
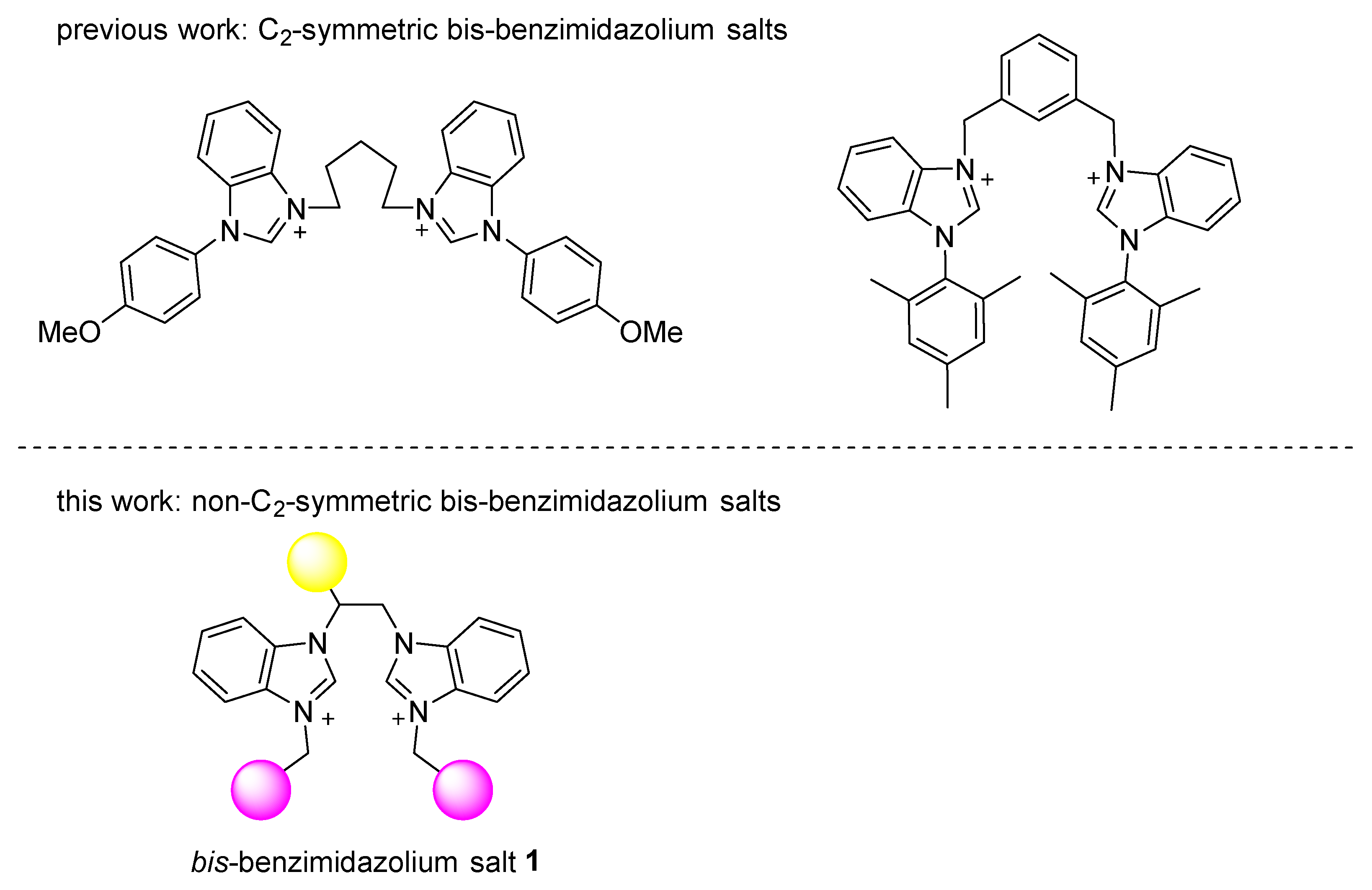
Non-C2-Symmetric Bis-Benzimidazolium Salt Applied in the Synthesis of Sterically Hindered Biaryls

 and
and
Abstract
:
1. Introduction
2. Results
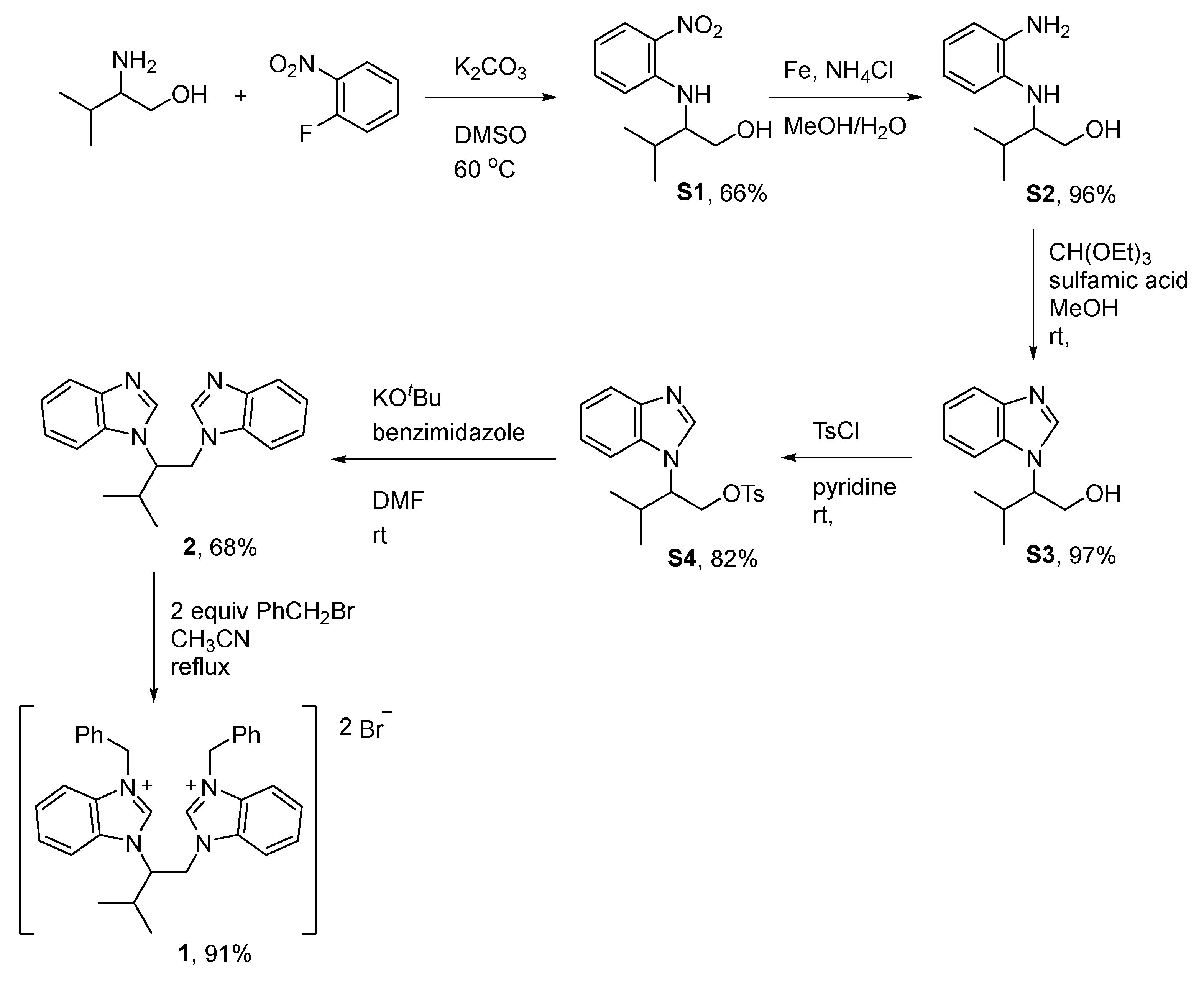
2.1. Synthesis and Characterization of the Bis-Benzimidazolium Salts 3
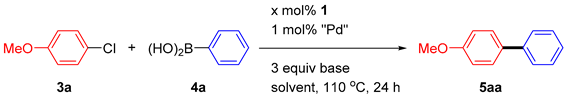
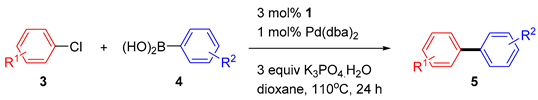
2.2. The Suzuki–Miyaura Cross-Coupling Reaction
3. Materials and Methods
3.1. General Methods
3.2. Experimental Procedures and Spectral Data
3.2.1. Synthesis of Non-C2-Symmetric Bis-Benzimidazolium Salt 1
3.2.2. General Procedures for Suzuki–Miyaura Cross-Coupling Reactions under N2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rao, A.V.R.; Gurjar, M.K.; Reddy, K.L.; Rao, A.S. Studies Directed toward the Synthesis of Vancomycin and Related Cyclic Peptides. Chem. Rev. 1995, 95, 2135–2167. [Google Scholar] [CrossRef]
- Williams, D.H.; Bardsley, B. The Vancimycin Group of Antibiotics and the Fight against Resistant Bacteria. Angew. Chem. Int. Ed. 1999, 38, 1172–1193. [Google Scholar]
- Nicolaou, K.C.; Boddy, C.N.C.; Brase, S.; Winssinger, N. Chemistry, Biology, and Medicine of the Glycopeptide Antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar]
- Hubbard, B.K.; Walsh, C.T. Vancomycin Assembly: Nature’s Way. Angew. Chem. Int. Ed. 2003, 42, 730–765. [Google Scholar] [CrossRef]
- Baudoin, O.; Gueritte, F. Natural Bridged Biaryls with Axial Chirality and Antimitotic Properties. In Studies in Natural Product Chemistry; Rahman, A.U., Ed.; Elseviser: Amsterdam, The Netherlands, 2003; Volume 29, pp. 355–417. [Google Scholar]
- Bringmann, G.; Pokorny, F. The Naphthylisoquinoline Alkaloids. In The Alkaloids: Chemistry and Pharmacology; Cordell, G.A., Ed.; Academic Press: New York, NY, USA, 1995; Volume 46, pp. 127–271. [Google Scholar]
- Rizzacasa, M.A. Total synthesis of naphthylisoquinoline alkaloids. In Studies in Natural Product Chemistry; Rahman, A.U., Ed.; Elseviser: Amsterdam, The Netherlands, 1998; Volume 20, pp. 407–455. [Google Scholar]
- Hillier, A.C.; Grasa, G.A.; Viciu, M.S.; Lee, H.M.; Yang, C.; Nolan, S.P. Catalytic cross-coupling reactions mediated by palladium/nucleophilic carbene systems. J. Organomet. Chem. 2002, 653, 69–82. [Google Scholar] [CrossRef]
- Littke, A.F.; Fu, G.C. Palladiu-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Corbet, J.P.; Mignani, G. Selected Patented Cross-Coupling Technologies. Chem. Rev. 2006, 106, 2651–2710. [Google Scholar] [CrossRef]
- Clavier, H.; Nolan, S.P. N-Heterocyclic carbenes: Advances in transition metal-mediated transformations and organocatalysis. Annu. Rep. Prog. Chem. Sect. B Org. Chem. 2007, 103, 193–222. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamamoto, Y. Cross-coupling Reactions of Organoboranes: An Easy Method for C–C Bonding. Chem. Lett. 2011, 40, 894–901. [Google Scholar]
- Wong, S.M.; Yuen, O.Y.; Choy, P.Y.; Kwong, F.Y. When Cross-Coupling Partners Meet Indolylphosphines. Coord. Chem. Rev. 2015, 293, 158–186. [Google Scholar]
- Biffis, A.; Centomo, P.; Zotto, D.A.; Zecca, M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Gou, X.X.; Han, Y.F. Chelating Bis(N-Heterocyclic Carbene) Palladium-Catalyzed Reactions. Chem.—Asian J. 2018, 13, 2257–2276. [Google Scholar] [CrossRef]
- Gardiner, M.G.; Ho, C.C. Recent advances in bidentate bis(N-heterocyclic carbene) transition metal complexes and their applications in metal-mediated reactions. Coord. Chem. Rev. 2018, 375, 373–388. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Reisinger, C.P.; Spiegler, M. Chelating N-heterocyclic carbene ligands in palladium-catalyzed heck-type reactions. J. Organomet. Chem. 1998, 557, 93–96. [Google Scholar] [CrossRef]
- Zhang, C.; Trudell, M.L. Palladiu-bisimidazol-2-ylidene complexes as catalysts for general and efficient Suzuki cross-coupling reactions of aryl chlorides with arylboronic acids. Tetrahedron Lett. 2000, 41, 595–598. [Google Scholar] [CrossRef]
- Shi, M.; Qian, H.X. A new dimeric bidentated NHC–Pd(II) complex from trans-cyclohexane-1,2-diamine for Suzuki reaction and Heck reaction. Tetrahedron 2005, 61, 4949–4955. [Google Scholar] [CrossRef]
- Xu, Q.; Duan, W.L.; Lei, Z.Y.; Zhu, Z.B.; Shi, M. A novel cis-chelated Pd(II)–NHC complex for catalyzing Suzuki and Heck-type cross-coupling reactions. Tetrahedron 2005, 61, 11225–11229. [Google Scholar]
- Demir, S.; Özdemir, I.; Çetinkaya, B. Use of bis(benzimidazolium)–palladium system as a convenient catalyst for Heck and Suzuki coupling reactions of aryl bromides and chlorides. Appl. Organometal. Chem. 2006, 20, 254–259. [Google Scholar]
- Liu, Z.; Zhang, T.; Shi, M. Cyclometalated cis-Chelated Bidentate N-Heterocyclic Carbene Palladium Complexes. Synthetic, Structural, and Catalytic Studies. Organometallics 2008, 27, 2668–2671. [Google Scholar] [CrossRef]
- Avery, K.B.; Devine, W.G.; Kormos, C.M.; Leadbeater, N.E. Use of a silicon carbide multi-well plate in conjunction with microwave heating for rapid ligand synthesis, formation of palladium complexes, and catalyst screening in a Suzuki coupling. Tetrahedron Lett. 2009, 50, 2851–2853. [Google Scholar] [CrossRef]
- Yilmaz, Ü.; Şireci, N.; Deniz, S.; Küçükbay, H. Synthesis and microwave-assisted catalytic activity of novel bis-benzimidazole salts bearing furfuryl and thenyl moieties in Heck and Suzuki cross-coupling reactions. Appl. Organometal. Chem. 2010, 24, 414–420. [Google Scholar]
- Micksch, M.; Strassner, T. Complexes with Chelating Biscarbene Ligands in the Catalytic Suzuki–Miyaura Cross-Coupling Reaction. Eur. J. Inorg. Chem. 2012, 2012, 5872–5880. [Google Scholar] [CrossRef]
- Li, Y.; Tang, J.; Gu, J.; Wang, Q.; Sun, P.; Zhang, D. Chiral 1,2-Cyclohexane-Bridged Bis-NHC Palladium Catalysts for Asymmetric Suzuki–Miyaura Coupling: Synthesis, Characterization, and Steric Effects in Enantiocontrol. Organometallics 2014, 33, 876–884. [Google Scholar] [CrossRef]
- Charbonneau, M.; Addoumieh, G.; Oguadinma, P.; Schmitzer, A.R. Support-Free Palladium-NHC Catalyst for Highly Recyclable Heterogeneous Suzuki-Miyaura Coupling in Neat Water. Organometallics 2014, 33, 6544–6549. [Google Scholar] [CrossRef]
- Li, X.; Zhang, G.; Chao, M.; Cao, C.; Pang, G.; Shi, Y. Synthesis and catalytic activity of chiral linker-bridged bis-N-heterocyclic carbene dipalladium complexes. J. Chem. Res. 2018, 42, 320–325. [Google Scholar] [CrossRef]
- Thapa, R.; Kilyanek, S.M. Synthesis and structural characterization of 20-mebered macrocyclic rings bearing trans-chelating bis(N-heterocyclic carbene) ligands and the catalytic activity of theirpalladium(II) complexes. Dalton Trans. 2019, 48, 12577–12589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yu, J. Fine Tuning of Chiral Bis(N-heterocyclic carbene) Palladium Catalysts for Asymmetric Suzuki–Miyaura Cross-Coupling Reactions: Exploring the Ligand Modification. Organometallics 2020, 39, 1269–1280. [Google Scholar]
- Nan, G.; Rao, B.; Luo, M. Cis-chelated palladium(II) complexes of biphenyl-linked bis(imidazolin-2-ylidene):synthesis and catalytic activity in the Suzuki-Miyaura reaction. Molecules 2011, 2, 29–40. [Google Scholar]
- Lin, Y.R.; Chiu, C.C.; Chiu, H.T.; Lee, D.S.; Lu, T.J. Bis-benzimidazolium-palladium system catalyzed Suzuki–Miyaura coupling reaction of aryl bromides under mild conditions. Appl. Organometal. Chem. 2018, 32, e3896. [Google Scholar]
- Chiu, C.C.; Chiu, H.T.; Lee, D.S.; Lu, T.J. An efficient class of bis-NHC salts: Applications in Pd-catalyzed reactions under mild reaction conditions. RSC Adv. 2018, 8, 26407–26415. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wei, T.R.; Huang, S.J.; Lai, Y.T.; Lee, D.S.; Lu, T.J. Synthesis of xylyl-linked bis-benzimidazolium salts and their application in the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction of aryl chlorides. Catalysts 2021, 11, 817. [Google Scholar] [CrossRef]
- McManus, H.A.; Guiry, P.J. Recent Developments in the Application of Oxazoline-Containing Ligands in Asymmetric Catalysis. Chem. Rev. 2004, 104, 4151–4202. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Ishida, Y.; Sasaki, D.; Saigo, K. Cyclophan-Type Imidazolium Salts with Planar Chirality as a New Class of N-Heterocyclic Carbene Precursors. Chem. Eur. J. 2008, 14, 9215–9222. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Kumpati, B.N.; Lei, X. Diaminophosphine oxides as preligands for Ni-catalyzed Suzuki cross-coupling reactions of aryl chlorides with arylboronic acids. Tetrahedron Lett. 2014, 55, 7215–7218. [Google Scholar] [CrossRef]
- Shahripour, A.B.; Plummer, M.S.; Lunney, E.A.; Albrecht, H.P.; Hays, S.J.; Kostlan, C.R.; Sawyer, T.K.; Walker, N.P.; Brady, K.D.; Allen, H.J. Structure-Based Design of Nonpeptide Inhibitors of Interleukin-1β Converting Enzyme (ICE, Caspase-1). Biorg. Med. Chem. 2002, 10, 31–40. [Google Scholar] [CrossRef]
- Cong, X.; Tang, H.; Zeng, X. Regio- Chemo selective Kumada–Tamao–Corriu Reaction of Aryl Alkyl Ethers Catalyzed by Chromium Under Mild Conditions. J. Am. Chem. Soc. 2015, 137, 14367–14372. [Google Scholar] [CrossRef]
- Jennings, W.B.; Farrel, B.M.; Malone, J.F. Stereodynamics and Edge-to-Face CH-π Aromatic Interactions in o-Phenethyl-Substituted Biaryls. J. Org. Chem. 2006, 71, 2277–2282. [Google Scholar]
- Yu, R.; Chen, X.; Martin, S.F.; Wang, Z. Differentially Substituted Phosphines via Decarbonylation of Acylphosphines. Org. Lett. 2017, 19, 1808–1811. [Google Scholar] [CrossRef]
- Wang, M.P.; Chiu, C.C.; Lu, T.J.; Lee, D.S. Indolylbenzimidazole-based ligands catalyze the coupling of arylboronic acids with aryl halides. Appl. Organometal. Chem. 2018, 32, e4348. [Google Scholar] [CrossRef]
- Beromi, M.M.; Nova, A.; Balcells, D.; Brasacchio, A.M.; Brudvig, G.W.; Guard, L.M.; Hazari, N.; Vinyard, D.J. Mechanistic Study of an Improved Ni Precatalyst for Suzuki-Miyaura Reactions of Aryl Sulfamates Understanding the Role of Ni(I) Species. J. Am. Chem. Soc. 2017, 139, 922–936. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.J.; Lee, D.H. A Practical Heterogeneous Catalyst for the Suzuki, Sonogashira, and Still Coupling Reactions of Unreactive Aryl Chlorides. Angew. Chem. Int. Ed. 2010, 49, 1119–1122. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Entry | Pd Source | 1 (mol%) | Solvent | Base | 3a (%)2 | 5aa (%) 2 |
|---|---|---|---|---|---|---|
| 1 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 11 | 87 |
| 2 | Pd(OAc)2 | 3.0 | Toluene | K3PO4·H2O | 77 | 17 |
| 3 | Pd(OAc)2 | 3.0 | CH3CN | K3PO4·H2O | 86 | 6 |
| 4 | Pd(OAc)2 | 3.0 | t-BuOH | K3PO4·H2O | 70 | 17 |
| 5 | Pd(OAc)2 | 3.0 | tBuOH/H2O 3 | K3PO4·H2O | 93 | 3 |
| 6 | Pd(OAc)2 | 2.0 | 1,4-Dioxane | K3PO4·H2O | 42 | 53 |
| 7 | Pd(OAc)2 | 1.0 | 1,4-Dioxane | K3PO4·H2O | 74 | 24 |
| 8 | Pd(OAc)2 | 0.5 | 1,4-Dioxane | K3PO4·H2O | 79 | 18 |
| 9 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | KOtBu | 80 | 4 |
| 10 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | KOAc | 99 | 0 |
| 11 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | K3PO4 | 65 | 32 |
| 12 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | K2CO3 | 82 | 14 |
| 13 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | Cs2CO3 | 87 | 10 |
| 14 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | CsF | 80 | 18 |
| 15 | Pd(OAc)2 | 3.0 | 1,4-Dioxane | KF | 81 | 17 |
| 16 | PdCl2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 66 | 31 |
| 17 | PdCl2(PPh3)2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 94 | 5 |
| 18 | PdCl2(CH3CN)2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 28 | 66 |
| 19 | [PdCl(C3H5)]2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 20 | 74 |
| 20 | Pd2(dba)3 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 4 | 88 |
| 21 | Pd(dba)2 | 3.0 | 1,4-Dioxane | K3PO4·H2O | 3 | 90 (83) 4 |
| 22 | Pd(dba)2 | – | 1,4-Dioxane | K3PO4·H2O | >99 | 0 |
 | |||
| Entry | ArCl 3 | Product 5 | Yield (%)2 |
|---|---|---|---|
| 1 |  |  | 83 |
| 2 | 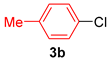 | 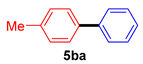 | 67 |
| 3 | 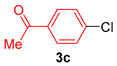 | 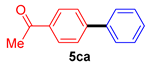 | 95 |
| 4 |  |  | 99 |
| 5 | 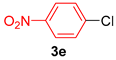 |  | 91 |
| 6 | 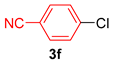 | 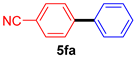 | 87 |
| 7 |  |  | 63 |
| 8 | 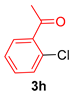 | 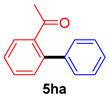 | 75 |
| 9 | 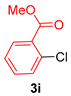 | 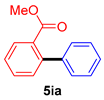 | 66 |
| 10 | 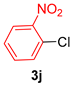 | 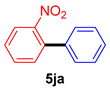 | 81 |
| 11 | 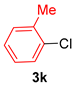 | 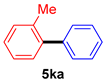 | 85 |
 | ||||
| Entry | ArCl 3 | ArB(OH)2 4 | Product 5 | Yield (%)2 |
|---|---|---|---|---|
| 1 |  | 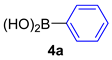 | 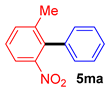 | 50 |
| 2 |  | 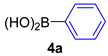 |  | 46 |
| 3 | 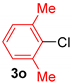 | 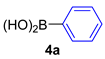 |  | 60 |
| 4 | 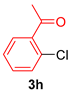 |  |  | 46 |
| 5 |  | 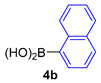 | 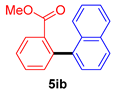 | 20 |
| 6 | 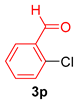 | 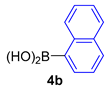 | 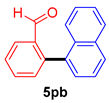 | 75 |
| 7 | 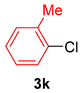 | 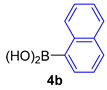 | 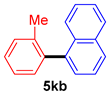 | 92 |
| 8 | 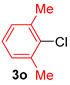 | 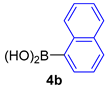 | 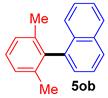 | 85 |
| 9 | 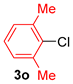 | 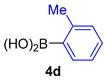 | 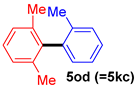 | 51 |
| 10 | 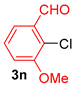 | 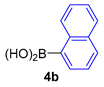 |  | 84 |
| 11 | 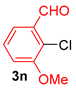 | 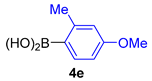 | 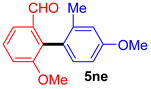 | 58 |
| 12 | 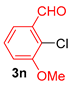 | 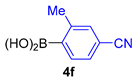 | 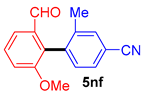 | 50 |
| 13 | 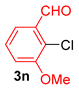 | 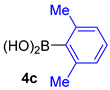 |  | ND 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.-H.; Huang, S.-J.; Hsu, T.-Y.; Hung, P.-Y.; Wei, T.-R.; Lee, D.-S.; Lu, T.-J. Non-C2-Symmetric Bis-Benzimidazolium Salt Applied in the Synthesis of Sterically Hindered Biaryls. Molecules 2021, 26, 6703. https://doi.org/10.3390/molecules26216703
Chen Y-H, Huang S-J, Hsu T-Y, Hung P-Y, Wei T-R, Lee D-S, Lu T-J. Non-C2-Symmetric Bis-Benzimidazolium Salt Applied in the Synthesis of Sterically Hindered Biaryls. Molecules. 2021; 26(21):6703. https://doi.org/10.3390/molecules26216703
Chicago/Turabian StyleChen, Yen-Hsin, Shu-Jyun Huang, Tung-Yu Hsu, Pei-Yu Hung, Ting-Rong Wei, Dong-Sheng Lee, and Ta-Jung Lu. 2021. "Non-C2-Symmetric Bis-Benzimidazolium Salt Applied in the Synthesis of Sterically Hindered Biaryls" Molecules 26, no. 21: 6703. https://doi.org/10.3390/molecules26216703
APA StyleChen, Y.-H., Huang, S.-J., Hsu, T.-Y., Hung, P.-Y., Wei, T.-R., Lee, D.-S., & Lu, T.-J. (2021). Non-C2-Symmetric Bis-Benzimidazolium Salt Applied in the Synthesis of Sterically Hindered Biaryls. Molecules, 26(21), 6703. https://doi.org/10.3390/molecules26216703