
Aqueous Solutions of Peptides and Trialkylamines Lead to Unexpected Peptide Modification


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
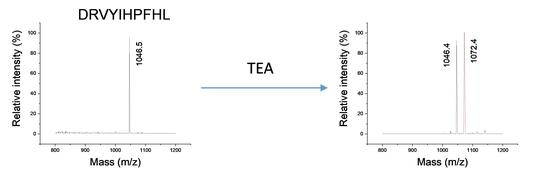
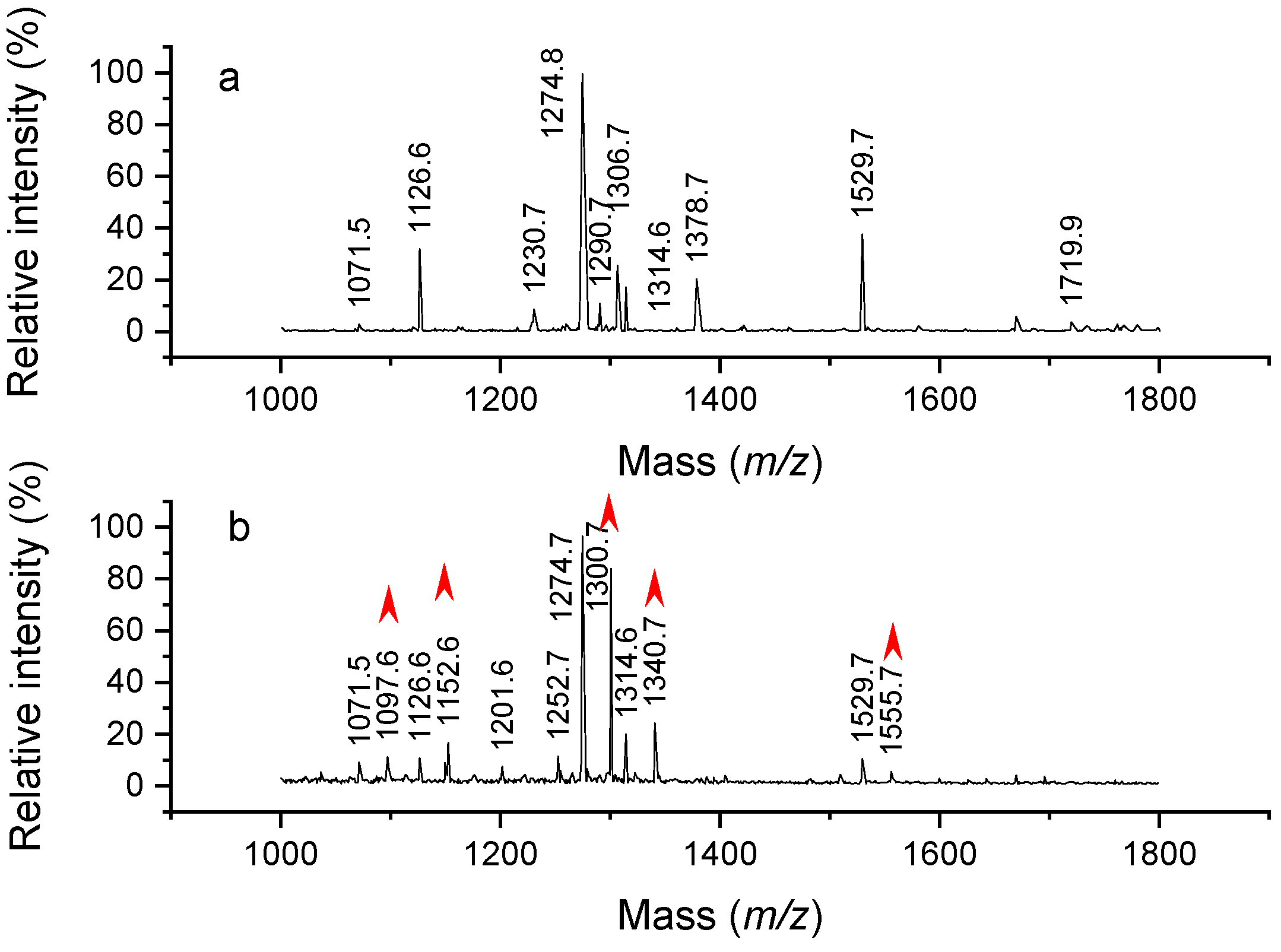
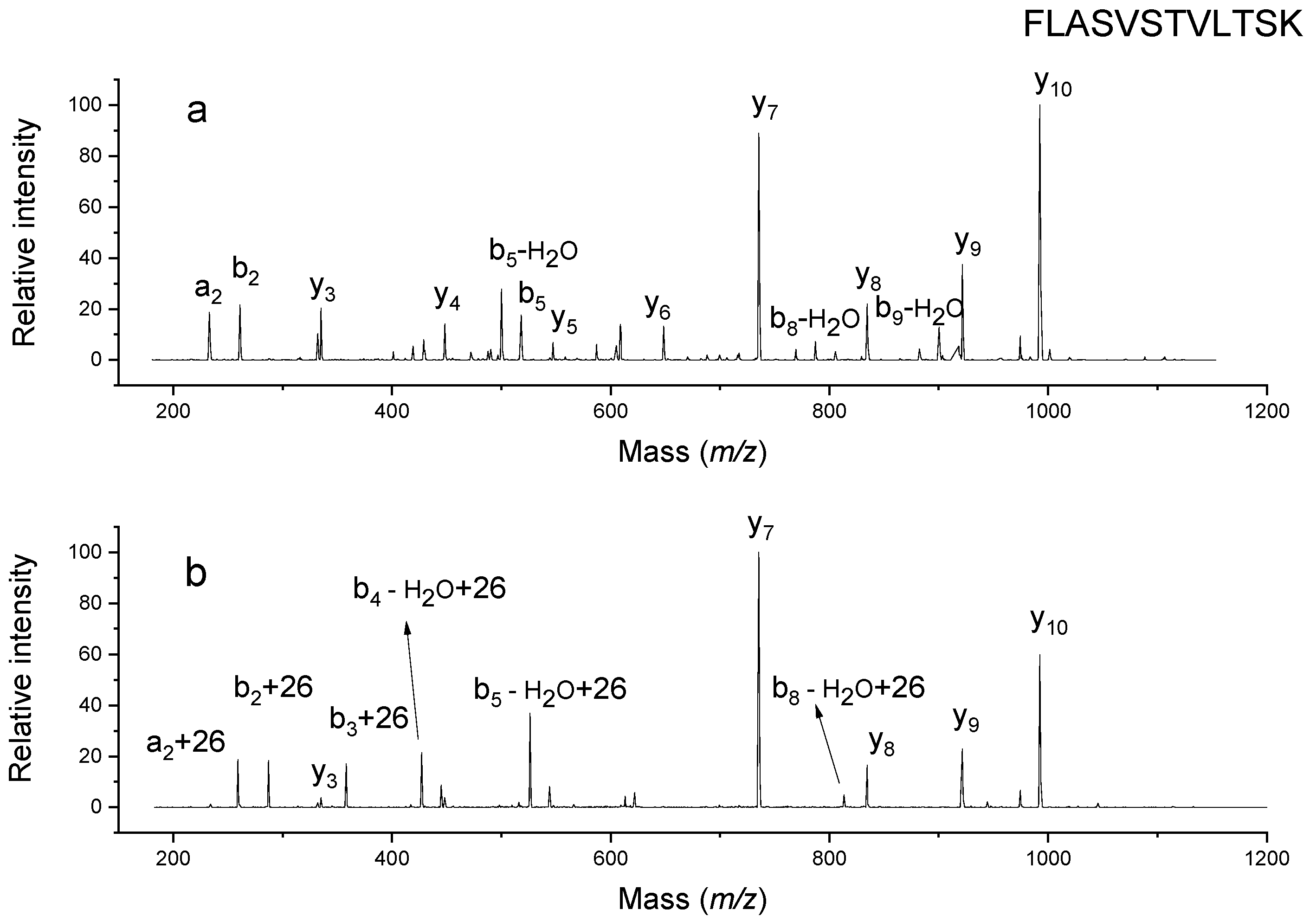
2.1. Analysis of Modified Hemoglobin Digest
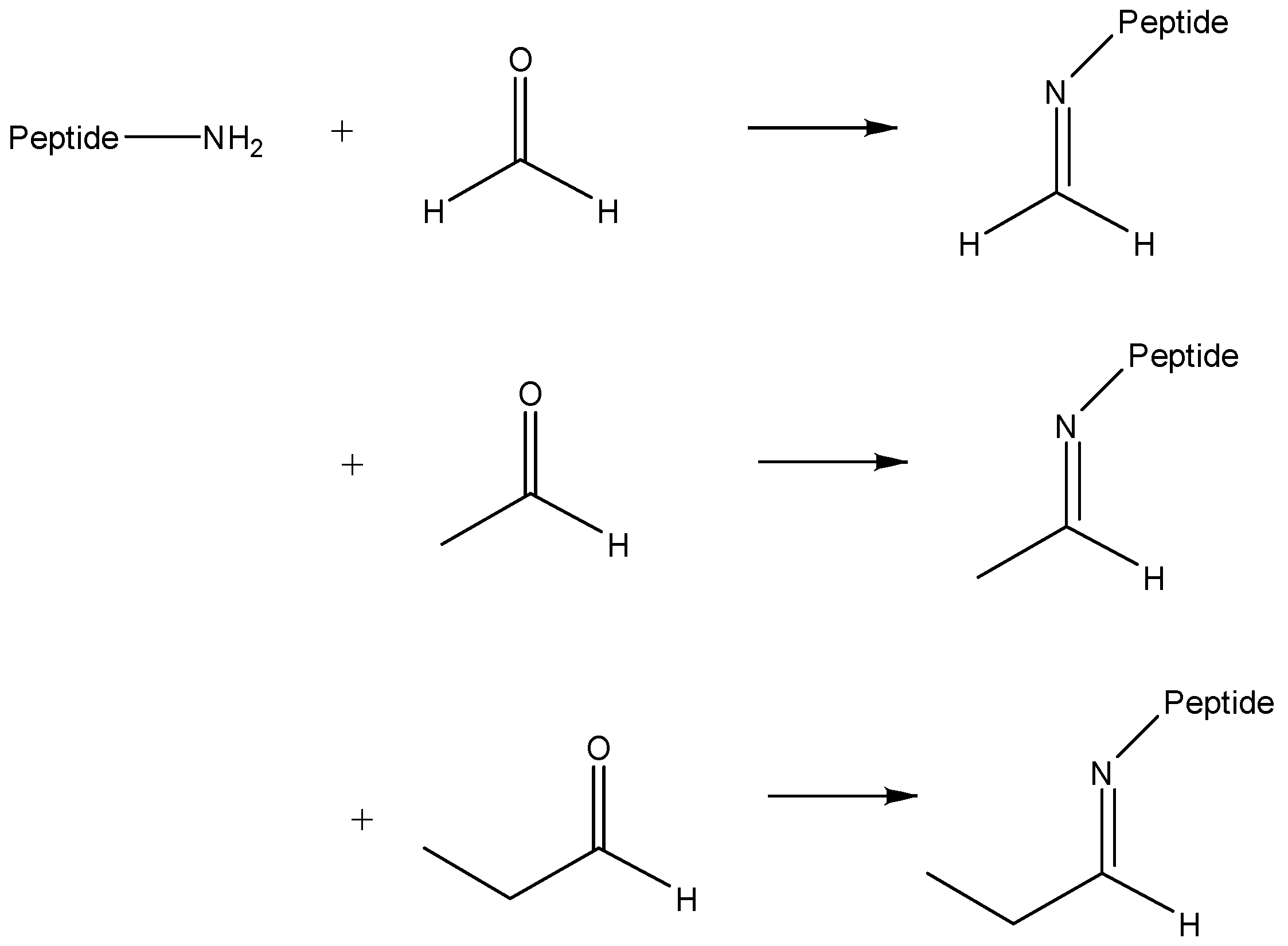
2.2. Confirmation of Chemical Composition of Modification
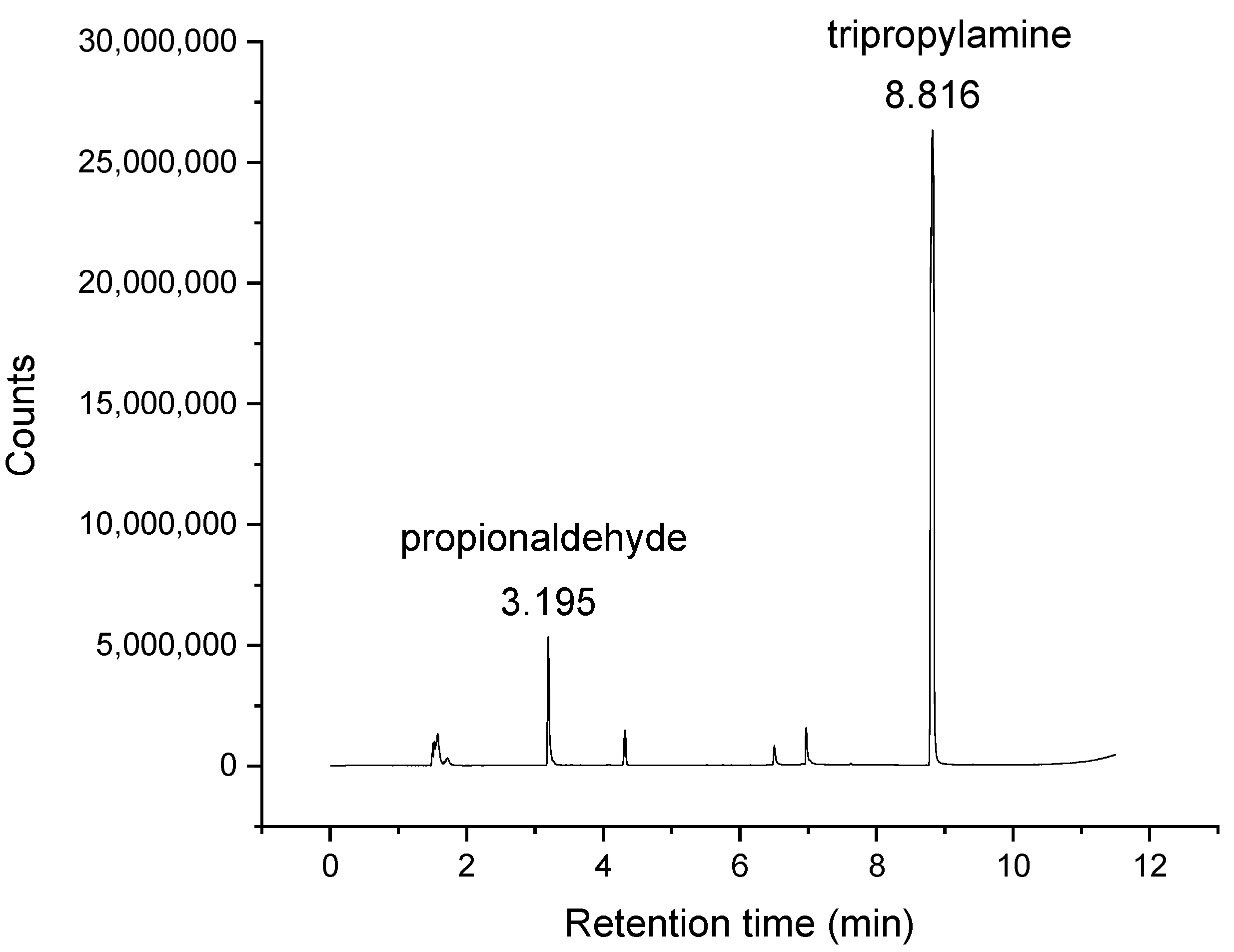
2.3. Verification of Presence of Aldehydes in Trialkylamines
2.4. Critical Conditions for Trialkylamine Modifications
2.5. Elimination of Undesirable Aldehyde-Induced Modifications with Propylamine
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jandera, P. Liquid Chromatography|Normal Phase. In Encyclopedia of Analytical Science; Elsevier: Amsterdam, The Netherlands, 2005; p. 142. [Google Scholar]
- Li, S.; Tian, M.; Row, K.H. Effect of Mobile Phase Additives on the Resolution of Four Bioactive Compounds by RP-HPLC. Int. J. Mol. Sci. 2010, 11, 2229–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, G.; Wei, X.; Liu, Z.; Liu, S.; Marcucci, G.; Chan, K.K. Characterization and quantification of Bcl-2 antisense G3139 and metabolites in plasma and urine by ion-pair reversed phase HPLC coupled with electrospray ion-trap mass spectrometry. J. Chromatogr. B 2005, 825, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Dubost, N.J.; Beelman, R.B.; Peterson, D.; Royse, D.J. Identification and Quantification of Ergothioneine in Cultivated Mushrooms by Liquid Chromatography-Mass Spectroscopy. Int. J. Med. Mushrooms 2006, 8, 215–222. [Google Scholar] [CrossRef]
- Bai, F.; Kirstein, M.N.; Hanna, S.K.; Iacono, L.C.; Johnston, B.; Stewart, C.F. Determination of plasma topotecan and its metabolite N-desmethyl topotecan as both lactone and total form by reversed-phase liquid chromatography with fluorescence detection. J. Chromatogr. B 2003, 784, 225–232. [Google Scholar] [CrossRef]
- Jurchen, J.C.; Williams, E.R. Origin of Asymmetric Charge Partitioning in the Dissociation of Gas-Phase Protein Homodimers. J. Am. Chem. Soc. 2003, 125, 2817–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Gough, P.C.; DeFelippis, M.R. Characterization of Poly(ethylene glycol) and PEGylated Products by LC/MS with Postcolumn Addition of Amines. Anal. Chem. 2008, 81, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Hogan, C.J., Jr.; De La Mora, J.F. Ion mobility measurements of nondenatured 12–150 kDa proteins and protein multimers by tandem differential mobility analysis-mass spectrometry (DMA-MS). J. Am. Soc. Mass Spectrom. 2011, 22, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yan, B. Improving Covalent Organic Frameworks Fluorescence by Triethylamine Pinpoint Surgery as Selective Biomarker Sensor for Diabetes Mellitus Diagnosis. Anal. Chem. 2019, 91, 13183–13190. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.P.; Moon, A.S.; Back, P.S.; Jami-Alahmadi, Y.; Vashisht, A.A.; Wohlschlegel, J.A.; Bradley, P.J. A photoactivatable crosslinking system reveals protein interactions in the Toxoplasma gondii inner membrane complex. PLoS Biol. 2019, 17, e3000475. [Google Scholar] [CrossRef]
- Tyler-Cross, R.; Schirch, V. Effects of amino acid sequence, buffers, and ionic strength on the rate and mechanism of deamidation of asparagine residues in small peptides. J. Biol. Chem. 1991, 266, 22549–22556. [Google Scholar] [CrossRef]
- Roose, P.; Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 1–55. [Google Scholar]
- Lemon, R.C.; Myerly, R.C. Production of Amines. U.S. Patent 3,022,349, 20 February 1962. [Google Scholar]
- Wall, P.E. Chromatography: Thin-layer (Planar)|Spray Reagents. In Encyclopedia of Separation Science; Academic Press: Cambridge, MA, USA, 2000; pp. 907–915. [Google Scholar]
- Fleischmann, G.; Jira, R.; Bolt, H.M.; Golka, K. Acetaldehyde. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 191–205. [Google Scholar]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Brown, S.M.; Urch, C.J. Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones: An Efficient, Aerobic Alternative. Science 1996, 274, 2044–2046. [Google Scholar] [CrossRef] [Green Version]
- DeLamater, E.D.; Mescon, H.; Barger, J.D. The Chemistry of the Feulgen Reaction and Related Histo- and Cytochemical Methods. J. Investig. Dermatol. 1950, 14, 133–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hormann, H.; Grassmany, W.; Frie, G. Uber Den Mechanismudse R Schiffschen Reaktion. Justus Liebigs Ann. Chem. 1958, 616, 125–147. [Google Scholar] [CrossRef]
- Robins, J.H.; Abrams, G.D.; Pincock, J.A. The structure of Schiff reagent aldehyde adducts and the mechanism of the Schiffreaction as determined by nuclear magnetic resonance spectroscopy. Can. J. Chem. 1980, 58, 339–347. [Google Scholar] [CrossRef]
- Farris, S.; Song, J.; Huang, Q. Alternative Reaction Mechanism for the Cross-Linking of Gelatin with Glutaraldehyde. J. Agric. Food Chem. 2010, 58, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, G.T. Bioconjugate Techniques, 2nd ed.; Academic Press: Cambridge, MA, USA, 2008; pp. 200–201. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Gandhi, P.J.; Reilly, J.P. Aqueous Solutions of Peptides and Trialkylamines Lead to Unexpected Peptide Modification. Molecules 2021, 26, 6481. https://doi.org/10.3390/molecules26216481
Ma Y, Gandhi PJ, Reilly JP. Aqueous Solutions of Peptides and Trialkylamines Lead to Unexpected Peptide Modification. Molecules. 2021; 26(21):6481. https://doi.org/10.3390/molecules26216481
Chicago/Turabian StyleMa, Yiran, Puja J. Gandhi, and James P. Reilly. 2021. "Aqueous Solutions of Peptides and Trialkylamines Lead to Unexpected Peptide Modification" Molecules 26, no. 21: 6481. https://doi.org/10.3390/molecules26216481