
Secondary Metabolites with Antimycobacterial Activities from One Actinobacteria: Herbidospora yilanensis

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
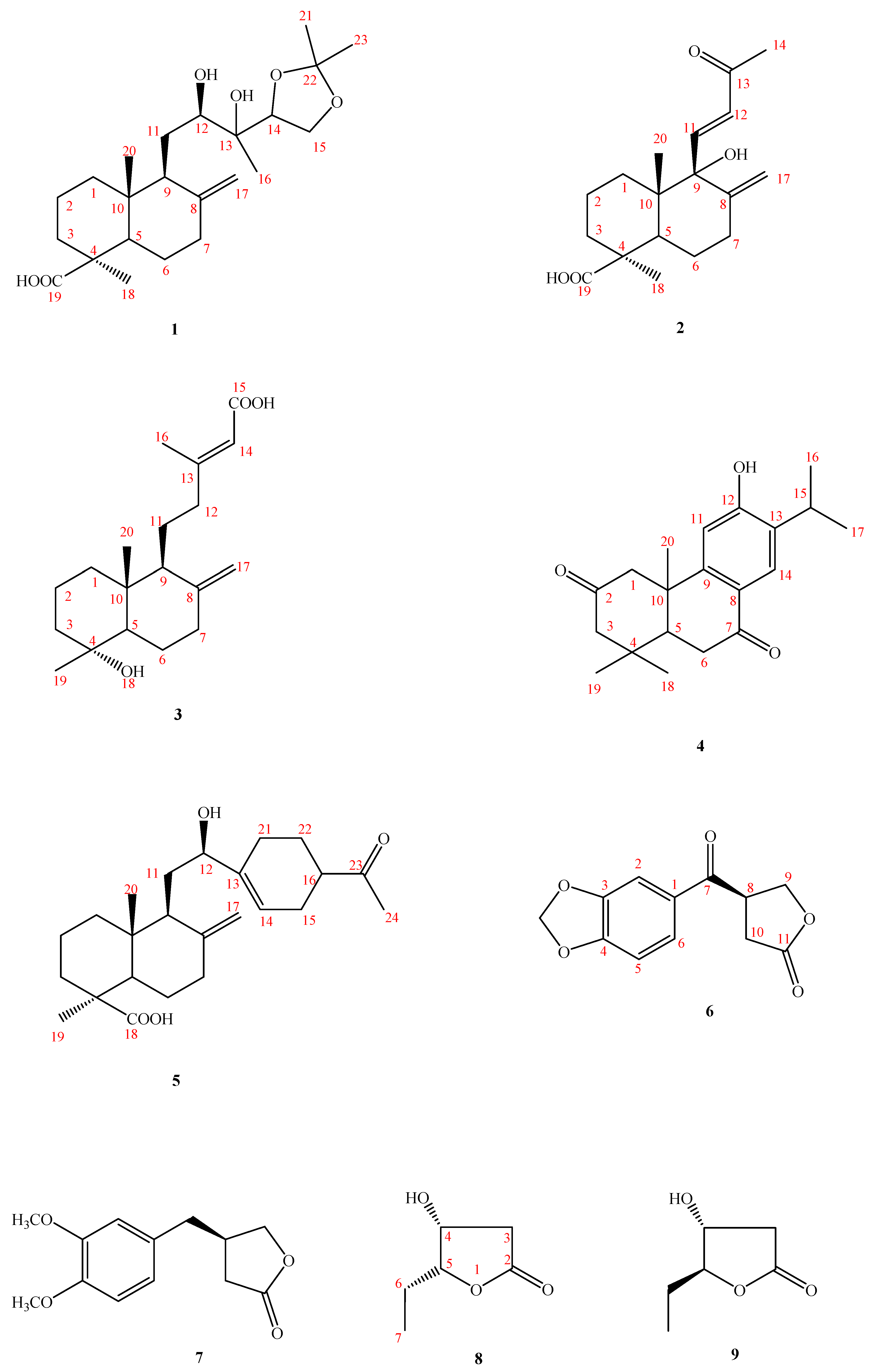
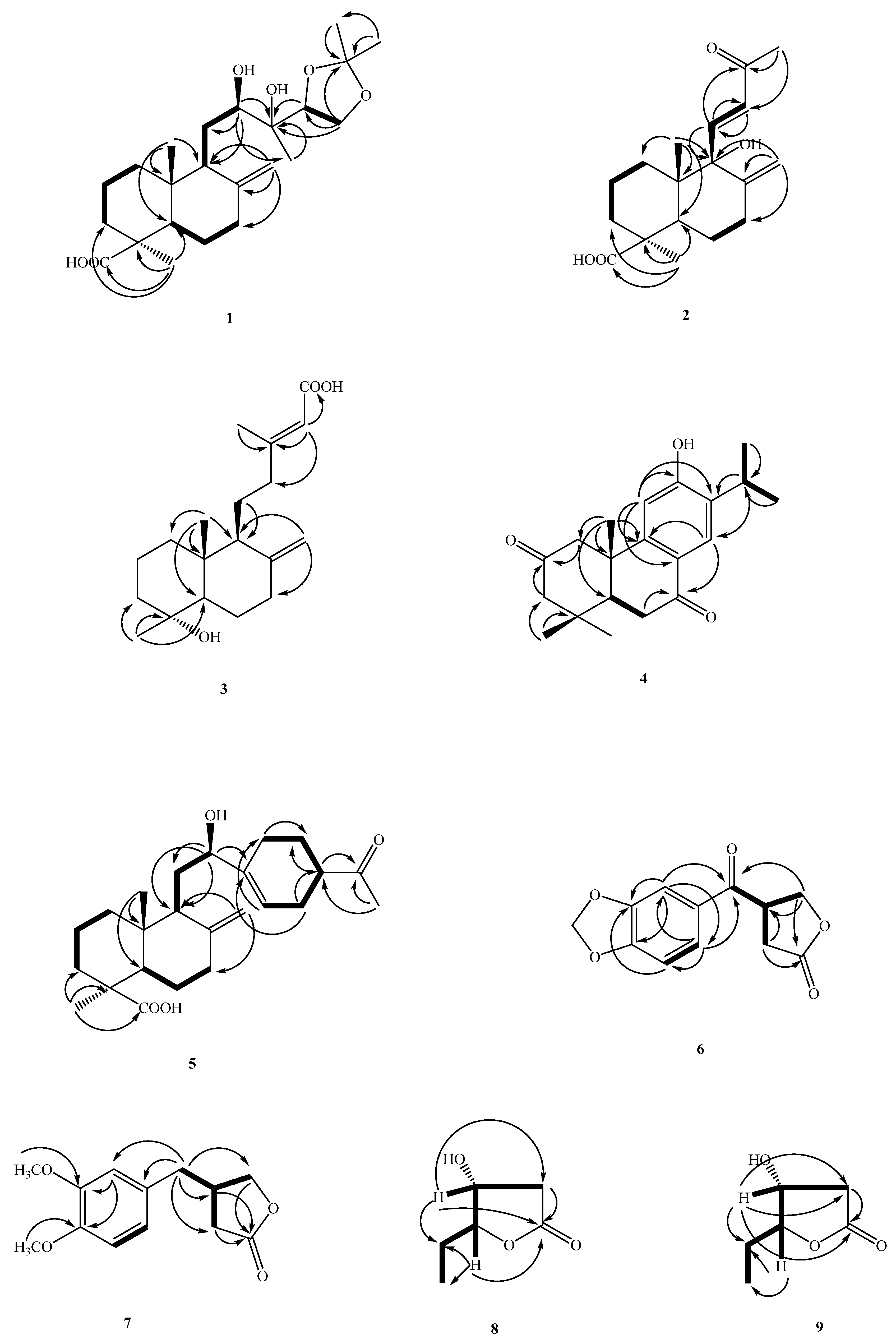
Structure Elucidation of Compounds
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Microorganism, Cultivation, and Preparation of the Actinobacteria Strain
3.3. Isolation and Characterization of Secondary Metabolites
3.4. Antitubercular Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osada, H. Fascinating bioactive compounds from actinomycetes. Actinomycetologica 1995, 9, 254–262. [Google Scholar] [CrossRef]
- Mythili, B.; Ayyappa Das, M.P. Studies on antimicrobial activity of Streptomyces spp. isolates from tea plantation soil. Res. J. Agric. Sci. 2011, 2, 104–106. [Google Scholar]
- Kuster, E. The Actinomycetes. In Soil Biology; Burges, A., Raw, F., Eds.; Academic Press: London, UK, 1968; pp. 111–124. [Google Scholar]
- Butler, M.S. The role of natural product chemistry in drug discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Kwon, O.W.; Park, J.S.; Kim, S.Y.; Kwon, H.C. Nocapyrones H–J, 3, 6-disubstituted α-pyrones from the marine actinomycete Nocardiopsis sp. KMF-001. Chem. Pharm. Bull. 2013, 61, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, M.; Yang, S.F.; Yuan, G.F. Herbidospora yilanensis sp. nov. and Herbidospora daliensis sp. nov., from sedimentInt. Int. J. Syst. Evol. Microbiol. 2010, 60, 1168–1172. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.L.; Fang, J.M.; Cheng, Y.S. Terpenoids and Flavonoids from Pseudotsuga wilsoniana. Phytochemistry 1998, 47, 845–850. [Google Scholar]
- Inoue, M.; Hasegawa, S.; Hirose, Y. Terpenoids from the seed of Platycladus orientalis. Phytochemistry 1985, 24, 1602–1604. [Google Scholar] [CrossRef]
- Carman, R.M.; Duffield, A.R. The Optical Rotation of Some C15-Oxygenated Labda-8(17),13-dienes. Aust. J. Chem. 1995, 48, 1357–1366. [Google Scholar] [CrossRef]
- Chang, H.M.; Cheng, K.P.; Chang, T.F.; Chow, H.F.; Chui, K.Y. Structure elucidation and total synthesis of new tanshinones isolated from Salvia miltiorrhiza Bunge (Danshen). J. Org. Chem. 1990, 55, 3537–3543. [Google Scholar] [CrossRef]
- Fang, J.M.; Sou, Y.C.; Chiu, Y.H.; Cheng, Y.S. Diterpenes from the bark of Juniperus chinensis. Phytochemistry 1993, 34, 1581–1584. [Google Scholar]
- Smith, S.M.; Hoang, G.L.; Pal, R.; Bani Khaled, M.O.; Pelter, L.S.W.; Zeng, X.C.; Takacs, J.M. γ-Selective Directed Catalytic Asymmetric Hydroboration of 1,1-Disubstituted Alkenes. Chem. Commun. 2012, 48, 12180–12182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, T.; Mizuguchi, H.; Tomioka, K.; Kora, K.S.R., VIII. Stereochemical Requirement for the Benzylic Oxidation of Lignan Lactone. A Highly Selective Synthesis of the Antitumor Lignan Lactone Steganacin by the Oxidation of Stegane. Chem. Pharm. Bull. 1985, 33, 609–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Sakurawi, K.; Ishii, H. Components of the Lauraceae family—I: New lactonic compounds from Litsea japoncia. Tetrahedron 1972, 28, 3757–3766. [Google Scholar] [CrossRef]
- Tanaka, H.; Nakamura, T.; Ichino, K.; Ito, K.; Tanaka, T. Butanolides from Litsea japonica. Phytochemistry 1990, 29, 857–859. [Google Scholar] [CrossRef]
- Wang, Z.M.; Zhang, X.L.; Barry Sharpless, K.; Sinha, S.C.; Sinha-Bagchi, A.; Keinan, E. A general approach to γ-lactones via osmium-catalyzed asymmetric dihydroxylation. Synthesis of (−)- and (+)-muricatacin. Tetrahedron Lett. 1992, 29, 6407–6410. [Google Scholar] [CrossRef]
- Inderlied, C.B.; Nash, K.A. Antibiotics in Laboratory Medicine, 4th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 1996; pp. 127–175. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| No | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 1.12 (m) 1.79 (m) | 1.11 (m) 1.63 (m) | 1.05 (d, J = 3.4) 1.74 (m) | 2.67 (d, J = 13.7, H-α) 2.90 (dd, J = 13.7, 2.0, H-β) | 1.12 (m) 1.80 (m) | ||
| 2 | 1.51 (m) 1.83 (m) | 1.46 (m) 1.77 (m) | 1.45 (td, J = 13.4, 3.4) 1.63 (m) | 1.51 (m) 1.82 (m) | 7.40 (d, J = 2.0) | 6.64 (d, J = 1.8) | |
| 3 | 1.04 (m) 2.13 (m) | 1.06 (m) 2.11 (m) | 1.35 (m) 1.77 (m) | 2.38 (dd, J = 13.1, 2.0, H-β) 2.43 (d, J = 13.1, H-α) | 1.05 (m) 2.14 (m) | ||
| 4 | |||||||
| 5 | 1.38 (dd, J = 12.0, 2.6) | 2.04 (dd, J = 12.6, 3.3) | 1.37 (m) | 2.42 (dd, J = 13.7, 3.8) | 1.38 (dd, J = 12.5, 2.6) | 6.88 (d, J = 8.0) | 6.79 (d, J = 8.1) |
| 6 | 1.86 (m) 2.00 (m) | 1.87 (m) 1.93 (m) | 1.36 (m) 1.95 (m) | 2.64 (dd, J = 17.9, 13.7, H-β) 2.79 (d, J = 17.9, 3.8, H-α) | 1.86 (m) 1.97 (m) | 7.48 (dd, J = 8.0, 2.0) | 6.67 (dd, J = 8.1, 1.8 |
| 7 | 1.94 (m) 2.39 (m) | 2.29 (m) 2.49 (m) | 1.98 (m) 2.41 (m) | 1.95 (m) 2.39 (m) | 2.70 (m) | ||
| 8 | 4.27 (m) | 2.81 (m) | |||||
| 9 | 1.98 (m) | 1.64 (m) | 1.99 (m) | 4.43 (dd, J = 9.2, 6.8) 4.57 (t, J = 9.2) | 4.02 (dd, J = 9.1, 6.0) 4.31 (dd, J = 9.1, 7.0) | ||
| 10 | 2.75 (dd, J = 17.6, 9.2) 3.00 (dd, J = 17.6, 7.6) | 2.27(dd, J = 17.5, 6.9) 2.58 (dd, J = 17.5, 8.2) | |||||
| 11 | 1.42 (m) 1.67 (m) | 7.17 (d, J = 15.8) | 1.52 (m) 1.64 (m) | 6.57 (s) | 1.52 (m) 1.55 (m) | ||
| 12 | 3.56 (d, J = 10.5) | 6.43 (d, J = 15.8) | 1.99 (m) 2.31 (m) | 6.37 (d, J = 16.0) | 3.99 (t, J = 5.6) | ||
| OH-12 | 6.18 (br s) | ||||||
| 13 | |||||||
| 14 | 4.08 (M) | 2.28 (s) | 5.66 (s) | 7.92 (s) | 5.65 (br s) | ||
| 15 | 3.92 (m) 3.99 (m) | 3.15 (sep, J = 6.8) | 2.19 (m) | ||||
| 16 | 1.25 (s) | 2.15 (s) | 1.25 (d, J = 6.8) | 2.56 (m) | |||
| 17 | 4.53 (br. s) 4.84 (br s) | 4.51 (br s) 4.87 (br s) | 1.25 (d, J = 6.8) | 4.44 (br s) 4.84 (br s) | |||
| 18 | 1.22 (s) | 1.27 (s) | 1.11 (s) | 1.22 (s) | |||
| 19 | 1.10 (s) | 1.01 (s) | |||||
| 20 | 0.59 (s) | 0.89 (s) | 0.64 (s) | 1.23 (s) | 0.57 (s) | ||
| 21 | 1.39 (s) | 2.07 (m) 2.12 (m) | |||||
| 22 | 1.57 (m) 2.00 (m) | ||||||
| 23 | 1.35 (s) | ||||||
| 24 | 2.16 (s) | ||||||
| OCH2O | 6.07 (s) | ||||||
| OMe-3 | 3.85 (s) | ||||||
| OMe-4 | 3.85 (s) |
| No | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| 1 | 39.0 | 32.8 | 38.2 | 53.2 | 39.1 | 129.8 | 130.7 |
| 2 | 19.9 | 19.2 | 20.5 | 209.2 | 19.8 | 108.1 | 111.8 |
| 3 | 37.9 | 37.6 | 42.9 | 56.0 | 37.9 | 148.8 | 149.1 |
| 4 | 44.2 | 44.1 | 72.5 | 38.7 | 44.1 | 152.8 | 147.9 |
| 5 | 56.2 | 74.4 | 56.9 | 49.0 | 56.2 | 108.3 | 111.4 |
| 6 | 26.0 | 24.2 | 23.4 | 35.9 | 26.0 | 124.9 | 120.0 |
| 7 | 38.7 | 32.5 | 37.8 | 196.9 | 38.6 | 194.1 | 38.6 |
| 8 | 148.0 | 149.5 | 147.6 | 124.0 | 148.5 | 41.9 | 37.3 |
| 9 | 51.6 | 79.9 | 55.9 | 153.5 | 51.8 | 69.2 | 72.6 |
| 10 | 40.1 | 43.1 | 40.3 | 42.8 | 40.1 | 31.1 | 34.2 |
| 11 | 25.4 | 148.9 | 21.8 | 109.6 | 30.5 | 175.3 | 176.8 |
| 12 | 74.4 | 130.2 | 40.0 | 158.9 | 73.9 | ||
| 13 | 73.9 | 197.8 | 163.7 | 133.8 | 140.9 | ||
| 14 | 79.5 | 28.2 | 114.3 | 126.9 | 119.4 | ||
| 15 | 65.1 | 169.8 | 26.8 | 26.5 | |||
| 16 | 20.7 | 19.2 | 22.2 | 47.3 | |||
| 17 | 107.0 | 107.1 | 22.4 | 106.6 | |||
| 18 | 29.0 | 29.0 | 32.4 | 29.0 | |||
| 19 | 182.6 | 183.1 | 23.1 | 22.5 | 182.6 | ||
| 20 | 12.9 | 16.5 | 13.9 | 24.3 | 12.9 | ||
| 21 | 26.4 | 23.9 | |||||
| 22 | 108.4 | 24.6 | |||||
| 23 | 25.4 | 211.4 | |||||
| 24 | 28.0 | ||||||
| OCH2O | 102.2 | ||||||
| OMe-3 | 55.9 | ||||||
| OMe-4 | 55.89 |
| Compounds | MIC (μM) a |
|---|---|
| herbidosporayilanensin A (1) | 16.6 |
| herbidosporayilanensin B (2) | 19.2 |
| herbidosporayilanensin C (3) | 40.8 |
| herbidosporayilanensin D (4) | 50.6 |
| herbidosporayilanensin E (5) | >621 |
| herbidosporayilanensin F (6) | 18.2 |
| herbidosporayilanensin G (7) | >543 |
| Ethambutol b | 30.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, M.-J.; Wu, M.-D.; Chen, J.-J.; Su, Y.-S.; Kuo, Y.-H. Secondary Metabolites with Antimycobacterial Activities from One Actinobacteria: Herbidospora yilanensis. Molecules 2021, 26, 6236. https://doi.org/10.3390/molecules26206236
Cheng M-J, Wu M-D, Chen J-J, Su Y-S, Kuo Y-H. Secondary Metabolites with Antimycobacterial Activities from One Actinobacteria: Herbidospora yilanensis. Molecules. 2021; 26(20):6236. https://doi.org/10.3390/molecules26206236
Chicago/Turabian StyleCheng, Ming-Jen, Ming-Der Wu, Jih-Jung Chen, Yung-Shun Su, and Yueh-Hsiung Kuo. 2021. "Secondary Metabolites with Antimycobacterial Activities from One Actinobacteria: Herbidospora yilanensis" Molecules 26, no. 20: 6236. https://doi.org/10.3390/molecules26206236