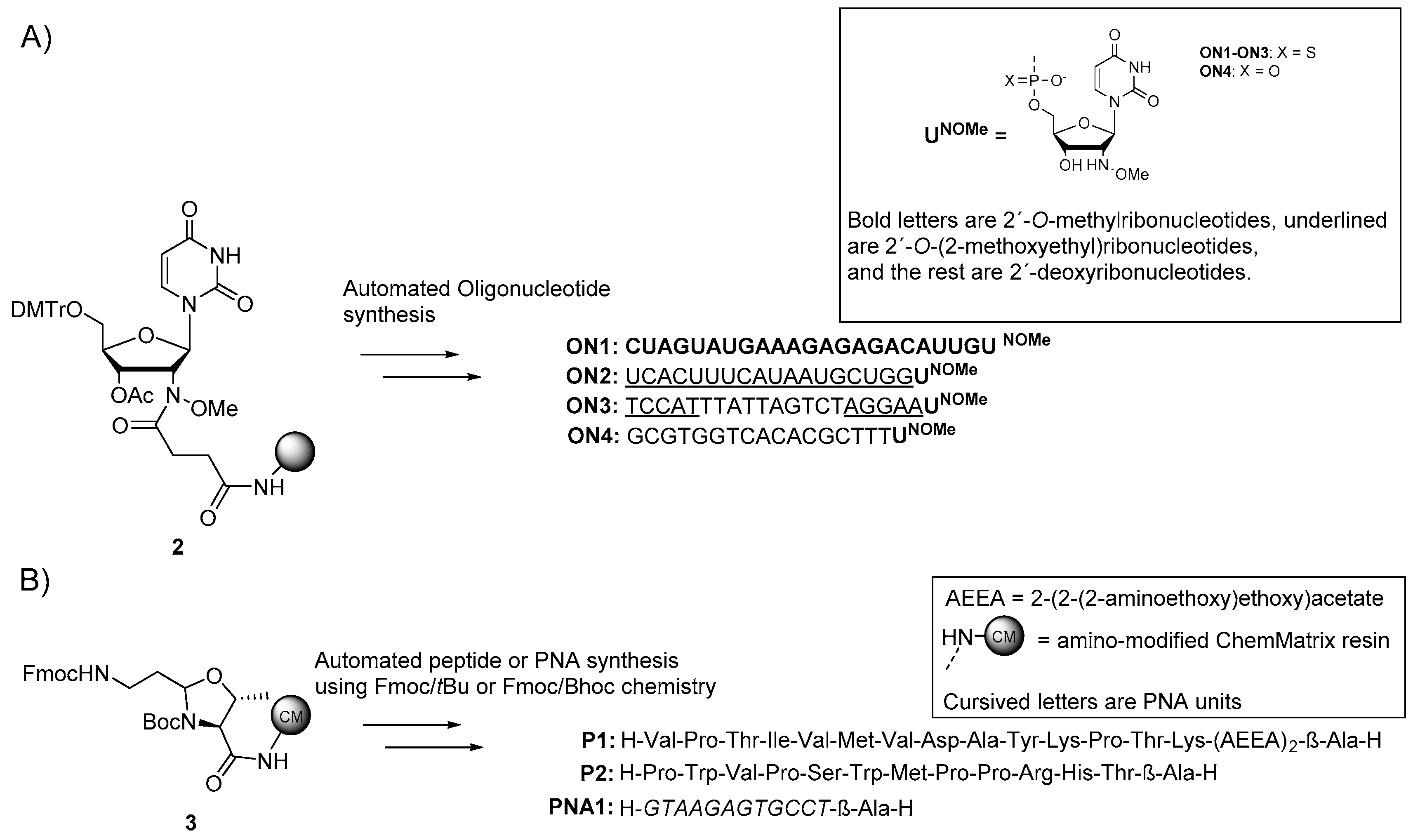
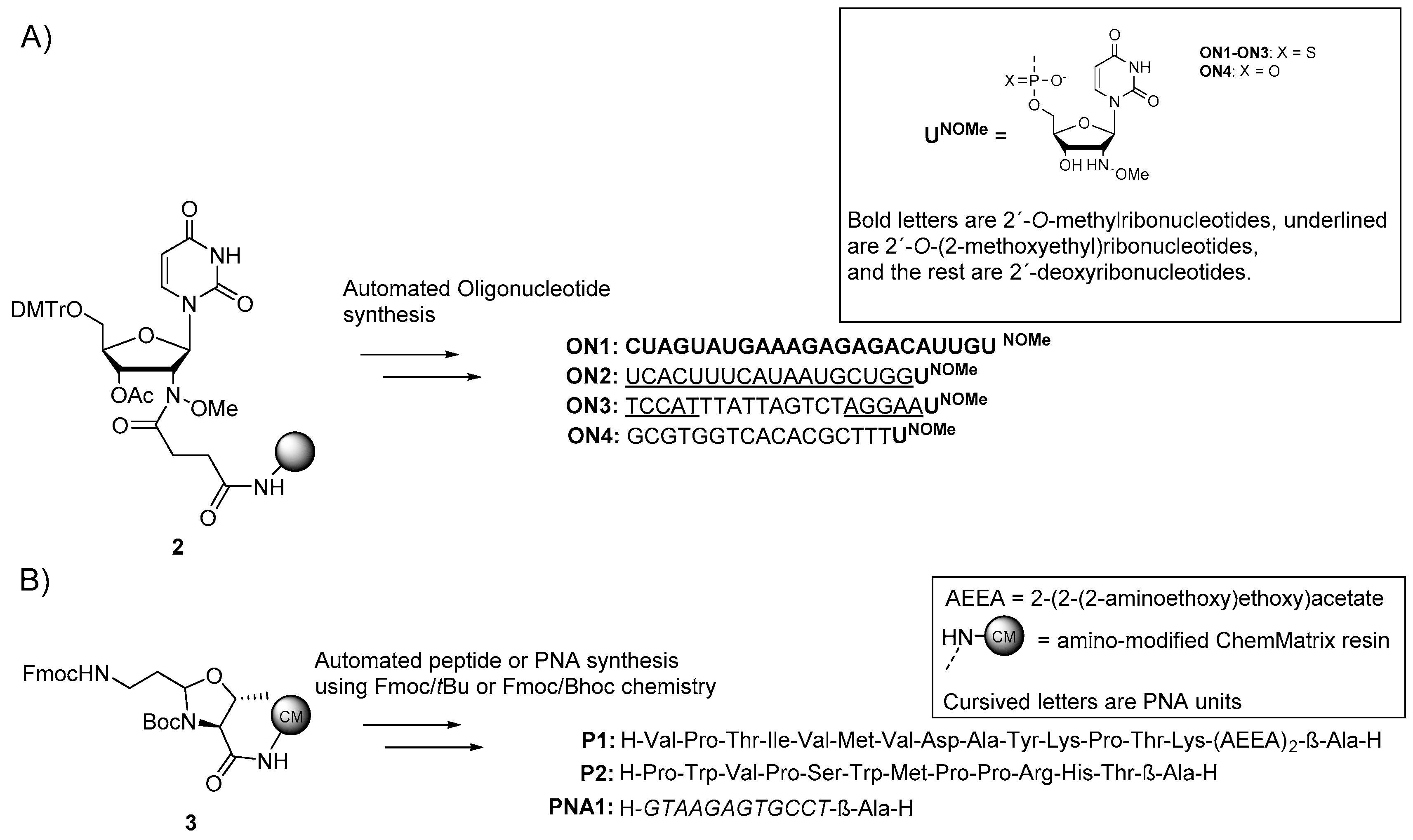
2.2. Synthesis of 2′-deoxy′-2′-(N-methoxyamino)uridine-Modified Oligonucleotides
2.3. Synthesis of β-Ala-H-Modified Biomolecules
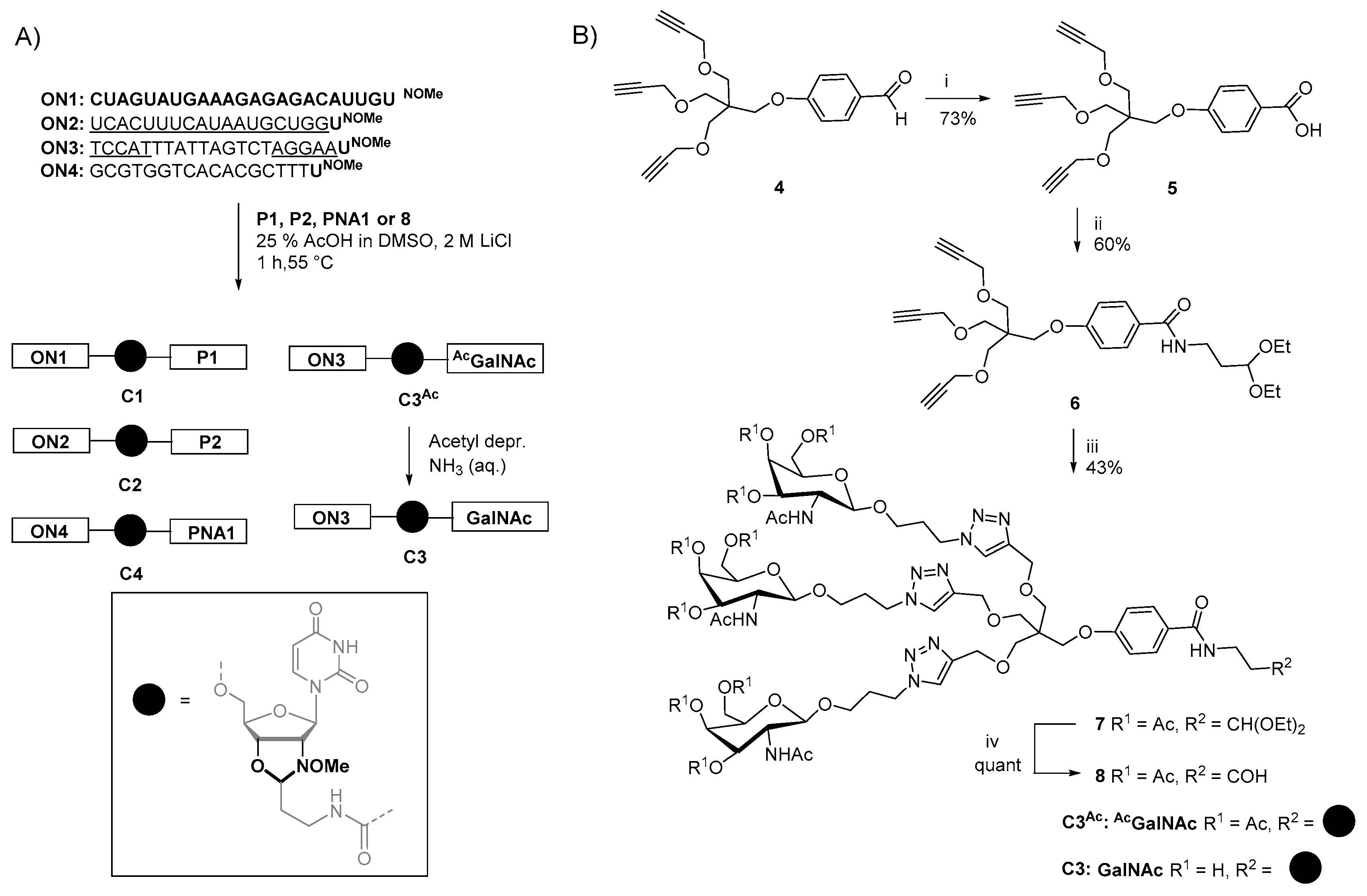
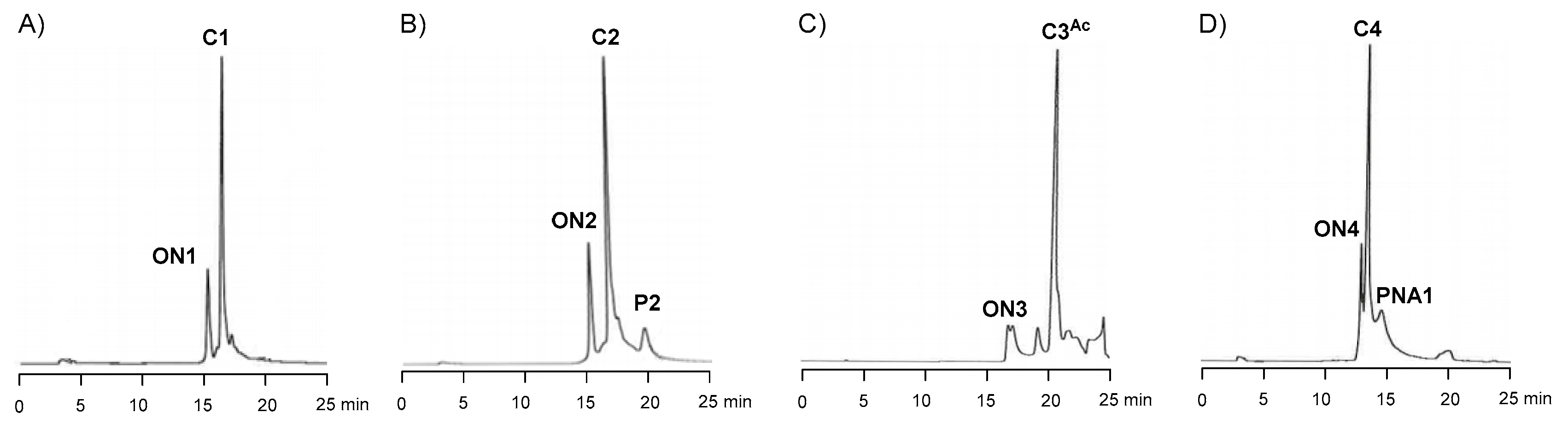
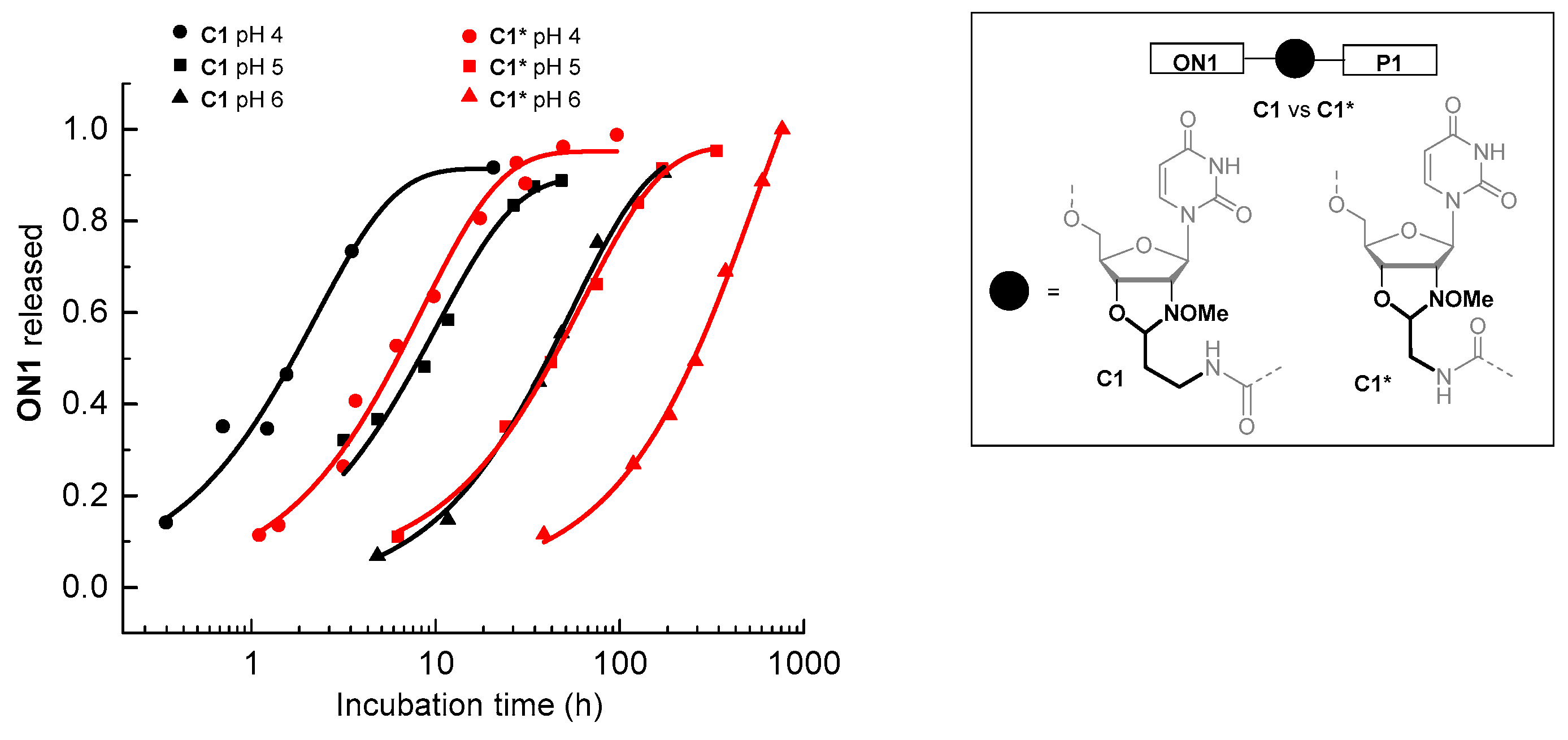
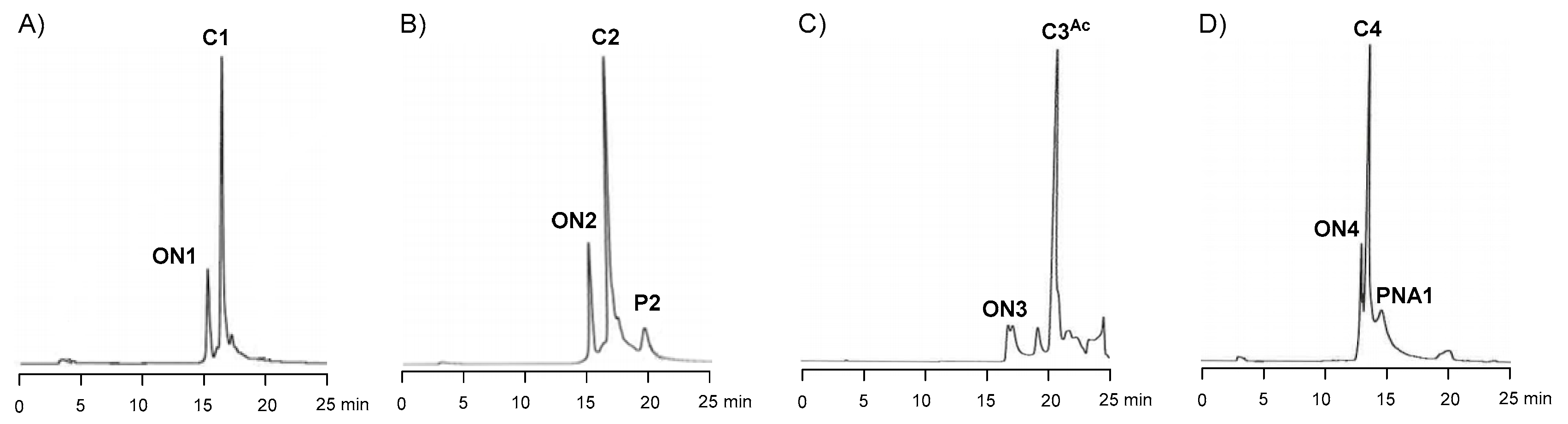
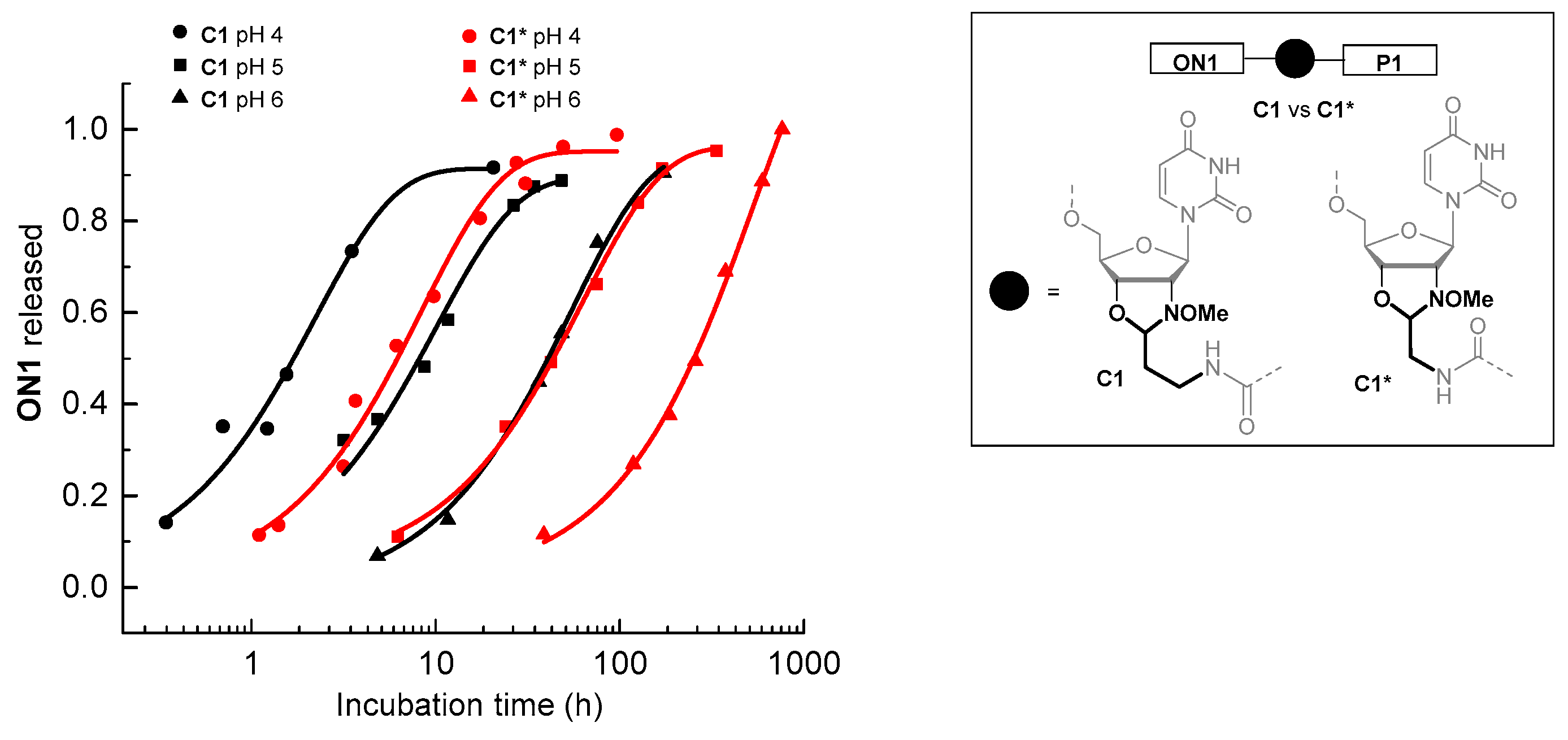
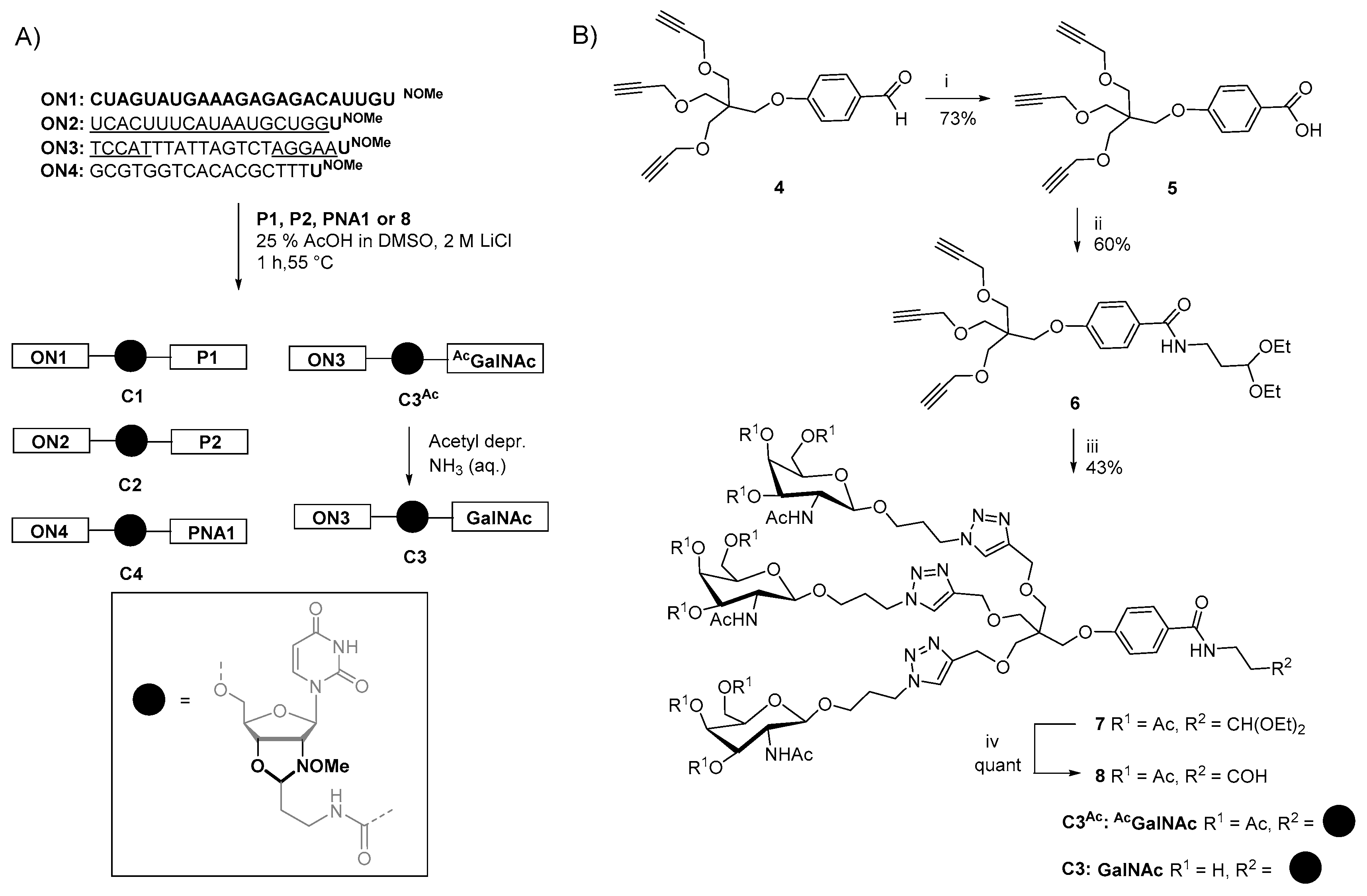
2.4. Synthesis of Oligonucleotide Conjugates C1-C4 Using N-(methoxy)oxazolidine Ligation
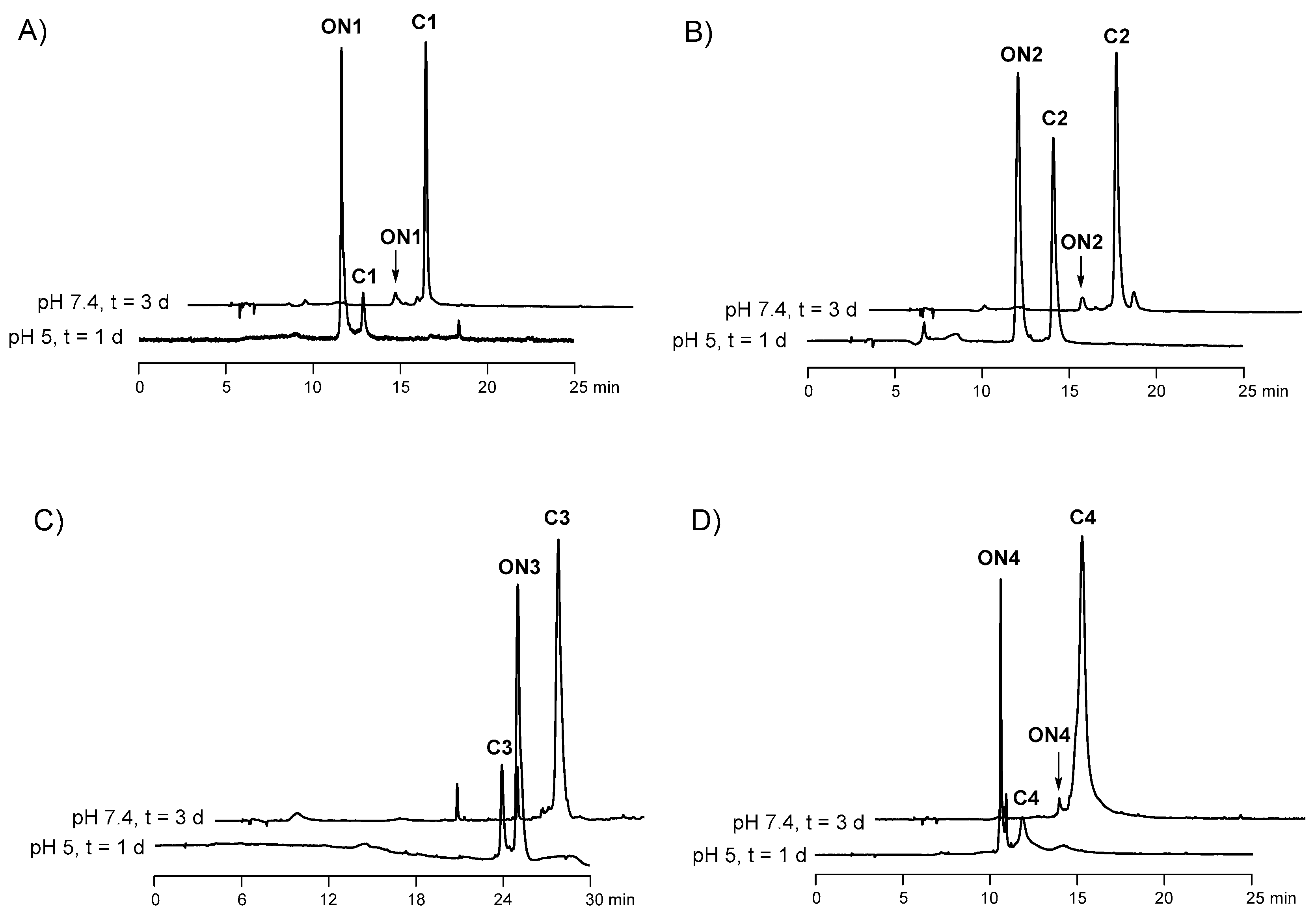
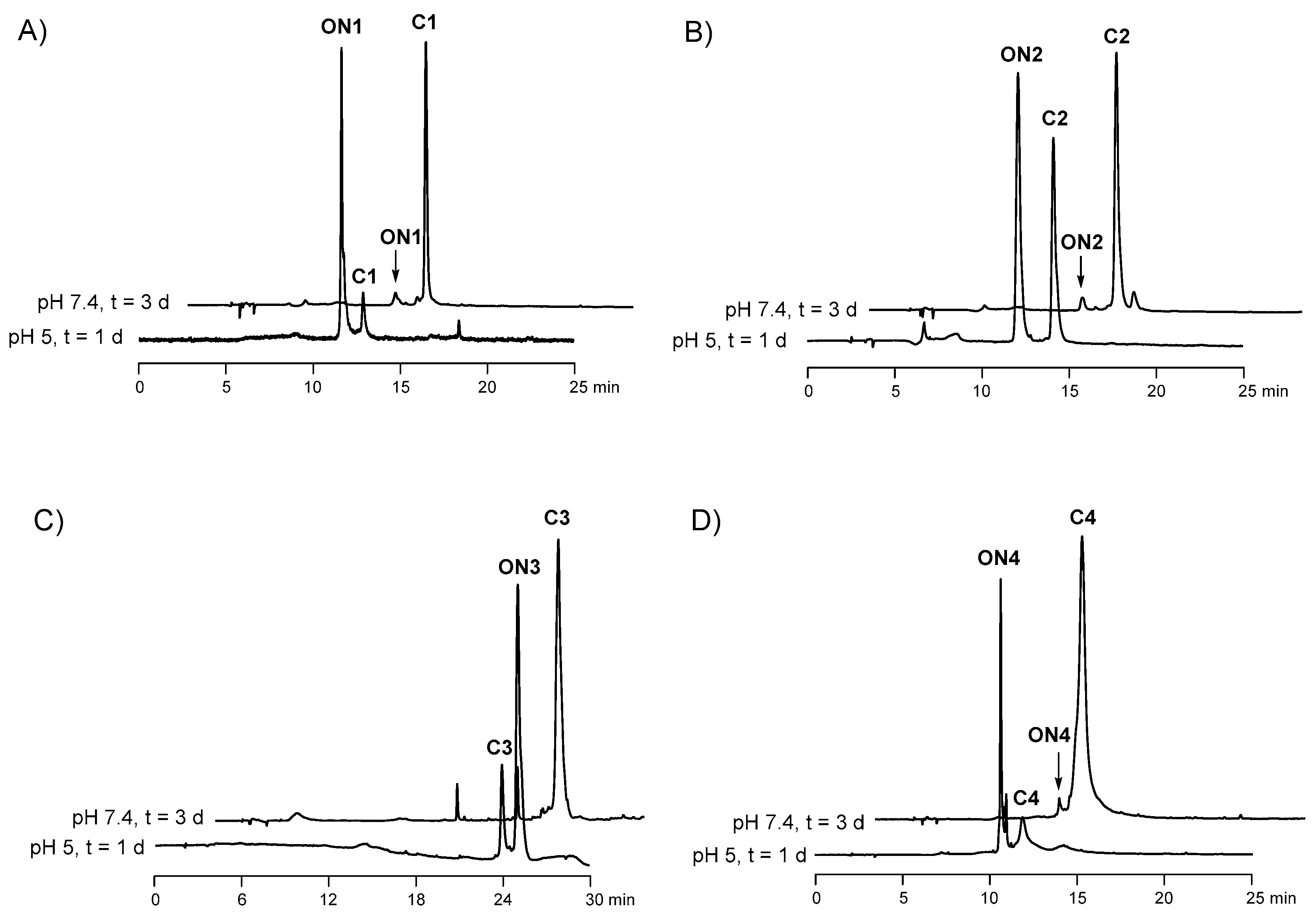
2.5. Hydrolysis of the Ligations Products
3. Discussion
4. Materials and Methods
4.1. 4-{3-(propynyloxy)-2,2-Bis [(propynyloxy)methyl]propoxy}benzoic Acid (5)
4.2. N-(3,3-diethoxypropyl)-4-{3-(propynyloxy)-2,2-bis [(propynyloxy)methyl]propoxy}benzamide (6)
4.3. Diethoxy Acetal-Protected Trivalent GalNAc Cluster (7)
4.4. Trivalent GalNAc Cluster (8)
4.5. Synthesis of Conjugates C1–C4 Using N-(methoxy)oxazolidine Ligation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ngamcherdtrakul, W.; Castro, D.J.; Gu, S.; Morry, J.; Reda, M.; Gray, J.W.; Yantasee, W. Current development of targeted oligonucleotide-based cancer therapies: Perspective on HER2-positive breast cancer treatment. Cancer Treat. Rev. 2016, 45, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinovich, K.M.; Shaw, N.C.; Kicic, A.; Schultz, A.; Fletcher, S.; Wilton, S.D.; Stick, S.M. The potential of antisense oligonucleotide therapies for inherited childhood lung diseases. Mol. Cell. Pediatr. 2018, 5, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Di Fusco, D.; Dinallo, V.; Marafini, I.; Figliuzzi, M.M.; Romano, B.; Monteleone, G. Antisense Oligonucleotide: Basic Concepts and Therapeutic Application in Inflammatory Bowel Disease. Front. Pharmacol. 2019, 10, 305. [Google Scholar] [CrossRef] [Green Version]
- Le, B.T.; Raguraman, P.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense Oligonucleotides Targeting Angiogenic Factors as Potential Cancer Therapeutics. Mol. Ther. Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef] [Green Version]
- Krichevsky, A.M.; Uhlmann, E.J. Oligonucleotide Therapeutics as a New Class of Drugs for Malignant Brain Tumors: Targeting mRNAs, Regulatory RNAs, Mutations, Combinations, and Beyond. Neurotherapeutics 2019, 16, 319–347. [Google Scholar] [CrossRef] [Green Version]
- Takakura, K.; Kawamura, A.; Torisu, Y.; Koido, S.; Yahagi, N.; Saruta, M. The Clinical Potential of Oligonucleotide Therapeutics against Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 3331. [Google Scholar] [CrossRef] [Green Version]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug. Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Alkhouri, N.; Reddy, G.K.; Lawitz, E. Oligonucleotide-Based Therapeutics: An Emerging Strategy for the Treatment of Chronic Liver Diseases. Hepatology 2020. [Google Scholar] [CrossRef]
- Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhu, P.; Lee, S.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; et al. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Dibirdik, I.; Myers, D.E. Rational design of an immunoconjugate for selective knock-down of leukemia-specific E2A–PBX1 fusion gene expression in human Pre-B leukemia. Integr. Biol. 2013, 5, 122–132. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of THIOMAB-siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type–specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J. Cell-Type–Specific Aptamer and Aptamer-Small Interfering RNA Conjugates for Targeted Human Immunodeficiency Virus Type 1 Therapy. J. Investig. Med. 2014, 62, 914. [Google Scholar] [CrossRef]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Yerneni, S.S.; Lathwal, S.; Shrestha, P.; Shirwan, H.; Matyjaszewski, K.; Weiss, L.; Yolcu, E.S.; Campbell, P.G.; Das, S.R. Rapid On-Demand Extracellular Vesicle Augmentation with Versatile Oligonucleotide Tethers. ACS Nano 2019, 13, 10555. [Google Scholar] [CrossRef]
- Zatsepin, T.S.; Oretskaya, T.S. Synthesis and applications of oligonucleotide-carbohydrate conjugates. Chem. Biodivers 2004, 1, 1401–1417. [Google Scholar] [CrossRef]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Godeau, G.; Staedel, C.; Barthélémy, P. Lipid-Conjugated Oligonucleotides via “Click Chemistry” Efficiently Inhibit Hepatitis C Virus Translation. J. Med. Chem. 2008, 51, 4374–4376. [Google Scholar] [CrossRef] [PubMed]
- Chernikov, I.V.; Gladkikh, D.V.; Meschaninova, M.I.; Ven’yaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-Containing Nuclease-Resistant siRNA Accumulates in Tumors in a Carrier-free Mode and Silences MDR1 Gene. Mol. Ther. Nucleic Acids 2017, 6, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Nishina, K.; Unno, T.; Uno, Y.; Kubodera, T.; Kanouchi, T.; Mizusawa, H.; Yokota, T. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol. Ther. 2008, 16, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, O.; Ming, X.; Huang, L.; Juliano, R.L. Targeted Intracellular Delivery of Antisense Oligonucleotides via Conjugation with Small-Molecule Ligands. J. Am. Chem. Soc. 2010, 132, 8848–8849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohmen, C.; Fröhlich, T.; Lächelt, U.; Röhl, I.; Vornlocher, H.; Hadwiger, P.; Wagner, E. Defined Folate-PEG-siRNA Conjugates for Receptor-specific Gene Silencing. Mol. Ther. Nucleic Acids 2012, 1, e7. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ming, X.; Nakagawa, O. Cellular Uptake and Intracellular Trafficking of Antisense and siRNA Oligonucleotides. Bioconjugate Chem. 2012, 23, 147–157. [Google Scholar]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef] [Green Version]
- Lönn, P.; Dowdy, S.F. Cationic PTD/CPP-mediated macromolecular delivery: Charging into the cell. Expert Opin. Drug Deliv. 2015, 12, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Gait, M.J.; Arzumanov, A.A.; McClorey, G.; Godfrey, C.; Betts, C.; Hammond, S.; Wood, M.J.A. Cell-Penetrating Peptide Conjugates of Steric Blocking Oligonucleotides as Therapeutics for Neuromuscular Diseases from a Historical Perspective to Current Prospects of Treatment. Nucleic Acid Ther. 2019, 29, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Kasuya, T.; Yokota, S.; Onishi, R.; Ikehara, T.; Kugimiya, A.; Watanabe, A. Gene Silencing Activity and Hepatic Accumulation of Antisense Oligonucleotides Bearing Cholesterol-Conjugated Thiono Triester at the Gap Region. Nucleic Acid Ther. 2017, 27, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, C.; Tang, X. Cholesterol-Modified Caged siRNAs for Photoregulating Exogenous and Endogenous Gene Expression. Bioconjugate Chem. 2018, 29, 1010–1015. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Zatsepin, T.S.; Stetsenko, D.A.; Arzumanov, A.A.; Romanova, E.A.; Gait, M.J.; Oretskaya, T.S. Synthesis of Peptide−Oligonucleotide Conjugates with Single and Multiple Peptides Attached to 2‘-Aldehydes through Thiazolidine, Oxime, and Hydrazine Linkages. Bioconjugate Chem. 2002, 13, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, N.; Olivier, C.; Gouyette, C.; Huynh-Dinh, T.; Gras-Masse, H.; Melnyk, O. Synthesis of oligonucleotide–peptide conjugates using hydrazone chemical ligation. Tetrahedron Lett. 2002, 43, 997–999. [Google Scholar] [CrossRef]
- Aho, A.; Sulkanen, M.; Korhonen, H.; Virta, P. Conjugation of Oligonucleotides to Peptide Aldehydes via a pH-Responsive N-Methoxyoxazolidine Linker. Org. Lett. 2020, 22, 6714–6718. [Google Scholar] [CrossRef] [PubMed]
- Luna Velez, M.V.; Verhaegh, G.W.; Smit, F.; Sedelaar, J.P.M.; Schalken, J.A. Suppression of prostate tumor cell survival by antisense oligonucleotide-mediated inhibition of AR-V7 mRNA synthesis. Oncogene 2019, 38, 3696–3709. [Google Scholar] [CrossRef] [Green Version]
- Li, Q. Nusinersen as a Therapeutic Agent for Spinal Muscular Atrophy. Yonsei Med. J. 2020, 61, 273–283. [Google Scholar] [CrossRef]
- Yu, X.X.; Murray, S.F.; Pandey, S.K.; Booten, S.L.; Bao, D.; Song, X.Z.; Kelly, S.; Chen, S.; McKay, R.; Monia, B.P.; et al. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42, 362–371. [Google Scholar] [CrossRef]
- Choi, C.S.; Savage, D.B.; Kulkarni, A.; Yu, X.X.; Liu, Z.X.; Morino, K.; Kim, S.; Distefano, A.; Samuel, V.T.; Neschen, S.; et al. Suppression of diacylglycerol acyltransferase-2 (DGAT2), but not DGAT1, with antisense oligonucleotides reverses diet-induced hepatic steatosis and insulin resistance. J. Biol. Chem. 2007, 282, 22678–22688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macdonald, J.; Houghton, P.; Xiang, D.; Duan, W.; Shigdar, S. Truncation and Mutation of a Transferrin Receptor Aptamer Enhances Binding Affinity. Nucleic Acid Ther. 2016, 26, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, 690. [Google Scholar] [CrossRef] [Green Version]
- Arranz-Gibert, P.; Ciudad, S.; Seco, J.; García, J.; Giralt, E.; Teixidó, M. Immunosilencing peptides by stereochemical inversion and sequence reversal: Retro-D-peptides. Sci. Rep. 2018, 8, 6446. [Google Scholar] [CrossRef]
- Lee, J.H.; Engler, J.A.; Collawn, J.F.; Moore, B.A. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur. J. Biochem. 2001, 268, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Prades, R.; Guerrero, S.; Araya, E.; Molina, C.; Salas, E.; Zurita, E.; Selva, J.; Egea, G.; López-Iglesias, C.; Teixidó, M.; et al. Delivery of gold nanoparticles to the brain by conjugation with a peptide that recognizes the transferrin receptor. Biomaterials 2012, 33, 7194–7205. [Google Scholar] [CrossRef] [PubMed]
- Rembach, A.; Turner, B.J.; Bruce, S.; Cheah, I.K.; Scott, R.L.; Lopes, E.C.; Zagami, C.J.; Beart, P.M.; Cheung, N.S.; Langford, S.J.; et al. Antisense peptide nucleic acid targeting GluR3 delays disease onset and progression in the SOD1 G93A mouse model of familial ALS. J. Neurosci. Res. 2004, 77, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Ede, N.J.; Bray, A.M. A simple linker for the attachment of aldehydes to the solid phase. Application to solid phase synthesis by the multipin™ method. Tetrahedron Lett. 1997, 38, 7119–7122. [Google Scholar] [CrossRef]
- Ede, N.J.; Eagle, S.N.; Wickham, G.; Bray, A.M.; Warne, B.; Shoemaker, K.; Rosenberg, S. Solid phase synthesis of peptide aldehyde protease inhibitors. Probing the proteolytic sites of hepatitis C virus polyprotein. J. Peptide Sci. 2000, 6, 11–18. [Google Scholar] [CrossRef]
- Kushnarova-Vakal, A.; Äärelä, A.; Huovinen, T.; Virta, P.; Lamminmäki, U. Site-Specific Linking of an Oligonucleotide to Mono- and Bivalent Recombinant Antibodies with SpyCatcher-SpyTag System for Immuno-PCR. ACS omega 2020, 5, 24927–24934. [Google Scholar] [CrossRef]
- Mäkilä, J.; Jadhav, S.; Kiviniemi, A.; Käkelä, M.; Liljenbäck, H.; Poijärvi-Virta, P.; Laitala-Leinonen, T.; Lönnberg, H.; Roivainen, A.; Virta, P. Synthesis of multi-galactose-conjugated 2′-O-methyl oligoribonucleotides and their in vivo imaging with positron emission tomography. Bioorg. Med. Chem. 2014, 22, 6806–6813. [Google Scholar] [CrossRef] [PubMed]
- Karskela, M.; Helkearo, M.; Virta, P.; Lönnberg, H. Synthesis of Oligonucleotide Glycoconjugates Using Sequential Click and Oximation Ligations. Bioconjugate Chem. 2010, 21, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szekely, T.; Roy, O.; Dériaud, E.; Job, A.; Lo-Man, R.; Leclerc, C. Design, Synthesis, and Immunological Evaluation of a Multicomponent Construct Based on a Glycotripeptoid Core Comprising B and T Cell Epitopes and a Toll-like Receptor 7 Agonist That Elicits Potent Immune Responses. J. Med. Chem. 2018, 61, 9568–9582. [Google Scholar] [CrossRef]
- Häring, A.P.; Biallas, P.; Kirsch, S.F. An Unconventional Reaction of 2,2-Diazido Acylacetates with Amines. Eur. J. Org. Chem. 2017, 2017, 1526–1539. [Google Scholar] [CrossRef]
- Laitar, D.S.; Kramer, J.W.; Whiting, B.T.; Lobkovsky, E.B.; Coates, G.W. β-Amidoaldehydes via oxazoline hydroformylation. Chem. Commun. 2009, 5704–5706. [Google Scholar] [CrossRef]
- Leyerer, K.; Koppermann, S.; Ducho, C. Solid Phase-Supported Synthesis of Muraymycin Analogues. Eur. J. Org. Chem. 2019, 2019, 7420–7431. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conjugate a | pH b | t0.5 (h) | ON Released at Equilibrium (%) |
|---|---|---|---|---|
| 1 c | C1* | 4 | 5.80 ± 0.52 | 95.2 ± 2.1 |
| 2 c | C1* | 5 | 41.7 ± 2.3 | 95.8 ± 1.6 |
| 3 c | C1* | 6 | 222 ± 20 | quant. |
| 4 | C1 | 4 | 1.53 ± 0.25 | 91.5 ± 5.1 |
| 5 | C1 | 5 | 7.17 ± 0.77 | 89.7 ± 2.9 |
| 6 | C1 | 6 | 37.3 ± 3.3 | 95.7 ± 3.4 |
| 7 | C2 | 5 | 4.41 ± 0.17 | 59.2 ± 0.6 |
| 8 | C3 | 5 | 10.2 ± 0.81 | 94.8 ± 2.4 |
| 9 | C4 | 5 | 7.40 ± 1.03 | 63.9 ± 2.5 |
| Entry | R b | pH | t0.5 Decay (h) c | Equilibrium Constant K (l mol−1) c | Equilibrium Yield (%) c |
|---|---|---|---|---|---|
| 1 a | BzNHCH2 | 4 | 16.1 ± 0.7 | 4958 ± 50 | 82 |
| 2 a | “ | 5 | 75.5 ± 16.3 | ||
| 3 a | “ | 6 | n/a | ||
| 4 | BzNHCH2CH2 | 4 | 5.28 ± 0.58 | 2398 ± 425 | 75 |
| 5 | “ | 5 | 28.5 ± 2.2 | ||
| 6 | “ | 6 | 310 ± 34 |
| Conjugate | Aldehyde | Aldehyde Excess (Equiv) | Isolated Yield d | Observed Molecular Mass | Calculated Molecular Mass |
|---|---|---|---|---|---|
| C1 | P1 | 8 | 25% | 9999.4 a | 9999.8 |
| C2 | P2 | 8 | 19% | 9006.2 b | 9006.3 |
| C3 | 8 | 5 | 47% | 8851.1 c | 8852.7 |
| C4 | PNA1 | 2 | 43% | 8860.4 c | 8859.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aho, A.; Äärelä, A.; Korhonen, H.; Virta, P. Expanding the Scope of the Cleavable N-(Methoxy)oxazolidine Linker for the Synthesis of Oligonucleotide Conjugates. Molecules 2021, 26, 490. https://doi.org/10.3390/molecules26020490
Aho A, Äärelä A, Korhonen H, Virta P. Expanding the Scope of the Cleavable N-(Methoxy)oxazolidine Linker for the Synthesis of Oligonucleotide Conjugates. Molecules. 2021; 26(2):490. https://doi.org/10.3390/molecules26020490
Chicago/Turabian StyleAho, Aapo, Antti Äärelä, Heidi Korhonen, and Pasi Virta. 2021. "Expanding the Scope of the Cleavable N-(Methoxy)oxazolidine Linker for the Synthesis of Oligonucleotide Conjugates" Molecules 26, no. 2: 490. https://doi.org/10.3390/molecules26020490
APA StyleAho, A., Äärelä, A., Korhonen, H., & Virta, P. (2021). Expanding the Scope of the Cleavable N-(Methoxy)oxazolidine Linker for the Synthesis of Oligonucleotide Conjugates. Molecules, 26(2), 490. https://doi.org/10.3390/molecules26020490