
Applying Active Learning to the Screening of Molecular Oxygen Evolution Catalysts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
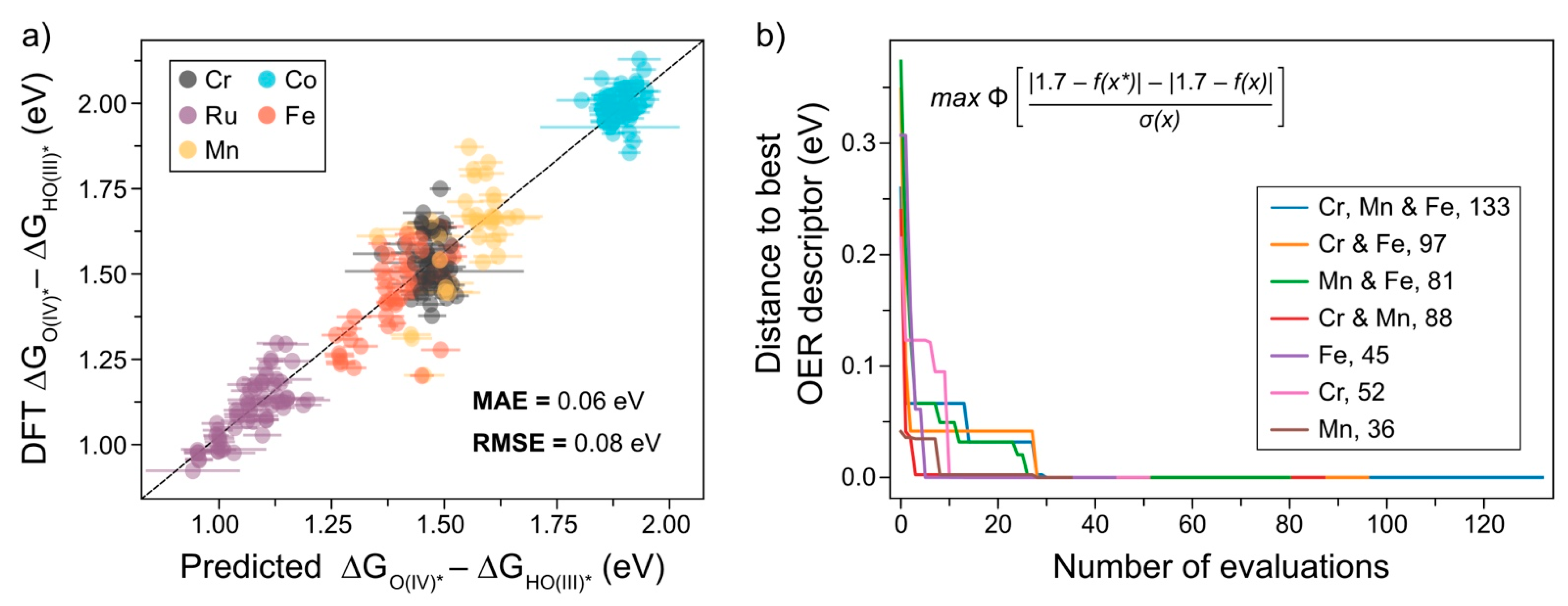
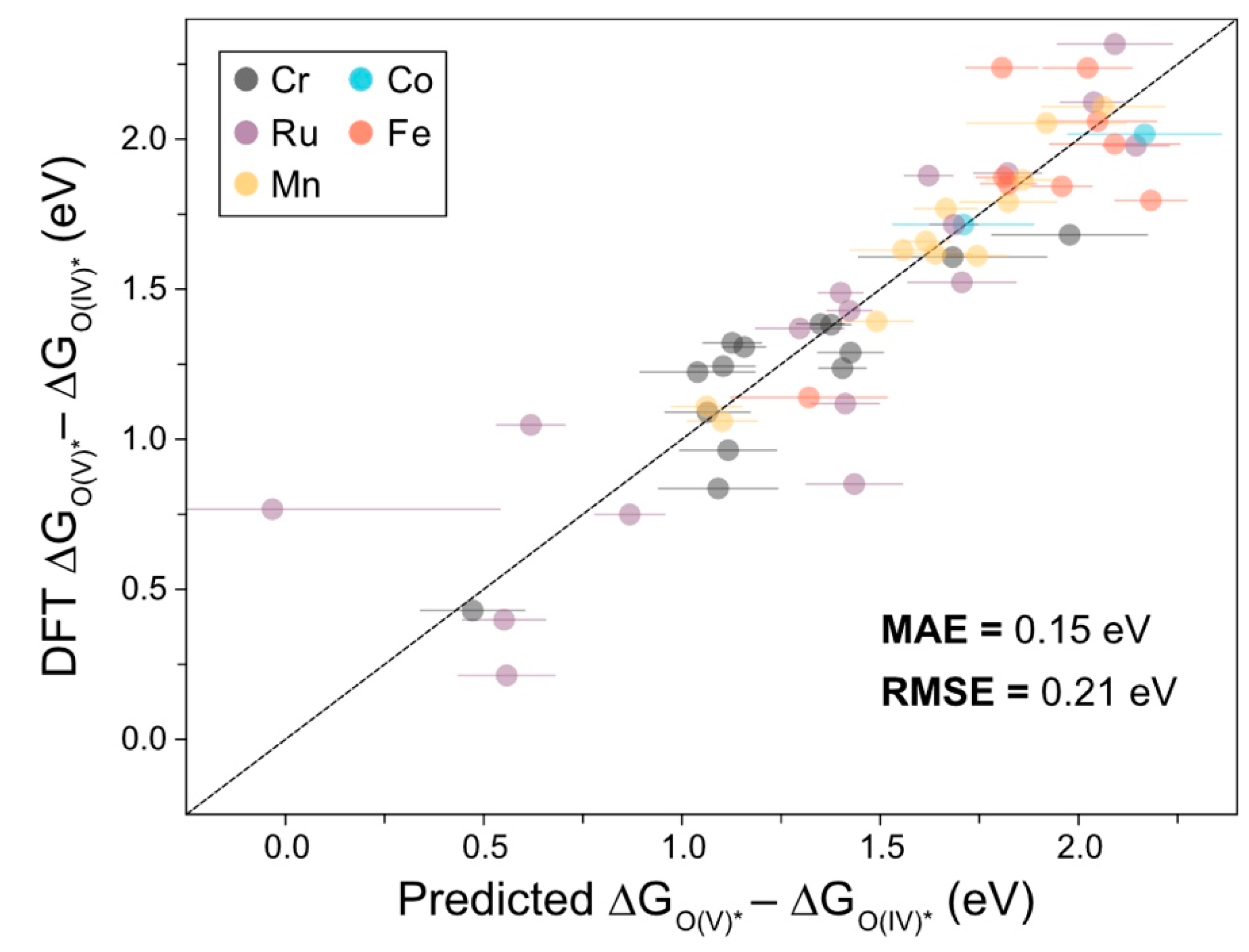
2.1. Machine Learning Models
2.2. Active Learning Applied to the OER Descriptor
2.3. AL for an Extra Oxidation Mechanism
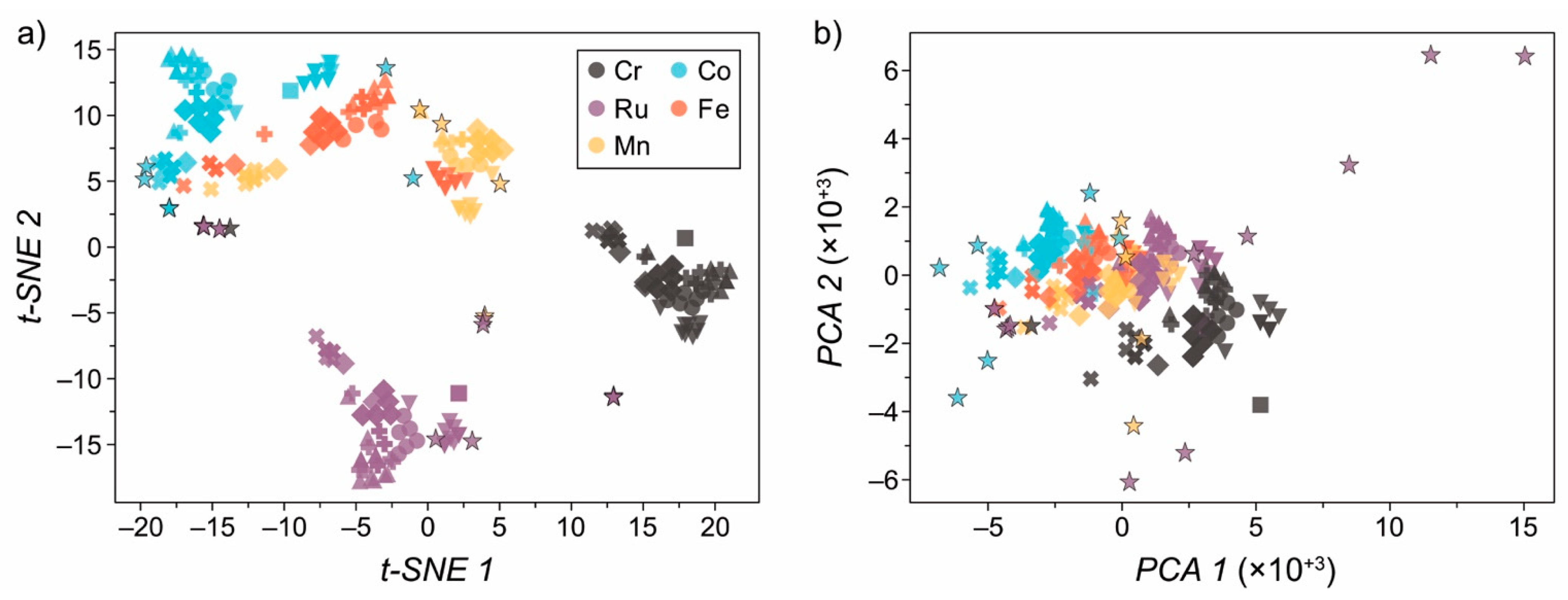
2.4. Dataset Bias
2.5. Outlook
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhong, M.; Tran, K.; Min, Y.; Wang, C.; Wang, Z.; Dinh, C.-T.; De Luna, P.; Yu, Z.; Rasouli, A.S.; Brodersen, P.; et al. Accelerated Discovery of CO2 Electrocatalysts Using Active Machine Learning. Nature 2020, 581, 178–183. [Google Scholar] [CrossRef]
- Sun, Y.; Liao, H.; Wang, J.; Chen, B.; Sun, S.; Ong, S.J.H.; Xi, S.; Diao, C.; Du, Y.; Wang, J.-O.; et al. Covalency Competition Dominates the Water Oxidation Structure–Activity Relationship on Spinel Oxides. Nat. Catal. 2020, 3, 554–563. [Google Scholar] [CrossRef]
- Craig, M.J.; García-Melchor, M. High-Throughput Screening and Rational Design to Drive Discovery in Molecular Water Oxidation Catalysis. Cell Rep. Phys. Sci. 2021, 2, 100492. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Ramírez, J.; López, N. Strategies to Break Linear Scaling Relationships. Nat. Catal. 2019, 2, 971–976. [Google Scholar] [CrossRef]
- Huang, Z.-F.; Song, J.; Dou, S.; Li, X.; Wang, J.; Wang, X. Strategies to Break the Scaling Relation toward Enhanced Oxygen Electrocatalysis. Matter 2019, 1, 1494–1518. [Google Scholar] [CrossRef] [Green Version]
- Vereshchuk, N.; Matheu, R.; Benet-Buchholz, J.; Pipelier, M.; Lebreton, J.; Dubreuil, D.; Tessier, A.; Gimbert-Suriñach, C.; Ertem, M.Z.; Llobet, A. Second Coordination Sphere Effects in an Evolved Ru Complex Based on Highly Adaptable Ligand Results in Rapid Water Oxidation Catalysis. J. Am. Chem. Soc. 2020, 142, 5068–5077. [Google Scholar] [CrossRef]
- Craig, M.J.; Coulter, G.; Dolan, E.; Soriano-López, J.; Mates-Torres, E.; Schmitt, W.; García-Melchor, M. Universal Scaling Relations for the Rational Design of Molecular Water Oxidation Catalysts with Near-Zero Overpotential. Nat. Commun. 2019, 10, 4993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido Torres, J.A.; Jennings, P.C.; Hansen, M.H.; Boes, J.R.; Bligaard, T. Low-Scaling Algorithm for Nudged Elastic Band Calculations Using a Surrogate Machine Learning Model. Phys. Rev. Lett. 2019, 122, 156001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Lengeling, B.; Roch, L.M.; Perea, J.D.; Langner, S.; Brabec, C.J.; Aspuru-Guzik, A. A Bayesian Approach to Predict Solubility Parameters. Adv. Theory Simul. 2019, 2, 1800069. [Google Scholar] [CrossRef]
- Jennings, P.C.; Lysgaard, S.; Hummelshøj, J.S.; Vegge, T.; Bligaard, T. Genetic Algorithms for Computational Materials Discovery Accelerated by Machine Learning. NPJ Comput. Mater. 2019, 5, 46. [Google Scholar] [CrossRef]
- Janet, J.P.; Ramesh, S.; Duan, C.; Kulik, H.J. Accurate Multiobjective Design in a Space of Millions of Transition Metal Complexes with Neural-Network-Driven Efficient Global Optimization. ACS Cent. Sci. 2020, 6, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Ulissi, Z.W.; Singh, A.R.; Tsai, C.; Nørskov, J.K. Automated Discovery and Construction of Surface Phase Diagrams Using Machine Learning. J. Phys. Chem. Lett. 2016, 7, 3931–3935. [Google Scholar] [CrossRef] [PubMed]
- Foscato, M.; Jensen, V.R. Automated in Silico Design of Homogeneous Catalysts. ACS Catal. 2020, 10, 2354–2377. [Google Scholar] [CrossRef]
- Murphy, K.P. Machine learning: A Probabilistic Perspective. In Machine Learning: A Probabilistic Perspective; MIT Press: Cambridge, MA, USA, 2012; Chapter 15. [Google Scholar]
- Rasmussen, C.E.; Williams, C.K.I. Gaussian Processes for Machine Learning; Adaptive Computation and Machine Learning; MIT Press: Cambridge, MA, USA, 2006; ISBN 978-0-262-18253-9. [Google Scholar]
- Ioannidis, E.I.; Gani, T.Z.H.; Kulik, H.J. MolSimplify: A Toolkit for Automating Discovery in Inorganic Chemistry. J. Comp. Chem. 2016, 37, 2106–2117. [Google Scholar] [CrossRef]
- Nandy, A.; Duan, C.; Janet, J.P.; Gugler, S.; Kulik, H.J. Strategies and Software for Machine Learning Accelerated Discovery in Transition Metal Chemistry. Ind. Eng. Chem. Res. 2018, 57, 13973–13986. [Google Scholar] [CrossRef]
- Nandy, A.; Zhu, J.; Janet, J.P.; Duan, C.; Getman, R.B.; Kulik, H.J. Machine Learning Accelerates the Discovery of Design Rules and Exceptions in Stable Metal–Oxo Intermediate Formation. ACS Catal. 2019, 9, 8243–8255. [Google Scholar] [CrossRef] [Green Version]
- Dickens, C.F.; Kirk, C.; Nørskov, J.K. Insights into the Electrochemical Oxygen Evolution Reaction with Ab Initio Calculations and Microkinetic Modeling: Beyond the Limiting Potential Volcano. J. Phys. Chem. C 2019, 123, 18960–18977. [Google Scholar] [CrossRef]
- Christensen, R.; Hansen, H.A.; Dickens, C.F.; Nørskov, J.K.; Vegge, T. Functional Independent Scaling Relation for ORR/OER Catalysts. J. Phys. Chem. C 2016, 120, 24910–24916. [Google Scholar] [CrossRef] [Green Version]
- Janet, J.P.; Duan, C.; Yang, T.; Nandy, A.; Kulik, H.J. A Quantitative Uncertainty Metric Controls Error in Neural Network-Driven Chemical Discovery. Chem. Sci. 2019, 10, 7913–7922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coggins, M.K.; Zhang, M.-T.; Vannucci, A.K.; Dares, C.J.; Meyer, T.J. Electrocatalytic Water Oxidation by a Monomeric Amidate-Ligated Fe(III)–Aqua Complex. J. Am. Chem. Soc. 2014, 136, 5531–5534. [Google Scholar] [CrossRef]
- Hunter, B.M.; Thompson, N.B.; Müller, A.M.; Rossman, G.R.; Hill, M.G.; Winkler, J.R.; Gray, H.B. Trapping an Iron(VI) Water-Splitting Intermediate in Nonaqueous Media. Joule 2018, 2, 747–763. [Google Scholar] [CrossRef] [Green Version]
- Van der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Van der Maaten, L. Accelerating T-SNE Using Tree-Based Algorithms. J. Mach. Learn. Res. 2014, 15, 3221–3245. [Google Scholar]
- Hotelling, H. Analysis of a Complex of Statistical Variables into Principal Components. J. Educ. Psychol. 1933, 24, 417–441. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal Component Analysis: A Review and Recent Developments. Phil. Trans. R. Soc. A 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Balcells, D.; Skjelstad, B.B. TmQM Dataset—Quantum Geometries and Properties of 86k Transition Metal Complexes. J. Chem. Inf. Model. 2020, 60, 6135–6146. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Thorarinsdottir, A.E.; Nocera, D.G. Energy Catalysis Needs Ligands with High Oxidative Stability. Chem Catal. 2021, 1, 32–43. [Google Scholar] [CrossRef]
- Collins, T.J. Designing Ligands for Oxidizing Complexes. Acc. Chem. Res. 1994, 27, 279–285. [Google Scholar] [CrossRef]
- Codolà, Z.; Gamba, I.; Acuña-Parés, F.; Casadevall, C.; Clémancey, M.; Latour, J.-M.; Luis, J.M.; Lloret-Fillol, J.; Costas, M. Design of Iron Coordination Complexes as Highly Active Homogenous Water Oxidation Catalysts by Deuteration of Oxidation-Sensitive Sites. J. Am. Chem. Soc. 2019, 141, 323–333. [Google Scholar] [CrossRef] [Green Version]
- Craig, M.; Garcia-Melchor, M. Discerning Activity and Inactivity in Earth-Abundant Molecular Water Oxidation Catalysts. ChemCatChem 2020, 12, 4775–4779. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 09, Revision A.02; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Ehlers, A.W.; Böhme, M.; Dapprich, S.; Gobbi, A.; Höllwarth, A.; Jonas, V.; Köhler, K.F.; Stegmann, R.; Veldkamp, A.; Frenking, G. A Set of F-Polarization Functions for Pseudo-Potential Basis Sets of the Transition Metals Sc-Cu, Y-Ag and La-Au. Chem. Phys. Lett. 1993, 208, 111–114. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-Learn: Machine Learning in Python. arXiv 2018, arXiv:1201.0490. [Google Scholar]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-Generation Hyperparameter Optimization Framework. In Proceedings of the Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar]
- Lannelongue, L.; Grealey, J.; Inouye, M. Green Algorithms: Quantifying the Carbon Footprint of Computation. Adv. Sci. 2021, 8, 2100707. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Craig, M.J.; García-Melchor, M. Applying Active Learning to the Screening of Molecular Oxygen Evolution Catalysts. Molecules 2021, 26, 6362. https://doi.org/10.3390/molecules26216362
Craig MJ, García-Melchor M. Applying Active Learning to the Screening of Molecular Oxygen Evolution Catalysts. Molecules. 2021; 26(21):6362. https://doi.org/10.3390/molecules26216362
Chicago/Turabian StyleCraig, Michael John, and Max García-Melchor. 2021. "Applying Active Learning to the Screening of Molecular Oxygen Evolution Catalysts" Molecules 26, no. 21: 6362. https://doi.org/10.3390/molecules26216362
APA StyleCraig, M. J., & García-Melchor, M. (2021). Applying Active Learning to the Screening of Molecular Oxygen Evolution Catalysts. Molecules, 26(21), 6362. https://doi.org/10.3390/molecules26216362