
Panduratin A Derivative Protects against Cisplatin-Induced Apoptosis of Renal Proximal Tubular Cells and Kidney Injury in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
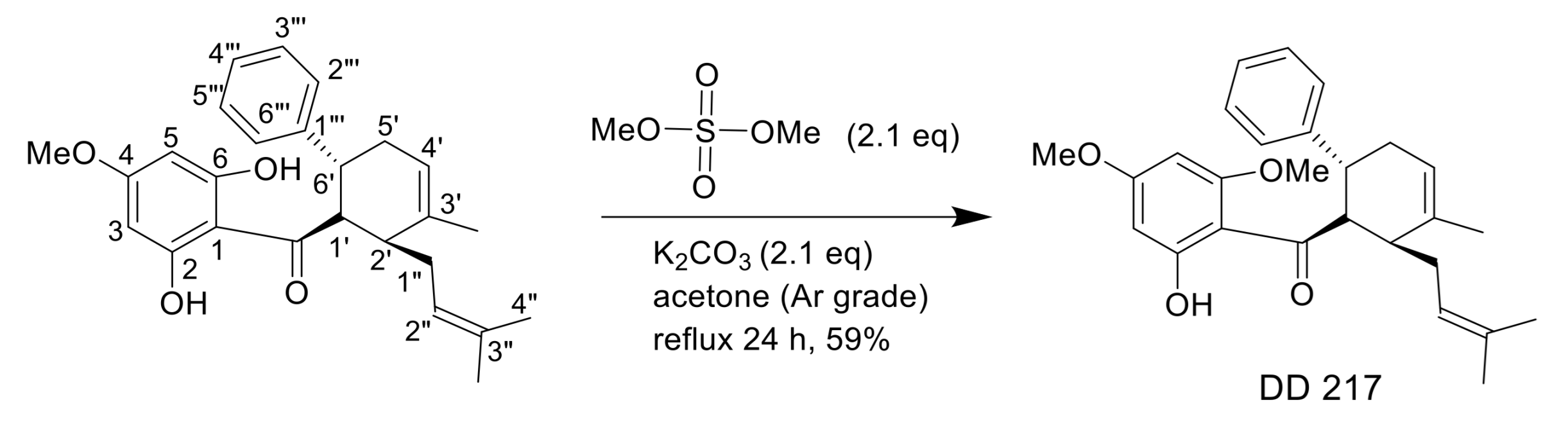
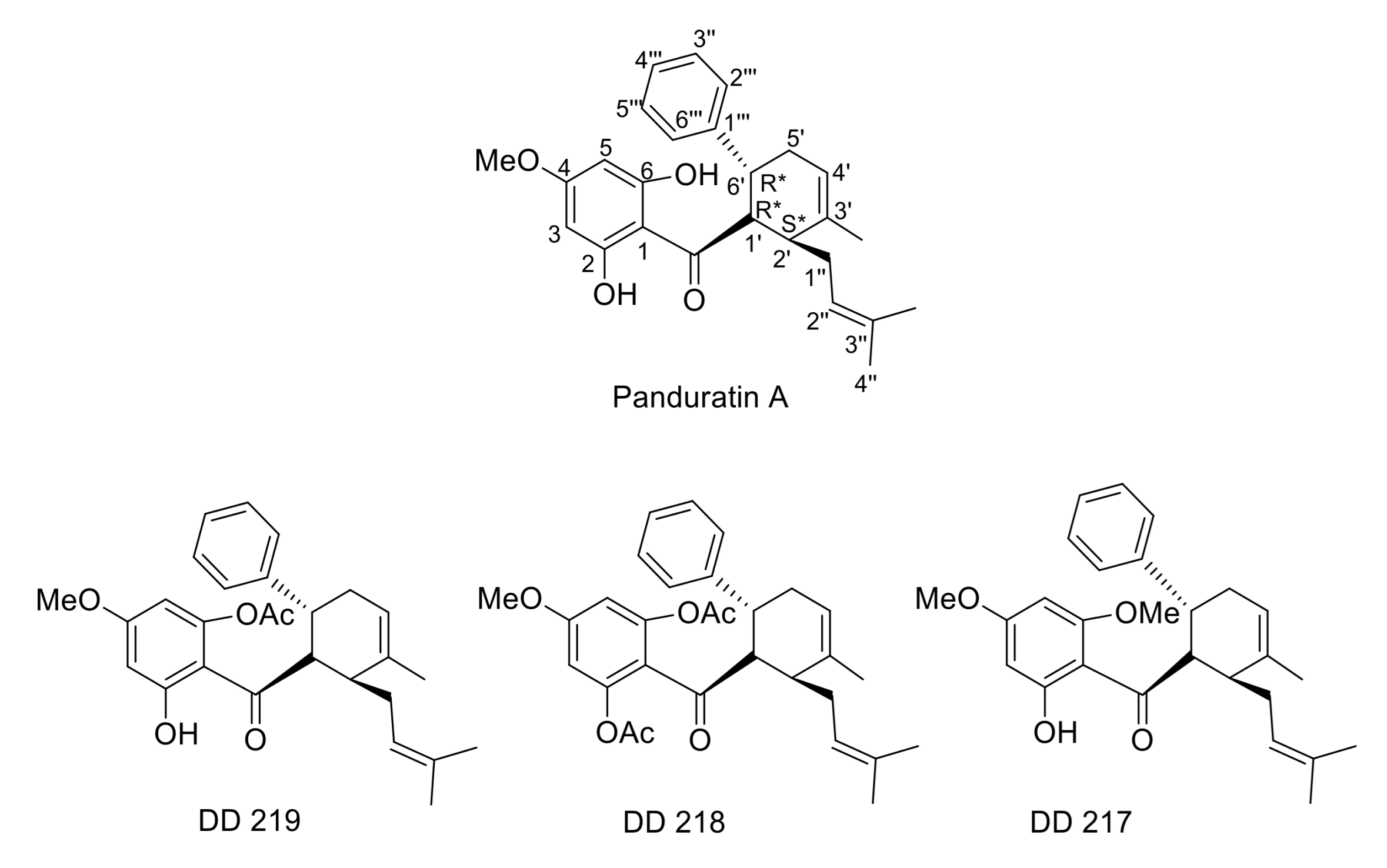
2.1. Synthesis of (±)-(1′R*,2′S*,6′R*)-(2-Hydroxy-4,6-Dimethoxyphenyl)[3′-Methyl-2′-(3″-Methylbut-2″-Enyl)-6-Phenylcyclohex-3′-Enyl]Methanone (DD-217)
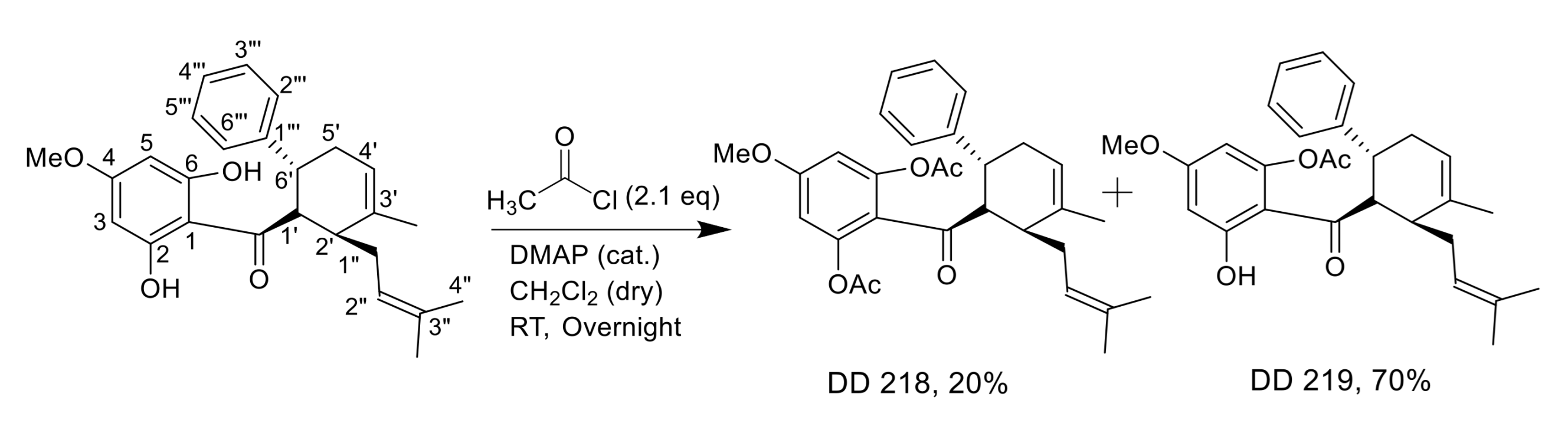
2.2. Synthesis of (±)-(1′R*,2′S*,6′R*)-(2,6-Diacetoxy-4-Methoxyphenyl)[3′-Methyl-2′-(3″-Methylbut-2″-Enyl)-6-Phenylcyclohex-3′-Enyl]Methanone (DD-218) and (±)-(1′R*,2′S*,6′R*)-(6-Acetoxy-2-Hydroxy-4-Methoxyphenyl)[3′-Methyl-2′-(3″-Methylbut-2″-Enyl)-6-Phenylcyclohex-3′-Enyl]Methanone (DD-219)
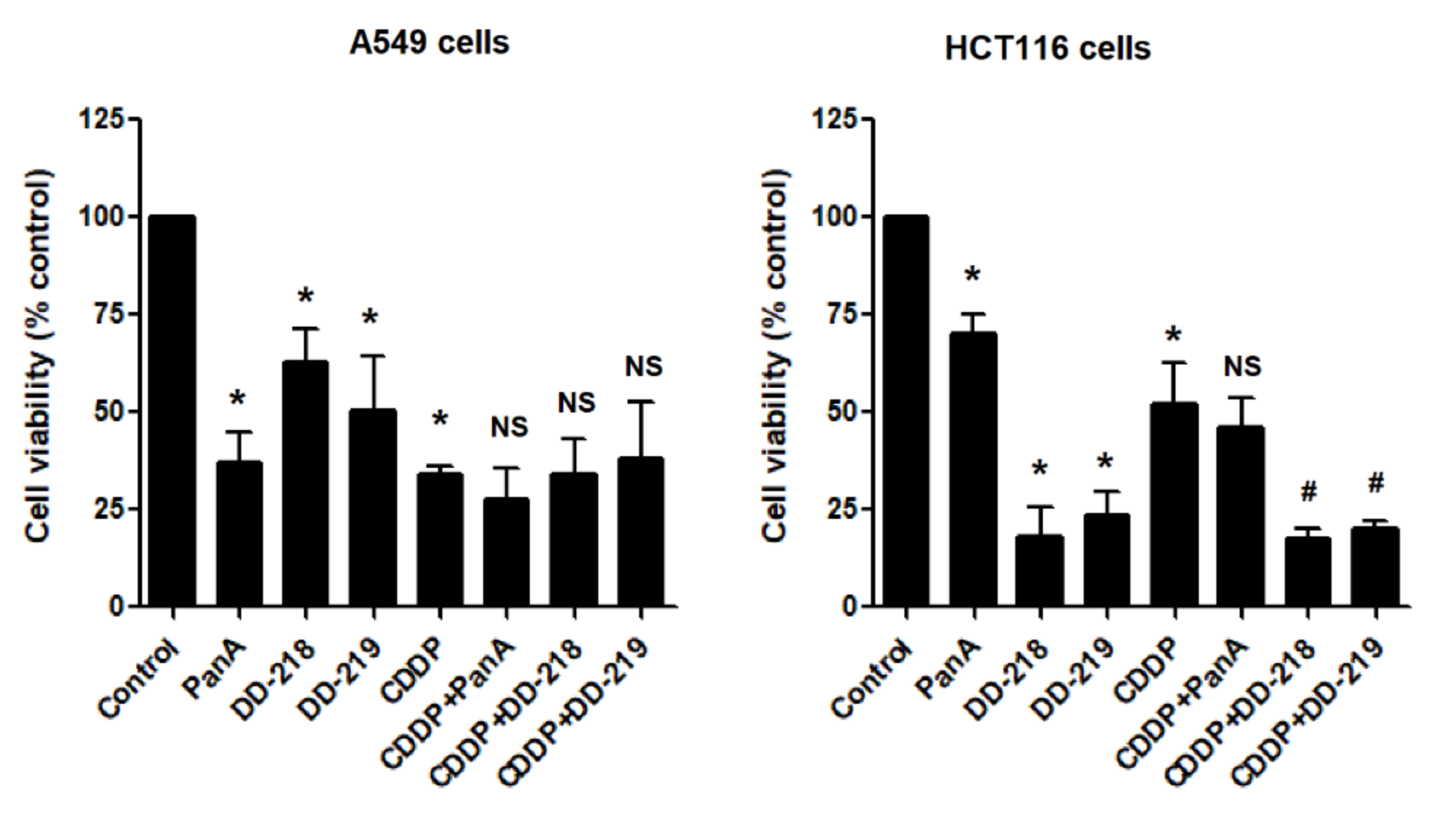
2.3. Derivatives of Panduratin A Mitigate CDDP-Induced Cytotoxicity in Human Renal Proximal Tubular Cells
2.4. The Derivatives of Panduratin A Do Not Limit the Anti-Cancer Activity of CDDP on Cancer Cells
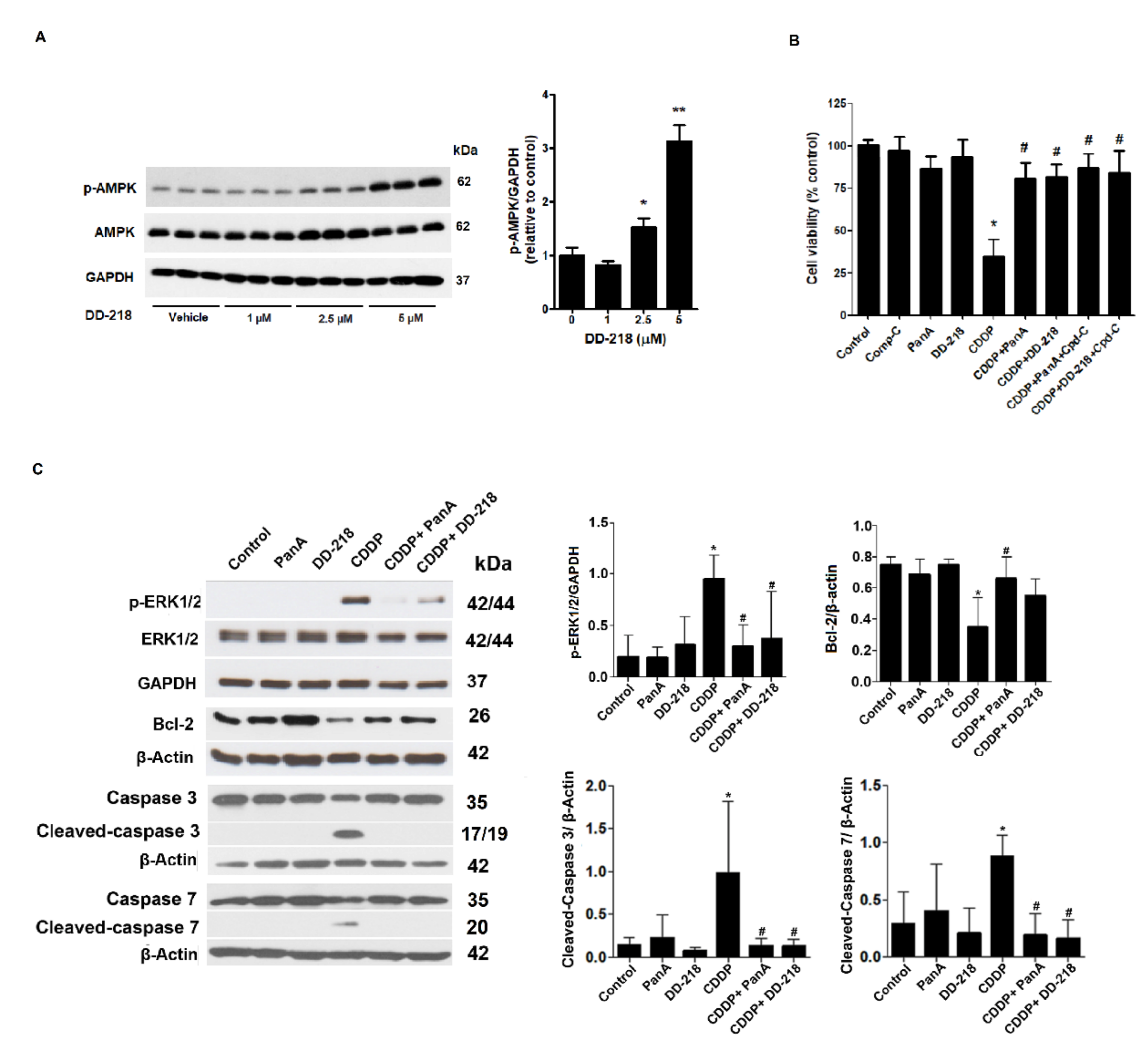
2.5. Underlying Mechanisms of the DD-218 Derivative Reduces the Cytotoxicity of CDDP in Human Renal Proximal Tubular Cells
2.6. DD-218 Reduces CDDP-Induced Oxidative Stress and Protects Mitochondria Function in Human Renal Proximal Tubular Cells
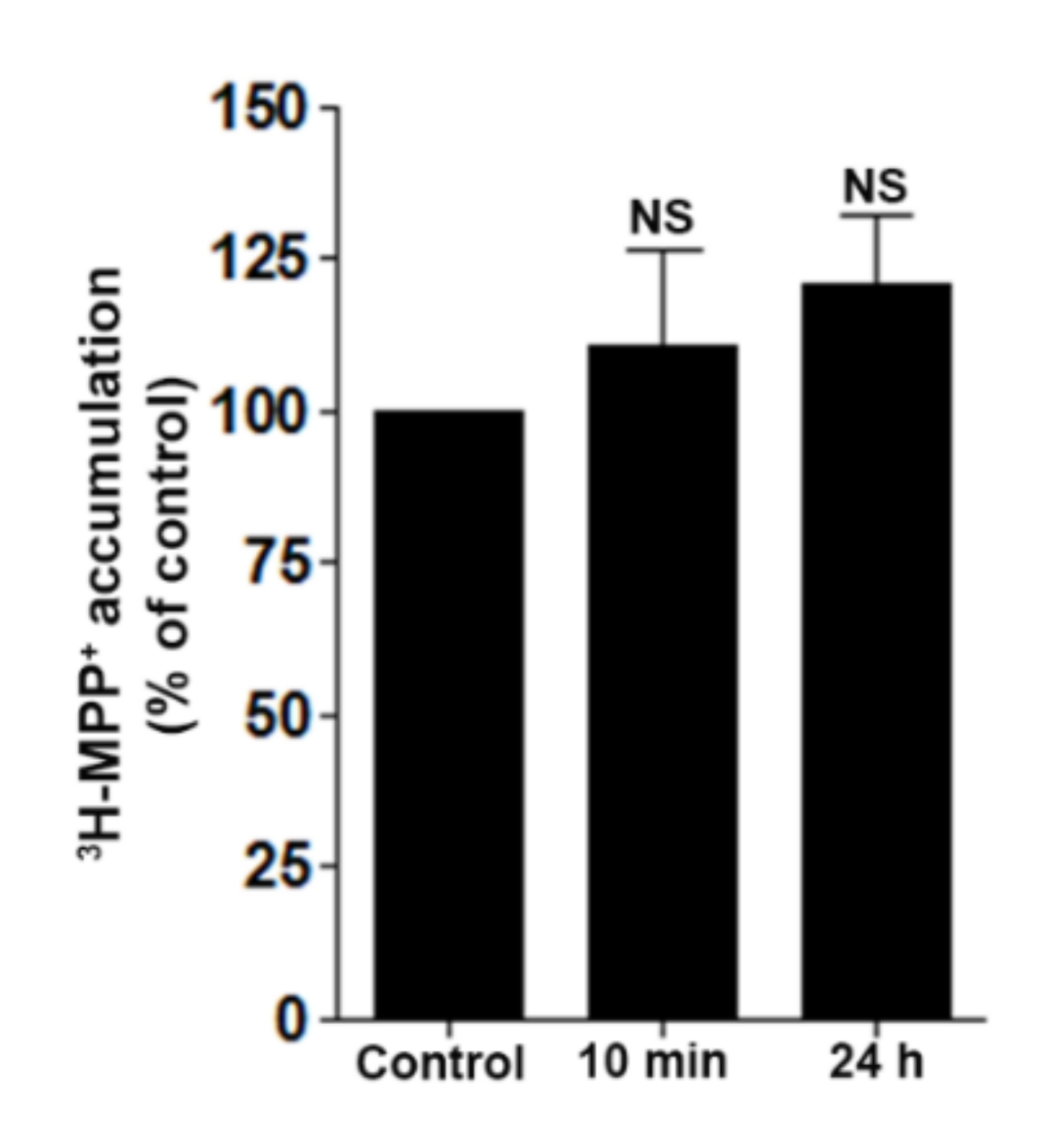
2.7. DD-218 Does Not Inhibit Transport Function of OCT2 in Renal Proximal Tubular Cells
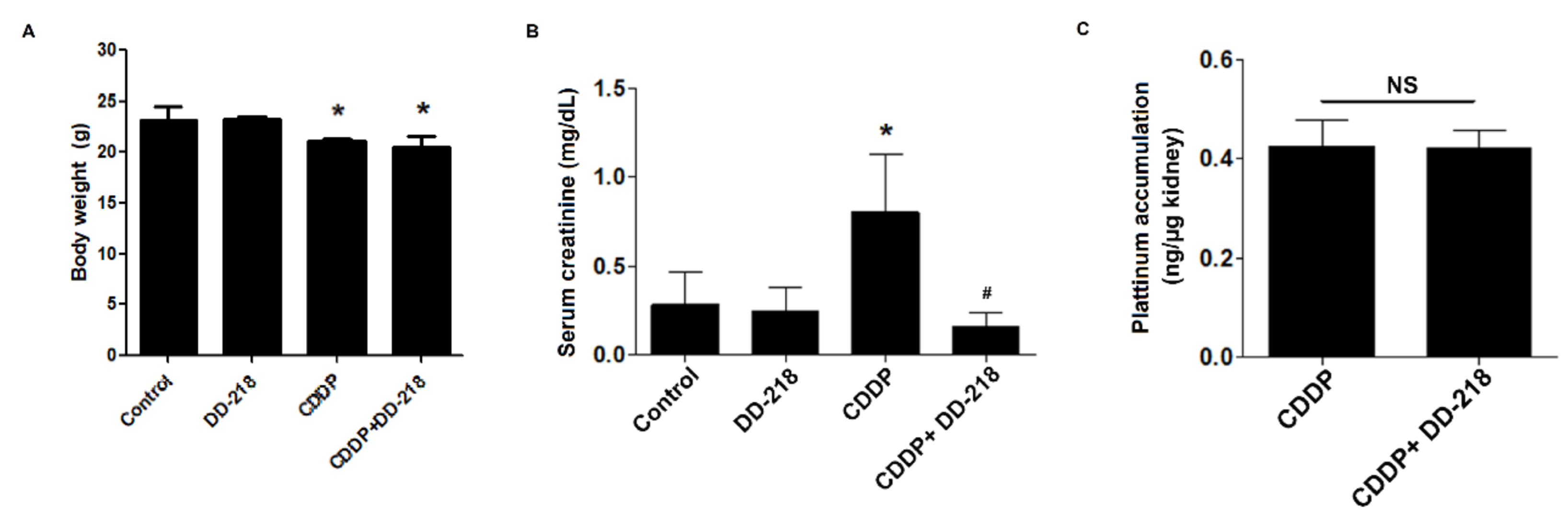
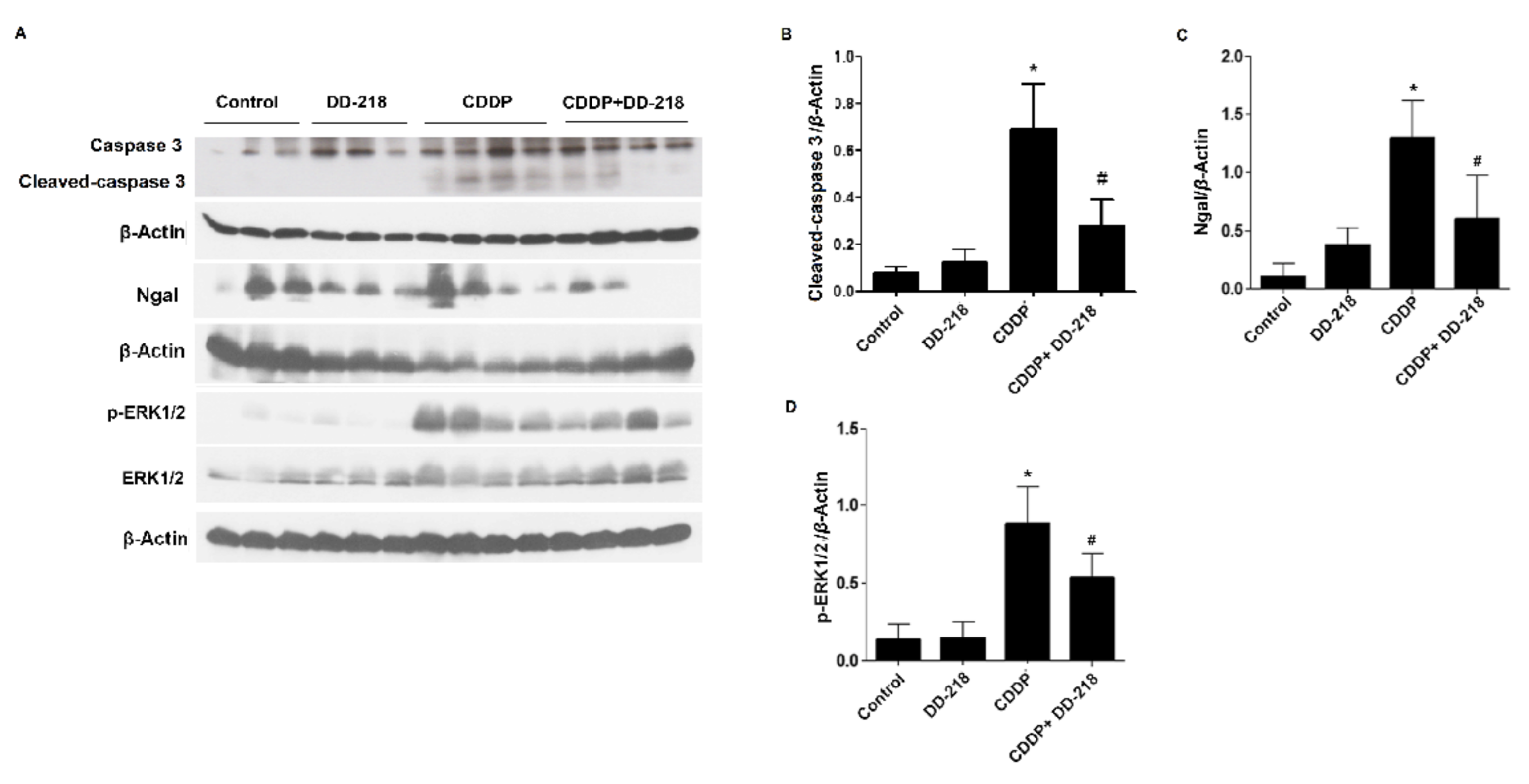
2.8. DD-218 Ameliorates CDDP-Induced Nephrotoxicity in Mice
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. General Experimental Procedures for Synthesis of Panduratin A Derivatives
4.3. Cell Lines and Culture
4.4. Animal
4.5. Cell Viability
4.6. Cell Apoptosis
4.7. Intracellular ROS Accumulation
4.8. Mitochondrial Membrane Potential (JC-1) Assay
4.9. 3H-MMP+ Uptake Assay
4.10. Platinum Accumulation in Renal Tissue
4.11. Immunoblotting
4.12. Renal Function
4.13. Data and Statistical Analysis
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Florea, A.; Büsselberg, D. Metals and metal compounds: Occurrence, use, benefits and toxic cellular effects. Biometals 2006, 19, 419–427. [Google Scholar] [CrossRef]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B.J.T. Mechanisms of cisplatin nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybak, L.P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Cisplatin ototoxicity and protection: Clinical and experimental studies. Tohoku J. Exp. Med. 2009, 219, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miura, K.; Goldstein, R.S.; Pasino, D.A.; Hook, J.B. Cisplatin nephrotoxicity: Role of filtration and tubular transport of cisplatin in isolated perfused kidneys. Toxicology 1987, 44, 147–158. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, Z. Cisplatin nephrotoxicity: Mechanisms and renoprotective strategies. Kidney Int. 2008, 73, 994–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarimboli, G.; Deuster, D.; Knief, A.; Sperling, M.; Holtkamp, M.; Edemir, B.; Pavenstädt, H.; Lanvers-Kaminsky, C.; am Zehnhoff-Dinnesen, A.; Schinkel, A.H.; et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010, 176, 1169–1180. [Google Scholar] [CrossRef]
- Pabla, N.; Murphy, R.F.; Liu, K.; Dong, Z. The copper transporter Ctr1 contributes to cisplatin uptake by renal tubular cells during cisplatin nephrotoxicity. Am. J. Physiol. Renal Physiol. 2009, 296, F505–F511. [Google Scholar] [CrossRef]
- McSweeney, K.R.; Gadanec, L.K.; Qaradakhi, T.; Ali, B.A.; Zulli, A.; Apostolopoulos, V. Mechanisms of Cisplatin-Induced Acute Kidney Injury: Pathological Mechanisms, Pharmacological Interventions, and Genetic Mitigations. Cancers 2021, 13, 1572. [Google Scholar] [CrossRef]
- Clark, J.S.; Faisal, A.; Baliga, R.; Nagamine, Y.; Arany, I. Cisplatin induces apoptosis through the ERK–p66shc pathway in renal proximal tubule cells. Cancer Lett. 2010, 297, 165–170. [Google Scholar] [CrossRef]
- Francescato, H.D.; Costa, R.S.; da Silva, C.G.; Coimbra, T.M. Treatment with a p38 MAPK inhibitor attenuates cisplatin nephrotoxicity starting after the beginning of renal damage. Life Sci. 2009, 84, 590–597. [Google Scholar] [CrossRef]
- Wei, S.Q.; Sui, L.H.; Zheng, J.H.; Zhang, G.M.; Kao, Y.L. Role of ERK1/2 kinase in cisplatin-induced apoptosis in human ovarian carcinoma cells. Chin. Med. Sci. J. 2004, 19, 125–129. [Google Scholar]
- Tsang, R.Y.; Al-Fayea, T.; Au, H.-J. Cisplatin overdose. Drug Saf. 2009, 32, 1109–1122. [Google Scholar] [CrossRef]
- Eng-Chong, T.; Yean-Kee, L.; Chin-Fei, C.; Choon-Han, H.; Sher-Ming, W.; Li-Ping, C.T.; Gen-Teck, F.; Khalid, N.; Abd Rahman, N.; Karsani, S.A.; et al. Boesenbergia rotunda: From Ethnomedicine to Drug Discovery. Evid. Based Complementa. Altern. Med. 2012, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongwisespaiboon, O.; Jiraungkoorskul, W. Fingerroot, Boesenbergia rotunda and its Aphrodisiac Activity. Pharmacogn. Rev. 2017, 11, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Rosdianto, A.M.; Puspitasari, I.M.; Lesmana, R.; Levita, J. Bioactive compounds of Boesenbergia sp. and their anti-inflammatory mechanism: A review. J. Appl. Pharma. Sci. 2020, 10, 116–126. [Google Scholar]
- Tuchinda, P.; Reutrakul, V.; Claeson, P.; Pongprayoon, U.; Sematong, T.; Santisuk, T.; Taylor, W.C.J.P. Anti-inflammatory cyclohexenyl chalcone derivatives in Boesenbergia pandurata. Phytochemistry 2002, 59, 169–173. [Google Scholar] [CrossRef]
- Salama, S.M.; AlRashdi, A.S.; Abdulla, M.A.; Hassandarvish, P.; Bilgen, M. Protective activity of Panduratin A against thioacetamide-induced oxidative damage: Demonstration with in vitro experiments using WRL-68 liver cell line. BMC Complement Altern. Med. 2013, 13, 279. [Google Scholar] [CrossRef] [Green Version]
- Park, K.-M.; Choo, J.-H.; Sohn, J.-H.; Lee, S.-H.; Hwang, J.-K. Antibacterial activity of panduratin A isolated from Kaempferia pandurata against Porphyromonas gingivalis. Food Sci. Biotechnol. 2005, 14, 286–289. [Google Scholar]
- Rukayadi, Y.; Lee, K.; Han, S.; Yong, D.; Hwang, J.-K. In vitro activities of panduratin A against clinical Staphylococcus strains. Antimicrob. Agents Chemother. 2009, 53, 4529–4532. [Google Scholar] [CrossRef] [Green Version]
- Kirana, C.; Jones, G.; Roland Record, I.; McIntosh, G. Anticancer properties of panduratin A isolated from Boesenbergia pandurata (Zingiberaceae). J. Nat. Med. 2007, 61, 131–137. [Google Scholar] [CrossRef]
- Liu, Q.; Cao, Y.; Zhou, P.; Gui, S.; Wu, X.; Xia, Y.; Tu, J. Panduratin A inhibits cell proliferation by inducing G0/G1 phase cell cycle arrest and induces apoptosis in breast cancer cells. Biomol. Ther. 2018, 26, 328. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.-M.; Kweon, M.-H.; Kwon, H.; Hwang, J.-K.; Mukhtar, H. Induction of apoptosis and cell cycle arrest by a chalcone panduratin A isolated from Kaempferia pandurata in androgen-independent human prostate cancer cells PC3 and DU145. Carcinogenesis 2006, 27, 1454–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Kim, M.S.; Hwang, J.-K. Inhibitory Effects of Panduratin A on Allergy-Related Mediator Production in Rat Basophilic Leukemia Mast Cells. Inflammation 2012, 35, 1904–1915. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, M.-S.; Jo, K.; Lee, K.-E.; Hwang, J.-K. Therapeutic potential of panduratin A, LKB1-dependent AMP-activated protein kinase stimulator, with activation of PPARα/δ for the treatment of obesity. Diabetes Obes. Metab. 2011, 13, 584–593. [Google Scholar] [CrossRef]
- Thongnuanjan, P.; Soodvilai, S.; Fongsupa, S.; Chabang, N.; Vivithanaporn, P.; Tuchinda, P.; Soodvilai, S. Protective Effect of Panduratin A on Cisplatin-Induced Apoptosis of Human Renal Proximal Tubular Cells and Acute Kidney Injury in Mice. Biol. Pharm. Bull. 2021, 44, 830–837. [Google Scholar] [CrossRef]
- Tuntiwachwuttikul, P.; Pancharoen, O.; Reutrakul, V.; Byrne, L. (1′RS, 2′SR, 6′RS)-(2, 6-Dihydroxy-4-methoxyphenyl)-[3′-methyl-2′-(3″-methylbut-2″-enyl)-6′-phenyl-cyclohex-3′-enyl] methanone (panduratin A)-a Constituent of the Red Rhizomers of a variety of Boesenbergia pandurata. Aust. J. Chem. 1984, 37, 449–453. [Google Scholar] [CrossRef]
- Cagnol, S.; Chambard, J.-C. ERK and cell death: Mechanisms of ERK-induced cell death–apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef]
- Kruidering, M.; Van de Water, B.; de Heer, E.; Mulder, G.J.; Nagelkerke, J.F. Cisplatin-induced nephrotoxicity in porcine proximal tubular cells: Mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997, 280, 638–649. [Google Scholar] [PubMed]
- Filipski, K.K.; Mathijssen, R.H.; Mikkelsen, T.S.; Schinkel, A.H.; Sparreboom, A. Contribution of organic cation transporter 2 (OCT2) to cisplatin-induced nephrotoxicity. Clin. Pharmacol. Ther. 2009, 86, 396–402. [Google Scholar] [CrossRef]
- Sprowl, J.A.; van Doorn, L.; Hu, S.; van Gerven, L.; de Bruijn, P.; Li, L.; Gibson, A.A.; Mathijssen, R.H.; Sparreboom, A. Conjunctive therapy of cisplatin with the OCT2 inhibitor cimetidine: Influence on antitumor efficacy and systemic clearance. Clin. Pharmacol. Ther. 2013, 94, 585–592. [Google Scholar] [CrossRef] [Green Version]
- Lai, S.-L.; Mustafa, M.R.; Wong, P.-F. Panduratin A induces protective autophagy in melanoma via the AMPK and mTOR pathway. Phytomedicine 2018, 42, 144–151. [Google Scholar] [CrossRef]
- Kim, D.-Y.; Kim, M.-S.; Sa, B.-K.; Kim, M.-B.; Hwang, J.-K. Boesenbergia pandurata Attenuates Diet-Induced Obesity by Activating AMP-Activated Protein Kinase and Regulating Lipid Metabolism. Int. J. Mol. Sci. 2012, 13, 994–1005. [Google Scholar] [CrossRef]
- Cassidy, H.; Radford, R.; Slyne, J.; O’Connell, S.; Slattery, C.; Ryan, M.P.; McMorrow, T. The role of MAPK in drug-induced kidney injury. J. Signal Transduct. 2012, 2012, 463617. [Google Scholar] [CrossRef] [Green Version]
- Nowak, G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Wei, Q.; Wang, J.; Du, Q.; Yu, J.; Zhang, L.; Dong, Z. Regulation of PUMA- by p53 in cisplatin-induced renal cell apoptosis. Oncogene 2006, 25, 4056–4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Kaushal, V.; Shah, S.V.; Kaushal, G.P. Mcl-1 is downregulated in cisplatin-induced apoptosis, and proteasome inhibitors restore Mcl-1 and promote survival in renal tubular epithelial cells. Am. J. Physiol. Renal Physiol. 2007, 292, F1710–F1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirino, Y.I.; Pedraza-Chaverri, J.J.E.; Pathology, T. Role of oxidative and nitrosative stress in cisplatin-induced nephrotoxicity. Exp. Toxicol. Pathol. 2009, 61, 223–242. [Google Scholar] [CrossRef]
- dos Santos, N.A.G.; Carvalho Rodrigues, M.A.; Martins, N.M.; dos Santos, A.C. Cisplatin-induced nephrotoxicity and targets of nephroprotection: An update. Arch. Toxicol. 2012, 86, 1233–1250. [Google Scholar] [CrossRef] [PubMed]
- Praetorius, N.P.; Mandal, T.K. Alternate delivery route for amifostine as a radio-/chemo-protecting agent. J. Pharm. Pharmacol. 2008, 60, 809–815. [Google Scholar] [CrossRef]
- Kemp, G.; Rose, P.; Lurain, J.; Berman, M.; Manetta, A.; Roullet, B.; Homesley, H.; Belpomme, D.; Glick, J. Amifostine pretreatment for protection against cyclophosphamide-induced and cisplatin-induced toxicities: Results of a randomized control trial in patients with advanced ovarian cancer. J. Clin. Oncol. 1996, 14, 2101–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.M.; Kim, H.K.; Shim, W.; Anwar, M.A.; Kwon, J.W.; Kwon, H.K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of Cisplatin-Induced Cytotoxicity Is Correlated to Impaired Metabolism Due to Mitochondrial ROS Generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [Green Version]
- Ciarimboli, G.J.S. Membrane transporters as mediators of cisplatin effects and side effects. Scientifica 2012, 2012, 1–8. [Google Scholar] [CrossRef]
- Filipski, K.K.; Loos, W.J.; Verweij, J.; Sparreboom, A. Interaction of Cisplatin with the Human Organic Cation Transporter 2. Clin. Cancer Res. 2008, 14, 3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongwan, T.; Kittayaruksakul, S.; Asavapanumas, N.; Chatsudthipong, V.; Soodvilai, S. Activation of liver X receptor inhibits OCT2-mediated organic cation transport in renal proximal tubular cells. Pflugers Arch. 2017, 469, 1471–1481. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thongnuanjan, P.; Soodvilai, S.; Fongsupa, S.; Thipboonchoo, N.; Chabang, N.; Munyoo, B.; Tuchinda, P.; Soodvilai, S. Panduratin A Derivative Protects against Cisplatin-Induced Apoptosis of Renal Proximal Tubular Cells and Kidney Injury in Mice. Molecules 2021, 26, 6642. https://doi.org/10.3390/molecules26216642
Thongnuanjan P, Soodvilai S, Fongsupa S, Thipboonchoo N, Chabang N, Munyoo B, Tuchinda P, Soodvilai S. Panduratin A Derivative Protects against Cisplatin-Induced Apoptosis of Renal Proximal Tubular Cells and Kidney Injury in Mice. Molecules. 2021; 26(21):6642. https://doi.org/10.3390/molecules26216642
Chicago/Turabian StyleThongnuanjan, Penjai, Sirima Soodvilai, Somsak Fongsupa, Natechanok Thipboonchoo, Napason Chabang, Bamroong Munyoo, Patoomratana Tuchinda, and Sunhapas Soodvilai. 2021. "Panduratin A Derivative Protects against Cisplatin-Induced Apoptosis of Renal Proximal Tubular Cells and Kidney Injury in Mice" Molecules 26, no. 21: 6642. https://doi.org/10.3390/molecules26216642
APA StyleThongnuanjan, P., Soodvilai, S., Fongsupa, S., Thipboonchoo, N., Chabang, N., Munyoo, B., Tuchinda, P., & Soodvilai, S. (2021). Panduratin A Derivative Protects against Cisplatin-Induced Apoptosis of Renal Proximal Tubular Cells and Kidney Injury in Mice. Molecules, 26(21), 6642. https://doi.org/10.3390/molecules26216642