
Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2 †

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
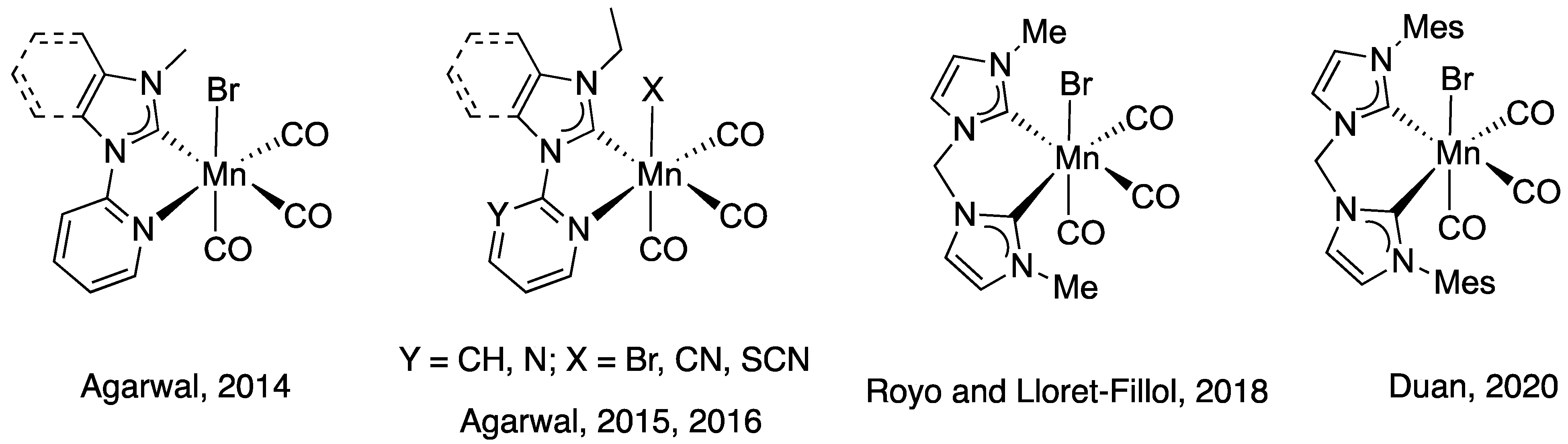
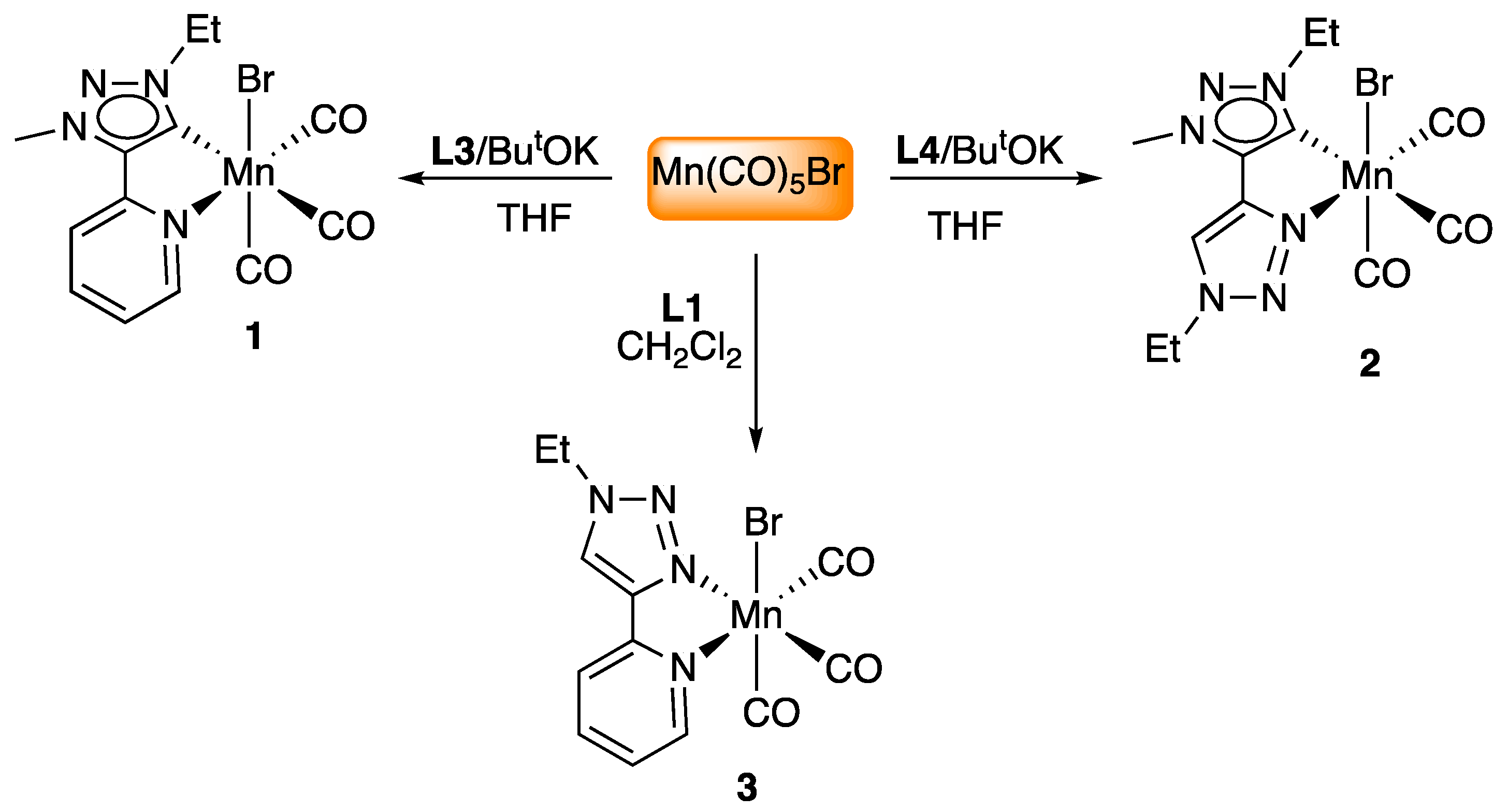
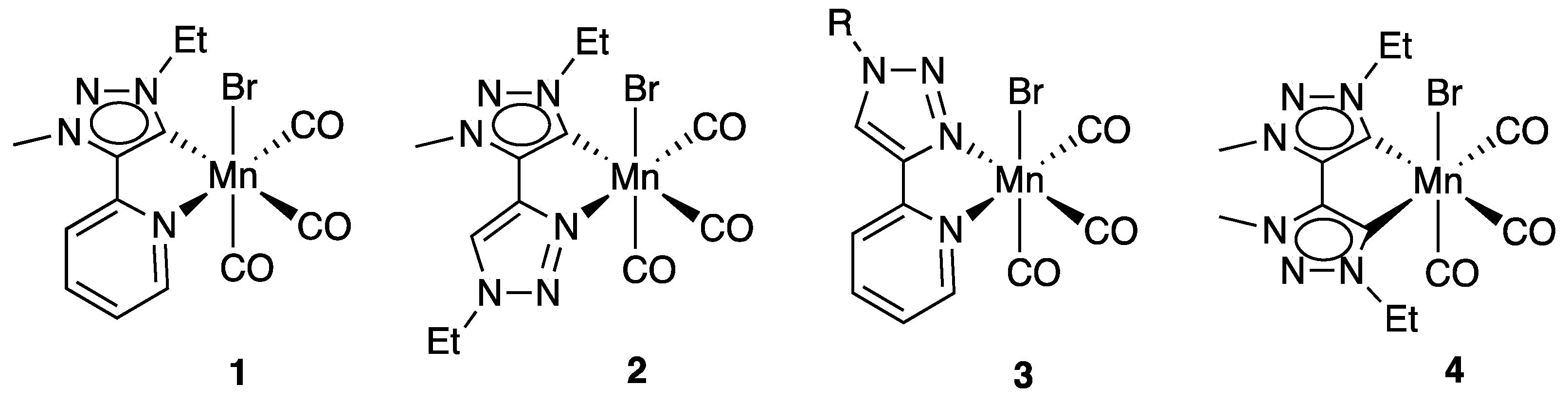
2.1. Synthesis and Characterization of Mn Complexes
2.2. Electrocatalytic Reduction of CO2 Mediated by Mn Complexes 1–4
2.2.1. CV Studies Performed under Nitrogen Atmosphere
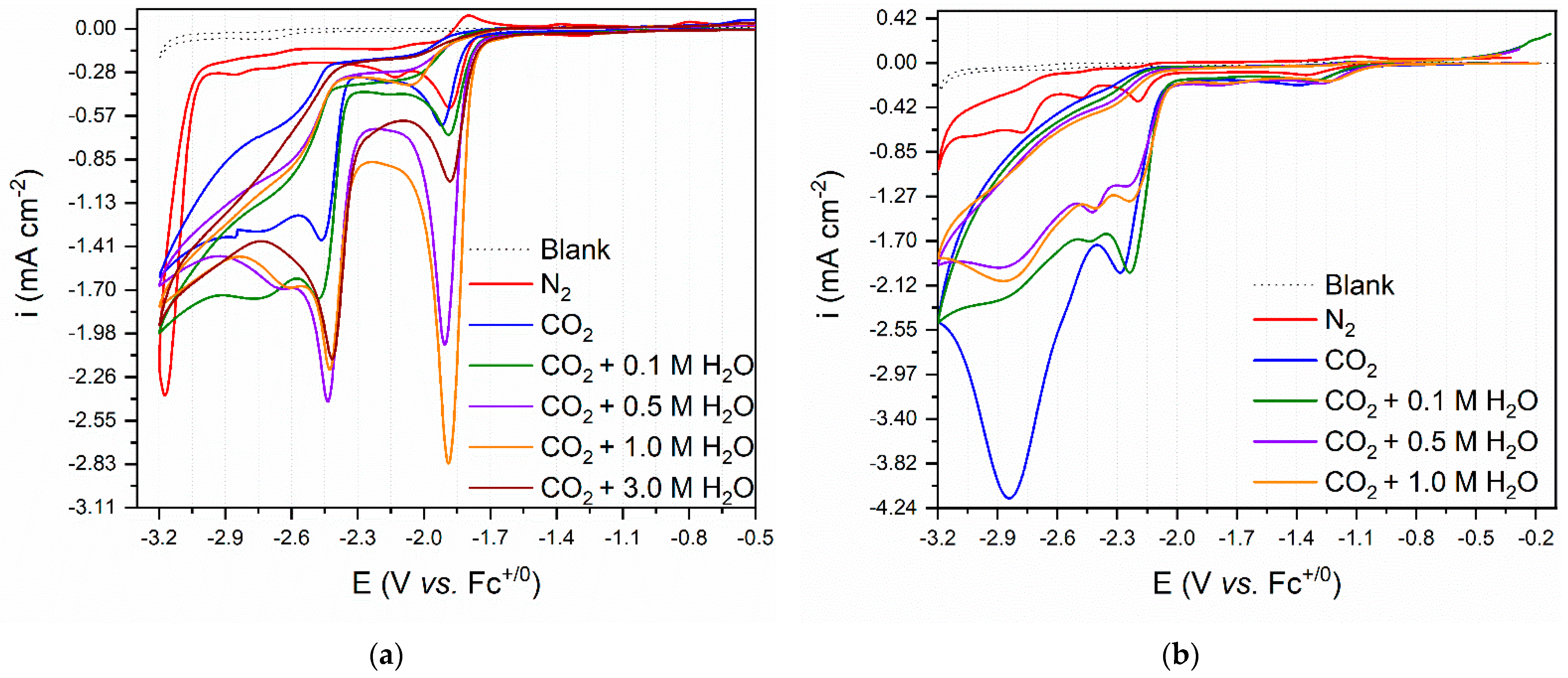
2.2.2. CV Studies Performed under CO2 Atmosphere
2.2.3. Bulk Electrolysis Experiments
3. Materials and Methods
3.1. General Considerations
3.2. Synthetic Procedures
3.2.1. Preparation of Triazolium Bromide Salt L3
3.2.2. Preparation of Triazolium Bromide Salt L4
3.2.3. Preparation of Complexes 1 and 2
3.2.4. Preparation of Complex 3
3.3. Electrochemical Studies
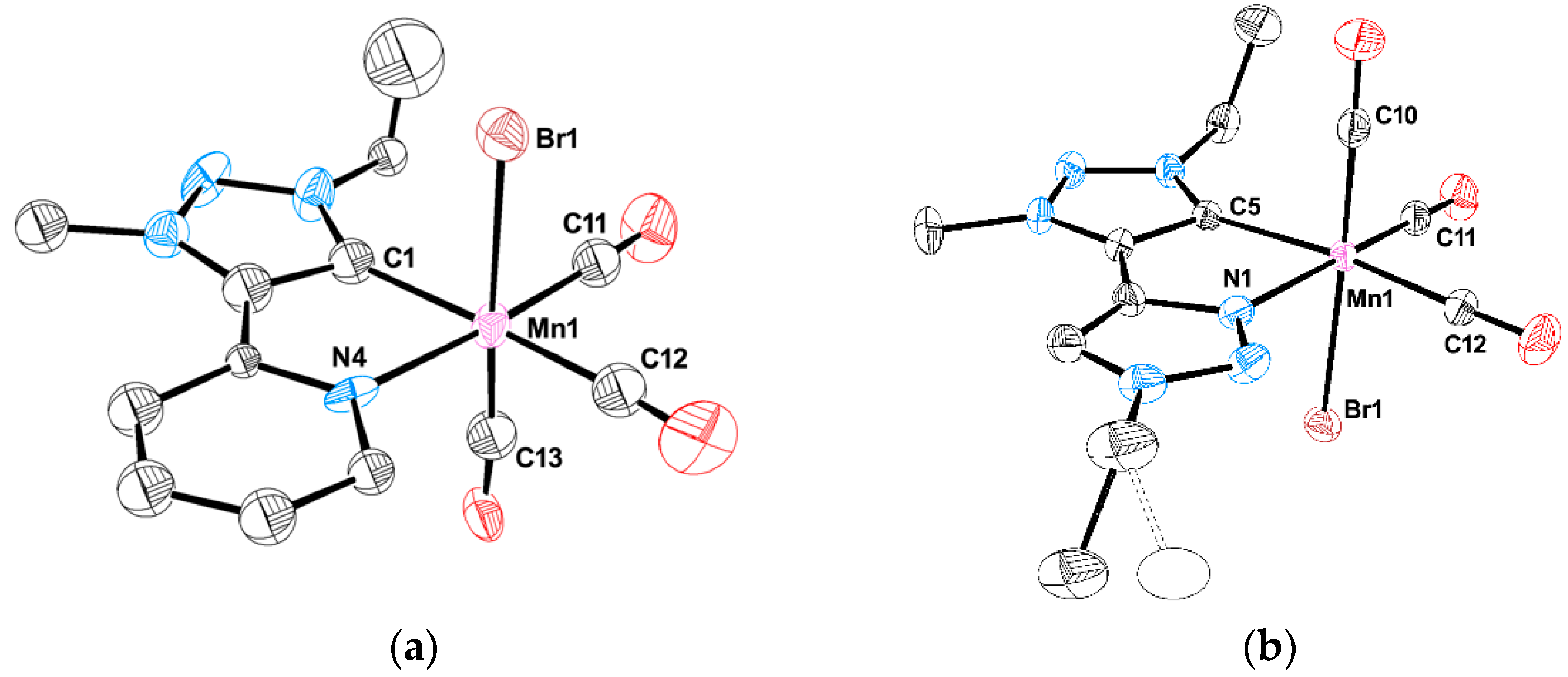
3.4. X-ray Diffraction Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aresta, M. Carbon Dioxide as Chemical Feedstock; Wiley-VCH: Weinheim, Germany, 2010; ISBN 9783527324750. [Google Scholar]
- Boutin, E.; Robert, M. Molecular Electrochemical Reduction of CO2 beyond Two Electrons. Trends Chem. 2021, 3, 359–372. [Google Scholar] [CrossRef]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Werlé, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef]
- Liu, D.-C.; Zhong, D.-C.; Lu, T.-B. Non-noble metal-based molecular complexes for CO2 reduction: From the ligand design perspective. EnergyChem 2020, 2, 100034–100075. [Google Scholar] [CrossRef]
- Steinlechner, C.; Roesel, A.F.; Oberem, E.; Päpcke, A.; Rockstroh, N.; Frédé, F.; Gloaguen, F.; Lochbrunner, S.; Ludwig, R.; Spannenberg, A.; et al. Selective Earth-Abundant System for CO2 Reduction: Comparing Photo-and Electrocatalytic Processes. ACS Catal. 2019, 9, 2091–2100. [Google Scholar] [CrossRef] [Green Version]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Franco, F.; Fernández, S.; Lloret-Fillol, J. Advances in the electrochemical catalytic reduction of CO2 with metal complexes. Curr. Opin. Electrochem. 2019, 15, 109–117. [Google Scholar] [CrossRef]
- Stanbury, M.; Compain, J.D.; Chardon-Noblat, S. Electro and photoreduction of CO2 driven by manganese-carbonyl molecular catalysts. Coord. Chem. Rev. 2018, 361, 120–137. [Google Scholar] [CrossRef]
- Roy, S.S.; Talukdar, K.; Jurss, J.W. Electro- and Photochemical Reduction of CO2 by Molecular Manganese Catalysts: Exploring the Positional Effect of Second-Sphere Hydrogen-Bond Donors. ChemSusChem 2021, 14, 662–670. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machan, C.W.; Stanton, C.J.; Vandezande, J.E.; Majetich, G.F.; Schaefer, H.F.; Kubiak, C.P.; Agarwal, J. Electrocatalytic Reduction of Carbon Dioxide by Mn(CN)(2,2′-bipyridine)(CO)3: CN Coordination Alters Mechanism. Inorg. Chem. 2015, 54, 8849–8856. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.D.; Nguyen, A.D.; Grice, K.A.; Moore, C.E.; Rheingold, A.L.; Kubiak, C.P. Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: Eliminating dimerization and altering catalysis. J. Am. Chem. Soc. 2014, 136, 5460–5471. [Google Scholar] [CrossRef] [PubMed]
- Rønne, M.H.; Cho, D.; Madsen, M.R.; Jakobsen, J.B.; Eom, S.; Escoudé, É.; Hammershøj, H.C.D.; Nielsen, D.U.; Pedersen, S.U.; Baik, M.-H.; et al. Ligand-Controlled Product Selectivity in Electrochemical Carbon Dioxide Reduction Using Manganese Bipyridine Catalysts. J. Am. Chem. Soc. 2020, 142, 4265–4275. [Google Scholar]
- Mukherjee, J.; Siewert, I. Manganese and Rhenium Tricarbonyl Complexes Equipped with Proton Relays in the Electrochemical CO2 Reduction Reaction. Eur. J. Inorg. Chem. 2020, 2020, 4319–4333. [Google Scholar] [CrossRef]
- Agarwal, J.; Shaw, T.W.; Stanton, C.J.; Majetich, G.F.; Bocarsly, A.B.; Schaefer, H.F. NHC-containing manganese(I) electrocatalysts for the two-electron reduction of CO2. Angew. Chem. Int. Ed. 2014, 53, 5152–5155. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Stanton III, C.J.; Shaw, T.W.; Vandezande, J.E.; Majetich, G.F.; Bocarsly, A.B.; Schaefer III, H.F. Exploring the effect of axial ligand substitution (X = Br, NCS, CN) on the photodecomposition and electrochemical activity of [MnX(N-C)(CO)3] complexes. Dalton Trans. 2015, 44, 2122–2131. [Google Scholar] [CrossRef] [PubMed]
- Stanton, C.J.; Vandezande, J.E.; Majetich, G.F.; Schaefer, H.F.; Agarwal, J. Mn-NHC Electrocatalysts: Increasing π Acidity Lowers the Reduction Potential and Increases the Turnover Frequency for CO2 Reduction. Inorg. Chem. 2016, 55, 9509–9512. [Google Scholar] [CrossRef]
- Franco, F.; Pinto, M.F.; Royo, B.; Lloret-Fillol, J. A Highly Active N-Heterocyclic Carbene Manganese(I) Complex for Selective Electrocatalytic CO2 Reduction to CO. Angew. Chem. Int. Ed. 2018, 57, 4603–4606. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Z.; Chang, X.; Zhang, Y.Q.; Liao, R.Z.; Duan, L. Highly Active Manganese-Based CO2 Reduction Catalysts with Bulky NHC Ligands: A Mechanistic Study. Inorg. Chem. 2020, 59, 10234–10242. [Google Scholar] [CrossRef]
- Pinto, M.; Friães, S.; Franco, F.; Lloret-Fillol, J.; Royo, B. Manganese N-Heterocyclic Carbene Complexes for Catalytic Reduction of Ketones with Silanes. ChemCatChem 2018, 10, 2734–2740. [Google Scholar] [CrossRef]
- Sousa, S.C.A.; Carrasco, C.J.; Pinto, M.F.; Royo, B. A Manganese N-Heterocyclic Carbene Catalyst for Reduction of Sulfoxides with Silanes. ChemCatChem 2019, 11, 3839–3843. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.C.A.; Realista, S.; Royo, B. Bench-Stable Manganese NHC Complexes for the Selective Reduction of Esters to Alcohols with Silanes. Adv. Synth. Catal. 2020, 362, 2437–2443. [Google Scholar] [CrossRef]
- Pinto, M.F.; Olivares, M.; Vivancos, Á.; Guisado-Barrios, G.; Albrecht, M.; Royo, B. (Di)triazolylidene manganese complexes in catalytic oxidation of alcohols to ketones and aldehydes. Catal. Sci. Technol. 2019, 9, 2421–2425. [Google Scholar] [CrossRef]
- Friães, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Veiros, L.F.; Albrecht, M.; Royo, B. Manganese complexes with chelating and bridging di-triazolylidene ligands: Synthesis and reactivity. Dalton Trans. 2021, 50, 5911–5920. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.F.; Petronilho, A.; Albrecht, M. Application of 1,2,3-triazolylidenes as versatile NHC-type ligands: Synthesis, properties, and application in catalysis and beyond. Chem. Commun. 2013, 49, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suntrup, L.; Klenk, S.; Klein, J.; Sobottka, S.; Sarkar, B. Gauging Donor/Acceptor Properties and Redox Stability of Chelating Click-Derived Triazoles and Triazolylidenes: A Case Study with Rhenium(I) Complexes. Inorg. Chem. 2017, 56, 5771–5783. [Google Scholar] [CrossRef] [PubMed]
- Schweinfurth, D.; Hettmanczyk, L.; Suntrup, L.; Sarkar, B. Metal Complexes of Click-Derived Triazoles and Mesoionic Carbenes: Electron Transfer, Photochemistry, Magnetic Bistability, and Catalysis. Z. Anorg. Allg. Chem. 2017, 643, 554–584. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, S.-X.; Mueller-Bunz, H.; Albrecht, M. Synthesis of Triazolylidene Nickel Complexes and Their Catalytic Application in Selective Aldehyde Hydrosilylation. ACS Catal. 2016, 6, 8192–8200. [Google Scholar] [CrossRef]
- Johnson, C.; Albrecht, M. Triazolylidene Iron(II) Piano-Stool Complexes: Synthesis and Catalytic Hydrosilylation of Carbonyl Compounds. Organometallics 2017, 36, 2902–2913. [Google Scholar] [CrossRef]
- Bertini, S.; Rahaman, M.; Dutta, A.; Schollhammer, P.; Rudnev, A.V.; Gloaguen, F.; Broekmann, P.; Albrecht, M. Oxo-functionalised mesoionic NHC nickel complexes for selective electrocatalytic reduction of CO2 to formate. Green Chem. 2021, 23, 3365–3373. [Google Scholar] [CrossRef]
- Vivancos, Á.; Segarra, C.; Albrecht, M. Mesoionic and Related Less Heteroatom-Stabilized N-Heterocyclic Carbene Complexes: Synthesis, Catalysis, and Other Applications. Chem. Rev. 2018, 118, 9493–9586. [Google Scholar] [CrossRef]
- Bernet, L.; Lalrempuia, R.; Ghattas, W.; Mueller-Bunz, H.; Vigara, L.; Llobet, A.; Albrecht, M. Tunable single-site ruthenium catalysts for efficient water oxidation. Chem. Commun. 2011, 47, 8058–8060. [Google Scholar] [CrossRef]
- Vivancos, Á.; Albrecht, M. Influence of the Linker Length and Coordination Mode of (Di)Triazolylidene Ligands on the Structure and Catalytic Transfer Hydrogenation Activity of Iridium(III) Centers. Organometallics 2017, 36, 1580–1590. [Google Scholar] [CrossRef]
- Bolje, A.; Košmrlj, J. A Selective Approach to Pyridine Appended 1,2,3-Triazolium Salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef] [PubMed]
- Hohloch, S.; Kaiser, S.; Duecker, F.L.; Bolje, A.; Maity, R.; Košmrlj, J.; Sarkar, B. Catalytic oxygenation of sp3 “C-H” bonds with Ir(III) complexes of chelating triazoles and mesoionic carbenes. Dalton Trans. 2015, 44, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SADABS 2016/2: Krause, L.; Herbst-Irmer, R.; Sheldrick, G.M.; Stalke, D.J. Comparison of silver and molybdenum microfocus X-ray sources for single-crystal structure determination. J. Appl. Crystallogr. 2015, 48, 3–10. [Google Scholar]
- SHELXL: Sheldrick, G.M. Acta Crystallogr. Sect. C-Struct. Chem. 2015, 71, 3–8. [Google Scholar]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Crystallogr. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrugia, L.J.J. Facile Preparation of 1,2-Diols from Chalcones: An NMR Spectroscopy and X-ray Crystallography Study. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
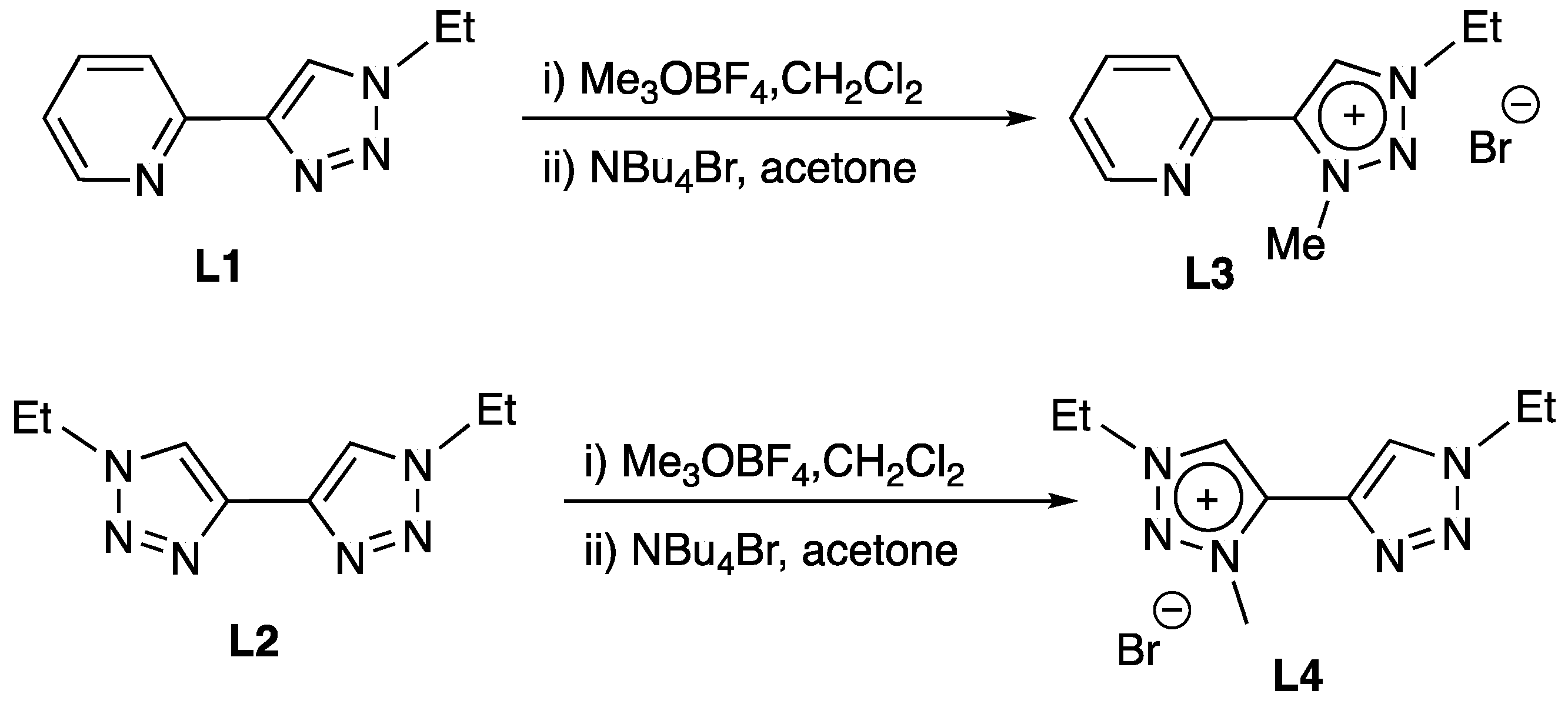

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ν (CO) (cm−1) | 13C NMR-Ccarbene (ppm) |
|---|---|---|
| 1 | 2009, 1907, 1880 | 188.65 |
| 2 | 2019, 1919 | 185.83 |
| 3 | 2025, 1963, 1917 | -- |
| Complex | [H2O] (M) a | Ecat/2 (V vs. Fc+/0) b | (icat/ip)2 |
|---|---|---|---|
| 1 | 0.1 | −2.35 | 2.62 |
| 2 | 0.1 | −2.58 | 4.80 |
| 3 | 1.0/0.5 | −1.86/−2.39 c | 30.1/22.1 c |
| 4 | 0 | −2.14/−2.48 c | 29.81/39.1 c |
| Entry | Complex | [acid] (M) | Eapplied (V vs. Fc+/0) a | FECO (%) b |
|---|---|---|---|---|
| 1 | 3 | 0 | −1.89 | 56 |
| 2 | 0.5 (H2O) | −1.85 | 72 | |
| 3 | 1.0 (H2O) | −1.89 | 62 | |
| 4 | 1.0 (TFE) | −1.92 | 61 | |
| 5 | 4 | 0 | −2.08 | 69 |
| 6 | 4 | 0 | −2.15 | 70 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friães, S.; Realista, S.; Gomes, C.S.B.; Martinho, P.N.; Royo, B. Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules 2021, 26, 6325. https://doi.org/10.3390/molecules26216325
Friães S, Realista S, Gomes CSB, Martinho PN, Royo B. Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules. 2021; 26(21):6325. https://doi.org/10.3390/molecules26216325
Chicago/Turabian StyleFriães, Sofia, Sara Realista, Clara S. B. Gomes, Paulo N. Martinho, and Beatriz Royo. 2021. "Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2" Molecules 26, no. 21: 6325. https://doi.org/10.3390/molecules26216325
APA StyleFriães, S., Realista, S., Gomes, C. S. B., Martinho, P. N., & Royo, B. (2021). Click-Derived Triazoles and Triazolylidenes of Manganese for Electrocatalytic Reduction of CO2. Molecules, 26(21), 6325. https://doi.org/10.3390/molecules26216325