
Sonochemical Reaction of Bifunctional Molecules on Silicon (111) Hydride Surface




Abstract
:1. Introduction
2. Materials and Methods
2.1. Thermal Reaction
2.2. Sonochemical Reaction
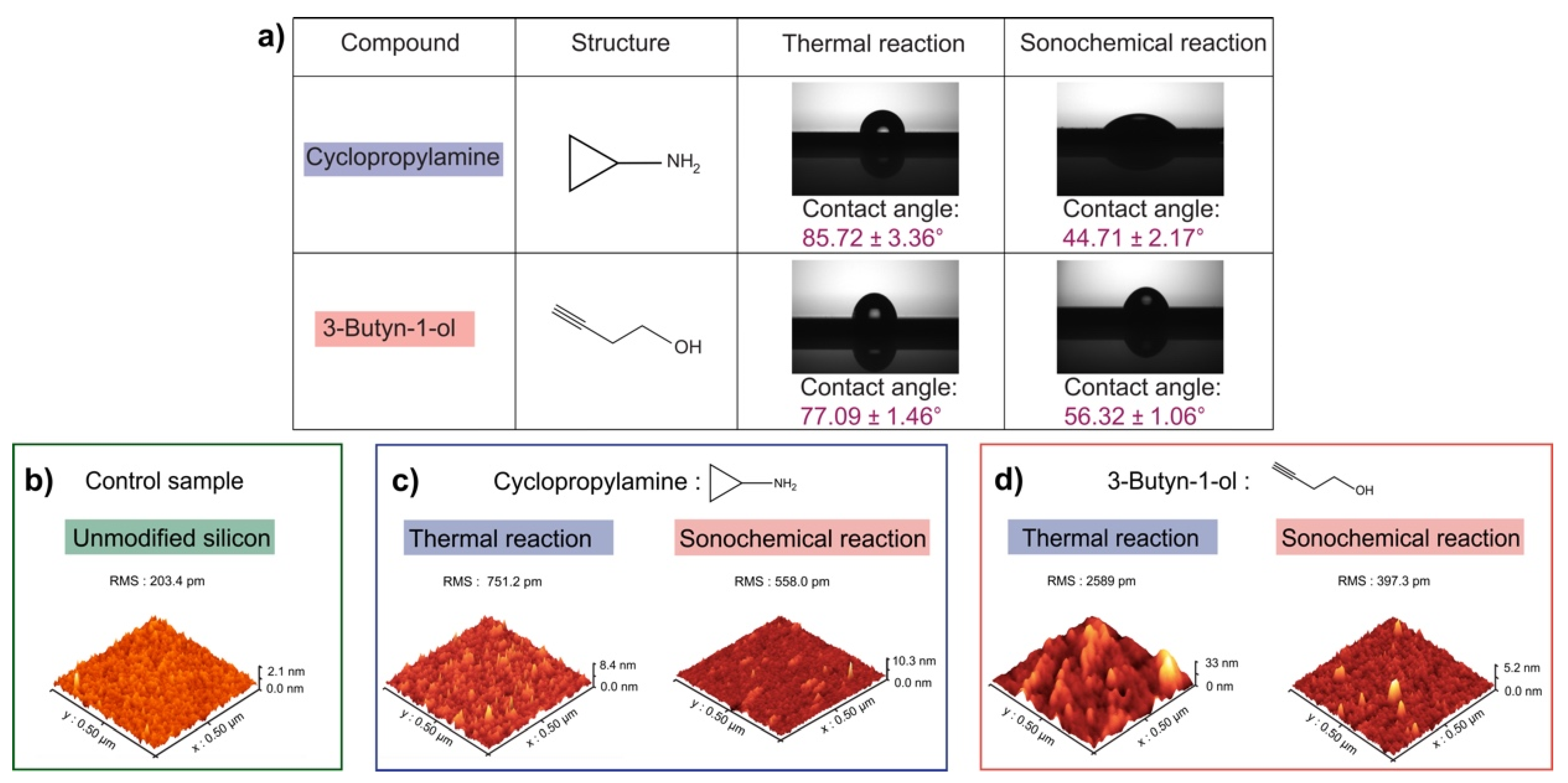
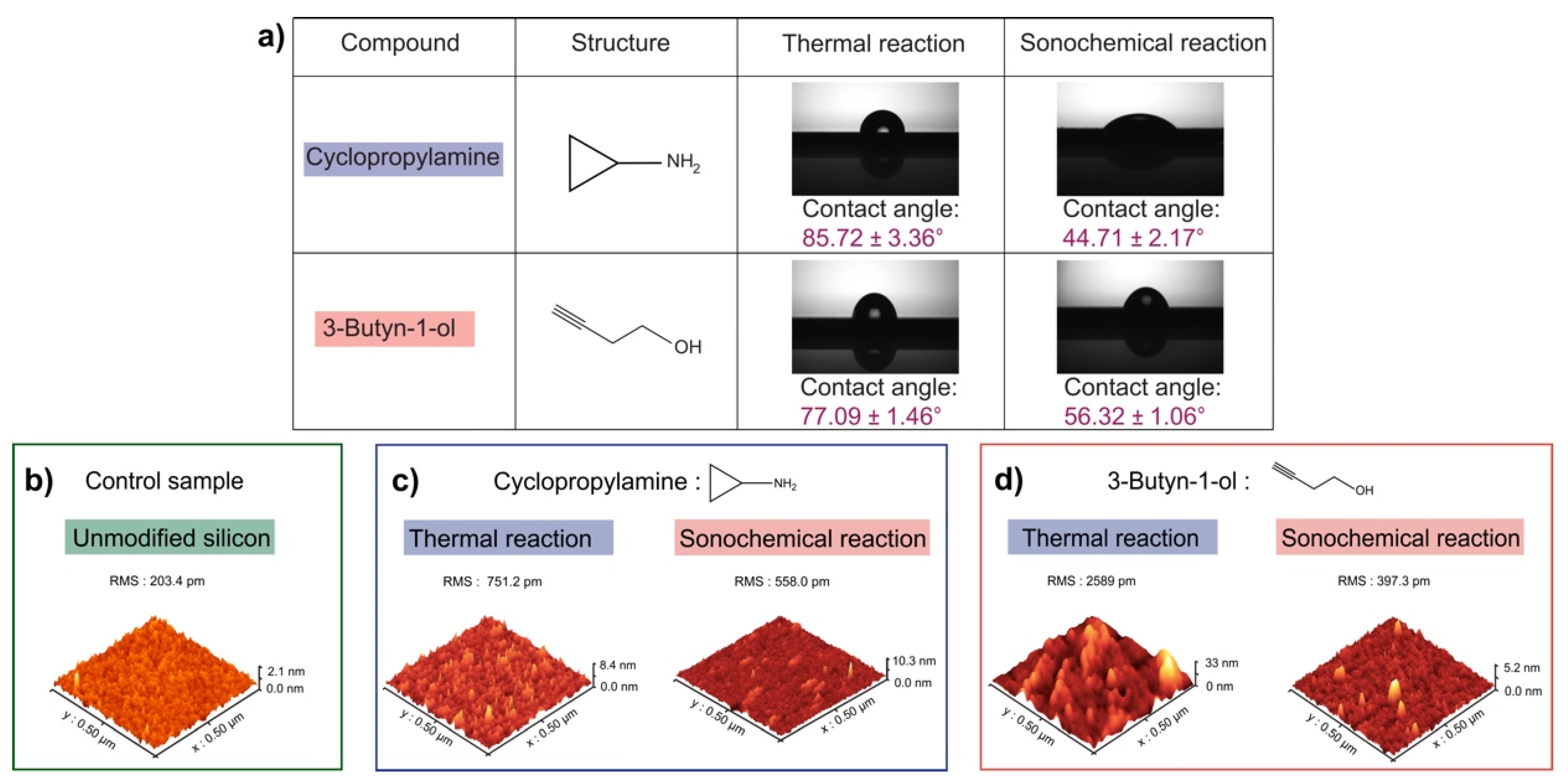
2.3. Contact Angle Measurements
2.4. Atomic Force Microscopy Measurements (AFM)
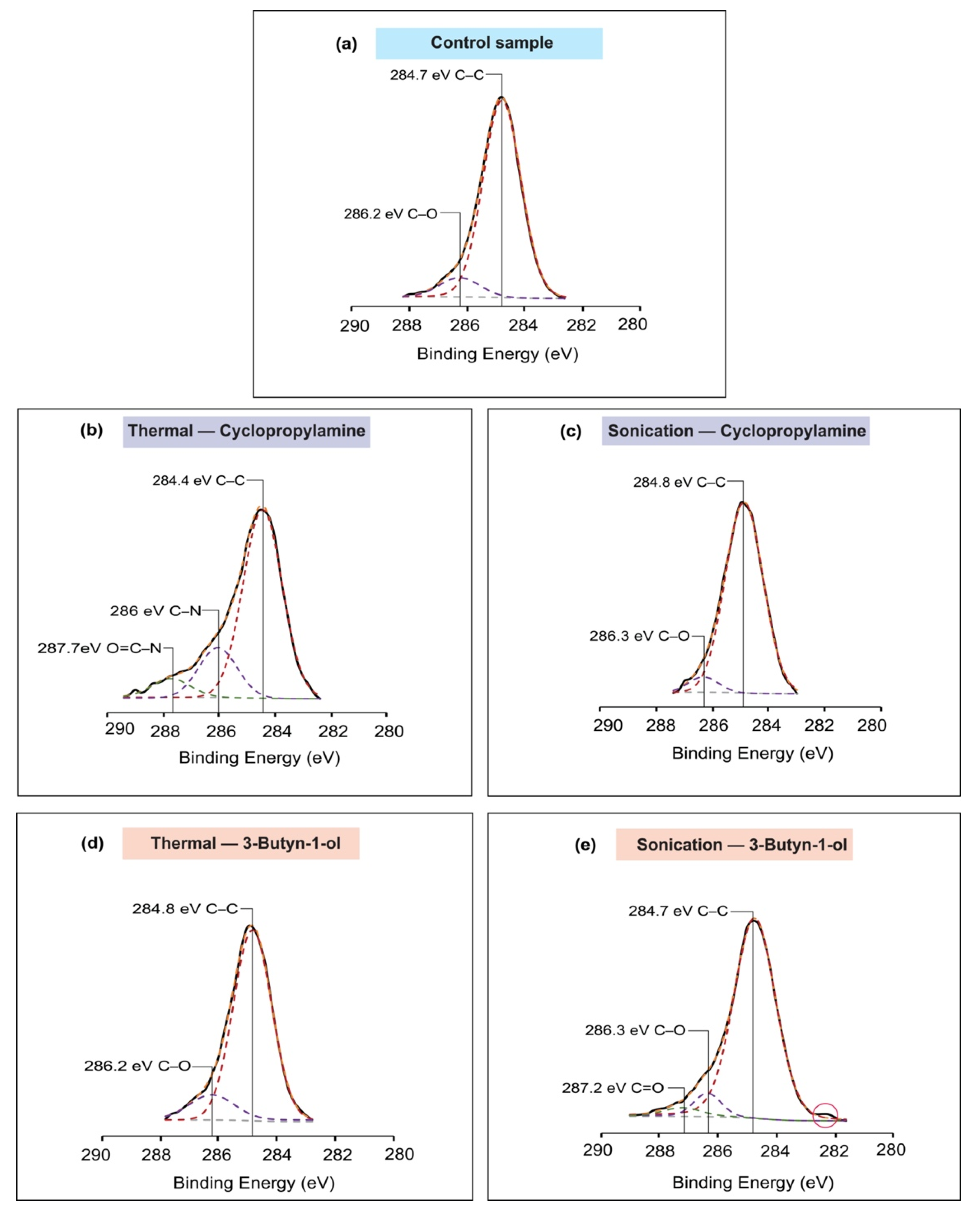
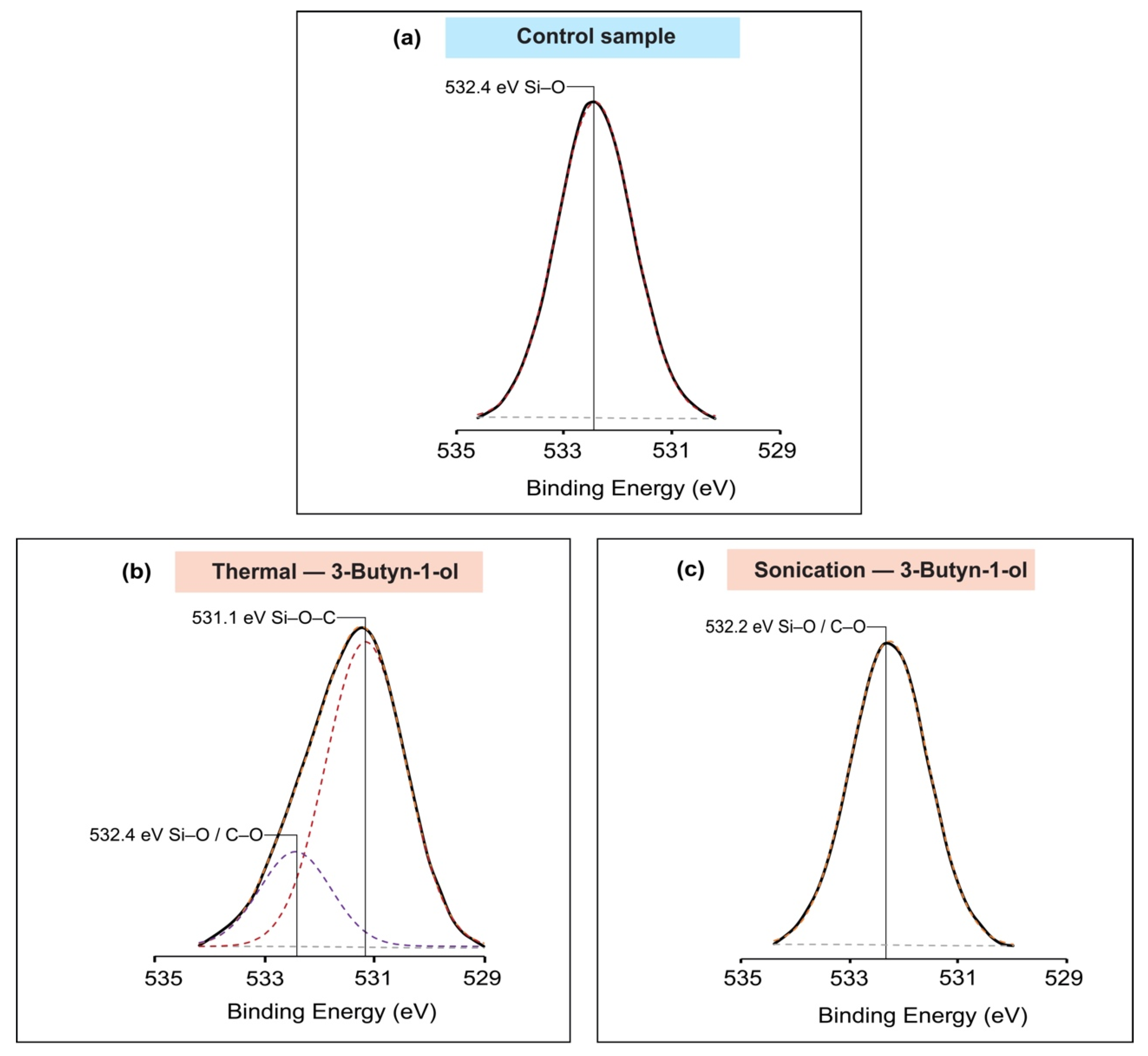
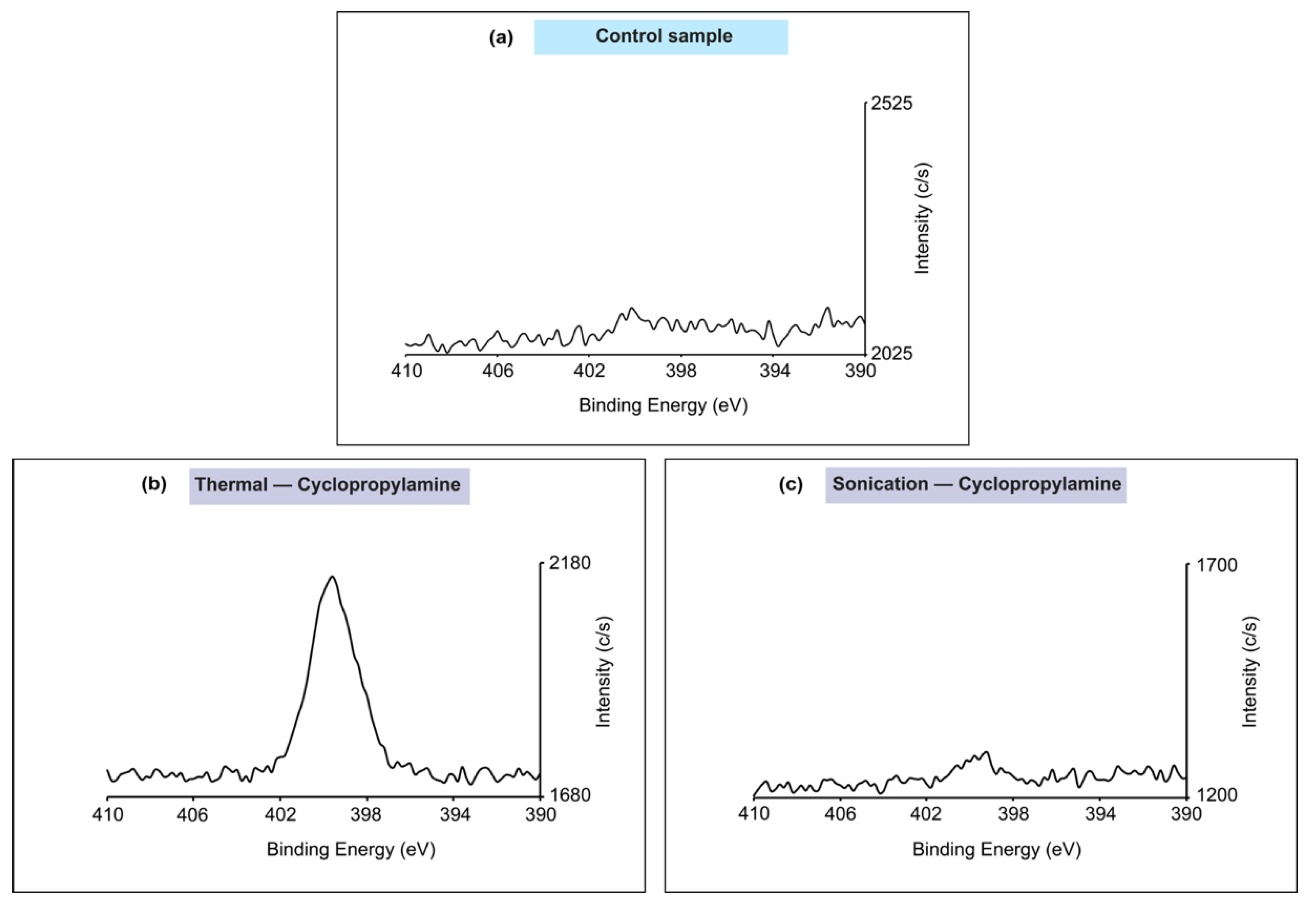
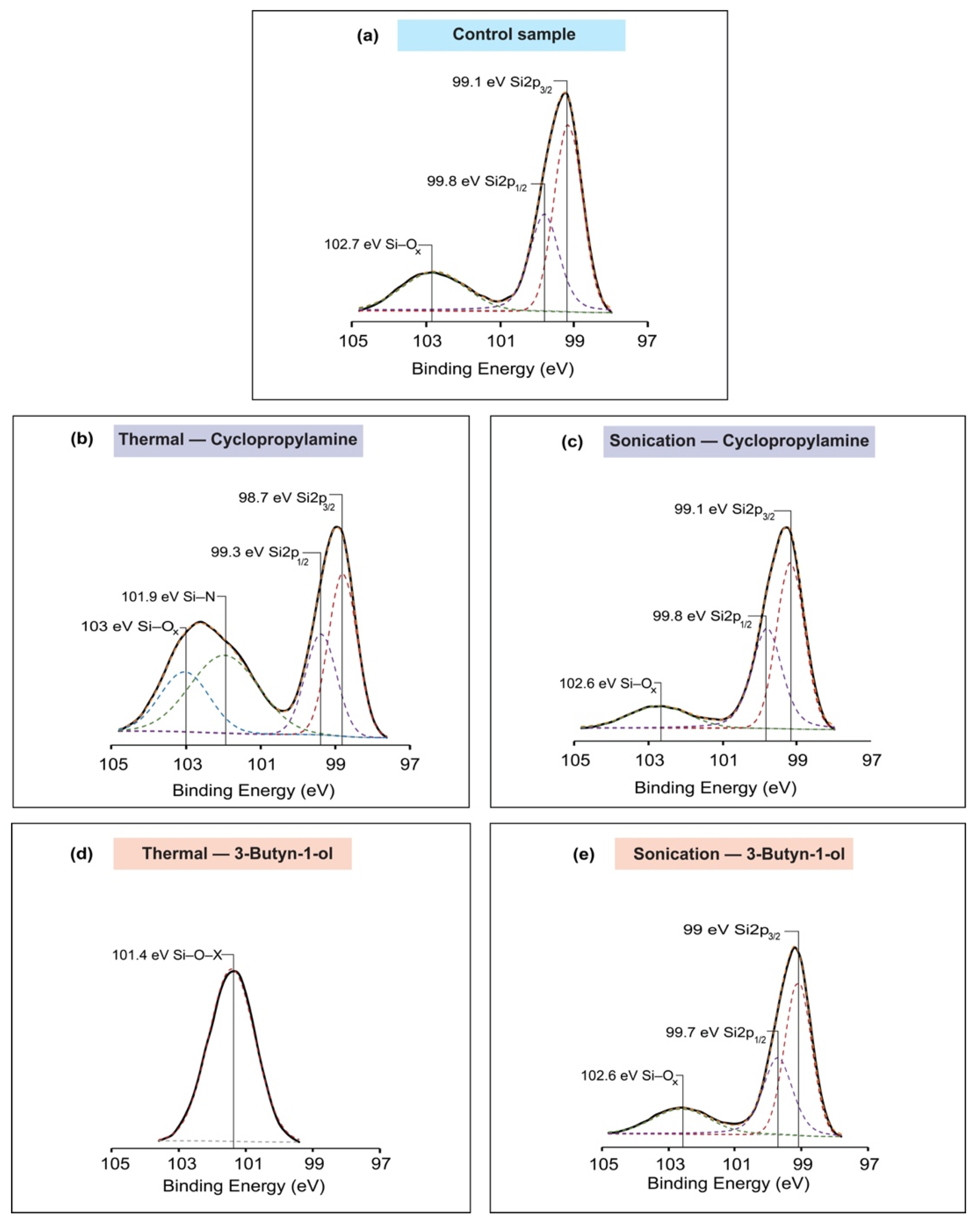
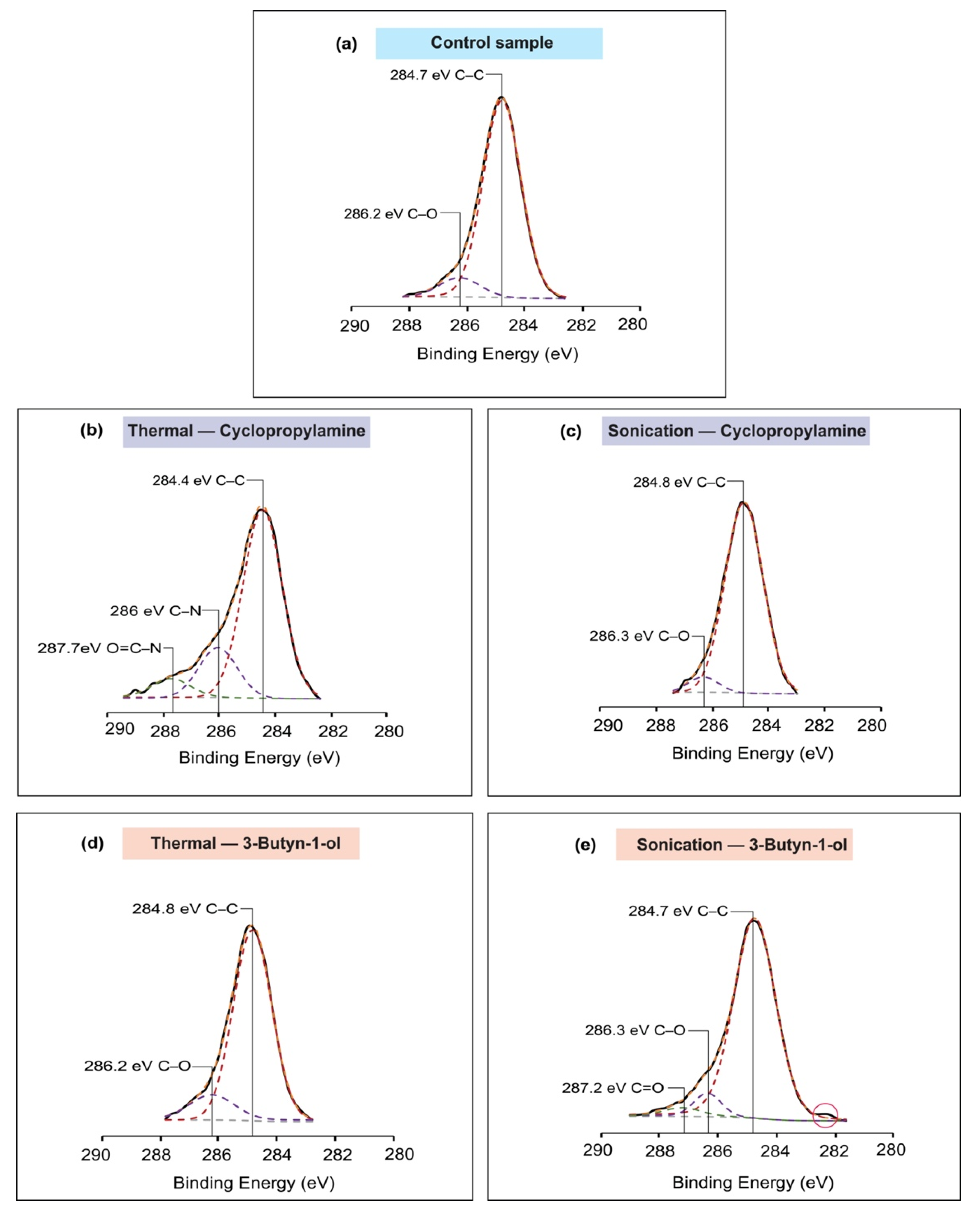
2.5. X-ray Photoelectron Spectroscopy (XPS)
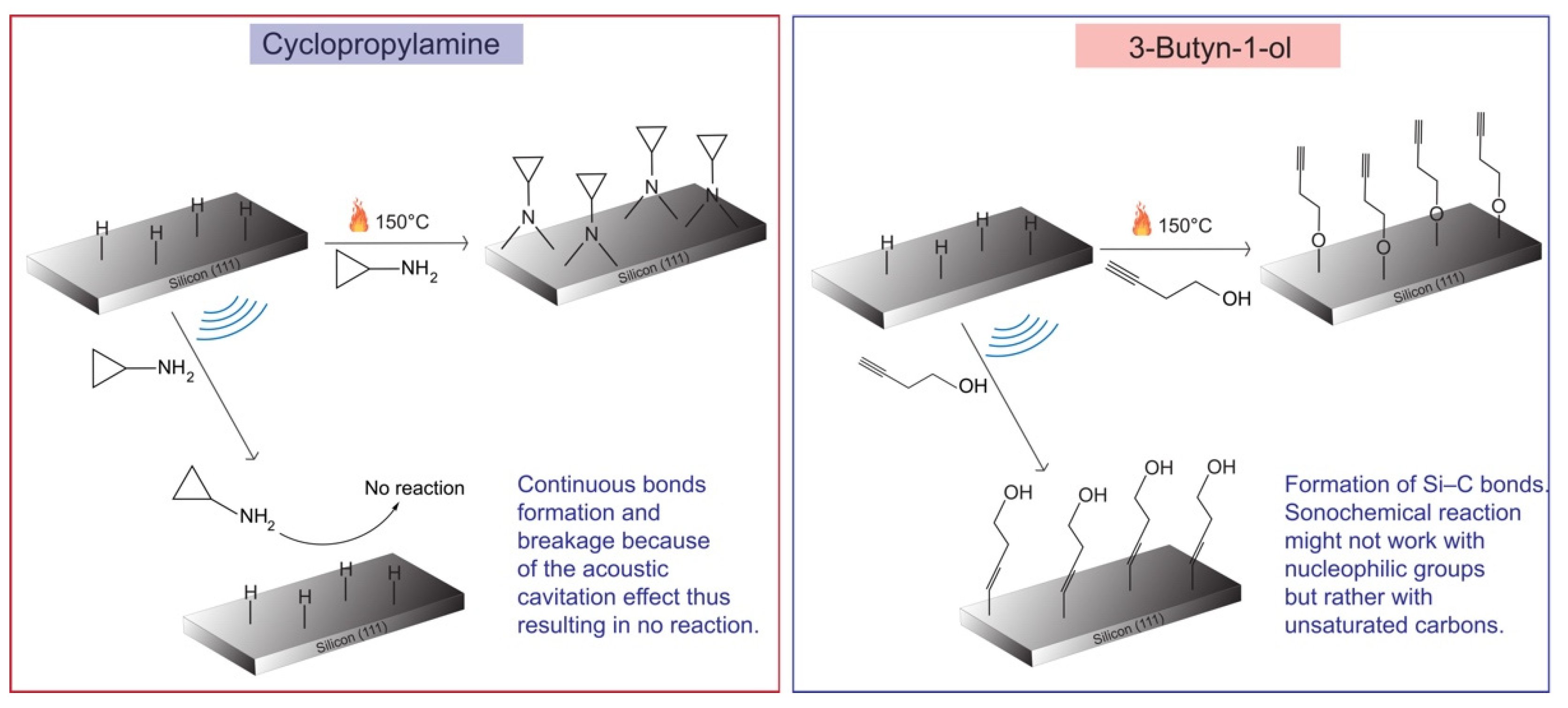
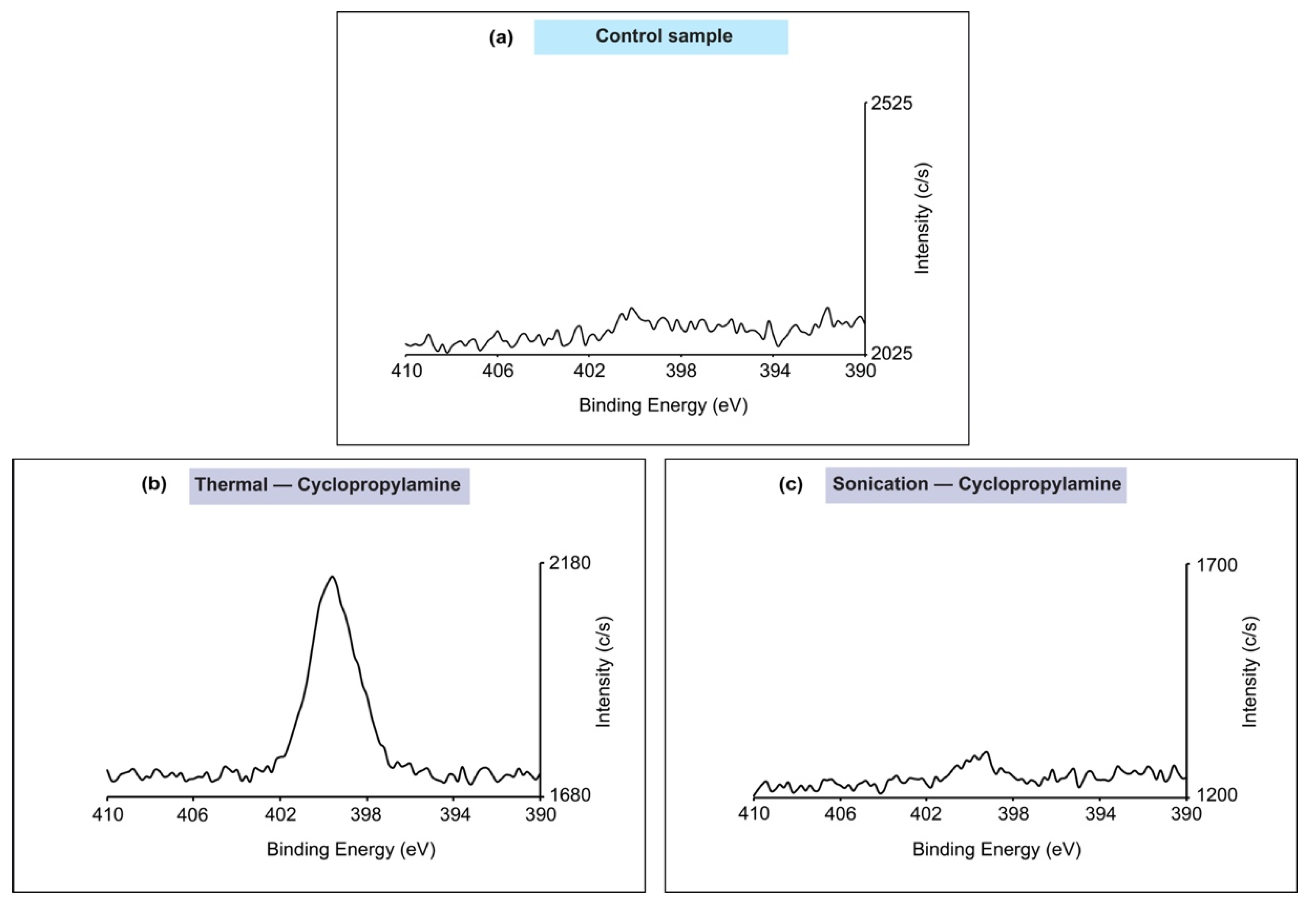
3. Results
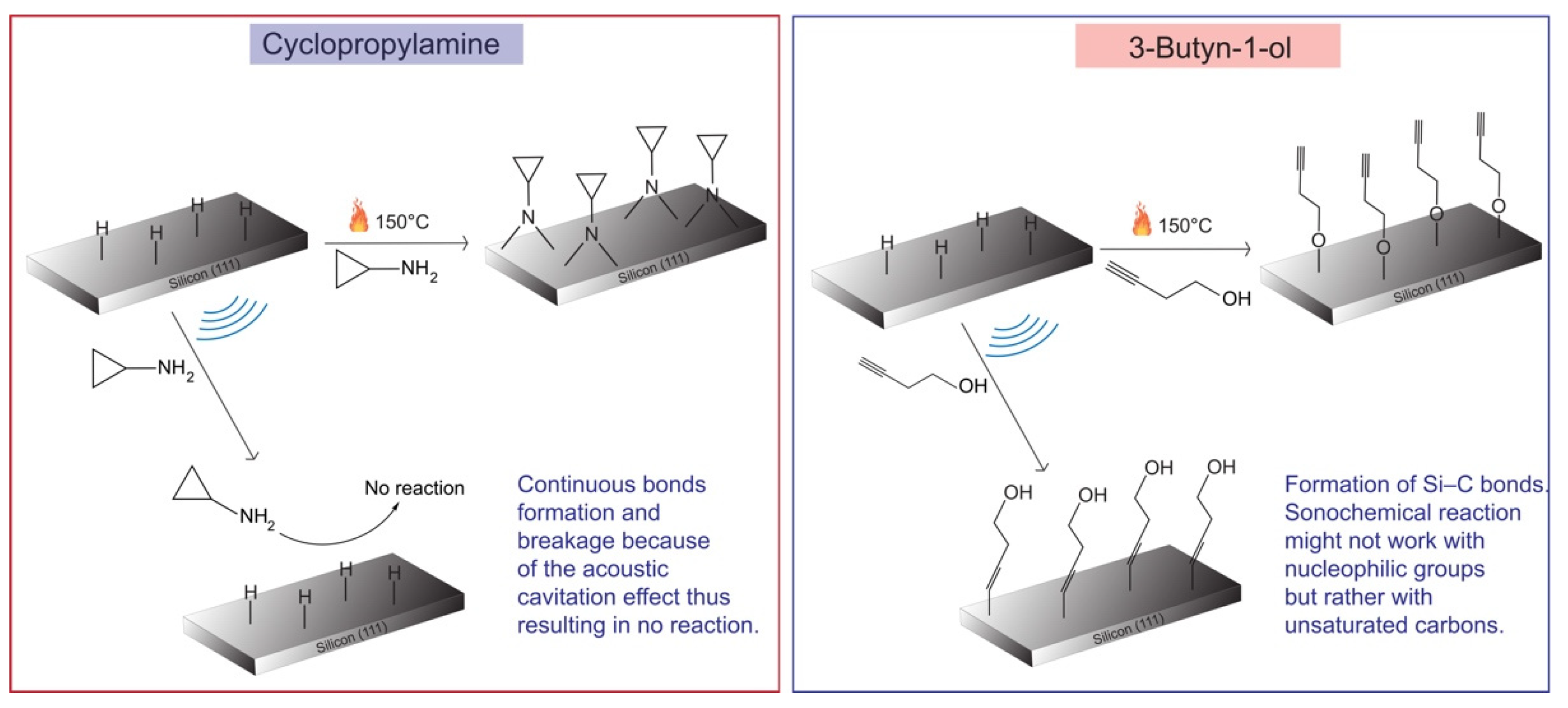
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Lang, W. Silicon microstructuring technology. Mater. Sci. Eng. R. Rep. 1996, 17, 1–55. [Google Scholar] [CrossRef]
- Ieong, M.; Doris, B.; Kedzierski, J.; Rim, K.; Yang, M. Silicon Device Scaling to the Sub-10-nm Regime. Science 2004, 306, 2057–2060. [Google Scholar] [CrossRef] [PubMed]
- Bond, R.; McAuliffe, J.C. Silicon biotechnology: New opportunities for carbohydrate science. Aust. J. Chem. 2003, 56, 7–11. [Google Scholar] [CrossRef]
- Morse, D.E. Silicon biotechnology: Harnessing biological silica production to construct new materials. TRENDS Biotechnol. 1999, 17, 230–232. [Google Scholar] [CrossRef]
- Zida, S.I.; Yang, C.-C.; Khung, Y.L.; Lin, Y.-D. Fabrication and Characterization of an Aptamer-Based N-type Silicon Nanowire FET Biosensor for VEGF Detection. J. Med. Biol. Eng. 2020, 40, 601–609. [Google Scholar] [CrossRef]
- Chhasatia, R.; Sweetman, M.J.; Prieto-Simon, B.; Voelcker, N.H. Performance optimisation of porous silicon rugate filter biosensor for the detection of insulin. Sens. Actuators B Chem. 2018, 273, 1313–1322. [Google Scholar] [CrossRef]
- Tran, D.P.; Pham, T.T.T.; Wolfrum, B.; Offenhäusser, A.; Thierry, B. CMOS-compatible silicon nanowire field-effect transistor biosensor: Technology development toward commercialization. Materials 2018, 11, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sieval, A.B.; Demirel, A.L.; Nissink, J.W.M.; Linford, M.R.; van der Maas, J.H.; de Jeu, W.H.; Zuilhof, H.; Sudhölter, E.J.R. Highly Stable Si−C Linked Functionalized Monolayers on the Silicon (100) Surface. Langmuir 1998, 14, 1759–1768. [Google Scholar] [CrossRef]
- Terry, J.; Linford, M.R.; Wigren, C.; Cao, R.; Pianetta, P.; Chidsey, C.E.D. Determination of the bonding of alkyl monolayers to the Si(111) surface using chemical-shift, scanned-energy photoelectron diffraction. Appl. Phys. Lett. 1997, 71, 1056–1058. [Google Scholar] [CrossRef]
- Vilan, A.; Yaffe, O.; Biller, A.; Salomon, A.; Kahn, A.; Cahen, D. Molecules on Si: Electronics with Chemistry. Adv. Mater. 2010, 22, 140–159. [Google Scholar] [CrossRef]
- Cerofolini, G.; Mascolo, D. A hybrid micro-nano-molecular route for nonvolatile memories. Semicond. Sci. Technol. 2006, 21, 1315. [Google Scholar] [CrossRef]
- Aswal, D.K.; Lenfant, S.; Guerin, D.; Yakhmi, J.V.; Vuillaume, D. Self assembled monolayers on silicon for molecular electronics. Anal. Chim. Acta 2006, 568, 84–108. [Google Scholar] [CrossRef]
- Roth, K.M.; Yasseri, A.A.; Liu, Z.; Dabke, R.B.; Malinovskii, V.; Schweikart, K.-H.; Yu, L.; Tiznado, H.; Zaera, F.; Lindsey, J.S.; et al. Measurements of Electron-Transfer Rates of Charge-Storage Molecular Monolayers on Si(100). Toward Hybrid Molecular/Semiconductor Information Storage Devices. J. Am. Chem. Soc. 2003, 125, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Bent, S.F. Organic functionalization of group IV semiconductor surfaces: Principles, examples, applications, and prospects. Surf. Sci. 2002, 500, 879–903. [Google Scholar] [CrossRef]
- Touahir, L.; Allongue, P.; Aureau, D.; Boukherroub, R.; Chazalviel, J.N.; Galopin, E.; Gouget-Laemmel, A.C.; de Villeneuve, C.H.; Moraillon, A.; Niedziółka-Jönsson, J.; et al. Molecular monolayers on silicon as substrates for biosensors. Bioelectrochemistry 2010, 80, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Masood, M.N.; Chen, S.; Carlen, E.T.; Berg, A.v.d. All-(111) Surface Silicon Nanowires: Selective Functionalization for Biosensing Applications. ACS Appl. Mater. Interfaces 2010, 2, 3422–3428. [Google Scholar] [CrossRef] [PubMed]
- Zazzera, L.A.; Evans, J.F.; Deruelle, M.; Tirrell, M.; Kessel, C.R.; McKeown, P. Bonding Organic Molecules to Hydrogen-Terminated Silicon Wafers. J. Electrochem. Soc. 1997, 144, 2184–2189. [Google Scholar] [CrossRef]
- Boukherroub, R.; Wojtyk, J.T.C.; Wayner, D.D.M.; Lockwood, D.J. Thermal Hydrosilylation of Undecylenic Acid with Porous Silicon. J. Electrochem. Soc. 2002, 149, H59. [Google Scholar] [CrossRef]
- Tung, J.; Tew, L.S.; Coluccini, C.; Lin, Y.-D.; Khung, Y.L. Grafting Behavior for the Resonating Variants of Ethynylaniline on Hydrogenated Silicon (100) Surfaces under Thermal Hydrosilylation. Chemistry 2018, 24, 13270–13277. [Google Scholar] [CrossRef]
- Huck, L.A.; Buriak, J.M. UV-Initiated Hydrosilylation on Hydrogen-Terminated Silicon (111): Rate Coefficient Increase of Two Orders of Magnitude in the Presence of Aromatic Electron Acceptors. Langmuir 2012, 28, 16285–16293. [Google Scholar] [CrossRef]
- Terry, J.; Mo, R.; Wigren, C.; Cao, R.; Mount, G.; Pianetta, P.; Linford, M.R.; Chidsey, C.E. Reactivity of the H–Si (111) surface. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 1997, 133, 94–101. [Google Scholar] [CrossRef]
- Boukherroub, R.; Petit, A.; Loupy, A.; Chazalviel, J.-N.; Ozanam, F. Microwave-Assisted Chemical Functionalization of Hydrogen-Terminated Porous Silicon Surfaces. J. Phys. Chem. B 2003, 107, 13459–13462. [Google Scholar] [CrossRef]
- Small, J.C.; Dam, H.M.; Siegel, J.L.; Crepinsek, A.J.; Neal, T.A.; Althoff, A.A.; Line, N.S.; Porter, L.A. Alkyl-functionalization of porous silicon via multimode microwave-assisted hydrosilylation. Polyhedron 2016, 114, 225–231. [Google Scholar] [CrossRef]
- Zhong, Y.L.; Bernasek, S.L. Mild and Efficient Functionalization of Hydrogen-Terminated Si(111) via Sonochemical Activated Hydrosilylation. J. Am. Chem. Soc. 2011, 133, 8118–8121. [Google Scholar] [CrossRef]
- Boukherroub, R.; Wayner, D.D.M.; Sproule, G.I.; Lockwood, D.J.; Canham, L.T. Stability Enhancement of Partially-Oxidized Porous Silicon Nanostructures Modified with Ethyl Undecylenate. Nano Lett. 2001, 1, 713–717. [Google Scholar] [CrossRef]
- Ciampi, S.; Harper, J.B.; Gooding, J.J. Wet chemical routes to the assembly of organic monolayers on silicon surfaces via the formation of Si–C bonds: Surface preparation, passivation and functionalization. Chem. Soc. Rev. 2010, 39, 2158–2183. [Google Scholar] [CrossRef] [Green Version]
- Cerofolini, G.F.; Galati, C.; Reina, S.; Renna, L. Functionalization of the (100) surface of hydrogen-terminated silicon via hydrosilation of 1-alkyne. Mater. Sci. Eng. C 2003, 23, 253–257. [Google Scholar] [CrossRef]
- De La Hoz, A.; Alcázar, J.; Carrillo, J.; Herrero, M.A.; Muñoz, J.D.M.; Prieto, P.; De Cózar, A.; Diaz-Ortiz, A. Reproducibility and Scalability of Microwave-Assisted Reactions; IntechOpen: London, UK, 2011. [Google Scholar]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Leighton, T.G. Bubble population phenomena in acoustic cavitation. Ultrason. Sonochem. 1995, 2, S123–S136. [Google Scholar] [CrossRef]
- Theerthagiri, J.; Madhavan, J.; Lee, S.J.; Choi, M.Y.; Ashokkumar, M.; Pollet, B.G. Sonoelectrochemistry for energy and environmental applications. Ultrason. Sonochem. 2020, 63, 104960. [Google Scholar] [CrossRef]
- Chatel, G.; Varma, R.S. Ultrasound and microwave irradiation: Contributions of alternative physicochemical activation methods to Green Chemistry. Green Chem. 2019, 21, 6043–6050. [Google Scholar] [CrossRef]
- Mason, T.J.; Peters, D. Practical Sonochemistry: Power Ultrasound Uses and Applications; Elsevier Science: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Colmenares, J.C. Sonication-induced pathways in the synthesis of light-active catalysts for photocatalytic oxidation of organic contaminants. ChemSusChem 2014, 7, 1512–1527. [Google Scholar] [CrossRef] [PubMed]
- Pinjari, D.; Prasad, K.; Gogate, P.; Mhaske, S.; Pandit, A. Synthesis of titanium dioxide by ultrasound assisted sol–gel technique: Effect of calcination and sonication time. Ultrason. Sonochem. 2015, 23, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Gedanken, A. Sonochemistry and its application to nanochemistry. Curr. Sci. 2003, 85, 1720–1722. [Google Scholar]
- Afreen, S.; Muthoosamy, K.; Manickam, S. Sono-nano chemistry: A new era of synthesising polyhydroxylated carbon nanomaterials with hydroxyl groups and their industrial aspects. Ultrason. Sonochem. 2019, 51, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Theerthagiri, J.; Lee, S.J.; Muthusamy, G.; Ashokkumar, M.; Choi, M.Y. Integrated technique of pulsed laser irradiation and sonochemical processes for the production of highly surface-active NiPd spheres. Chem. Eng. J. 2021, 411, 128486. [Google Scholar] [CrossRef]
- Tuziuti, T. Influence of sonication conditions on the efficiency of ultrasonic cleaning with flowing micrometer-sized air bubbles. Ultrason. Sonochem. 2016, 29, 604–611. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.; Collings, A.; Jameson, G. Effect of ultrasound on surface cleaning of silica particles. Int. J. Mineral. Process. 2000, 60, 101–113. [Google Scholar] [CrossRef]
- Theerthagiri, J.; Lee, S.J.; Karuppasamy, K.; Arulmani, S.; Veeralakshmi, S.; Ashokkumar, M.; Choi, M.Y. Application of advanced materials in sonophotocatalytic processes for the remediation of environmental pollutants. J. Hazard. Mater. 2021, 412, 125245. [Google Scholar] [CrossRef]
- Frederick, E.; Dickerson, P.N.; Zhong, Y.L.; Bernasek, S.L. Substituent Effects on the Kinetics of Bifunctional Styrene SAM Formation on H-Terminated Si. Langmuir 2014, 30, 7687–7694. [Google Scholar] [CrossRef]
- Khung, Y.L.; Ngalim, S.H.; Scaccabarozi, A.; Narducci, D. Thermal and UV Hydrosilylation of Alcohol-Based Bifunctional Alkynes on Si (111) surfaces: How surface radicals influence surface bond formation. Sci. Rep. 2015, 5, 11299. [Google Scholar] [CrossRef] [Green Version]
- Khung, Y.L.; Ngalim, S.H.; Meda, L.; Narducci, D. Preferential Formation of Si–O–C over Si–C Linkage upon Thermal Grafting on Hydrogen-Terminated Silicon (111). Chemistry 2014, 20, 15151–15158. [Google Scholar] [CrossRef]
- Lee, C.H.; Khung, Y.L. Molecular geometry influencing thermal-based nucleophilic reactions on silicon (111) hydride surfaces. Appl. Surf. Sci. 2020, 527, 146697. [Google Scholar] [CrossRef]
- Nonhebel, D.C. The chemistry of cyclopropylmethyl and related radicals. Chem. Soc. Rev. 1993, 22, 347–359. [Google Scholar] [CrossRef]
- Tung, J.; Ching, J.Y.; Ng, Y.M.; Tew, L.S.; Khung, Y.L. Grafting of Ring-Opened Cyclopropylamine Thin Films on Silicon (100) Hydride via UV Photoionization. ACS Appl. Mater. Interfaces 2017, 9, 31083–31094. [Google Scholar] [CrossRef] [PubMed]
- Ching, J.Y.; Huang, B.J.; Hsu, Y.-T.; Khung, Y.L. Anti-Adhesion Behavior from Ring-Strain Amine Cyclic Monolayers Grafted on Silicon (111) Surfaces. Sci. Rep. 2020, 10, 8758. [Google Scholar] [CrossRef] [PubMed]
- Mathioudaki, S.; Vandenabeele, C.R.; Tonneau, R.; Pflug, A.; Tennyson, J.; Lucas, S. Plasma polymerization of cyclopropylamine in a low-pressure cylindrical magnetron reactor: A PIC-MC study of the roles of ions and radicals. J. Vac. Sci. Technol. A 2020, 38, 033003. [Google Scholar] [CrossRef]
- Wang, Y.H.; Moitreyee, M.R.; Kumar, R.; Shen, L.; Zeng, K.Y.; Chai, J.W.; Pan, J.S. A comparative study of low dielectric constant barrier layer, etch stop and hardmask films of hydrogenated amorphous Si-(C, O, N). Thin Solid Film. 2004, 460, 211–216. [Google Scholar] [CrossRef]
- Laminack, W.; Gole, J.L.; White, M.G.; Ozdemir, S.; Ogden, A.G.; Martin, H.J.; Fang, Z.; Wang, T.-H.; Dixon, D.A. Synthesis of nanoscale silicon oxide oxidation state distributions: The transformation from hydrophilicity to hydrophobicity. Chem. Phys. Lett. 2016, 653, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Song, Y.; Zhu, C.; Fu, S.; Shi, Q.; Sun, Y.-M.; Jia, B.; Du, D.; Xu, Z.-L.; Lin, Y. Fluorescent silicon nanoparticles-based ratiometric fluorescence immunoassay for sensitive detection of ethyl carbamate in red wine. Sens. Actuators B Chem. 2018, 255, 2742–2749. [Google Scholar] [CrossRef]
- Eickhoff, T.; Medicherla, V.; Drube, W. Final state contribution to the Si 2p binding energy shift in SiO2/Si(100). J. Electron. Spectrosc. Relat. Phenom. 2004, 137–140, 85–88. [Google Scholar] [CrossRef]
- Seal, S.; Barr, T.L.; Krezoski, S.; Petering, D. Surface modification of silicon and silica in biological environment: An X-ray photoelectron spectroscopy study. Appl. Surf. Sci. 2001, 173, 339–351. [Google Scholar] [CrossRef]
- Yoon, S.J.; Ryu, J.-H.; Ismail, M.; Chen, Y.-C.; Chang, Y.-F.; Yun, M.J.; Kim, H.-D.; Kim, S. Compliance current and temperature effects on non-volatile memory switching and volatile switching dynamics in a Cu/SiOx/p++-Si device. Appl. Phys. Lett. 2019, 115, 212102. [Google Scholar] [CrossRef]
- Biniak, S.; Szymański, G.; Siedlewski, J.; Świątkowski, A. The characterization of activated carbons with oxygen and nitrogen surface groups. Carbon 1997, 35, 1799–1810. [Google Scholar] [CrossRef]
- Kubala-Kukuś, A.; Banaś, D.; Stabrawa, I.; Szary, K.; Sobota, D.; Majewska, U.; Wudarczyk-Moćko, J.; Braziewicz, J.; Pajek, M. Analysis of Ti and TiO2 nanolayers by total reflection X-ray photoelectron spectroscopy. Spectrochim. Acta Part B At. Spectrosc. 2018, 145, 43–50. [Google Scholar] [CrossRef]
- Ganguly, A.; Sharma, S.; Papakonstantinou, P.; Hamilton, J. Probing the Thermal Deoxygenation of Graphene Oxide Using High-Resolution In Situ X-ray-Based Spectroscopies. J. Phys. Chem. C 2011, 115, 17009–17019. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Scudiero, L.; Espinal, J.; McEwen, J.-S.; Garcia-Perez, M. Improving the deconvolution and interpretation of XPS spectra from chars by ab initio calculations. Carbon 2016, 110, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Effenberger, F.; Götz, G.; Bidlingmaier, B.; Wezstein, M. Photoactivated preparation and patterning of self-assembled monolayers with 1-alkenes and aldehydes on silicon hydride surfaces. Angew. Chem. Int. Ed. 1998, 37, 2462–2464. [Google Scholar] [CrossRef]
- Indrawirawan, S.; Sun, H.; Duan, X.; Wang, S. Low temperature combustion synthesis of nitrogen-doped graphene for metal-free catalytic oxidation. J. Mater. Chem. A 2015, 3, 3432–3440. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, X.; Gu, H.; Li, F.; Huang, W.; Liang, L.; Ye, Z. Highly selective colorimetric sensing of Cu2+ using a Schiff base derivative immobilized on polyvinyl alcohol microspheres. New J. Chem. 2018, 42, 11682–11688. [Google Scholar] [CrossRef]
- Kumar, B.; Asadi, M.; Pisasale, D.; Sinha-Ray, S.; Rosen, B.A.; Haasch, R.; Abiade, J.; Yarin, A.L.; Salehi-Khojin, A. Renewable and metal-free carbon nanofibre catalysts for carbon dioxide reduction. Nat. Commun. 2013, 4, 2819. [Google Scholar] [CrossRef]
- Yan, Y.; Lin, J.; Jiang, J.; Wang, H.; Qi, J.; Zhong, Z.; Cao, J.; Fei, W.; Feng, J. A general strategy to construct N-doped carbon-confined MoO2 and MnO for high-performance hybrid supercapacitors. Vacuum 2019, 165, 179–185. [Google Scholar] [CrossRef]
- Vallée, A.; Humblot, V.; Méthivier, C.; Pradier, C.M. Glutathione adsorption from UHV to the liquid phase at various pH on gold and subsequent modification of protein interaction. Surf. Interface Anal. 2008, 40, 395–399. [Google Scholar] [CrossRef]
- Gu, S.; Hsieh, C.-T.; Lin, T.-W.; Yuan, C.-Y.; Ashraf Gandomi, Y.; Chang, J.-K.; Li, J. Atomic layer oxidation on graphene sheets for tuning their oxidation levels, electrical conductivities, and band gaps. Nanoscale 2018, 10, 15521–15528. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Y.; Song, L.; Xing, W.; Lu, H.; Lv, P.; Jie, G. Flame retardancy and thermal degradation mechanism of epoxy resin composites based on a DOPO substituted organophosphorus oligomer. Polymer 2010, 51, 2435–2445. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Kusumoto, K.; Li, C.J. XPS Analysis of SiC Films Prepared by Radio Frequency Plasma Sputtering. Phys. Procedia 2012, 32, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Dai, Y.; Pan, K.; Jiang, B.; Tian, C.; Tian, G.; Zhou, W.; Wang, L.; Wang, X.; Fu, H. Recovery of silicon from sewage sludge for production of high-purity nano-SiO2. Chemosphere 2013, 90, 2332–2339. [Google Scholar] [CrossRef] [PubMed]
- Khung, Y.L.; Ngalim, S.H.; Scaccabarozzi, A.; Narducci, D. Formation of stable Si-O-C submonolayers on hydrogen-terminated silicon(111) under low-temperature conditions. Beilstein J. Nanotechnol. 2015, 6, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehner, A.; Steinhoff, G.; Brandt, M.S.; Eickhoff, M.; Stutzmann, M. Hydrosilylation of crystalline silicon (111) and hydrogenated amorphous silicon surfaces: A comparative x-ray photoelectron spectroscopy study. J. Appl. Phys. 2003, 94, 2289–2294. [Google Scholar] [CrossRef]
- López, G.P.; Castner, D.G.; Ratner, B.D. XPS O 1s binding energies for polymers containing hydroxyl, ether, ketone and ester groups. Surf. Interface Anal. 1991, 17, 267–272. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lee, H.S.; Yeom, H.W.; Ryoo, D.-Y.; Huh, S.-B.; Lee, J.-G. Nitrogen bonding structure in ultrathin silicon oxynitride films on Si(100) prepared by plasma nitridation. Phys. Rev. B 2004, 70, 165320. [Google Scholar] [CrossRef]
- Chang, J.P.; Green, M.L.; Donnelly, V.M.; Opila, R.L.; Eng, J., Jr.; Sapjeta, J.; Silverman, P.J.; Weir, B.; Lu, H.C.; Gustafsson, T.; et al. Profiling nitrogen in ultrathin silicon oxynitrides with angle-resolved x-ray photoelectron spectroscopy. J. Appl. Phys. 2000, 87, 4449–4455. [Google Scholar] [CrossRef]
- Ando, T.; Sumi, S.; Kawate, T.; Ichihara, J.; Hanafusa, T. Sonochemical switching of reaction pathways in slid–liquid two-phase reactions. J. Chem. Soc. Chem. Commun. 1984, 439–440. [Google Scholar] [CrossRef]
- Luche, J.L.; Einhorn, C.; Einhorn, J.; Sinisterra-Gago, J.V. Organic sonochenistry: A new interpretation and its consequences. Tetrahedron Lett. 1990, 31, 4125–4128. [Google Scholar] [CrossRef]
- Kegelaers, Y.; Eulaerts, O.; Reisse, J.; Segebarth, N. On the quantitative measure of a sonochemical effect in heterogeneous sonochemistry. Eur. J. Org. Chem. 2001, 2001, 3683–3688. [Google Scholar] [CrossRef]
- Martina, K.; Calsolaro, F.; Zuliani, A.; Berlier, G.; Chávez-Rivas, F.; Moran, M.J.; Luque, R.; Cravotto, G. Sonochemically-Promoted Preparation of Silica-Anchored Cyclodextrin Derivatives for Efficient Copper Catalysis. Molecules 2019, 24, 2490. [Google Scholar] [CrossRef] [Green Version]
- Dickens, M.J.; Luche, J.-L. Further evidence for the effect of ultrasonic waves on electron transfer processes—the case of the kornblum-russell reaction. Tetrahedron Lett. 1991, 32, 4709–4712. [Google Scholar] [CrossRef]
- Anto, T.; Bauchat, P.; Foucaud, A.; Fujita, M.; Kimura, T.; Sohmiya, H. Sonochemical switching from ionic to radical pathways in the reactions of styrene and trans-β-Methylstyrene with lead tetraacetate. Tetrahedron Lett. 1991, 32, 6379–6382. [Google Scholar] [CrossRef]
- Ando, T.; Kimura, T.; Levêque, J.-M.; Lorimer, J.P.; Luche, J.-L.; Mason, T.J. Sonochemical Reactions of Lead Tetracarboxylates with Styrene. J. Org. Chem. 1998, 63, 9561–9564. [Google Scholar] [CrossRef]
- Hickenboth, C.R.; Moore, J.S.; White, S.R.; Sottos, N.R.; Baudry, J.; Wilson, S.R. Biasing reaction pathways with mechanical force. Nature 2007, 446, 423–427. [Google Scholar] [CrossRef]
- Cravotto, G.; Cintas, P. Power ultrasound in organic synthesis: Moving cavitational chemistry from academia to innovative and large-scale applications. Chem. Soc. Rev. 2006, 35, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Vinatoru, M.; Mason, T.J. Jean-Louis Luche and the interpretation of sonochemical reaction mechanisms. Molecules 2021, 26, 755. [Google Scholar] [CrossRef] [PubMed]
- Paulusse, J.M.; Sijbesma, R.P. Reversible mechanochemistry of a PdII coordination polymer. Angew. Chem. Int. Ed. 2004, 43, 4460–4462. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Hessel, C.M.; Bogart, T.D.; Panthani, M.G.; Rasch, M.R.; Korgel, B.A. Room temperature hydrosilylation of silicon nanocrystals with bifunctional terminal alkenes. Langmuir 2013, 29, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, D.R. Ultrasonics and Acoustics. In Encyclopedia of Physical Science and Technology, 3rd ed.; Meyers, R.A., Ed.; Academic Press: New York, NY, USA, 2003; pp. 269–287. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Model | Binding Energy (eV) | Assignment | FWHM | Area under the Peak | ||
|---|---|---|---|---|---|---|
| Control Sample | Si2p | 99.1 | Si2p 3/2 | 0.9 | 6997.69 | |
| 99.8 | Si2p 1/2 | 0.93 | 4361.18 | |||
| 102.7 | Si–Ox | 2.02 | 3225.73 | |||
| C1s | 284.7 | C–C | 1.53 | 4289.18 | ||
| 286.2 | C–O | 1.65 | 458.30 | |||
| O1s | 532.4 | Si–O | 1.67 | 17,751.58 | ||
| Cyclopropylamine | Thermal reaction | Si2p | 98.7 | Si2p 3/2 | 0.91 | 2767.26 |
| 99.3 | Si2p 1/2 | 0.98 | 1855.55 | |||
| 101.9 | Si–N | 2.17 | 3135.52 | |||
| 103 | Si–Ox | 1.55 | 1730.05 | |||
| C1s | 284.4 | C–C | 1.64 | 3814.62 | ||
| 286 | C–N | 1.62 | 1000.39 | |||
| 287.7 | O=C–N | 1.66 | 439.55 | |||
| Sonochemical reaction | Si2p | 99.1 | Si2p 3/2 | 0.88 | 6271.15 | |
| 99.8 | Si2p 1/2 | 0.95 | 4823.99 | |||
| 102.6 | Si–Ox | 1.89 | 1720.59 | |||
| C1s | 284.8 | C–C | 1.57 | 2897.68 | ||
| 286.3 | C–O | 1.27 | 185.57 | |||
| 3-Butyn-1-ol | Thermal reaction | Si2p | 101.4 | Si–O–X | 1.64 | 3279.78 |
| C1s | 284.8 | C–C | 1.54 | 3182.46 | ||
| 286.2 | C–O | 1.78 | 469.47 | |||
| O1s | 531.1 | Si–O–C | 1.72 | 10,290.74 | ||
| 532.4 | Si–O/C–O | 1.53 | 2879.55 | |||
| Sonochemical reaction | Si2p | 99 | Si2p 3/2 | 0.95 | 5912.40 | |
| 99.7 | Si2p 1/2 | 1.05 | 3856.94 | |||
| 102.6 | Si–Ox | 1.96 | 1979.99 | |||
| C1s | 284.7 | C–C | 1.68 | 4057.23 | ||
| 286.3 | C–O | 1.07 | 280.74 | |||
| 287.2 | C=O | 1.46 | 140.44 | |||
| O1s | 532.2 | Si–O/C–O | 1.68 | 10,898.10 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zida, S.I.; Lin, Y.-D.; Khung, Y.L. Sonochemical Reaction of Bifunctional Molecules on Silicon (111) Hydride Surface. Molecules 2021, 26, 6166. https://doi.org/10.3390/molecules26206166
Zida SI, Lin Y-D, Khung YL. Sonochemical Reaction of Bifunctional Molecules on Silicon (111) Hydride Surface. Molecules. 2021; 26(20):6166. https://doi.org/10.3390/molecules26206166
Chicago/Turabian StyleZida, Serge Ismael, Yue-Der Lin, and Yit Lung Khung. 2021. "Sonochemical Reaction of Bifunctional Molecules on Silicon (111) Hydride Surface" Molecules 26, no. 20: 6166. https://doi.org/10.3390/molecules26206166
APA StyleZida, S. I., Lin, Y.-D., & Khung, Y. L. (2021). Sonochemical Reaction of Bifunctional Molecules on Silicon (111) Hydride Surface. Molecules, 26(20), 6166. https://doi.org/10.3390/molecules26206166