
Efficient Degradation of Iopromide by Using Sulfite Activated with Mackinawite

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Characterization of FeS
2.2. Control Experiments
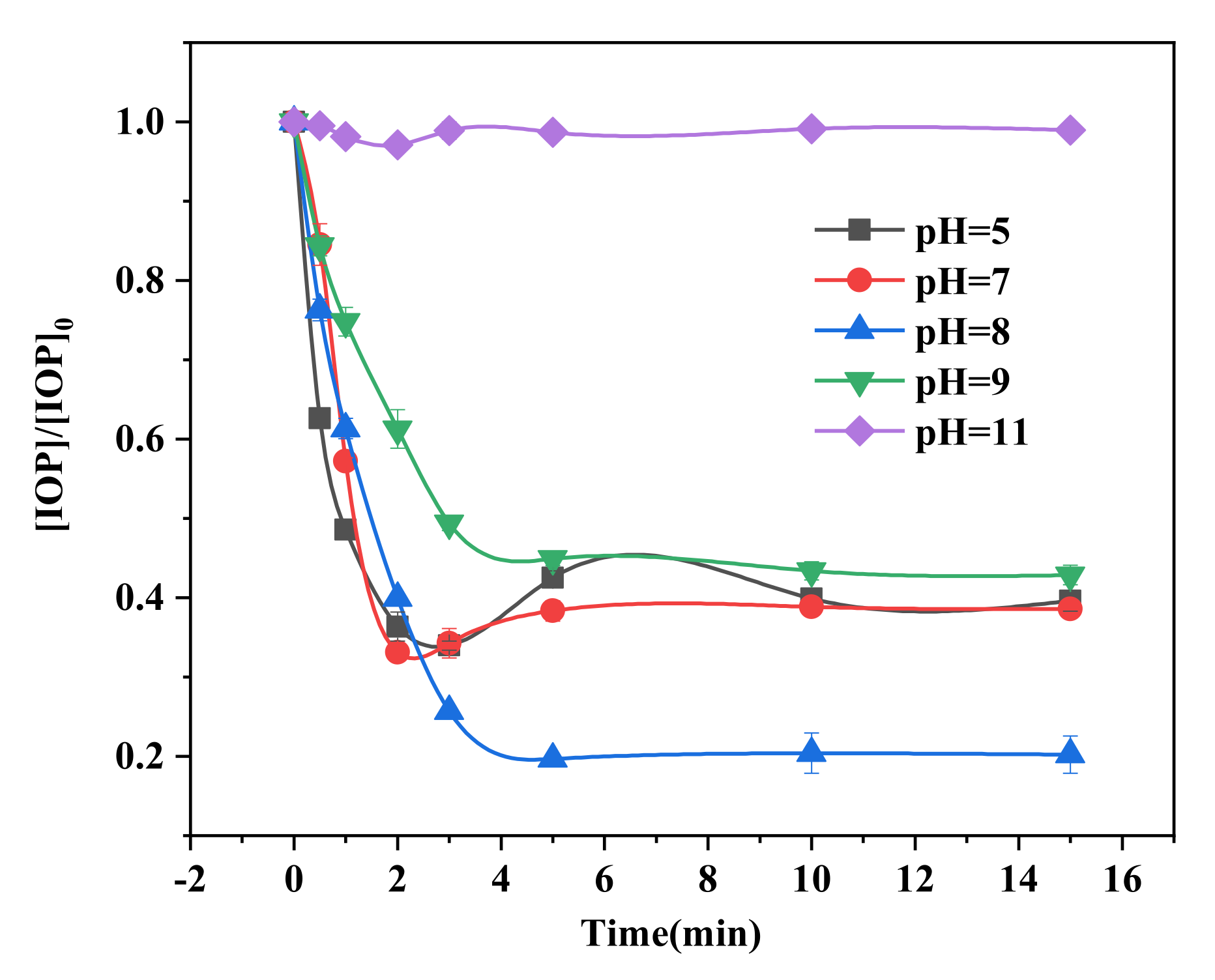
2.3. Effects of Initial pH
2.4. Effects of FeS Dosage
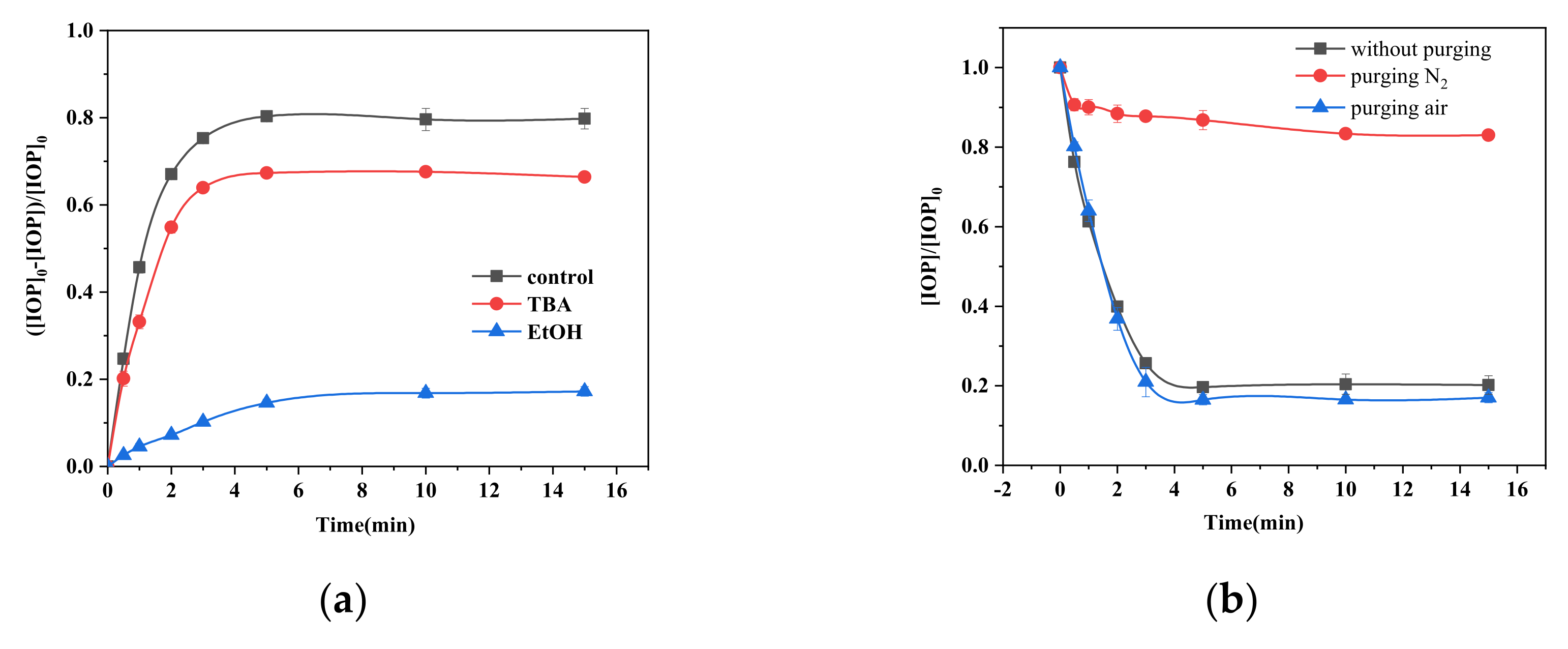
2.5. Role of Radicals and Dissolved Oxygen
2.6. Effects of Inorganic Anions
2.7. Removal of TOC
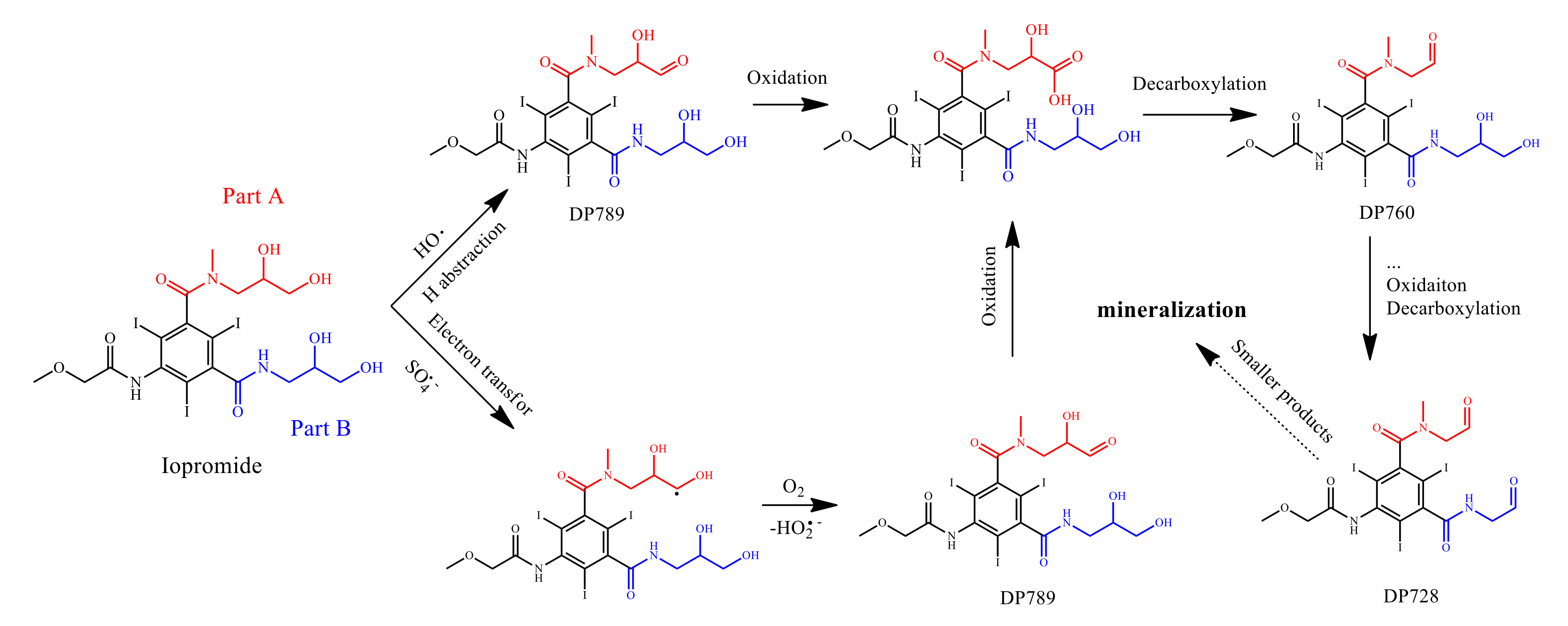
2.8. Determination of Degradation Products
3. Materials and Methods
3.1. Materials
3.2. Experiment
3.3. Analytical Methods
3.3.1. Characterization
3.3.2. High-Performance Liquid Chromatography (HPLC)
3.3.3. Liquid Chromatography-MS
3.3.4. The Other Analytical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nowak, A.; Pacek, G.; Mrozik, A. Transformation and ecotoxicological effects of iodinated X-ray contrast media. Rev. Environ. Sci. Bio 2020, 19, 337–354. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Yuan, R.; Xiao, D. Resolving the kinetic and intrinsic constraints of heat-activated peroxydisulfate oxidation of iopromide in aqueous solution. J. Hazard. Mater. 2020, 384. [Google Scholar] [CrossRef]
- Zhang, W.; Soutrel, I.; Amrane, A.; Fourcade, F.; Geneste, F. Electro-reductive deiodination of iohexol catalyzed by vitamin B12 and biodegradability investigation. J. Electroanal. Chem. 2021, 897. [Google Scholar] [CrossRef]
- Sengar, A.; Vijayanandan, A. Comprehensive review on iodinated X-ray contrast media: Complete fate, occurrence, and formation of disinfection byproducts. Sci. Total Environ. 2021, 769. [Google Scholar] [CrossRef]
- Eversloh, C.L.; Henning, N.; Schulz, M.; Ternes, T.A. Electrochemical treatment of iopromide under conditions of reverse osmosis concentrates–Elucidation of the degradation pathway. Water Res. 2014, 48, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Bichsel, Y.; von Gunten, U. Formation of Iodo-Trihalomethanes during disinfection and oxidation of Iodide-Containing waters. Environ. Sci. Technol. 2000, 34, 2784–2791. [Google Scholar] [CrossRef]
- Lopez-Prieto, I.J.; Wu, S.; Ji, W.; Daniels, K.D.; Snyder, S.A. A direct injection liquid chromatography tandem mass spectrometry method for the kinetic study on iodinated contrast media (ICMs) removal in natural water. Chemosphere 2020, 243. [Google Scholar] [CrossRef] [PubMed]
- Westerhoff, P.; Yoon, Y.; Snyder, S.; Wert, E. Fate of Endocrine-Disruptor, pharmaceutical, and personal care product chemicals during simulated drinking water treatment processes. Environ. Sci. Technol. 2005, 39, 6649–6663. [Google Scholar] [CrossRef]
- Xu, H.; Wang, L.; Li, X.; Chen, Z.; Zhang, T. Thiourea dioxide coupled with trace Cu(II): An effective process for the reductive degradation of diatrizoate. Environ. Sci. Technol. 2021, 55, 12009–12018. [Google Scholar] [CrossRef] [PubMed]
- Durán-álvarez, J.C.; Hernández-Morales, V.A.; Rodríguez-Varela, M.; Guerrero-Araque, D.; Ramirez-Ortega, D.; Castillón, F.; Acevedo-Peña, P.; Zanella, R. Ag2O/TiO2 nanostructures for the photocatalytic mineralization of the highly recalcitrant pollutant iopromide in pure and tap water. Catal. Today 2020, 341, 71–81. [Google Scholar] [CrossRef]
- Zhang, T.; Zhu, H.; Croué, J. Production of sulfate radical from peroxymonosulfate induced by a magnetically separable CuFe2O4 spinel in water: Efficiency, stability, and mechanism. Environ. Sci. Technol. 2013, 47, 2784–2791. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Hanna, K.; Mailhot, G.; Brigante, M. A review of Manganese(III) (Oxyhydr)Oxides use in advanced oxidation processes. Molecules (Basel, Switzerland) 2021, 26, 5748. [Google Scholar] [CrossRef]
- Hu, C.; Hou, Y.; Lin, Y.; Deng, Y.; Hua, S.; Du, Y.; Chen, C.; Wu, C. Investigation of iohexol degradation kinetics by using heat-activated persulfate. Chem. Eng. J. 2020, 379. [Google Scholar] [CrossRef]
- Chan, T.W.; Graham, N.J.D.; Chu, W. Degradation of iopromide by combined UV irradiation and peroxydisulfate. J. Hazard. Mater. 2010, 181, 508–513. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Pan, F.; Qin, Y.; Xia, J.; Li, J.; Wu, F. Synthesis of the mesoporous carbon-nano-zero-valent iron composite and activation of sulfite for removal of organic pollutants. Chem. Eng. J. 2018, 353, 542–549. [Google Scholar] [CrossRef]
- Anipsitakis, G.P.; Dionysiou, D.D. Radical generation by the interaction of transition metals with common oxidants. Environ. Sci. Technol. 2004, 38, 3705–3712. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Vione, D.; Rivoira, L.; Carena, L.; Castiglioni, M.; Bruzzoniti, M.C. A review on the degradation of pollutants by Fenton-Like systems based on Zero-Valent iron and persulfate: Effects of reduction potentials, pH, and anions occurring in waste waters. Molecules 2021, 26, 4584. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Choi, J.; Kim, M.; Kim, M.S.; Lee, D.; Bang, W.H.; Yun, E.; Lee, H.; Lee, J.; Lee, C.; et al. Chloride-Mediated enhancement in Heat-Induced activation of peroxymonosulfate: New reaction pathways for oxidizing radical production. Environ. Sci. Technol. 2021, 55, 5382–5392. [Google Scholar] [CrossRef]
- Berruti, I.; Nahim-Granados, S.; Abeledo-Lameiro, M.J.; Oller, I.; Polo-Lopez, M.I. UV-C peroxymonosulfate activation for wastewater regeneration: Simultaneous inactivation of pathogens and degradation of contaminants of emerging concern. Molecules 2021, 26, 5748. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; von Gunten, U.; Kim, J. Persulfate-Based advanced oxidation: Critical assessment of opportunities and roadblocks. Environ. Sci. Technol. 2020, 54, 3064–3081. [Google Scholar] [CrossRef] [PubMed]
- Fazli, A.; Khataee, A.; Brigante, M.; Mailhot, G. Cubic cobalt and zinc co-doped magnetite nanoparticles for persulfate and hydrogen peroxide activation towards the effective photodegradation of Sulfalene. Chem. Eng. J. 2021, 404. [Google Scholar] [CrossRef]
- Chen, Y.; Li, M.; Tong, Y.; Liu, Z.; Fang, L.; Wu, Y.; Fang, Z.; Wu, F.; Huang, L. Radical generation via sulfite activation on NiFe2O4 surface for estriol removal: Performance and mechanistic studies. Chem. Eng. J. 2019, 368, 495–503. [Google Scholar] [CrossRef]
- Bäckström, H.L.J. Der Kettenmechanismus bei der Autoxydation von Aldehyden. Zeitschrift für Physikalische Chemie 1934, 25, 122–138. [Google Scholar] [CrossRef]
- Yu, Y.; Li, S.; Peng, X.; Yang, S.; Zhu, Y.; Chen, L.; Wu, F.; Mailhot, G. Efficient oxidation of bisphenol a with oxysulfur radicals generated by iron-catalyzed autoxidation of sulfite at circumneutral pH under UV irradiation. Environ. Chem. Lett. 2016, 14, 527–532. [Google Scholar] [CrossRef]
- Zuo, Y.; Zhan, J.; Wu, T. Effects of monochromatic UV-Visible light and sunlight on Fe(III)-Catalyzed oxidation of dissolved sulfur dioxide. J. Atmos. Chem. 2005, 50, 195–210. [Google Scholar] [CrossRef]
- Yang, Y.; Sun, M.; Zhou, J.; Ma, J.; Komarneni, S. Degradation of orange II by Fe@Fe2O3 core shell nanomaterials assisted by NaHSO3. Chemosphere 2020, 244, 125588. [Google Scholar] [CrossRef]
- Zhu, C.; Fang, G.; Dionysiou, D.D.; Liu, C.; Gao, J.; Qin, W.; Zhou, D. Efficient transformation of DDTs with persulfate activation by zero-valent iron nanoparticles: A mechanistic study. J. Hazard. Mater. 2016, 316, 232–241. [Google Scholar] [CrossRef]
- Yuan, Y.; Tao, H.; Fan, J.; Ma, L. Degradation of p-chloroaniline by persulfate activated with ferrous sulfide ore particles. Chem. Eng. J. 2015, 268, 38–46. [Google Scholar] [CrossRef]
- Fan, J.; Gu, L.; Wu, D.; Liu, Z. Mackinawite (FeS) activation of persulfate for the degradation of p-chloroaniline: Surface reaction mechanism and sulfur-mediated cycling of iron species. Chem. Eng. J. 2018, 333, 657–664. [Google Scholar] [CrossRef]
- Rickard, D. Kinetics of FeS precipitation: Part 1. Competing reaction mechanisms. Geochim. Cosmochim. Ac. 1995, 59, 4367–4379. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Feng, M.; Liu, W.; Wang, W.; Yang, Q.; Hu, Y. Degradation of 2,4-dichlorophenoxyacetic acid in water by persulfate activated with FeS (mackinawite). Chem. Eng. J. 2017, 313, 498–507. [Google Scholar] [CrossRef]
- Fan, J.; Cai, Y.; Shen, S.; Gu, L. New insights into FeS/persulfate system for tetracycline elimination: Iron valence, homogeneous-heterogeneous reactions and degradation pathways. J. Environ. Sci.-China 2022, 112, 48–58. [Google Scholar] [CrossRef]
- Chen, Y.; Tong, Y.; Xue, Y.; Liu, Z.; Tang, M.; Huang, L.; Shao, S.; Fang, Z. Degradation of the β-blocker propranolol by sulfite activation using FeS. Chem. Eng. J. 2020, 385, 123884. [Google Scholar] [CrossRef]
- Wolthers, M.; Van Der Gaast, S.J.; Rickard, D. The structure of disordered mackinawite. Am. Mineral. 2003, 88, 2007–2015. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Yang, Z.; Yang, Q.; Li, B.; Bai, Z. Heterogeneous fenton-like catalytic degradation of 2,4-dichlorophenoxyacetic acid in water with FeS. Chem. Eng. J. 2015, 273, 481–489. [Google Scholar] [CrossRef]
- Schmidt, T.; Leemann, A.; Gallucci, E.; Scrivener, K. Physical and microstructural aspects of iron sulfide degradation in concrete. Cement Concrete Res. 2011, 41, 263–269. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, J.; Dong, F. Spectroscopic study on biological mackinawite (FeS) synthesized by ferric reducing bacteria (FRB) and sulfate reducing bacteria (SRB): Implications for in-situ remediation of acid mine drainage. Spectrochim. Acta A 2017, 173, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Khabbaz, M.; Entezari, M.H. Simple and versatile one-step synthesis of FeS2 nanoparticles by ultrasonic irradiation. J. Colloid Interf. Sci. 2016, 470, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Jia, S.; Yu, B.; Wu, S.; Han, X. Formation of iron (hydr)oxides during the abiotic oxidation of Fe(II) in the presence of arsenate. J. Hazard. Mater. 2015, 294, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Herbst, J.; Heyne, K.; Diller, R. Femtosecond infrared spectroscopy of bacteriorhodopsin chromophore isomerization. Science (New York N.Y.) 2002, 297, 822–825. [Google Scholar] [CrossRef]
- He, Y.T.; Wilson, J.T.; Wilkin, R.T. Impact of iron sulfide transformation on trichloroethylene degradation. Geochim. Cosmochim. Ac. 2010, 74, 2025–2039. [Google Scholar] [CrossRef]
- Gong, Y.; Tang, J.; Zhao, D. Application of iron sulfide particles for groundwater and soil remediation: A review. Water Res. 2016, 89, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Chen, L.; Li, J.; Wu, F. Transition metal catalyzed sulfite auto-oxidation systems for oxidative decontamination in waters: A state-of-the-art minireview. Chem. Eng. J. 2018, 346, 726–738. [Google Scholar] [CrossRef]
- Chen, L.; Peng, X.; Liu, J.; Li, J.; Wu, F. Decolorization of orange II in aqueous solution by an Fe(II)/sulfite system: Replacement of persulfate. Ind. Eng. Chem. Res. 2012, 51, 13632–13638. [Google Scholar] [CrossRef]
- Brandt, C.; Fabian, I.; van Eldik, R. Kinetics and mechanism of the Iron(III)-catalyzed autoxidation of Sulfur(IV) oxides in aqueous solution. Evidence for the redox cycling of iron in the presence of oxygen and modeling of the overall reaction mechanism. Inorg. Chem. 1994, 33, 687–701. [Google Scholar] [CrossRef]
- Duinea, M.I.; Costas, A.; Baibarac, M.; Chirita, P. Mechanism of the cathodic process coupled to the oxidation of iron monosulfide by dissolved oxygen. J. Colloid Interf. Sci. 2016, 467, 51–59. [Google Scholar] [CrossRef]
- Tamura, H.; Goto, K.; Yotsuyanagi, T.; Nagayama, M. Spectrophotometric determination of iron(II) with 1,10-phenanthroline in the presence of large amounts of iron(III). Talanta 1974, 21, 314–318. [Google Scholar] [CrossRef]
- Gupta, S.S.; Gupta, Y.K. Hydrogen ion dependence of the oxidation of iron(II) with peroxydisulfate in acid perchlorate solutions. Cheminform 1981, 12, 454–457. [Google Scholar] [CrossRef]
- Thomas, J.E.; Jones, C.F.; Skinner, W.M.; Smart, R.S.C. The role of surface sulfur species in the inhibition of pyrrhotite dissolution in acid conditions. Geochim. Cosmochim. Ac. 1998, 62, 1555–1565. [Google Scholar] [CrossRef]
- Wine, P.H.; Tang, Y.; Thorn, R.P.; Wells, J.R.; Davis, D.D. Kinetics of aqueous phase reactions of the SO4−radical with potential importance in cloud chemistry. J. Geophys. Res. Atmos. 1989, 94, 1085–1094. [Google Scholar] [CrossRef]
- Zhang, W.; Singh, P.; Muir, D. Iron(II) oxidation by SO2/O2 in acidic media: Part, I. Kinetics and mechanism. Hydrometallurgy 2000, 55, 229–245. [Google Scholar] [CrossRef]
- Sun, M.; Huang, W.; Cheng, H.; Ma, J.; Kong, Y.; Komarneni, S. Degradation of dye in wastewater by homogeneous Fe(VI)/NaHSO3 system. Chemosphere 2019, 228, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (⋅OH/⋅o−) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef] [Green Version]
- Hayon, E.; Treinin, A.; Wilf, J. Electronic spectra, photochemistry, and autoxidation mechanism of the sulfite-bisulfite-pyrosulfite systems. SO2−, SO3−, SO4−, and SO5− radicals. J. Am. Chem. Soc. 1972, 94, 47–57. [Google Scholar] [CrossRef]
- Gordon, A.D.; Smirnov, A.; Shumlas, S.L.; Singireddy, S.; Decesare, M.; Schoonen, M.A.A.; Strongin, D.R. Reduction of Nitrite and Nitrate on Nano-dimensioned FeS. Origins Life Evol. B. 2013, 43, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Guo, Y.; Chen, Y.; Wang, Z.; Shang, R.; Wang, S.; Ding, J.; Wan, Y.; Jiang, W.; Ma, J. Application of a novel advanced oxidation process using sulfite and zero-valent iron in treatment of organic pollutants. Chem. Eng. J. 2017, 314, 240–248. [Google Scholar] [CrossRef]
- Fabbri, D.; Calza, P.; Dalmasso, D.; Chiarelli, P.; Santoro, V.; Medana, C. Iodinated X-ray contrast agents: Photoinduced transformation and monitoring in surface water. Sci. Total Environ. 2016, 572, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Aschmann, S.M.; Arey, J.; Atkinson, R. Kinetics and products of the reaction of OH radicals with 3-methoxy-3-methyl-1-butanol. Environ. Sci. Technol. 2011, 45, 6896–6901. [Google Scholar] [CrossRef]
- Zhao, H.; Ji, Y.; Kong, D.; Lu, J.; Yin, X.; Zhou, Q. Degradation of iohexol by Co2+ activated peroxymonosulfate oxidation: Kinetics, reaction pathways, and formation of iodinated byproducts. Chem. Eng. J. 2019, 373, 1348–1356. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Y.; Lyu, Y.; Zhang, T.; Liu, L.; Fan, B.; Wang, J.; Zhang, C. Efficient Degradation of Iopromide by Using Sulfite Activated with Mackinawite. Molecules 2021, 26, 6527. https://doi.org/10.3390/molecules26216527
Yu Y, Lyu Y, Zhang T, Liu L, Fan B, Wang J, Zhang C. Efficient Degradation of Iopromide by Using Sulfite Activated with Mackinawite. Molecules. 2021; 26(21):6527. https://doi.org/10.3390/molecules26216527
Chicago/Turabian StyleYu, Yingtan, Ying Lyu, Ting Zhang, Lin Liu, Bing Fan, Jian Wang, and Chaoxing Zhang. 2021. "Efficient Degradation of Iopromide by Using Sulfite Activated with Mackinawite" Molecules 26, no. 21: 6527. https://doi.org/10.3390/molecules26216527
APA StyleYu, Y., Lyu, Y., Zhang, T., Liu, L., Fan, B., Wang, J., & Zhang, C. (2021). Efficient Degradation of Iopromide by Using Sulfite Activated with Mackinawite. Molecules, 26(21), 6527. https://doi.org/10.3390/molecules26216527