
Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition


Abstract
:
1. Introduction
2. Theoretical Methods
3. Results and Discussion
3.1. Selection of Basis Sets
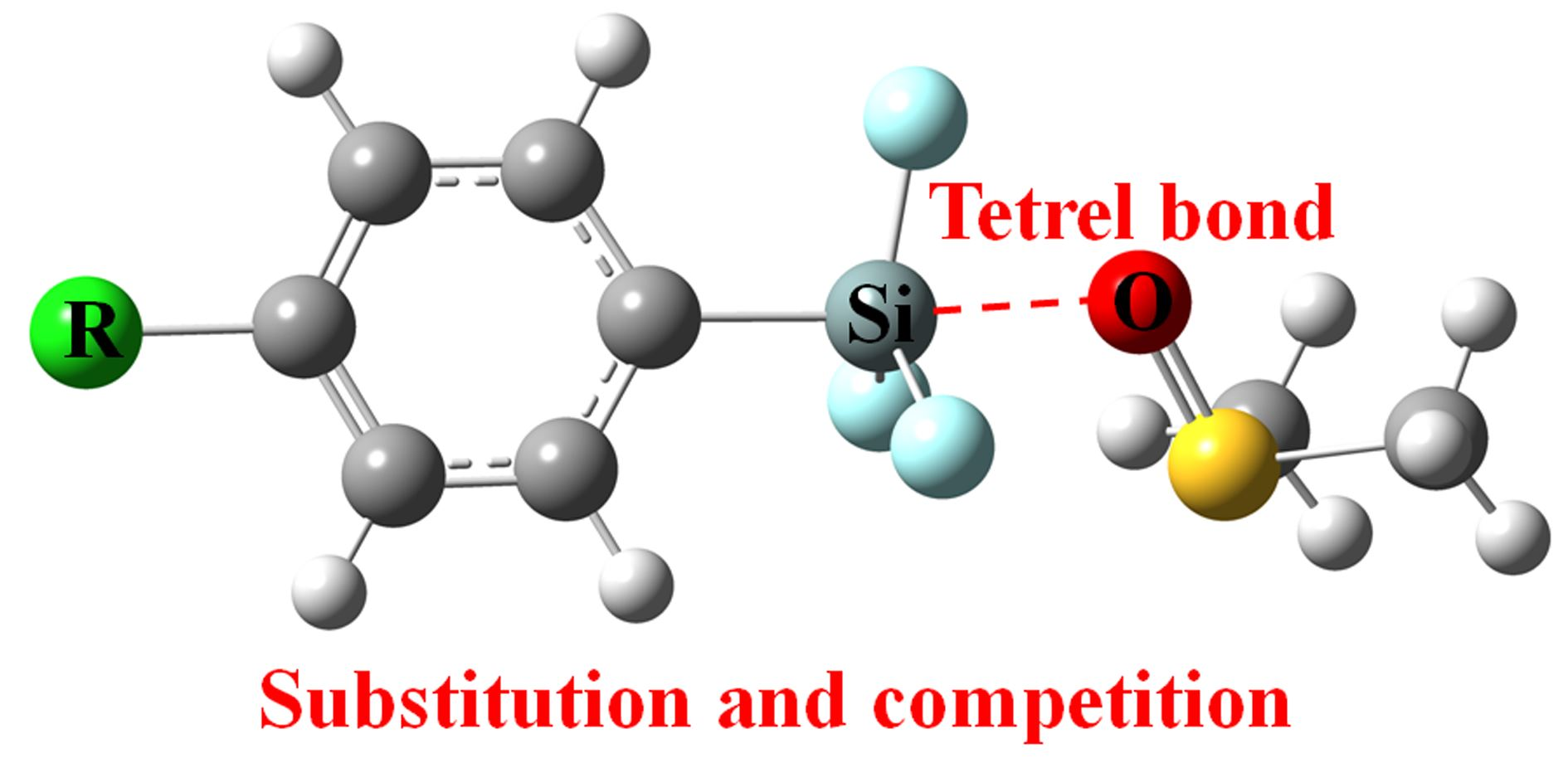
3.2. Interaction Energies and Geometries
3.3. NBO Analyses
3.4. AIM Analyses
3.5. Competition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, G.; Bauzá, A.; Amini, M.; Molins, E.; Mague, J.T.; Frontera, A. On the importance of tetrel bonding interactions in lead (II) complexes with (iso) nicotinohydrazide based ligands and several anions. Dalton Trans. 2016, 45, 10708–10716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gargari, M.S.; Stilinović, V.; Bauzá, A.; Frontera, A.; McArdle, P.; Derveer, D.V.; Ng, S.W.; Mahmoudi, G. Design of lead (II) metal–organic frameworks based on covalent and tetrel bonding. Chem. Eur. J. 2015, 21, 17951–17958. [Google Scholar] [CrossRef]
- Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J.X.; Hase, W.L.; Weidemüller, M.; Wester, R. Imaging nucleophilic substitution dynamics. Science 2008, 319, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Cheng, J.B.; Li, W.Z.; Li, H.B. Tetrel bond of pseudohalide anions with XH3F (X = C, Si, Ge, and Sn) and its role in SN2 reaction. J. Chem. Phys. 2016, 145, 224310. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C···π (X = F, Cl, Br, CN) carbon bond. J. Phys. Chem. A 2014, 118, 10081–10089. [Google Scholar] [CrossRef]
- Li, Q.Z.; Guo, X.; Yang, X.; Li, W.Z.; Cheng, J.B.; Li, H.B. A σ-hole interaction with radical species as electron donors: Does single-electron tetrel bonding exist? Phys. Chem. Chem. Phys. 2014, 16, 11617–11625. [Google Scholar] [CrossRef]
- Li, Q.Z.; Zhuo, H.Y.; Li, H.B.; Liu, Z.B.; Li, W.Z.; Cheng, J.B. Tetrel–hydride interaction between XH3F (X = C, Si, Ge, Sn) and HM (M = Li, Na, BeH, MgH). J. Phys. Chem. A 2015, 119, 2217–2224. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Li, W.Z.; Cheng, J.B. Carbene tetrel-bonded complexes. Struct. Chem. 2017, 28, 823–831. [Google Scholar] [CrossRef]
- Rossi, A.R.; Jasinski, J.M. Theoretical studies of neutral silane-ammonia adducts. Chem. Phys. Lett. 1990, 169, 399–404. [Google Scholar] [CrossRef]
- Ruoff, R.S.; Emilsson, T.; Jaman, A.I.; Germann, T.C.; Gutowsky, H.S. Rotational spectra, dipole moment, and structure of the SiF4–NH3 dimer. J. Chem. Phys. 1992, 96, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Keith, T.A.; Bader, R.F.W. Origin of dipole moment enhancement in the formation of SiF4–NH3 dimer. J. Chem. Phys. 1992, 96, 3447–3451. [Google Scholar] [CrossRef]
- Ignatov, S.K.; Sennikov, P.G.; Ault, B.S.; Bagatur’yants, A.A.; Simdyanov, I.V.; Razuvaev, A.G.; Klimov, E.J.; Gropen, O. Water complexes and hydrolysis of silicon tetrafluoride in the gas phase: An ab initio study. J. Phys. Chem. A 1999, 103, 8328–8336. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Guo, X.; Liu, Y.W.; Li, Q.Z.; Li, W.Z.; Cheng, J.B. Competition and cooperativity between tetrel bond and chalcogen bond in complexes involving F2CX (X = Se and Te). Chem. Phys. Lett. 2015, 620, 7–12. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Li, W.Z.; Cheng, J.B. Tetrel bonds between PySiX3 and some nitrogenated bases: Hybridization, substitution, and cooperativity. J. Mol. Graph. Model. 2016, 65, 35–42. [Google Scholar] [CrossRef]
- Liu, M.X.; Yang, L.; Li, Q.Z.; Li, W.Z.; Cheng, J.B.; Xiao, B.; Yu, X.F. Modulating the strength of tetrel bonding through beryllium bonding. J. Mol. Model. 2016, 22, 192. [Google Scholar] [CrossRef]
- Tang, Q.J.; Li, Q.Z. Interplay between tetrel bonding and hydrogen bonding interactions in complexes involving F2XO (X = C and Si) and HCN. Comput. Theor. Chem. 2014, 1050, 51–57. [Google Scholar] [CrossRef]
- McDowell, S.A.C. Sigma-hole cooperativity in anionic [FX⋯CH3⋯YF]−(X, Y = Cl, Br) complexes. Chem. Phys. Lett. 2014, 598, 1–4. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadirad, N.; Solimannejad, M. Tetrel bond cooperativity in open-chain (CH3CN)n and (CH3NC)n clusters (n = 2–7): An ab initio study. Chem. Phys. Lett. 2015, 628, 16–20. [Google Scholar] [CrossRef]
- Solimannejad, M.; Orojloo, M.; Amani, S.J. Effect of cooperativity in lithium bonding on the strength of halogen bonding and tetrel bonding: (LiCN)n···ClYF3 and (LiCN)n···YF3Cl (Y = C, Si and n = 1–5) complexes as a working model. Mol. Model. 2015, 21, 183. [Google Scholar] [CrossRef]
- Yourdkhani, S.; Korona, T.; Hadipour, N.L. Interplay between tetrel and triel bonds in RC6H4CN⋯MF3CN⋯BX3 complexes: A combined symmetry-adapted perturbation theory, Møller-Plesset, and quantum theory of atoms-in-molecules study. J. Comput. Chem. 2015, 36, 2412–2428. [Google Scholar] [CrossRef]
- Marín-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in tetrel bonds. J. Phys. Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Esrafili, M.D.; Nurazar, R.; Mohammadian-Sabet, F. Cooperative effects between tetrel bond and other σ–hole bond interactions: A comparative investigation. Mol. Phys. 2015, 113, 3703–3711. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F. Cooperativity of tetrel bonds tuned by substituent effects. Mol. Phys. 2016, 114, 1528–1538. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadian-Sabet, F. Tuning tetrel bonds via cation–π interactions: An ab initio study on concerted interaction in M+–C6H5XH3–NCY complexes (M = Li, Na, K.; X = Si, Ge; Y = H, F, OH). Mol. Phys. 2016, 114, 83–91. [Google Scholar] [CrossRef]
- Rezaei, Z.; Solimannejad, M.; Esrafili, M.D. Interplay between hydrogen bond and single-electron tetrel bond: H3C⋯ COX2⋯ HY and H3C⋯ CSX2⋯ HY (X= F, Cl; Y= CN, NC) complexes as a working model. Comput. Theor. Chem. 2015, 1074, 101–106. [Google Scholar] [CrossRef]
- Vatanparast, M.; Parvini, E.; Bahadori, A. Computational study of the cooperative effects between tetrel bond and halogen bond in XCN⋅⋅⋅F2CO⋅⋅⋅YCN complexes (X = H, F, Cl, Br; Y = F, Cl, Br). Mol. Phys. 2016, 114, 1478–1484. [Google Scholar] [CrossRef]
- Liu, M.X.; Li, Q.Z.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furanTF3 (T= C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.C.; Li, Q.Z.; Cheng, J.B.; Li, W.Z.; Li, H.B. Comparison of tetrel bonds and halogen bonds in complexes of DMSO with ZF3X (Z = C and Si; X = halogen). RSC Adv. 2016, 6, 79245–79253. [Google Scholar] [CrossRef]
- Nziko, V.D.P.; Scheiner, S. Comparison of π-hole tetrel bonding with σ-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.X.; Li, Q.Z.; Li, W.Z.; Cheng, J.B.; McDowell, S.A.C. Comparison of hydrogen, halogen, and tetrel bonds in the complexes of HArF with YH3X (X = halogen, Y = C and Si). RSC Adv. 2016, 6, 19136–19143. [Google Scholar] [CrossRef]
- Voronkov, A.M.G.; Grebneva, E.A.; Trofimova, O.M.; Chernov, N.F.; Albanov, A.I.; Chipanina, N.N. The unusual reaction of phenyltrifluorosilane with 2-aminoethanol and its N-methyl derivatives. Doklady Chem. 2006, 409, 139–141. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Trofimova, O.M.; Grebneva, E.A.; Chernov, N.F.; Abzaeva, K.A. Phenyltrifluorosilane in organoelemental and organic synthesis. Rus. J. Gen. Chem. 2011, 81, 2391–2411. [Google Scholar] [CrossRef]
- Martin, D.; Hauthal, H.G. Dimethyl Sulphoxide; John Wiley & Sons: New York, NY, USA, 1975. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, RevisionD.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Serebryanskaya, T.V.; Novikov, A.S.; Gushchin, P.V.; Haukka, M.; Asfin, R.E.; Tolstoy, P.M.; Kukushkin, V.Y. Identification and H(D)-bond energies of C–H(D)⋯Cl interactions in chloride–haloalkane clusters: A combined X-ray crystallographic, spectroscopic, and theoretical study. Phys. Chem. Chem. Phys. 2016, 18, 14104–14112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, D.M.; Kinzhalov, M.A.; Novikov, A.S.; Ananyev, I.V.; Romanova, A.A.; Boyarskiy, V.P.; Haukka, M.; Kukushkin, V.Y. H2C(X)–X···X− (X = Cl, Br) halogen bonding of dihalomethanes. Cryst. Growth Des. 2017, 17, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.S.; Gushchin, A. Trinuclear molybdenum clusters with sulfide bridges as potential anionic receptors via chalcogen bonding. Cryst. Eng. Comm. 2021, 23, 4607–4614. [Google Scholar] [CrossRef]
- Ivanov, D.M.; Kirina, Y.V.; Novikov, A.S.; Starova, G.L.; Kukushkin, V.Y. Efficient π-stacking with benzene provides 2D assembly of trans-[PtCl2(p-CF3C6H4CN)2]. J. Mol. Struct. 2016, 1104, 19–23. [Google Scholar] [CrossRef]
- Abramov, P.A.; Novikov, A.S.; Sokolov, M.N. Interactions of aromatic rings in the crystal structures of hybrid polyoxometalates and Ru clusters. Cryst. Eng. Comm. 2021, 23, 6409–6417. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. In A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Bader, R.F.W. AIM2000 Program, v. 2.0; McMaster University: Hamilton, ON, Canada, 2000. [Google Scholar]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Pnicogen-bonded complexes HnF5–nP:N-base, for n = 0–5. J. Phys. Chem. A 2014, 118, 10144–10154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Influence of substituent effects on the formation of P···Cl pnicogen bonds or halogen bonds. J. Phys. Chem. A 2014, 118, 2360–2366. [Google Scholar] [CrossRef] [Green Version]
- Alkorta, I.; Sánchez-Sanz, G.; Elguero, J.; Del Bene, J.E. Exploring (NH2F)2, H2FP:NFH2, and (PH2F)2 potential surfaces: Hydrogen bonds or pnicogen bonds? J. Phys. Chem. A 2013, 117, 183–191. [Google Scholar] [CrossRef]
- Schütz, M.; Brdarski, S.; Widmark, P.O.; Lindh, R.; Karlström, G. The water dimer interaction energy: Convergence to the basis set limit at the correlated level. J. Chem. Phys. 1997, 107, 4597–4605. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Non-covalent sp3 carbon bonding with ArCF3 is analogous to CH–π interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef]
- Bauzá, A.; Quiñonero, D.; Frontera, A.; Deyà, P.M. Substituent effects in halogen bonding complexes between aromatic donors and acceptors: A comprehensive ab initio study. Phys. Chem. Chem. Phys. 2011, 13, 20371–20379. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Lipkowski, P.; Grabowski, S.J.; Robinson, T.L.; Leszczynski, J. Properties of the C−H⋯H dihydrogen bond: An ab initio and topological analysis. J. Phys. Chem. A 2004, 108, 10865–10872. [Google Scholar] [CrossRef]
- Shchavlev, A.E.; Pankratov, A.N.; Borodulin, V.B.; Chaplygina, O.A. DFT study of the monomers and dimers of 2-pyrrolidone: Equilibrium structures, vibrational, orbital, topological, and NBO analysis of hydrogen-bonded interactions. J. Phys. Chem. A 2005, 109, 10982–10996. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Mück-Lichtenfeld, C.; Erker, G.; Kehr, G.; Wang, H.; Beckers, H.; Willner, H. When do interacting atoms form a chemical bond? Spectroscopic measurements and theoretical analyses of dideuteriophenanthrene. Angew. Chem. Int. Ed. 2009, 48, 2592–2595. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Bond paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, E.; Novoa, J.J. On the hydrogen bond nature of the C–H⋯F interactions in molecular crystals. An exhaustive investigation combining a crystallographic database search and ab initio theoretical calculations. Cryst. Eng. Comm. 2008, 10, 423–436. [Google Scholar] [CrossRef]
- Arnold, W.D.; Oldfield, E. The chemical nature of hydrogen bonding in proteins via NMR: J-couplings, chemical shifts, and AIM theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Pnicogen bonded complexes of PO2X (X = F, Cl) with nitrogen bases. J. Phys. Chem. A 2013, 117, 10497–10503. [Google Scholar] [CrossRef]
- Bauzá, A.; Ramis, R.; Frontera, A. A combined theoretical and cambridge structural database study of π-hole pnicogen bonding complexes between electron rich molecules and both nitro compounds and inorganic bromides (YO2Br, Y = N, P, and As). J. Phys. Chem. A 2014, 118, 2827–2834. [Google Scholar] [CrossRef]
- Solimannejad, M.; Ramezani, V.; Trujillo, C.; Alkorta, I.; Sánchez−Sanz, G.; Elguero, J. Competition and interplay between σ-hole and π-hole interactions: A computational study of 1:1 and 1:2 complexes of nitryl halides (O2NX) with ammonia. J. Phys. Chem. A 2012, 116, 5199–5206. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Trujillo, C.; Solimannejad, M.; Alkorta, I.; Elguero, J. Orthogonal interactions between nitryl derivatives and electron donors: Pnictogen bonds. Phys. Chem. Chem. Phys. 2013, 15, 14310–14318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Li, Q.Z.; Xiao, B.; Yang, X.; Li, W.Z.; Cheng, J.B. Influence of F and Se substitution on the structures, stabilities and nature of the complexes between F2CSe and HOX (X= F, Cl, Br, and I). RSC Adv. 2015, 5, 52667–52675. [Google Scholar] [CrossRef]
- Kirchner, B.; Reiher, M. The secret of dimethyl sulfoxide−water mixtures. A quantum chemical study of 1DMSO−nwater clusters. J. Am. Chem. Soc. 2002, 124, 6206–6215. [Google Scholar] [CrossRef]
- Cumming, J.B.; Kebarle, P. Summary of gas phase acidity measurements involving acids AH. Entropy changes in proton transfer reactions involving negative ions. Bond dissociation energies D(A-H) and electron affinities EA(A). Can. J. Chem. 1978, 56, 1–9. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]


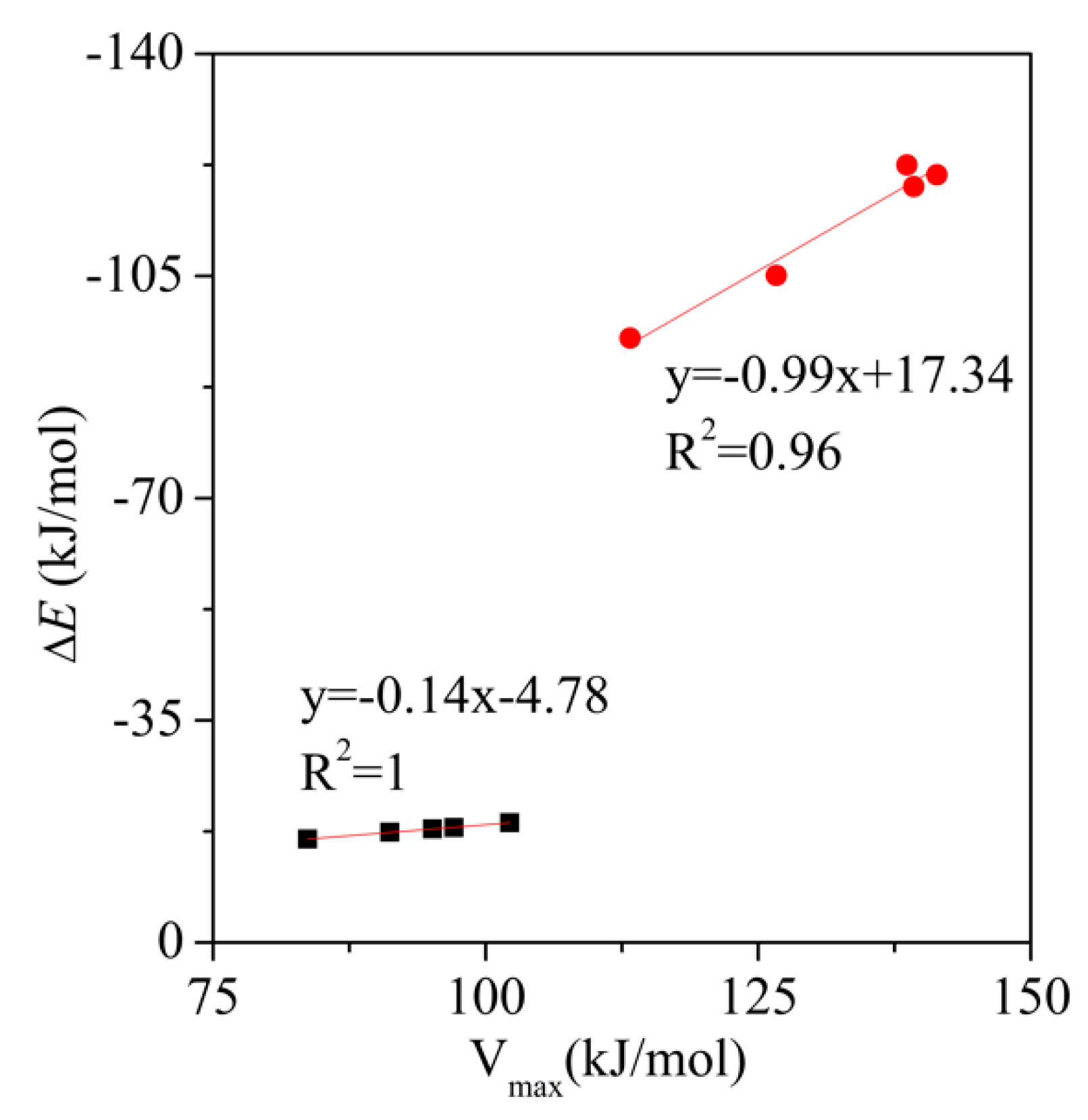
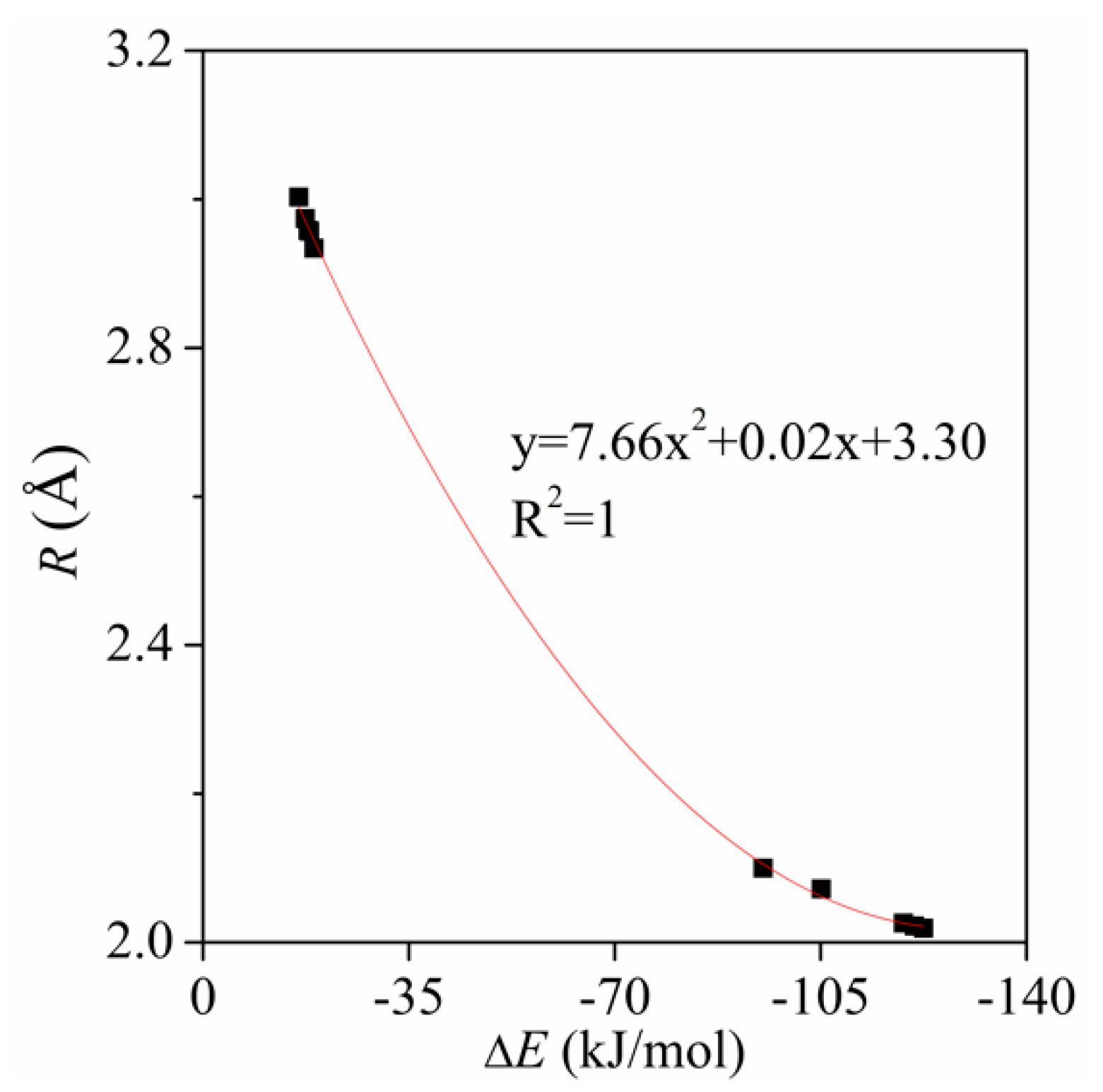
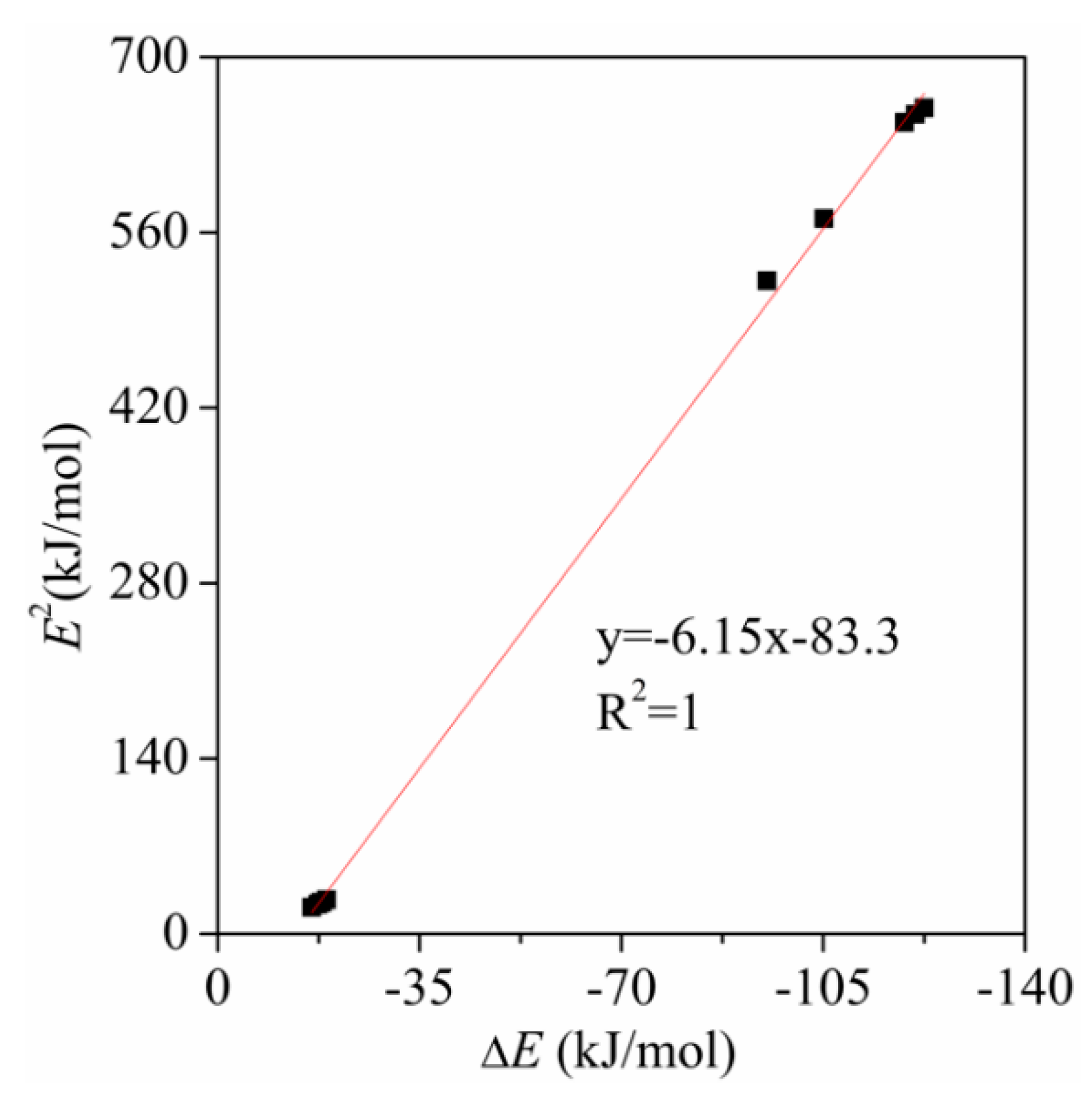
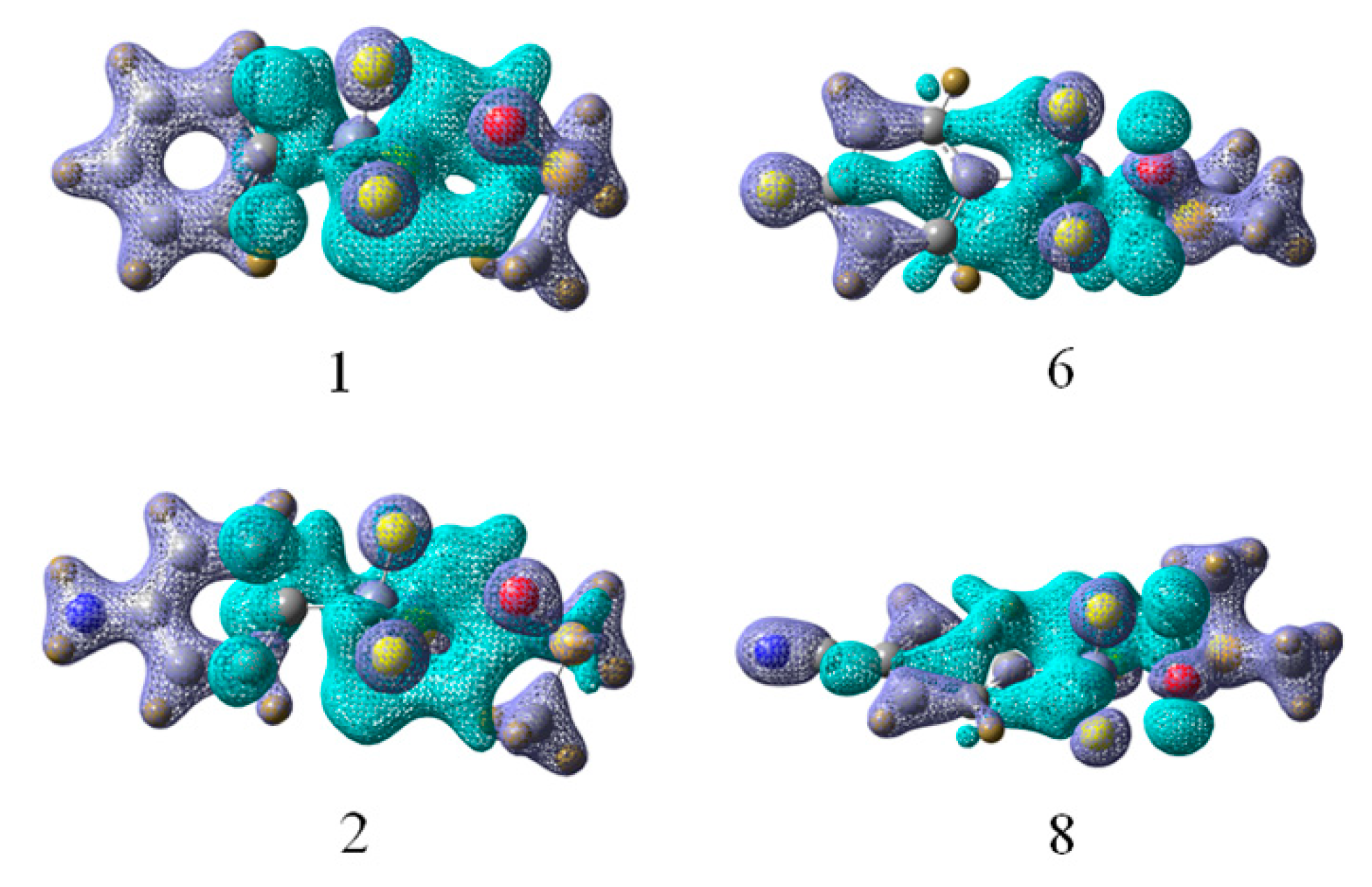



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X= | ΔE1 | ΔE2 | ΔE3 | ΔE4 | ΔE5 | |
|---|---|---|---|---|---|---|
| 1 | H | −16.62 | −15.41 | −22.07 | −19.58 | −18.87 |
| 2 | NH2 | −16.45 | −12.26 | −18.76 | −16.43 | −16.29 |
| 3 | OCH3 | −13.96 | −13.15 | −19.69 | −17.31 | −17.38 |
| 4 | CH3 | −112.42 | −13.06 | −19.66 | −17.31 | −17.89 |
| 5 | OH | −20.95 | −14.57 | −21.17 | −18.73 | −18.10 |
| 6 | F | −136.22 | −102.08 | −112.33 | −107.64 | −95.23 |
| 7 | CHO | −136.72 | −95.25 | −112.48 | −107.42 | −105.10 |
| 8 | CN | non | −116.59 | −135.46 | −130.46 | −119.10 |
| 9 | NO2 | −152.15 | −119.47 | −138.19 | −133.21 | −120.95 |
| 10 | SO3H | non | −119.00 | −137.67 | −132.56 | −122.51 |
| X= | R | r1 | Δr1 | Δr2 | Δα | |
|---|---|---|---|---|---|---|
| 1 | H | 2.9336 | 1.8453 | 0.0095 | 0.0035 | −3.0 |
| 2 | NH2 | 3.0032 | 1.8372 | 0.0088 | 0.0028 | −2.6 |
| 3 | OCH3 | 2.9740 | 1.8395 | 0.0090 | 0.0031 | −2.7 |
| 4 | CH3 | 2.9581 | 1.8429 | 0.0092 | 0.0032 | −2.8 |
| 5 | OH | 2.9566 | 1.8405 | 0.0093 | 0.0033 | −2.8 |
| 6 | F | 2.0996 | 1.8790 | 0.0439 | 0.0333 | −12.3 |
| 7 | CHO | 2.0715 | 1.8861 | 0.0460 | 0.0268 | −12.8 |
| 8 | CN | 2.0265 | 1.8910 | 0.0504 | 0.0384 | −13.4 |
| 9 | NO2 | 2.0221 | 1.8915 | 0.0501 | 0.0388 | −13.4 |
| 10 | SO3H | 2.0188 | 1.8926 | 0.0506 | 0.0392 | −13.5 |
| X= | E2(1) | E2(2) | Δq | |
|---|---|---|---|---|
| 1 | H | 23.70 | 2.76 | −0.0100 |
| 2 | NH2 | 18.85 | 2.30 | −0.0078 |
| 3 | OCH3 | 20.69 | 2.47 | −0.0086 |
| 4 | CH3 | 21.82 | 2.55 | −0.0092 |
| 5 | OH | 21.94 | 2.55 | −0.0092 |
| 6 | F | 469.04 | 20.06 | −0.1233 |
| 7 | CHO | 483.83 | 22.40 | −0.1329 |
| 8 | CN | 583.90 | 23.83 | −0.1440 |
| 9 | NO2 | 590.55 | 24.16 | −0.1455 |
| 10 | SO3H | 595.44 | 24.20 | −0.1465 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, X.; Yang, X.; Li, Q. Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition. Molecules 2021, 26, 7231. https://doi.org/10.3390/molecules26237231
An X, Yang X, Li Q. Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition. Molecules. 2021; 26(23):7231. https://doi.org/10.3390/molecules26237231
Chicago/Turabian StyleAn, Xiulin, Xin Yang, and Qingzhong Li. 2021. "Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition" Molecules 26, no. 23: 7231. https://doi.org/10.3390/molecules26237231
APA StyleAn, X., Yang, X., & Li, Q. (2021). Tetrel Bonds between Phenyltrifluorosilane and Dimethyl Sulfoxide: Influence of Basis Sets, Substitution and Competition. Molecules, 26(23), 7231. https://doi.org/10.3390/molecules26237231