
Excellent Room-Temperature Thermoelectricity of 2D GeP3: Mexican-Hat-Shaped Band Dispersion and Ultralow Lattice Thermal Conductivity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Methods
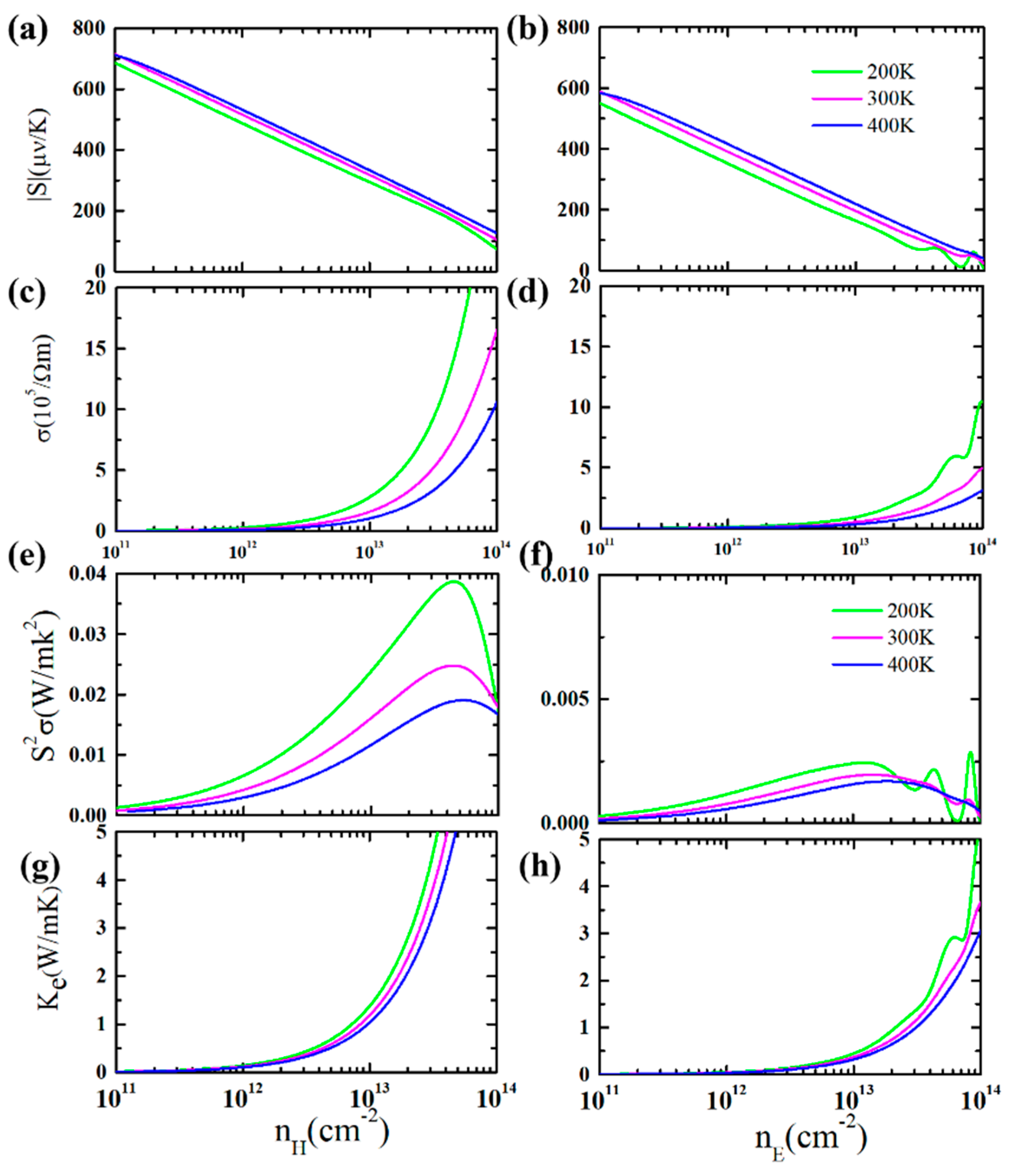
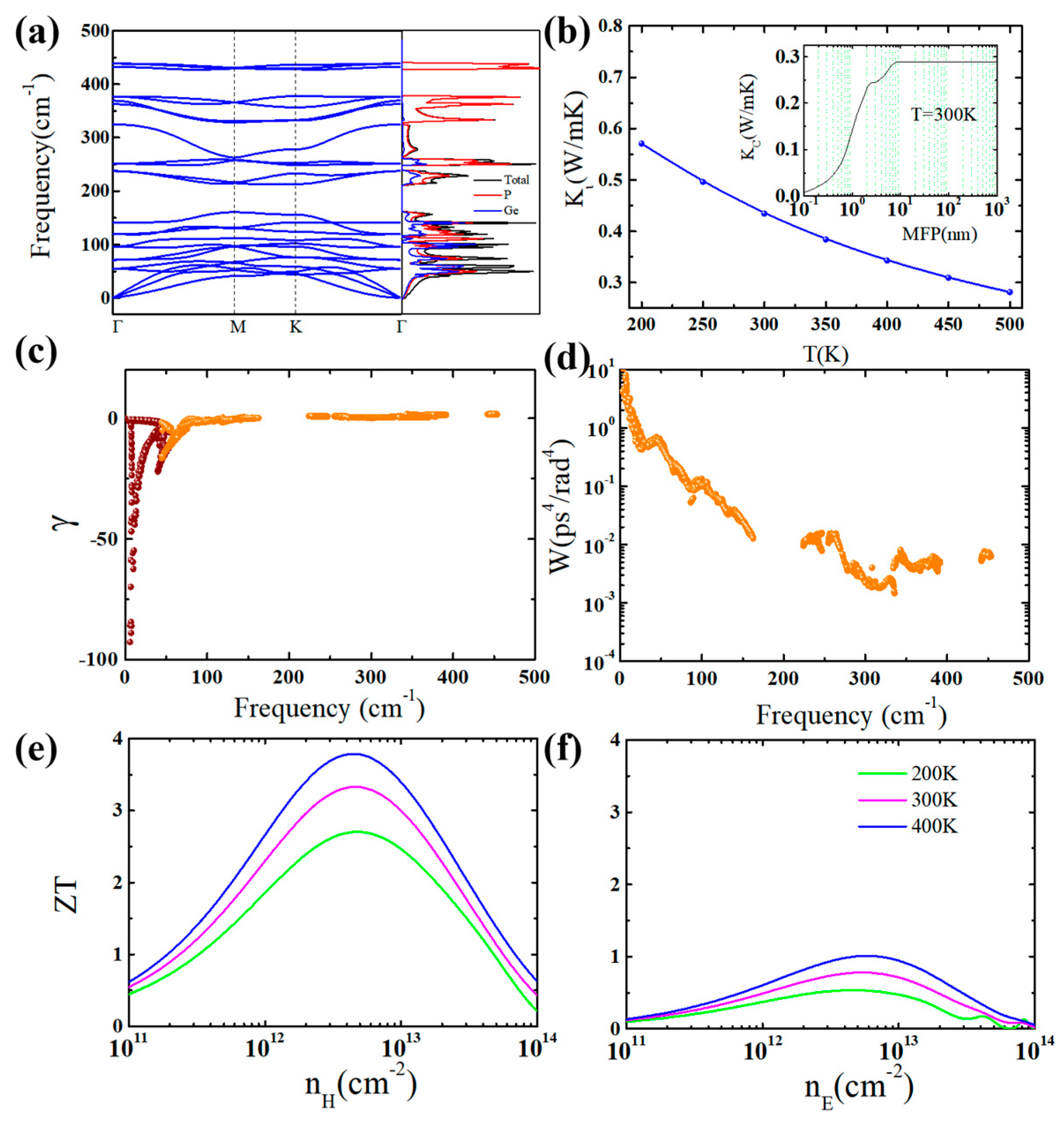
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bell, L.E. Cooling, heating, generating power, and recovering waste heat with thermoelectric systems. Science 2008, 321, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Liu, Z.; Zhou, J.; Zhu, H.; Zhang, Q.; Chen, G.; Ren, Z. Advances in thermoelectrics. Adv. Phys. 2018, 67, 69–147. [Google Scholar] [CrossRef]
- Dong, Z.; Xu, H.; Liang, F.; Luo, C.; Wang, C.; Cao, Z.-Y.; Chen, X.-J.; Zhang, J.; Wu, X. Raman characterization on two-dimensional materials-based thermoelectricity. Molecules 2019, 24, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, N.T.; Nugraha, A.R.T.; Yang, T.; Saito, R. Confinement effect in thermoelectric properties of two-dimensional materials. MRS Adv. 2020, 5, 469–479. [Google Scholar] [CrossRef]
- Hung, N.T.; Saito, R. The origin of quantum effects in low-dimensional thermoelectric materials. Adv. Quantum Technol. 2021, 4, 2000115. [Google Scholar] [CrossRef]
- Wang, C.; Zheng, C.; Gao, G. Bulk and monolayer ZrS3 as promising anisotropic thermoelectric materials: A comparative study. J. Phys. Chem. C 2020, 124, 6536–6543. [Google Scholar] [CrossRef]
- Wang, X.-M.; Mo, D.-C.; Lu, S.-S. On the thermoelectric transport properties of graphyne by the first-principles method. J. Chem. Phys. 2013, 138, 204704. [Google Scholar] [CrossRef]
- Kumar, S.; Schwingenschlogl, U. Thermoelectric response of bulk and monolayer MoSe2 and WSe2. Chem. Mater. 2015, 27, 1278–1284. [Google Scholar] [CrossRef]
- Yun, W.S.; Lee, J.D. Single-layer CdPSe3: A promising thermoelectric material persisting in high temperatures. Appl. Phys. Lett. 2019, 115, 193105. [Google Scholar] [CrossRef]
- Li, Y.; Wu, M.N.; Ding, T.; Ma, K.; Liu, F.S.; Ao, W.Q.; Li, J.Q. Promising thermoelectric properties and anisotropic electrical and thermal transport of monolayer SnTe. Appl. Phys. Lett. 2019, 114, 083901. [Google Scholar] [CrossRef]
- Patel, A.; Singh, D.; Sonvane, Y.; Thakor, P.B.; Ahuja, R. High thermoelectric performance in two-dimensional Janus monolayer material WS-X (X= Se and Te). ACS Appl. Mater. Interfaces 2020, 12, 46212–46219. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Wu, H.; Zhu, T.; Fu, C.; He, J.; Ying, P.; Zhao, X. Tuning multiscale microstructures to enhance thermoelectric performance of n-type Bismuth-Telluride-based solid solutions. Adv. Energy Mater. 2015, 5, 1500411. [Google Scholar] [CrossRef]
- Jing, Y.; Ma, Y.; Li, Y.; Heine, T. GeP3: A small indirect band gap 2D crystal with high carrier mobility and strong interlayer quantum confinement. Nano Lett. 2017, 17, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Gong, P.-L.; Zhang, F.; Huang, L.-F.; Zhang, H.; Li, L.; Xiao, R.-C.; Deng, B.; Pan, H.; Shi, X.-Q. Multifunctional two-dimensional semiconductors SnP3: Universal mechanism of layer-dependent electronic phase transition. J. Phys. Condens. Matter 2018, 30, 475702. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Meng, F.; Wang, H.; Wang, H.; Ni, Y. Novel two-dimensional semiconductor SnP3: High stability, tunable bandgaps and high carrier mobility explored using first-principles calculations. J. Mater. Chem. A 2018, 6, 11890–11897. [Google Scholar] [CrossRef]
- Miao, N.; Xu, B.; Bristowe, N.C.; Zhou, J.; Sun, Z. Tunable magnetism and extraordinary sunlight absorbance in indium triphosphide monolayer. J. Am. Chem. Soc. 2017, 139, 11125–11131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Jiao, Y.; He, T.; Ma, F.; Kou, L.; Liao, T.; Bottle, S.; Du, A. Two-dimensional GeP3 as a high capacity electrode material for Li-ion batteries. Phys. Chem. Chem. Phys. 2017, 19, 25886–25890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.-H.; Huang, H.; Zhong, J.; Yu, S.; Zhang, Q.; Zeng, X.C. Monolayer triphosphates MP3(M=Sn, Ge) with excellent basal catalytic activity for hydrogen evolution reaction. Nanoscale 2019, 11, 12210–12219. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, T.; Jiang, E.; Tang, C.; Li, J.; He, C.; Zhong, J. Thermal and thermoelectric properties of monolayer indium triphosphide (InP3): A first-principles study. J. Mater. Chem. A 2018, 6, 21532–21541. [Google Scholar] [CrossRef]
- Sun, Z.; Yuan, K.; Chang, Z.; Bi, S.; Zhang, X.; Tang, D. Ultra-low thermal conductivity and high thermoelectric performance of two-dimensional triphosphides (InP3, GaP3, SbP3 and SnP3): A comprehensive first-principles study. Nanoscale 2020, 12, 3330–3342. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.-L.; Liu, P.-F.; Zhang, J.; Zhang, P.; Zhou, W.-X.; Xie, G.; Wang, B.-T. Monolayer SnP3: An excellent p-type thermoelectric material. Nanoscale 2019, 11, 19923–19932. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef] [Green Version]
- Madsen, G.K.H.; Singh, D.J. BoltzTraP. A code for calculating band-structure dependent quantities. Comput. Phys. Commun. 2006, 175, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Jonson, M.; Mahan, G.D. Mott′s formula for the thermopower and the Wiedemann-Franz law. Phys. Rev. B 1980, 21, 4223. [Google Scholar] [CrossRef]
- Zhang, W.; Huang, Z.; Zhang, W.; Li, Y. Two-dimensional semiconductors with possible high room temperature mobility. Nano Res. 2014, 7, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wei, S.; Gao, G. Theoretical investigation of metal-shrouded Tl2O monolayers: Pudding-mold-type band structure and thermoelectric performance. ACS Appl. Nano Mater. 2019, 2, 4061–4066. [Google Scholar] [CrossRef]
- Zeng, B.; Long, M.; Zhang, X.; Dong, Y.; Li, M.; Yi, Y.; Duan, H. Strain engineering on electronic structure and carrier mobility in monolayer GeP3. J. Phys. D Appl. Phys. 2018, 51, 235302. [Google Scholar] [CrossRef]
- Li, W.; Carrete, J.; Katcho, N.A.; Mingo, N. ShengBTE: A solver of the Boltzmann transport equation for phonons. Comput. Phys. Commun. 2014, 185, 1747–1758. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Wen, Y.; Shi, L.; Chen, R.; Liu, H.; Shan, B. Titanium trisulfide monolayer as a potential thermoelectric material: A first-principles-based Boltzmann transport study. ACS Appl. Mater. Interfaces 2017, 9, 2509–2515. [Google Scholar] [CrossRef] [PubMed]
- Rashid, Z.; Nissimagoudar, A.S.; Li, W. Phonon transport and thermoelectric properties of semiconducting Bi2Te2X (X=S, Se, Te) monolayers. Phys. Chem. Chem. Phys. 2019, 21, 5679–5688. [Google Scholar] [CrossRef]
- Kozinsky, B.; Singh, D.J. Thermoelectrics by Computational Design: Progress and Opportunities. Ann. Rev. Mater. Res. 2021, 51, 565–590. [Google Scholar] [CrossRef]
- Wickramaratne, D.; Zahid, F.; Lake, R.K. Electronic and thermoelectric properties of van der Waals materials with ring-shaped valence bands. J. Appl. Phys. 2015, 118, 075101. [Google Scholar] [CrossRef] [Green Version]
- Çınar, M.N.; Sargın, G.Ö.; Sevim, K.; Özdamar, B.; Kurt, G.; Sevinçli, H. Ballistic thermoelectric transport properties of two-dimensional group III-VI monolayers. Phys. Rev. B 2021, 103, 165422. [Google Scholar] [CrossRef]
- Zhu, H.; He, R.; Mao, J.; Zhu, Q.; Li, C.; Sun, J.; Ren, W.; Wang, Y.; Liu, Z.; Tang, Z.; et al. Discovery of ZrCoBi based half Heuslers with high thermoelectric conversion efficiency. Nat. Commun. 2018, 9, 2497. [Google Scholar] [CrossRef]
- Ding, G.; Wang, C.; Gao, G.; Yao, K.; Dun, C.; Feng, C.; Li, D.; Zhang, G. Engineering of charge carriers via a two-dimensional heterostructure to enhance the thermoelectric figure of merit. Nanoscale 2018, 10, 7077–7084. [Google Scholar] [CrossRef]
- Cheng, L.; Liu, H.J.; Zhang, J.; Wei, J.; Liang, J.H.; Shi, J.; Tang, X.F. Effects of van der Waals interactions and quasiparticle corrections on the electronic and transport properties of Bi2Te3. Phys. Rev. B 2014, 90, 085118. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Chang, C.; Pei, Y.; Wu, D.; Peng, K.; Zhou, X.; Gong, S.; He, J.; Zhang, Y.; Zeng, Z.; et al. Origin of low thermal conductivity in SnSe. Phys. Rev. B 2016, 94, 125203. [Google Scholar] [CrossRef]
- Nian, T.; Wang, Z.; Dong, B. Thermoelectric properties of α-In2Se3 monolayer. Appl. Phys. Lett. 2021, 118, 033103. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Xu, Z.; Xu, K.; Gao, G. Excellent Room-Temperature Thermoelectricity of 2D GeP3: Mexican-Hat-Shaped Band Dispersion and Ultralow Lattice Thermal Conductivity. Molecules 2021, 26, 6376. https://doi.org/10.3390/molecules26216376
Wang C, Xu Z, Xu K, Gao G. Excellent Room-Temperature Thermoelectricity of 2D GeP3: Mexican-Hat-Shaped Band Dispersion and Ultralow Lattice Thermal Conductivity. Molecules. 2021; 26(21):6376. https://doi.org/10.3390/molecules26216376
Chicago/Turabian StyleWang, Cong, Zhiyuan Xu, Ke Xu, and Guoying Gao. 2021. "Excellent Room-Temperature Thermoelectricity of 2D GeP3: Mexican-Hat-Shaped Band Dispersion and Ultralow Lattice Thermal Conductivity" Molecules 26, no. 21: 6376. https://doi.org/10.3390/molecules26216376
APA StyleWang, C., Xu, Z., Xu, K., & Gao, G. (2021). Excellent Room-Temperature Thermoelectricity of 2D GeP3: Mexican-Hat-Shaped Band Dispersion and Ultralow Lattice Thermal Conductivity. Molecules, 26(21), 6376. https://doi.org/10.3390/molecules26216376