Abstract
The dissociative photodetachment dynamics of the oxalate anion, C2O4H− + hν → CO2 + HOCO + e−, were theoretically studied using the on-the-fly path-integral and ring-polymer molecular dynamics methods, which can account for nuclear quantum effects at the density-functional theory level in order to compare with the recent experimental study using photoelectron–photofragment coincidence spectroscopy. To reduce computational time, the force acting on each bead of ring-polymer was approximately calculated from the first and second derivatives of the potential energy at the centroid position of the nuclei beads. We find that the calculated photoelectron spectrum qualitatively reproduces the experimental spectrum and that nuclear quantum effects are playing a role in determining spectral widths. The calculated coincidence spectrum is found to reasonably reproduce the experimental spectrum, indicating that a relatively large energy is partitioned into the relative kinetic energy between the CO2 and HOCO fragments. This is because photodetachment of the parent anion leads to Franck–Condon transition to the repulsive region of the neutral potential energy surface. We also find that the dissociation dynamics are slightly different between the two isomers of the C2O4H− anion with closed- and open-form structures.
1. Introduction
Anion photodetachment spectroscopy is a powerful experimental technique for studying the nuclear dynamics on the potential energy surface of the corresponding neutral molecular system [1,2,3,4]. For example, if the stable structure of a monoanion is similar to the transition state structure of the neutral system, this technique can be employed to obtain detailed information on the properties of the transient transition state structure, including vibrational structures and energy distributions of the dissociated species. Thus, anion photodetachment spectroscopy can elucidate the nuclear dynamics on the potential energy surface region away from the stable potential energy well. Continetti et al. recently applied this technique to understanding the dissociative photodetachment dynamics of the oxalate anion (C2O4H−) and observed HOCO + CO2 products along with the relative translational energy distributions [5]. The oxalate anion is known to be stable with respect to electron loss, and this is in high contrast with the fact that the free oxalate dianion (C2O42−) is unstable [6]. Thus, this is a simple example of the anion stabilization by solvation. The photodetachment experiment is one of the useful tools to understand the stabilization mechanism of such charged species. In the present work, we simulate the photodetachment dynamics of C2O4H− theoretically to compare the computed results with their experimental results and to understand in greater detail the dissociation mechanisms and dynamics, which cannot be obtained from experiments alone.
The oxalate anion is a molecular system that contains CO2 molecular units with various charge states, which are important for understanding the activation of carbon dioxide [6]. As mentioned previously, the oxalate anion can be regarded as a complex of a proton (H+) and an oxalate dianion (C2O42−) and has been previously studied experimentally and theoretically due to its unique structure [7,8,9,10,11,12,13,14,15]. The oxalate anion has two stable minima on the potential energy surface. One is a closed-form, in which the proton is asymmetrically bound between the two oxygen atoms, forming a planar structure with a five-membered cyclic ionic hydrogen bond, and this structure has an ideal geometry for studying proton-transfer dynamics between the two oxygen atoms [7,8,9,10,11,13]. The other is an open-form, in which the proton in the OH bond points out of the molecular framework with an O-C-C-O angle of 90°. Quantum chemical calculations show that the closed-form is about 0.4 eV more thermodynamically stable compared with the open-form and that the isomerization barrier via rotation of the OH group measured from the closed-form energy to the open-form energy of about 0.9 eV is high [12,14]. Thus, Continetti et al. concluded that the stable closed-form is preferentially produced under their molecular beam conditions [5]. They found that the detachment of C2O4H− leads only to the two-body dissociation into HOCO + CO2 with a large kinetic energy release and that no other dissociation processes, including three-body dissociation, such as 2CO2 + H or CO2 + CO + OH, are observed. This result suggests that the dissociative potential energy surface along the C-C stretching coordinate is steeply repulsive for the neutral state. However, their experimental study could not identify the structure of the dissociated HOCO molecule, although HOCO can exist as two isomers, namely trans-HOCO and cis-HOCO. They speculated that trans-HOCO may be formed preferentially, based on their quantum chemistry calculation, in which geometry optimization starting from the minimum structure of the closed-form anion spontaneously produced CO2 and the trans-HOCO fragment. This also leads to an additional question on the correlation between the initial anion structure and the structure of the dissociated HOCO fragment. Therefore, it is interesting to study the dissociation dynamics also from the open-form C2O4H− anion, although the open-form is thermodynamically unstable compared to the closed-form.
In this work, we report the results of quantum dynamics calculations using path-integral molecular dynamics (PIMD) and ring-polymer molecular dynamics (RPMD) [16,17,18,19,20], in which the anionic and neutral potential energy surfaces are directly obtained from on-the-fly quantum chemical calculations at the density-functional theory (DFT) level. Since both the PIMD and RPMD methods can describe nuclear quantum effects, these methods can reveal the importance of nuclear quantum effects in the photodetachment dynamics of C2O4H−. It is worth mentioning that the importance of nuclear quantum effects has been frequently pointed out in describing the photoabsorption and photoelectron spectra for various chemical systems [21,22,23,24]. In particular, it has been found that nuclear quantum effects are playing an essential role in hydrogen-bonded systems, where hydrogen atom motions behave quantum mechanically. Thus, it should be reasonable to apply the path-integral methods to understand the photodetachment dynamics of C2O4H−.
2. Computational Procedure
In the path-integral formalism, the classical isomorphic ring-polymer Hamiltonian for the chemical system consisting of n atoms is expressed as [16]
where N is the number of imaginary time discretization steps (beads) and β is the reciprocal temperature with β = 1/kBT. mi is the atomic mass of the i-th nuclei. Qis and Pis are position and momentum vectors of the s-th bead for the i-th atom, respectively. V is the potential energy surface. Classical sampling of the ring-polymer Hamiltonian preserves quantum Boltzmann statistics, where the corresponding equations of motion associated with the ring-polymer Hamiltonian are numerically solved using the first derivatives of the potential energy at each bead. In the sampling scheme, we use the standard PIMD approach, where the equations of motion are solved by controlling the system temperature with the massive Nóse–Hoover chain thermostat technique combined with the velocity Verlet algorithm. In general, the equations of motion are solved using the potential energy derivatives at each bead position.
To save computational time, the first derivatives, which are needed to solve the equations of motion, are approximately calculated with the second-order Taylor expansion at the centroid position of the beads as
where Qs and Qc are the s-th bead position and centroid position, respectively. This approximation works well for T > 200 K, since the spread of quantum nuclei around the centroid is small. In this work, the PIMD calculations are performed at T = 300 K with 32 beads (Figure S1). The choice of these numerical parameters is coming from the benchmarking calculations performed with a different chemical system. Details are presented in the Supplementary Material. The total time step is set to 105 with a time increment of Δt = 0.3 fs. Then, the PIMD results are used to calculate the photoelectron spectrum. We use the following semiclassical approximation [25],
where Eb is the electron binding energy and Q collectively denotes the structural coordinates. Vn(Q) and Va(Q) are the potential energy surfaces for the neutral and anionic states, respectively. ρa(Q) is the quantum nuclear density obtained from the PIMD simulation.
We subsequently perform RPMD simulations to obtain the real-time nuclear dynamics on the neutral potential energy surface with the same bead number (32 beads). The initial structures and momenta are chosen from the results of the PIMD simulations, and then, the equations of motion are solved similar to the PIMD simulation, but without the Nóse–Hoover thermostat. We choose 500 different initial conditions. At the end of each RPMD trajectory, the relative transitional energy between the HOCO and CO2 were calculated using the atomic centroid velocities. Similarly, we have calculated the H-O-C-O dihedral angle using the atomic centroid coordinates at the end of each RPMD trajectory to identify if the product HOCO molecule takes trans or cis configuration. The classical molecular dynamics calculations were also performed with the same scheme as the PIMD/RPMD calculations, except that the number of beads was set to one. In this case, vibrational quantization and quantum nuclear fluctuations are completely ignored. All the PIMD and RPMD calculations in this work are performed using the open-source PIMD code [26].
3. Results and Discussion
All the on-the-fly PIMD and RPMD calculations presented in this paper are performed at the B3LYP/aug-cc-pVDZ level theory including GD3 dispersion correction implemented in the Gaussian09 programs [27]. This DFT functional is chosen from the comparison to the CCSD(T)/aug-cc-pVDZ results [5]. More specifically, the energy difference between the most stable closed-form C2O4H− anion and the neutral minimum with the open-form structure is calculated as 4.41 eV at the CCSD(T)/aug-cc-pVDZ level (without vibrational zero-point correction). The B3LYP/aug-cc-pVDZ level calculation provided this quantity as 4.45 eV. Notice that this energy is corresponding to the adiabatic electron affinity. In addition, the energy difference between the most stable closed-form anion and the open-form anion is calculated as 0.39 eV (without vibrational zero-point correction) at the CCSD(T)/aug-cc-pVDZ level, while this value was calculated as 0.38 eV at the B3LYP/aug-cc-pVDZ level. However, the B3LYP calculations yield slightly larger values for the energy levels of the asymptotic cis-HOCO + CO2 and trans-HOCO + CO2 fragments, which are 3.61 and 3.53 eV, respectively. The corresponding CCSD(T) values are reported to be 3.38 and 3.30 eV, respectively [6]. Thus, there is a possibility that the B3LYP potential energy surface gives smaller available energies for the dissociated fragments compared to the CCSD(T) potential energy surface.
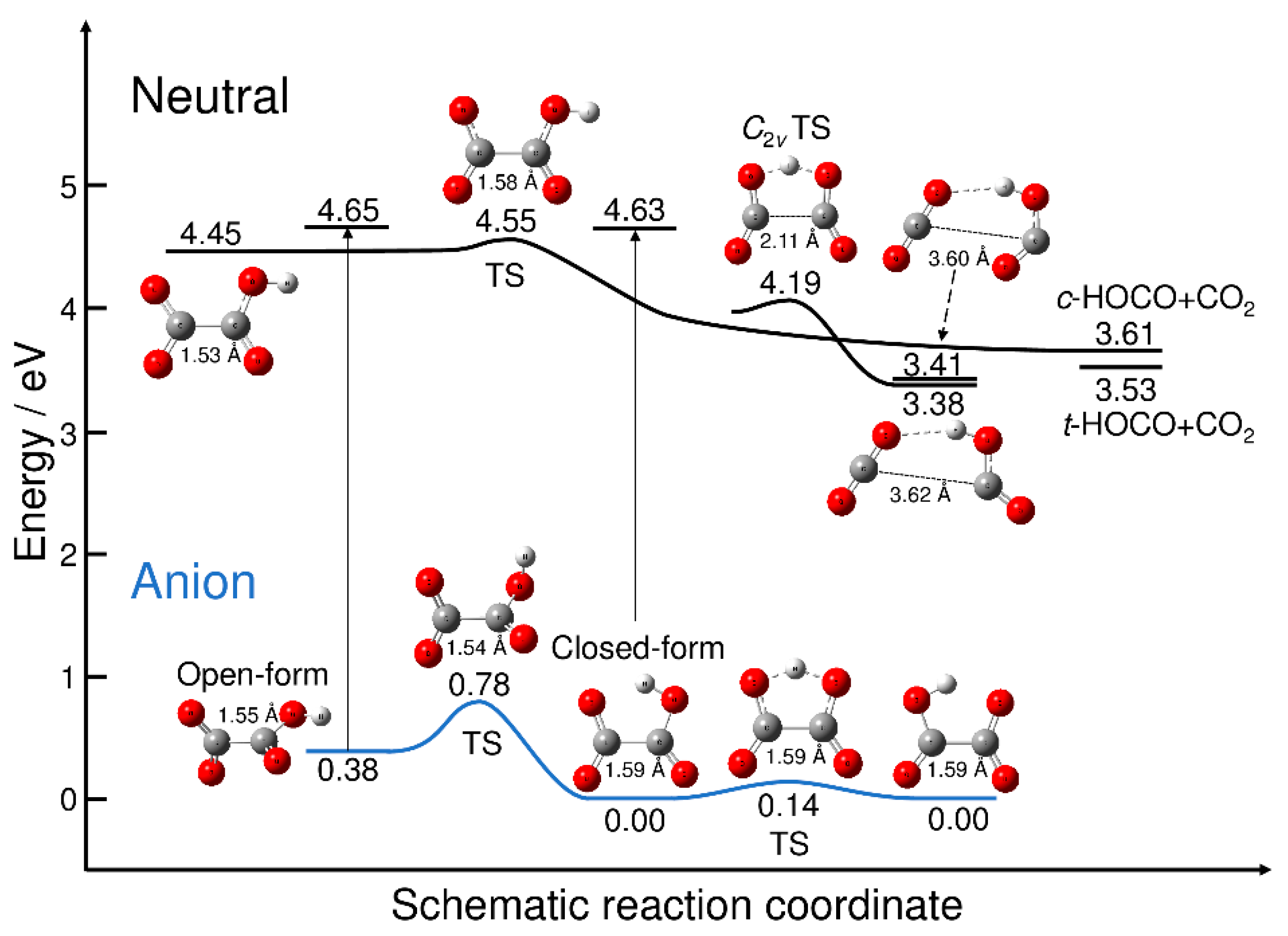
Figure 1 displays the energy levels of the stationary points on the potential energy surfaces of C2O4H− and C2O4H along with the energy levels of the neutral dissociated fragments. All the energy values are measured from the energy level of the most stable closed-form C2O4H− anion. Figure 1 also shows the energy levels of the neutral state at the two anion minimum structures, which yield the vertical electron detachment energies. The neutral potential energy surface has three equilibrium structures. The first structure is the planar open-form with a C-C distance of 1.53 Å that can dissociate into CO2 + cis-HOCO via the transition state structure with a C-C distance of 1.58 Å. All the reaction pathways associated with the transition states found in this work were confirmed from the intrinsic reaction coordinate (IRC) calculations. The barrier height measured from the well of the planar open-form is calculated to be only 0.093 eV. The second minimum structure on the neutral potential energy surface corresponds to the hydrogen-bonded complex between CO2 and trans-HOCO with a long C-C distance (3.62 Å). The C2v transition state is present, which is smoothly connected to the hydrogen-bonded minimum structure; there are two equivalent reaction paths leading to the hydrogen-bonded complex from the C2v transition state. The third minimum structure on the neutral potential energy surface is another hydrogen-bonded complex between CO2 and cis-HOCO with a long C-C distance (3.60 Å). The energy levels of these two hydrogen-bonded complexes are just below the dissociation energy levels.
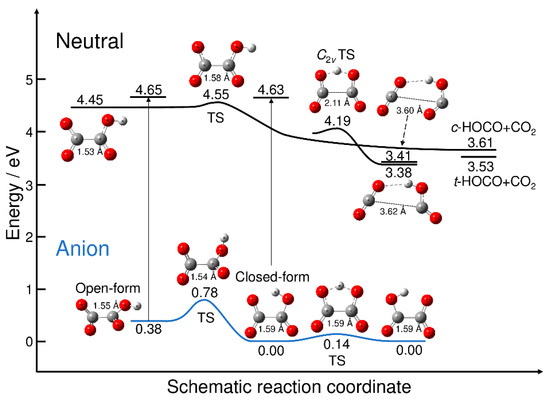
Figure 1.
Schematic potential energy surfaces for C2O4H− and C2O4H. Energy values were obtained at the B3LYP/aug-cc-pVDZ (with GD3 dispersion correction) level of theory. The energy was measured from the energy level of the most stable isomer of closed-form C2O4H−. The vertical thin lines with arrows indicate the vertical electron detachment process, and the corresponding energy values are the neutral energy levels at the anion minima. TS: transition state.
The anionic potential energy surface has two minima, corresponding to the open- and closed-forms. The C-C distance for the open-form (1.55 Å) is slightly shorter than that for the closed-form (1.59 Å), despite the presence of the five-membered cyclic structure. The results presented in Figure 1 qualitatively show the nuclear dynamics associated with the electron detachment process. When the excess electron of the most stable closed-form C2O4H− is detached, direct dissociation may occur to form CO2 and trans-HOCO fragments. This is because the electron-detached Franck–Condon structure is at an energy much higher than the neutral C2v transition state structure, and the electron detachment should be the transition to the almost repulsive region of the neutral potential energy surface. This scenario has already been discussed by Continetti et al. [5]. In contrast, if the excess electron is detached from metastable open-form C2O4H−, the dissociation can also occur, but via the transition state structure with the small barrier (Figure 1). This suggests the existence of an indirect dissociation mechanism, in which the nuclear trajectory may spend some time around the Franck–Condon region, which is followed by dissociation into CO2 + HOCO. Thus, there may be an important difference in the photodetachment nuclear dynamics between the closed- and open-form anions, although it may be difficult to preferentially produce metastable open-form C2O4H− under experimental conditions. In fact, if we assume a Boltzmann equilibrium, the concentration ratio of the closed-form and open-form would roughly be 1:10–6 at T = 300 K.
Figure 2 shows the three-dimensional perspective plots of the nuclear distributions obtained from the PIMD calculations at T = 300 K for both the closed- and open-form C2O4H− structures. Since there is a large barrier between these two forms (Figure 1), these two configurations can be independently sampled using different initial structural conditions. No isomerization between these two structures is observed within the present PIMD simulation time. For closed-form C2O4H− (Figure 2a), the nuclear configuration is distributed around the planar structure. This observation indicates that the intra-molecular hydrogen bond is sufficiently strong to maintain the planar structure, and that electron detachment from closed-form C2O4H− mainly produces the planar structure on the neutral potential energy surface. In addition, the proton is almost equally distributed around the two O atoms, indicating that proton transfer occurs between the two O atoms due to the small barrier, although the nuclear density in the proton-transfer transition state is small. For the open-form anion (Figure 2b), the O-C-C-O dihedral angle is broadly distributed around 90°, which is presumably due to the missing intramolecular hydrogen bond. This result suggests that electron detachment of the open-form anion yields neutral structures with a wide range of O-C-C-O dihedral angles.
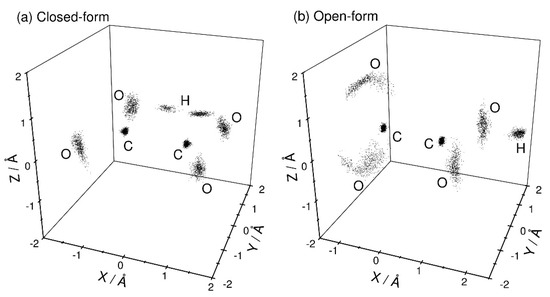
Figure 2.
Three-dimensional perspective plot of the nuclear probability distribution functions of (a) closed-form and (b) open-form C2O4H− calculated from the PIMD simulations at T = 300 K. The midpoint of the two carbon atoms is fixed to the coordinate origin, and the midpoint of the two oxygen atoms is always placed on the X-axis. In addition, the hydrogen atom is placed on the X-Y plane.
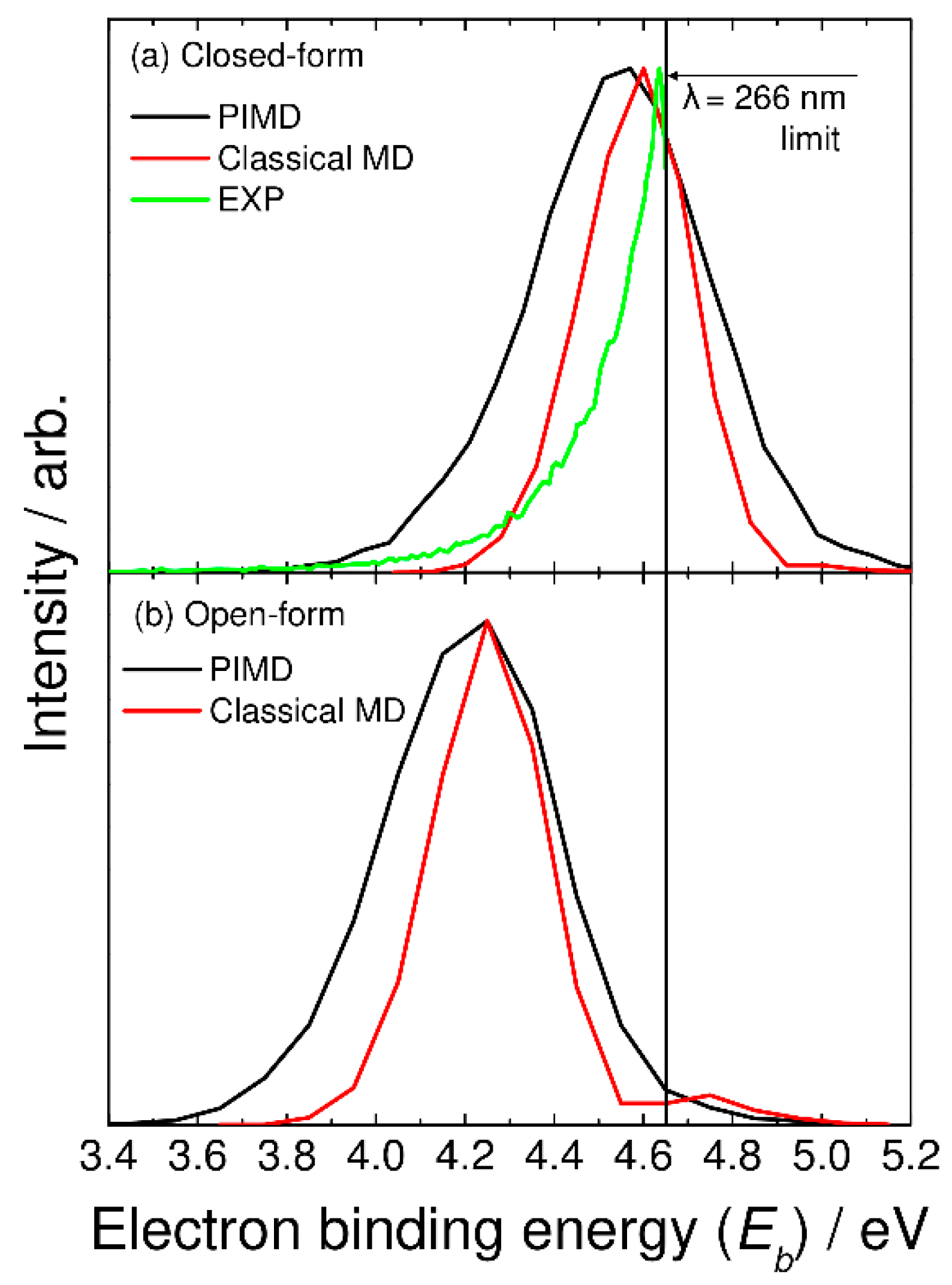
In Figure 3a, we compare the photoelectron spectrum calculated with the semiclassical method based on the PIMD nuclear density with the experimentally measured spectrum taken from Ref. [5]. These spectra are plotted as a function of the electron binding energy with Eb = hν − eKE, where hν and eKE are the excitation photon energy and kinetic energy of the photodetached electron, respectively. Here, Eb corresponds to the energy level of the neutral state measured from the energy level of the most stable closed-form C2O4H− (Figure 1). The experimental photoelectron spectrum is measured at a laser wavelength of 266 nm (hν = 4.66 eV) [5], and thus, the photoelectron spectrum in the higher electron binding energy region cannot be obtained. The spectrum calculated with the PIMD method shows a single broad peak about 0.5 eV wide at Eb = 4.6 eV. Although the experimental peak position cannot be determined due to the limited photodetachment laser energy, our calculated spectrum qualitatively reproduces the experimental spectral features. Continetti et al. analyzed their measured spectrum with the bound-continuum Franck–Condon factor model calculated using two one-dimensional potential energy curves for the anionic and neutral states along the C-C stretch coordinate [5]. The simulated spectrum shows a spectral peak at Eb ≈ 4.8 eV. If the one-dimensional analysis performed by Continetti et al. was correct, the peak position of our calculated spectrum should be shifted forward by about 0.2 eV. This indicates that the energy level of the neutral state calculated with the present DFT-B3LYP level is low; i.e., the electron affinity value is underestimated in the B3LYP calculations. Notice that good agreement in the vertical electron detachment energy is obtained between the CCSD(T) and B3LYP results, as mentioned previously. This indicates that more accurate calculations containing higher-order electron correlation must be performed to obtain more accurate electron affinity values.
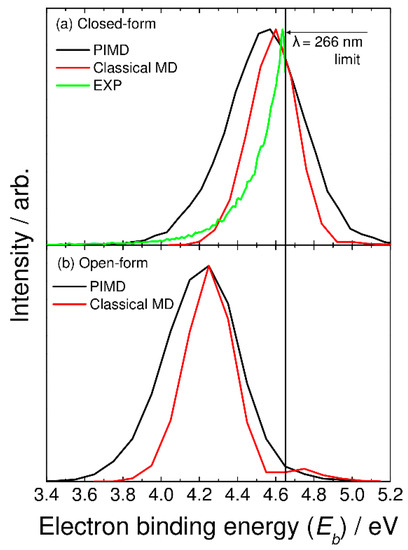
Figure 3.
Photoelectron spectra for (a) closed-form and (b) open-form C2O4H− calculated using the semiclassical method based on the PIMD nuclear density (black solid lines). In panel (a), the photoelectron spectrum calculated with the classical MD method is shown by the red solid line, and the experimental photoelectron spectrum measured at λ = 266 nm (taken from Ref. [5]) is also shown by the green solid line.
In Figure 3, we also show the spectra calculated from the classical molecular dynamics, where the bead number was set to one. In this case, vibrational quantization is completely ignored. The calculated spectra show a narrower feature compared to the PIMD results. This indicates that nuclear quantum effects including quantum vibrational amplitudes are playing an essential role in determining the spectral shape. The present results are in line with the previous theoretical studies of the nuclear quantum effects on photoabsorption and photoelectron spectra [21,22,23,24].
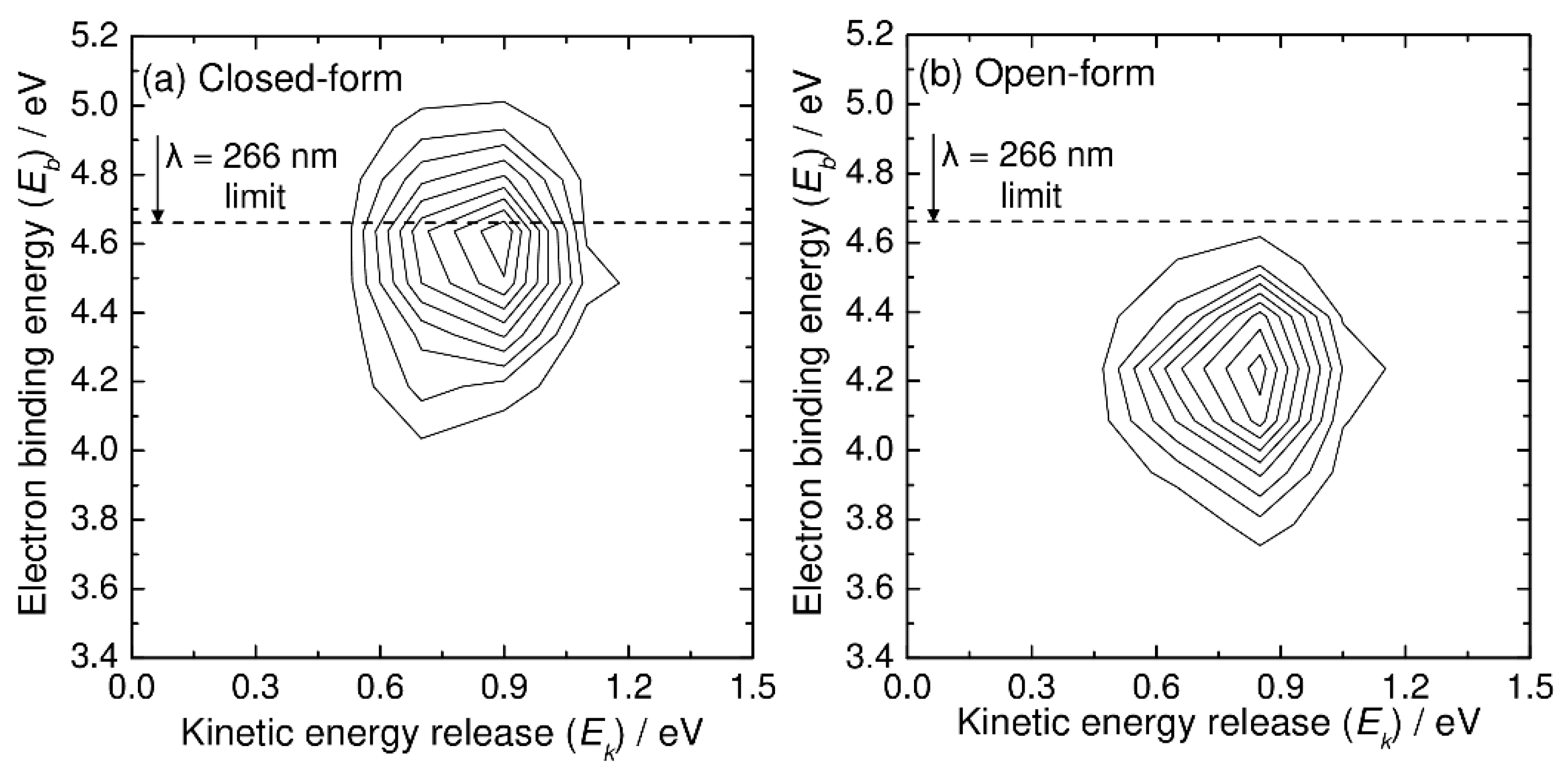
Next, we present the simulated photoelectron–photofragment coincidence spectra. Figure 4 shows the simulated two-dimensional coincidence spectra plotted as a function of the kinetic energy release (Ek) between CO2 and HOCO and the electron binding energy (Eb). The simulated coincidence spectrum of Figure 4a can be compared with Figure 5 of Ref. [5], although the experimental spectrum is plotted as a function of eKE instead of Eb. The simulated coincidence spectrum is obtained from 500 RPMD trajectories, and the kinetic energy release is calculated from the centroid velocities of the CO2 and HOCO dissociated fragments, as mentioned previously. There is a single broad Gaussian-like feature centered at Ek = 0.9 eV and Eb = 4.6 eV, which is qualitatively consistent with the experimental spectrum, although the experimental peak position for the kinetic energy release is observed at slightly higher energies of Ek = 1.1 eV and Eb = 4.7–4.8 eV. This difference in the peak position is originally coming from the fact that the B3LYP calculations give a lower energy level for the neutral state by about 0.2 eV at the Franck–Condon region. As mentioned previously, this energy difference affects the peak position (thus Eb) of the photoelectron spectrum. In addition to this, it is emphasized that the energy level at the Franck–Codon region also affects the available energy for the HOCO and CO2 fragments; the vertical transition to a higher neutral state would lead to an increase in the available energy for the dissociation fragments. Another important reason for the difference in the peak position in the two-dimensional coincidence spectrum between theory and experiment is the inaccurate energy levels of the asymptotic HOCO and CO2 fragments. As mentioned previously, the B3LYP calculations give somewhat larger energy levels for both the trans-HOCO + CO2 and cis-HOCO + CO2 channels compared to the CCSD(T) energetics (see also Figure 1) [5]. This can lead to the decrease in Ek in the simulated spectrum compared to the experimental result.
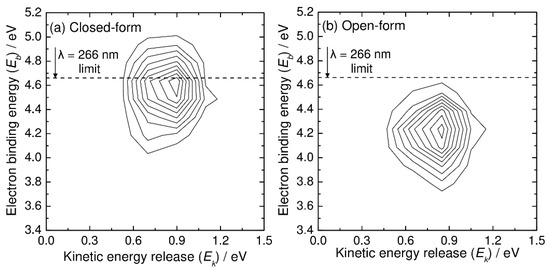
Figure 4.
N(Ek, Eb) two-dimensional correlation spectra for C2O4H− + hν → CO2 + HOCO + e−, where Ek and Eb are the relative kinetic energy between the CO2 and HOCO fragments and electron binding energy, respectively. Results from the RPMD calculations at T = 300 K for (a) closed-form and (b) open-form C2O4H−.
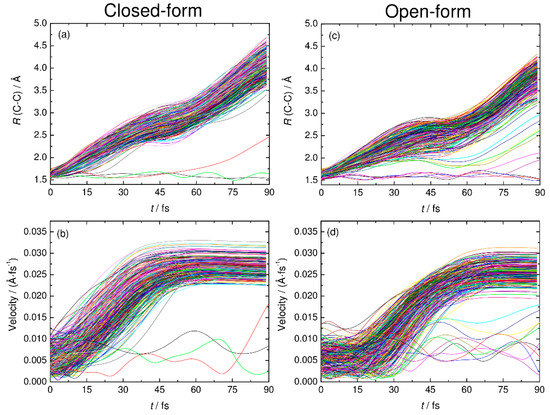
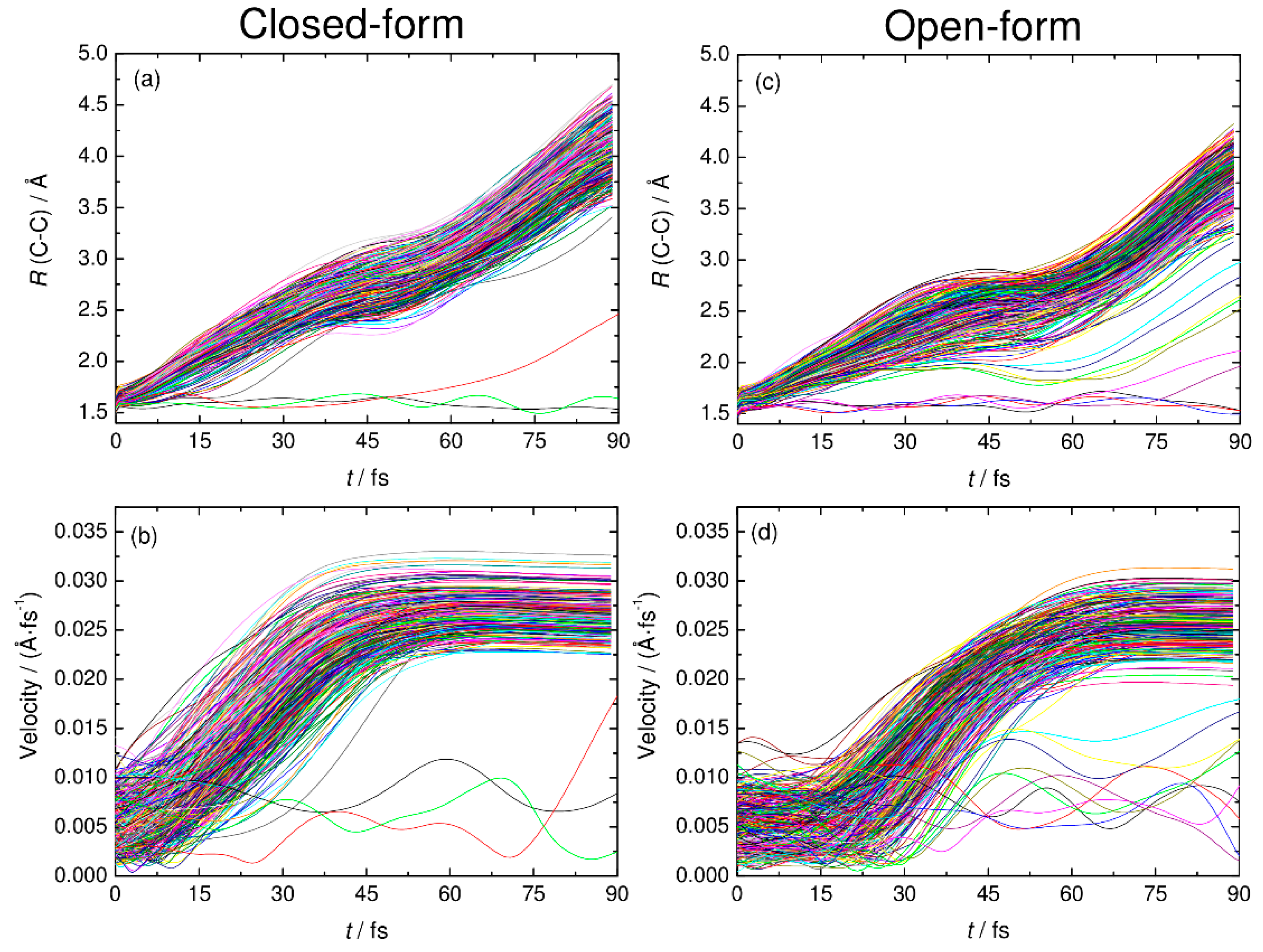
Figure 5.
The C-C distance and relative velocity between CO2 and HOCO plotted as function time for all the RPMD trajectories examined in this work. Panels (a) and (c) show the results of the C-C distance for closed-form and open-form, respectively. Panels (b) and (d) show the results of relative velocity for closed-form and open-form, respectively.
The simulated coincidence spectrum for the open-form anion obtained from 500 RPMD trajectories is also presented in Figure 4b, and it has similar features to that for the closed-form anion, although the peak is at a lower Eb value.
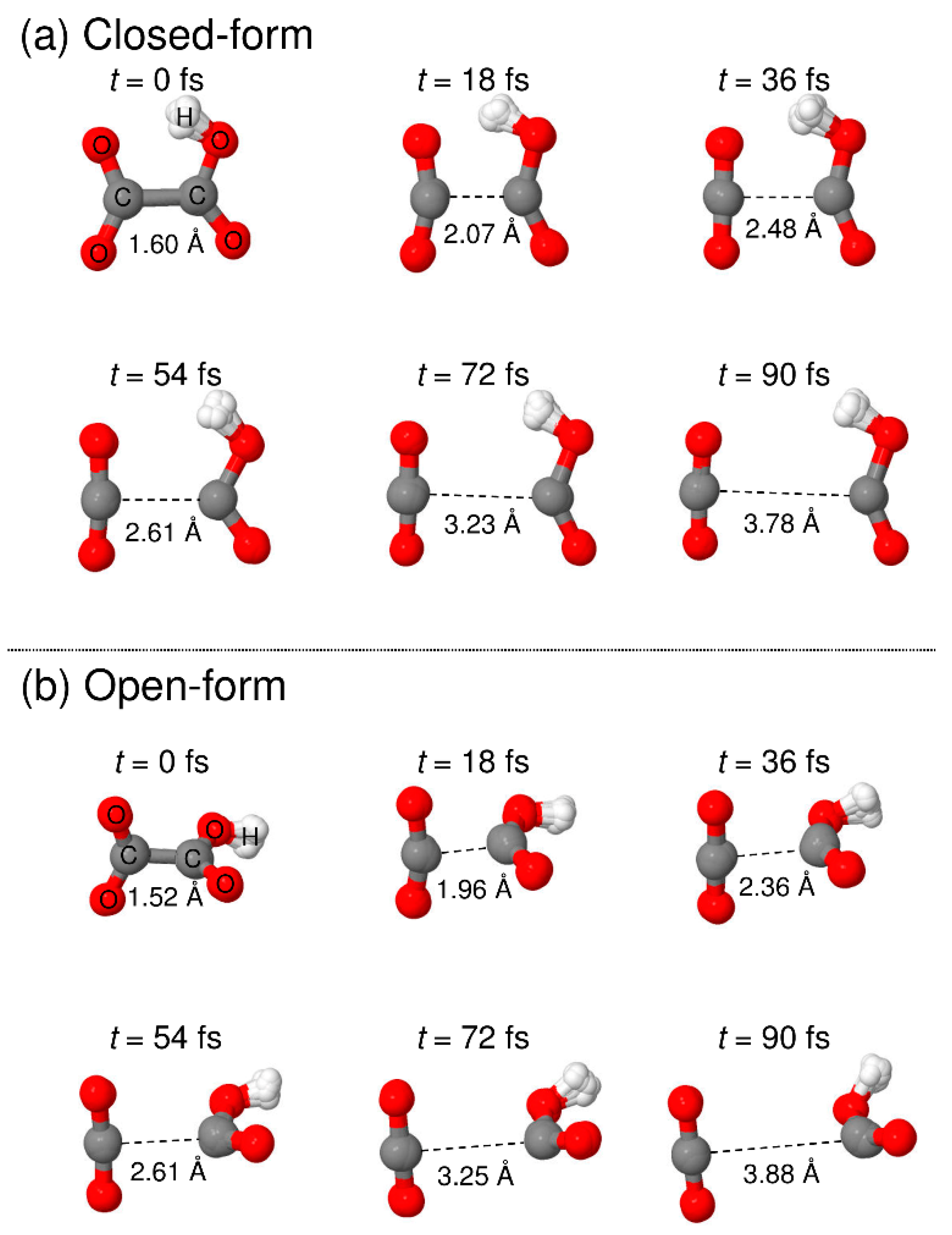
To understand the detailed photodetachment dissociation mechanism of C2O4H−, we present the time dependence of the C-C distance and the relative velocity between CO2 and HOCO for all the RPMD trajectories examined in this work. The left and right panels of Figure 5 correspond to the results for the closed- and open-form anions, respectively. The results in Figure 5a,b show that the dissociation process for the closed-form anion has a strongly repulsive character because both the C-C distance and the relative velocity rapidly increase just after the electron detachment. In addition, the starting time of the flat region of the relative velocity shows that most of the dissociation processes are complete within t ≈ 60 fs. Only three RPMD trajectories out of 500 show an indirect, delayed dissociation mechanism. In contrast to the closed-form anion results, the open-form anion has different dynamics features. The relative velocity is nearly constant at an early stage up to t = 20–30 fs, and then, it increases rapidly after this region followed by a flat region at t > 75 fs (Figure 5d). This indicates that many trajectories spend time around the Franck–Condon region before fast dissociation occurs. These nuclear dynamics are consistent with the potential energy surface features of the neutral state. In addition, 10 RPMD trajectories out of 500 show the indirect mechanism, which is in contrast to the closed-form results. For the closed- and open-form anion, there is a step-like feature in the time dependence of the C-C distance at t ≈ 40 and 50 fs, respectively. These regions are close to the starting region of the flat velocity feature; thus, the step-like feature suggests that the trajectories reach the flat region of the potential energy surface at longer C-C distances. To understand the dissociation mechanisms in more detail, we present snapshots of typical RPMD trajectories in Figure 6. We did not observe the formation of hydrogen-bonded complexes, at least for the trajectories showing the direct dissociation mechanisms.
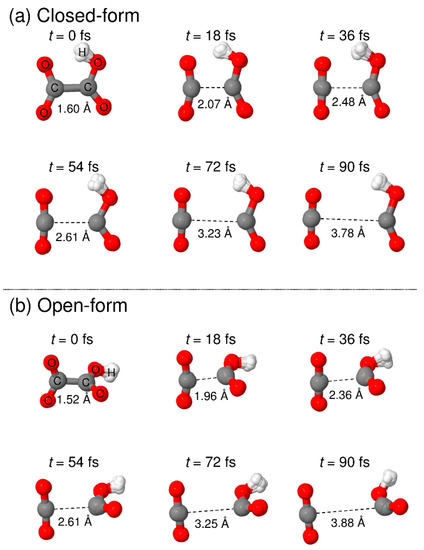
Figure 6.
Snapshots of typical RPMD trajectories calculated starting from the (a) closed-form and (b) open-form C2O4H− isomers.
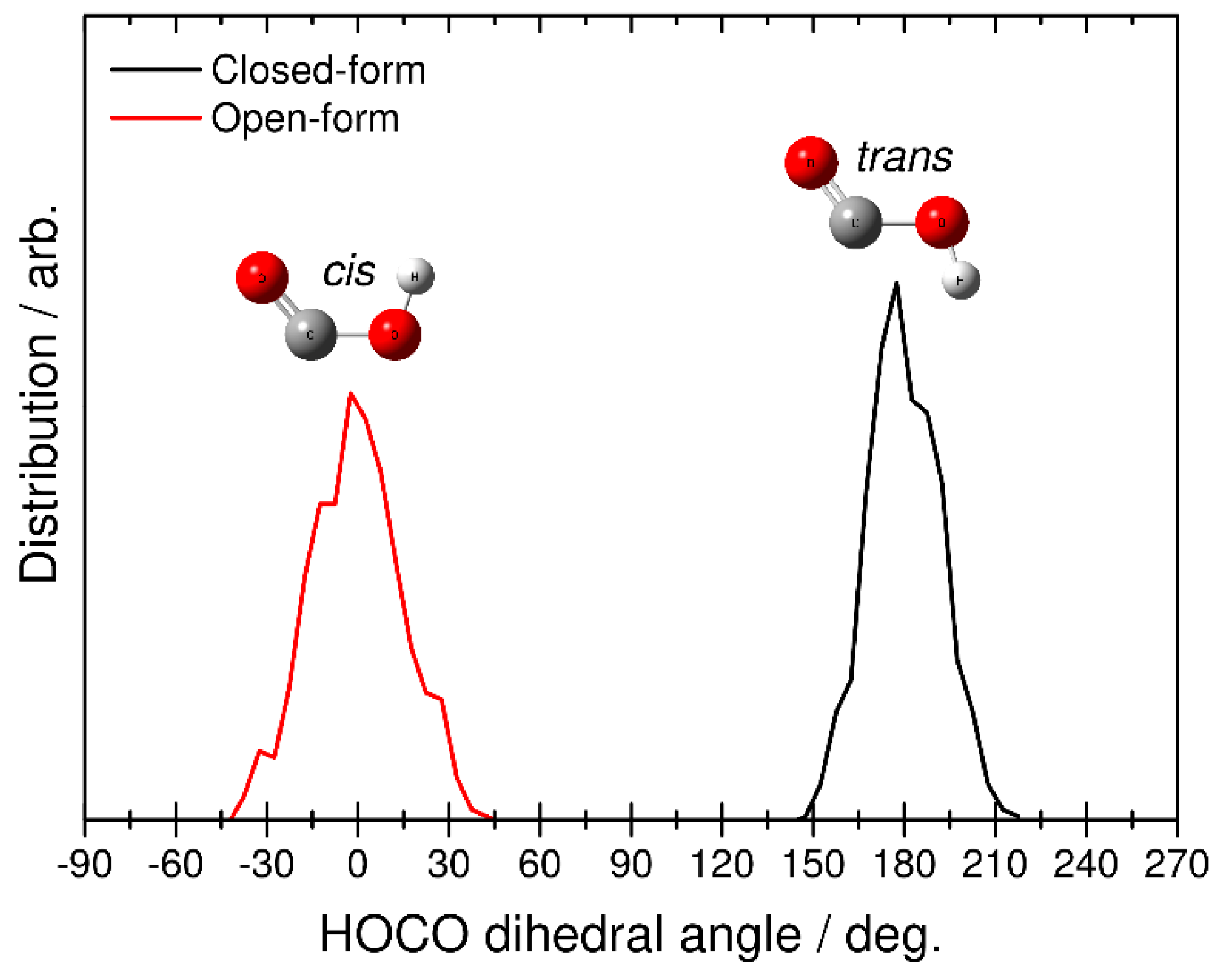
Finally, we identified the structure of the HOCO fragment by analyzing the H-O-C-O dihedral angular distributions. This analysis was performed at the end of each RPMD trajectory, where the relative velocity between CO2 and HOCO is almost constant (see also Figure 5). The black line in Figure 7 corresponds to the RPMD result calculated from the closed-form anion, while the red line corresponds to the result from the open-form anion. The former shows the angular distribution in the range of 150–210 degrees, and the latter shows the distribution from −45 to 45 degrees. From this result, it is found that dissociative photodetachment of the closed-form anion produces the trans-HOCO fragment and that photodetachment of the open-form anion gives the cis-HOCO product. In addition, these two angular distributions show a relatively narrow behavior. This result is consistent with fact that the dissociation dynamics is a fast direct process. In fact, from the results presented in Figure 5, dissociation of the closed-form completes within t < 60, fs while dissociation of the open-form completes within t < 75 fs. These results suggest that the available energy is not largely partitioned into the out-of-plane (internal rotational) motion of the OH bond in HOCO, and the cis−trans isomerization process does not occur in the dissociative photodetachment process. In other words, the trans-HOCO fragment can be formed preferentially from the closed-form anion, while cis-HOCO can be formed from the open-form anion. It is interesting that one of the two neutral HOCO isomers can be preferentially produced by choosing the initial anion structure through the dissociative photodetachment process.
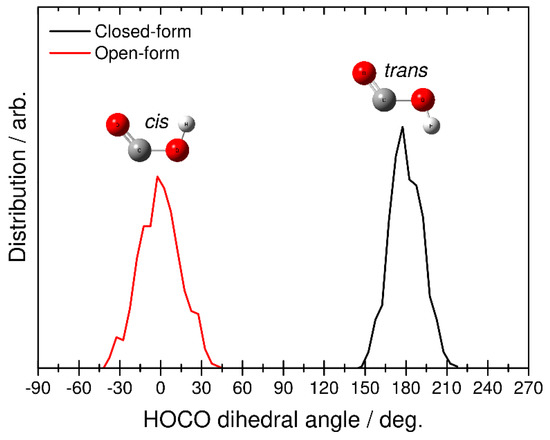
Figure 7.
Distributions for the final HOCO dissociation product plotted as a function of the H-O-C-O dihedral angle obtained from the RPMD calculations. Black and red solid lines correspond to the RPMD results obtained from initial closed- and open-form C2O4H−, respectively.
4. Conclusions
We investigated the dissociative photodetachment of the oxalate anion, C2O4H− + hν → CO2 + HOCO + e−, using the quantum PIMD and RPMD simulation methods, which can account for nuclear quantum effects. We used the on-the-fly technique, in which the potential energy derivatives are directly obtained from the DFT calculations at each time step. To reduce the computational time, the force acting on each bead was approximately calculated from the first and second derivatives of the potential energy at the centroid position of the nuclei beads. Since the anion potential energy surface has two isomers with closed- and open-form structures and the isomerization barrier between the two isomers is high, the PIMD and RPMD calculations for the two anion structures were independently performed. Only the results for the closed-form anion could be compared with the experimental results because the closed-form anion was more stable than the open-form anion. The photoelectron spectrum calculated using the PIMD method qualitatively agreed with the previous experimental spectrum [5], although the calculated spectral peak position was at a slightly lower energy compared with the experimental analysis. This is primarily due to the underestimation of the neutral energy level at the Franck–Condon region. The photoelectron spectra calculated using classical molecular dynamics method, which cannot describe quantized vibrational motions, show a narrower spectral feature compared to the PIMD results. This indicates that nuclear quantum effects are playing an essential role in determining the spectral shape.
We also compared the two-dimensional photoelectron–photofragment kinetic energy correlation spectrum calculated using the RPMD method with the previous experimental spectrum [5]. The calculated spectrum qualitatively reproduced the single Gaussian-like feature in the experimental spectrum, although the peak position is somewhat deviated from the experimental observation. Similar to the photoelectron spectrum case, this is due the inaccuracy of the DFT calculation, where the neutral energy level measured from the anion energy level is somewhat underestimated.
We discussed the difference in the photodetachment dynamics between the closed- and open-form anions, although no experimental data are available for the open-form anion. It is found that dissociative photodetachment of the closed-form anion preferentially leads to the trans-HOCO production and photodetachment of the open-form anion yields the cis-HOCO fragment. Detailed analysis of the RPMD trajectories shows that these dissociation processes are fast direct processes, which complete within t < 60–75 fs, and that the available energy is not largely partitioned into the out-of-plane motion of the OH bond in the HOCO fragment. We hope that the present computational study will stimulate future experimental studies of the open-form anion.
Supplementary Materials
The following are available online, Figure S1: Angular distribution functions for the F−·H2O (F−·D2O) cluster obtained from the two different PIMD schemes.
Author Contributions
Conceptualization, T.T.; Formal analysis, Y.T., Y.H. and K.S.; Investigation, Y.T., Y.H., K.S. and T.T.; Supervision, T.T.; Writing—original draft, Y.T. and T.T.; Writing—review and editing, Y.H., K.S. and T.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant No. 20H05847).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Neumark, D.M. Transition-state spectroscopy via negative ion photodetachment. Accounts Chem. Res. 1993, 26, 33–40. [Google Scholar] [CrossRef]
- Neumark, D.M. Spectroscopy of reactive potential energy surfaces. PhysChemComm 2002, 5, 76–81. [Google Scholar] [CrossRef]
- Neumark, D.M. Probing the transition state with negative ion photodetachment: Experiment and theory. Phys. Chem. Chem. Phys. 2005, 7, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Lineberger, W.C. Once upon Anion: A Tale of Photodetachment. Annu. Rev. Phys. Chem. 2013, 64, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Gibbard, J.A.; Castracane, E.; Shin, A.J.; Continetti, R.E. Dissociative photodetachment dynamics of the oxalate monoanion. Phys. Chem. Chem. Phys. 2020, 22, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Dodson, L.G.; Thompson, M.C.; Weber, J.M. Characterization of Intermediate Oxidation States in CO2 Activation. Annu. Rev. Phys. Chem. 2018, 69, 231–252. [Google Scholar] [CrossRef]
- Bosch, E.; Moreno, M.; Lluch, J.M.; Bertrán, J. Bidimensional tunneling dynamics of malonaldehyde and hydrogenoxalate anion. A comparative study. J. Chem. Phys. 1990, 93, 5685–5692. [Google Scholar] [CrossRef]
- Truong, T.N.; McCammon, J.A. Direct dynamics study of intramolecular proton transfer in hydrogenoxalate anion. J. Am. Chem. Soc. 1991, 113, 7504–7508. [Google Scholar] [CrossRef]
- Bosch, E.; Moreno, M.; Lluch, J.M. The role of coupling in intramolecular proton transfer reactions. The hydrogen oxalate anion as an example. Can. J. Chem. 1992, 70, 100–106. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Rodríguez-Otero, J.; Ríos, M.A. High Level and Dual Level Direct Dynamics in the Intramolecular Proton Transfer of Hydrogenoxalate Anion. Influence of Tunneling and Isotopic Effect. J. Phys. Chem. A 1998, 102, 2954–2961. [Google Scholar] [CrossRef]
- Wolke, C.T.; DeBlase, A.F.; Leavitt, C.M.; McCoy, A.B.; Johnson, M.A. Diffuse Vibrational Signature of a Single Proton Embedded in the Oxalate Scaffold, HO2CCO2–. J. Phys. Chem. A 2015, 119, 13018–13024. [Google Scholar] [CrossRef]
- Kroutil, O.; Minofar, B.; Kabeláč, M. Structure and dynamics of solvated hydrogenoxalate and oxalate anions: A theoretical study. J. Mol. Model. 2016, 22, 210. [Google Scholar] [CrossRef]
- Xu, Z.-H.; Meuwly, M. Vibrational Spectroscopy and Proton Transfer Dynamics in Protonated Oxalate. J. Phys. Chem. A 2017, 121, 5389–5398. [Google Scholar] [CrossRef]
- Kroutil, O.; Předota, M.; Kabeláč, M. Force field parametrization of hydrogenoxalate and oxalate anions with scaled charges. J. Mol. Model. 2017, 23, 327. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.M. The interaction of negative charge with carbon dioxide—Insight into solvation, speciation and reductive activation from cluster studies. Int. Rev. Phys. Chem. 2014, 33, 489–519. [Google Scholar] [CrossRef]
- Habershon, S.; Manolopoulos, D.E.; Markland, T.E.; Miller, T.F., III. Ring-Polymer Molecular Dynamics: Quantum Effects in Chemical Dynamics from Classical Trajectories in an Extended Phase Space. Annu. Rev. Phys. Chem. 2013, 64, 387–413. [Google Scholar] [CrossRef] [PubMed]
- Shiga, M.; Tachikawa, M.; Miura, S. Ab initio molecular orbital calculation considering the quantum mechanical effect of nuclei by path integral molecular dynamics. Chem. Phys. Lett. 2000, 332, 396–402. [Google Scholar] [CrossRef]
- Shiga, M.; Tachikawa, M.; Miura, S. A unified scheme for ab initio molecular orbital theory and path integral molecular dynamics. J. Chem. Phys. 2001, 115, 9149–9159. [Google Scholar] [CrossRef]
- Craig, I.R.; Manolopoulos, D. Quantum statistics and classical mechanics: Real time correlation functions from ring polymer molecular dynamics. J. Chem. Phys. 2004, 121, 3368–3373. [Google Scholar] [CrossRef]
- Craig, I.R.; Manolopoulos, D. Chemical reaction rates from ring polymer molecular dynamics. J. Chem. Phys. 2005, 122, 084106. [Google Scholar] [CrossRef] [PubMed]
- Law, Y.K.; Hassanali, A.A. The importance of nuclear quantum effects in spectral line broadening of optical spectra and electrostatic properties in aromatic chromophores. J. Chem. Phys. 2018, 148, 102331. [Google Scholar] [CrossRef] [PubMed]
- Hollas, D.; Muchová, E.; Slavíček, P. Modeling Liquid Photoemission Spectra: Path-Integral Molecular Dynamics Combined with Tuned Range-Separated Hybrid Functionals. J. Chem. Theory Comput. 2016, 12, 5009–5017. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Xu, J.; Qiu, D.Y.; Wu, X. Nuclear quantum effects on the quasiparticle properties of the chloride anion aqueous solution within the GW approximation. Phys. Rev. B 2021, 104, 035117. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Saito, K.; Takayanagi, T.; Tachikawa, H. Theoretical study of the dissociative photodetachment dynamics of the hydrated superoxide anion cluster. Phys. Chem. Chem. Phys. 2021, 23, 16958–16965. [Google Scholar] [CrossRef]
- Lax, M. The Franck-Condon Principle and Its Application to Crystals. J. Chem. Phys. 1952, 20, 1752–1760. [Google Scholar] [CrossRef]
- Shiga, M. PIMD Program, Version 2.4. Available online: ccse.jaea.go.jp/software/PIMD/download.jp.html (accessed on 7 January 2020).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).