
N-Acetyl Cysteine, Selenium, and Ascorbic Acid Rescue Diabetic Cardiac Hypertrophy via Mitochondrial-Associated Redox Regulators

 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
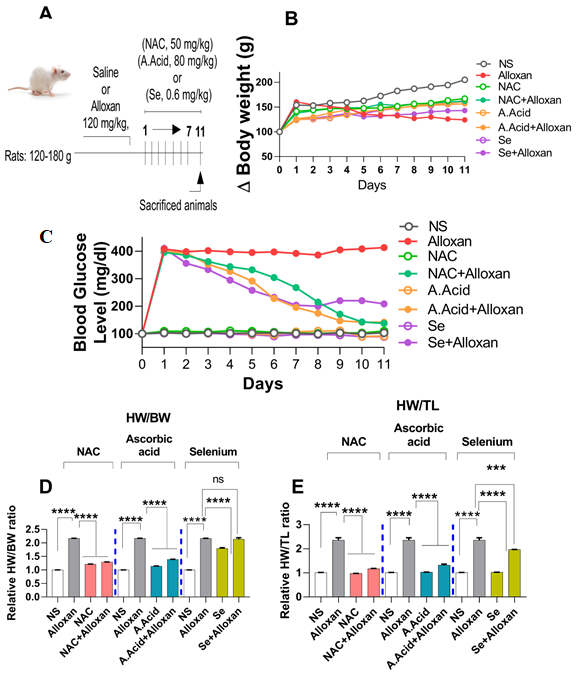
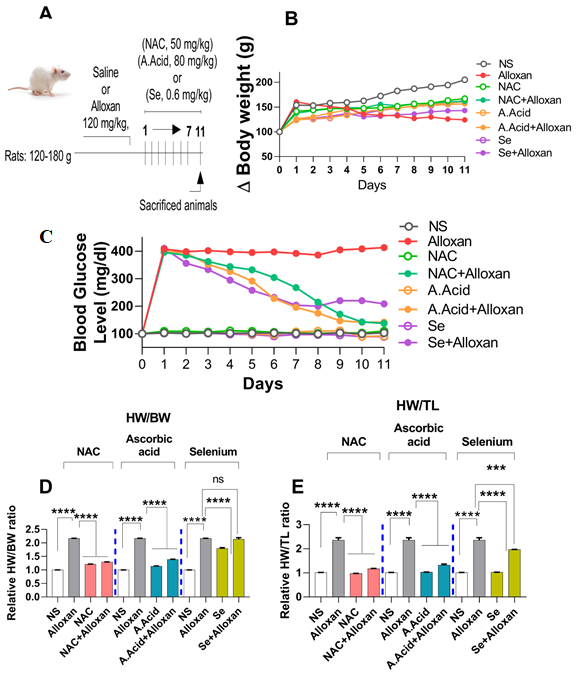
2.1. Body Weights and Effects of Antioxidants
2.2. Changes in Blood Glucose Levels and Morphology in Diabetes Induced Cardiac Hypertrophy Model
Blood Glucose Levels and Effects of N-acetyl Cysteine
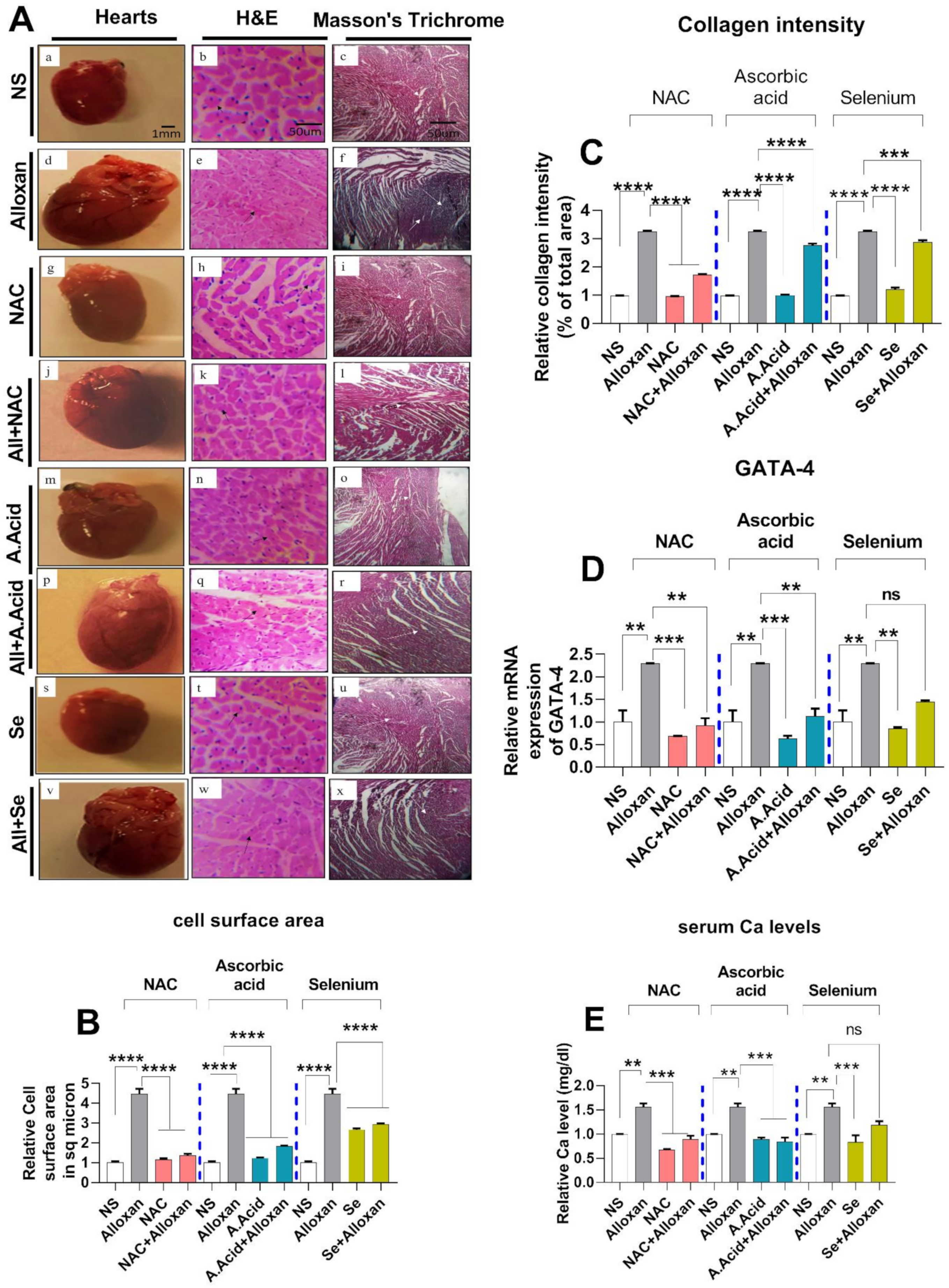
2.3. Cellular Organization, Structural Changes, and Tissue Damage in Diabetes-Induced Cardiac Pathology Model
N-acetyl Cysteine Treatment Restores Myocardial Integrity
2.4. Cardiac Fibrosis in Diabetes-Induced Cardiac Hypertrophy Model
2.5. Serum Calcium Levels and Transcription Factor GATA 4 Expressions in the Diabetic Cardiac Hypertrophy Model
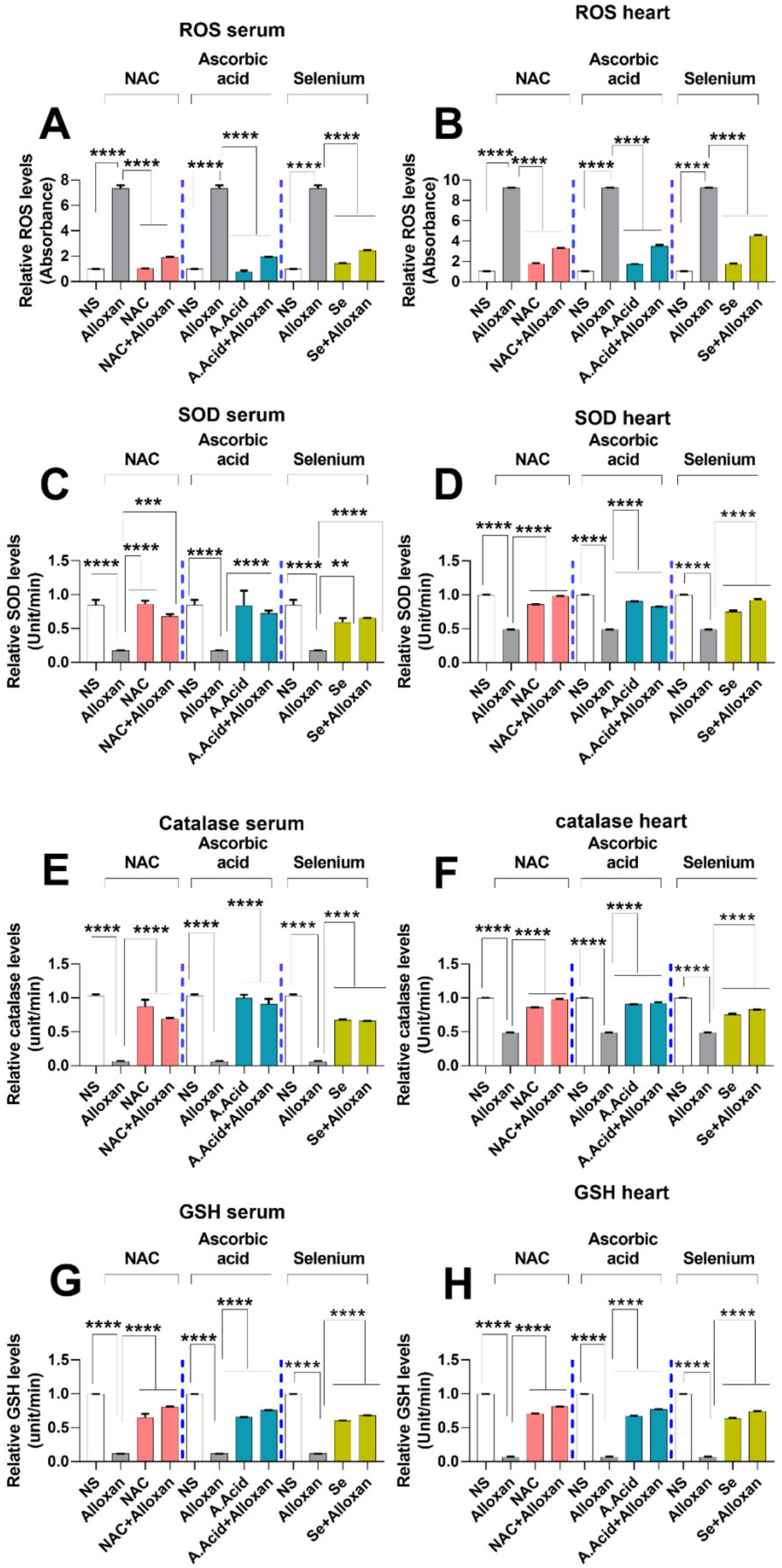
2.6. Production of Reactive Oxygen Species (ROS) Is Increased in Diabetic Cardiac Hyper-Trophy
2.7. Anti-Oxidative Defense Enzymes Are Downregulated in the Diabetic Cardiac Condition Which Is Restored by N-Acetyl Cysteine
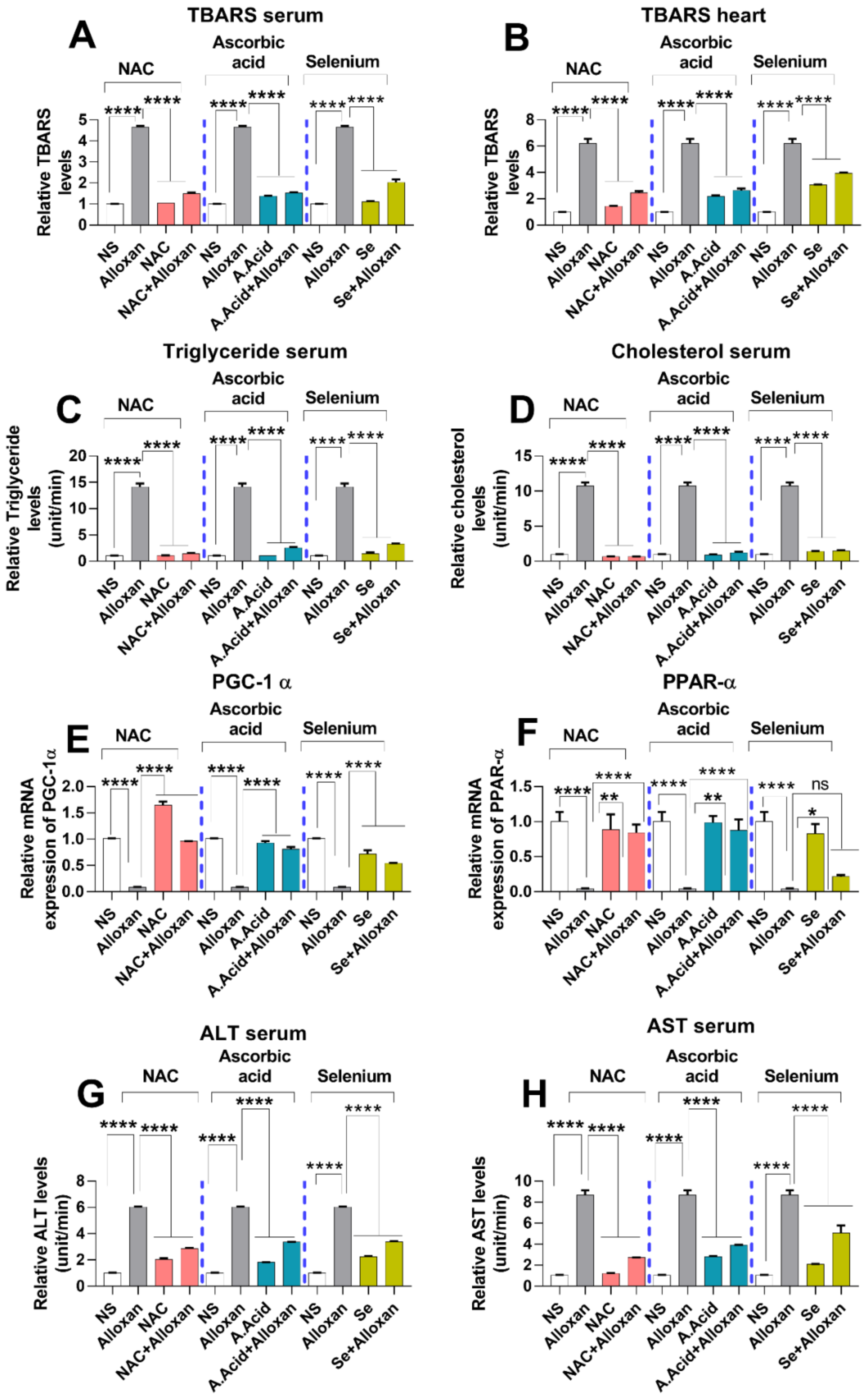
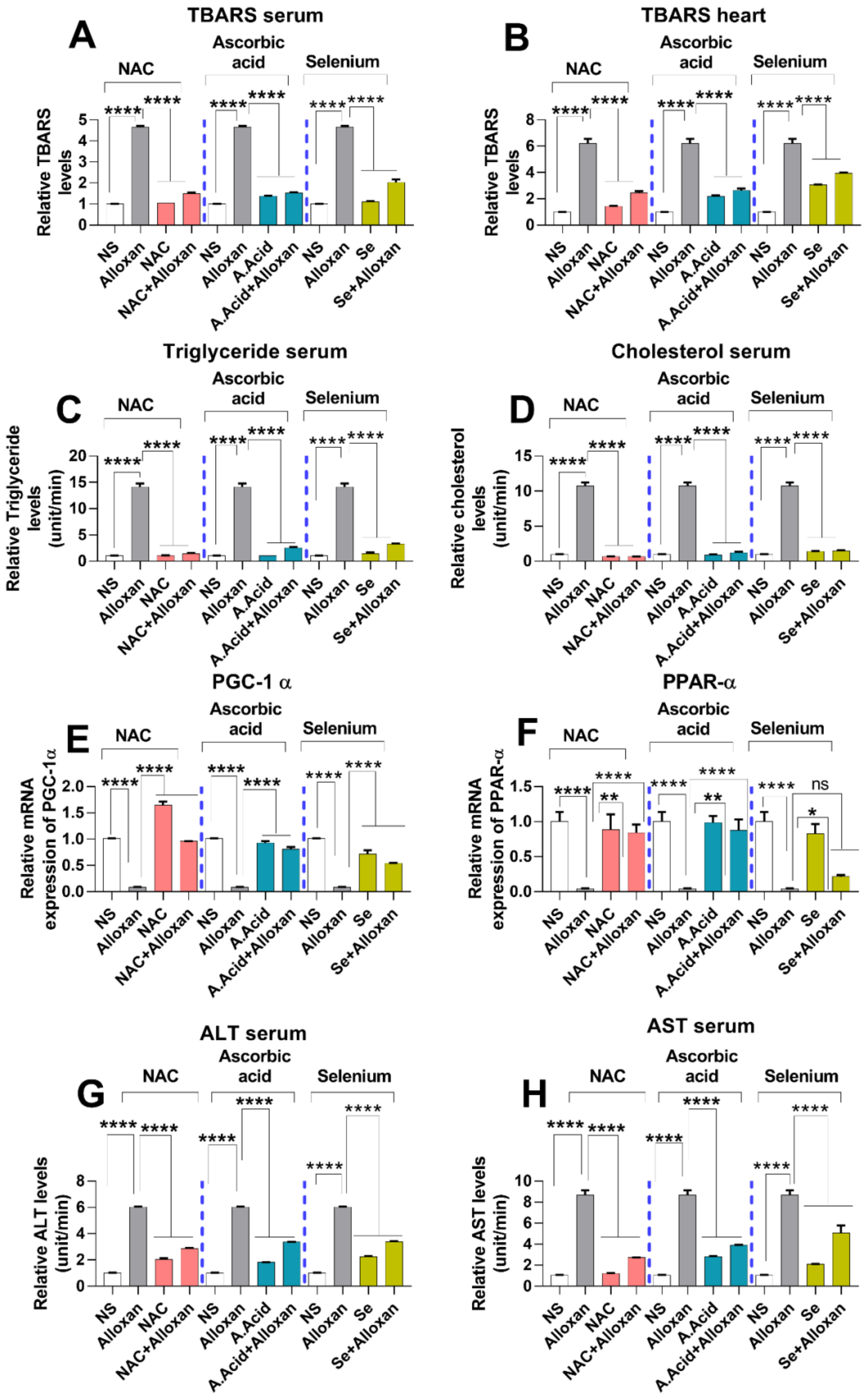
2.8. Analysis of Lipid Peroxidation and Lipid Profile in Cardiac Pathology
2.9. Expression of Peroxisome Proliferator-Activated Receptor-Gamma Coactivator (PGC)-1 Alpha and Peroxisome Proliferator-Activated Receptor PPARα
2.10. Toxicity Markers Show Safer dose Administration in Diabetes-Induced Cardiac Hypertrophy Model
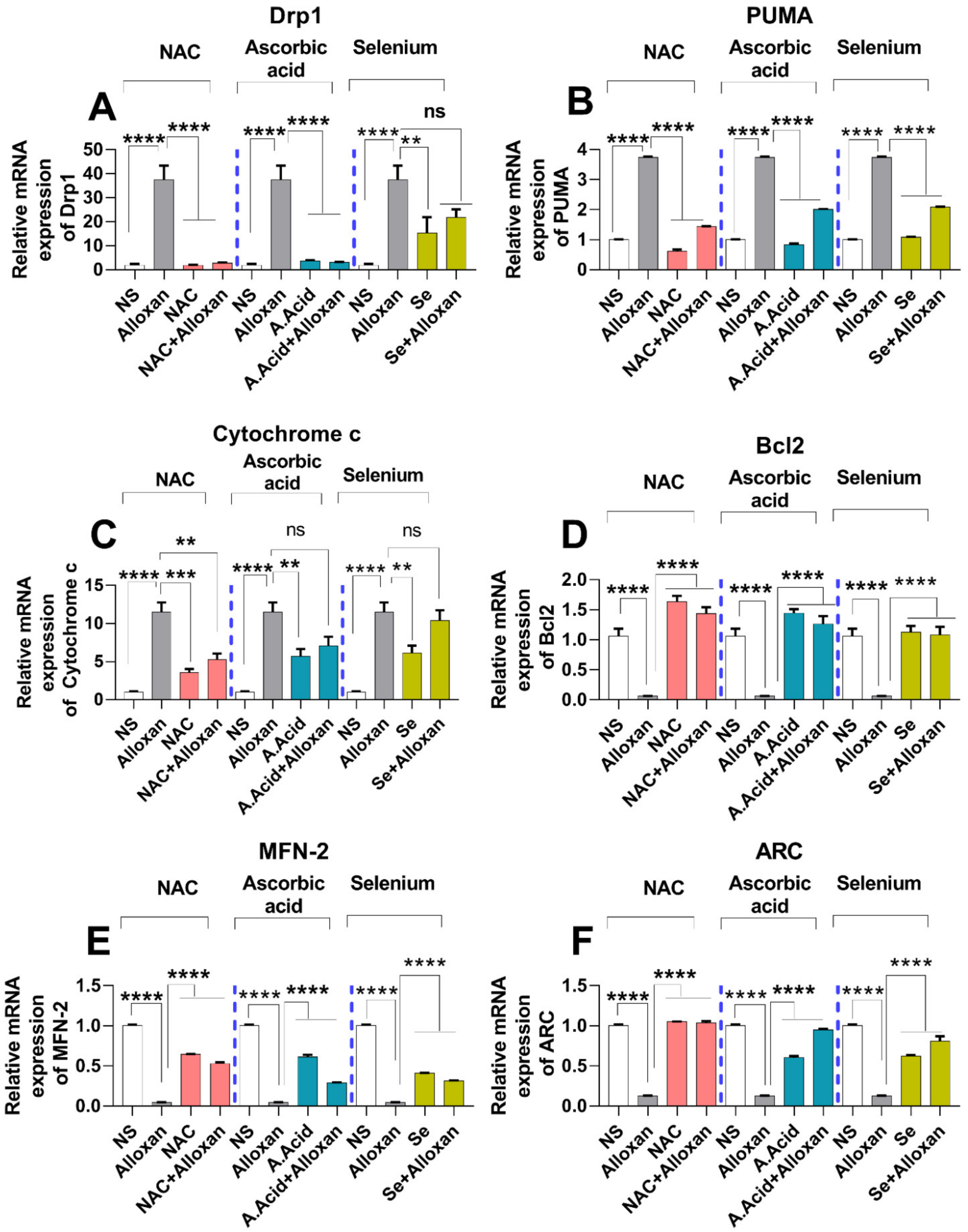
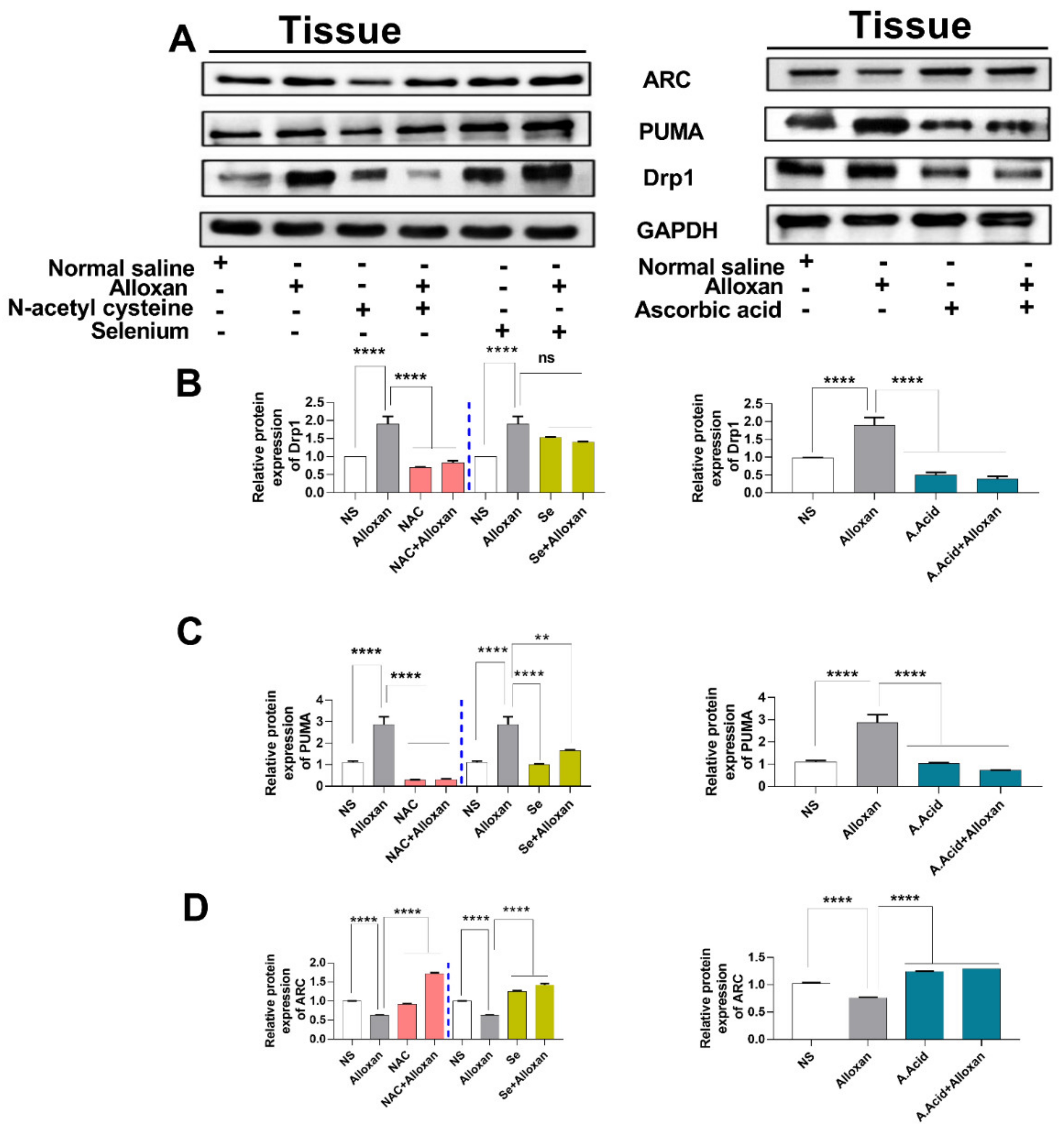
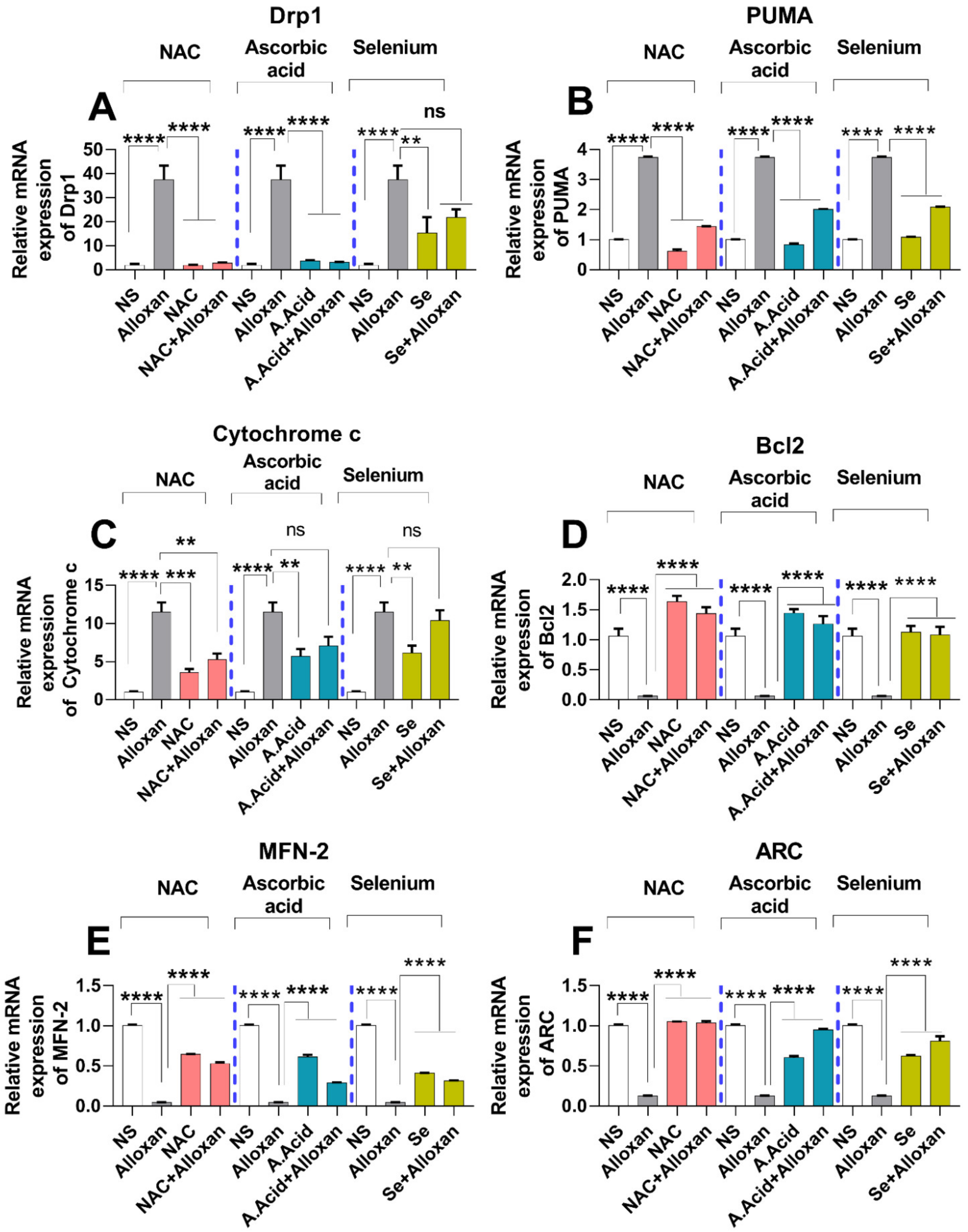
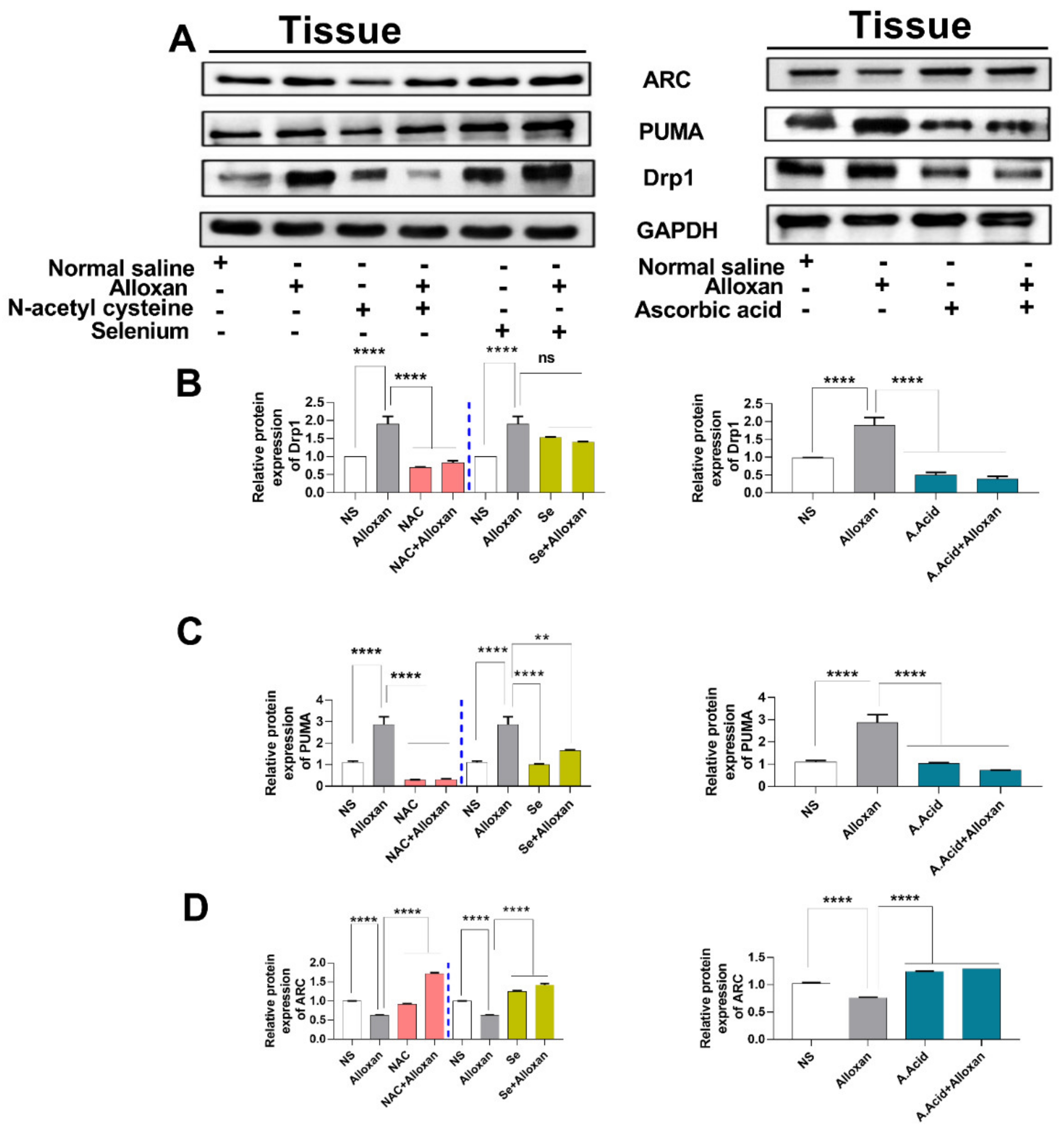
2.11. Expression of Drp1, PUMA, and cytochrome C Is Increased in Diabetic Induced Cardiac Pathology Model
2.12. Anti-Apoptotic Markers Bcl-2, MFN-2, and ARC Are Downregulated in Diabetic Cardiac Hypertrophy
2.13. Correlation between Diabetic, Oxidative, and Hypertrophic Parameters
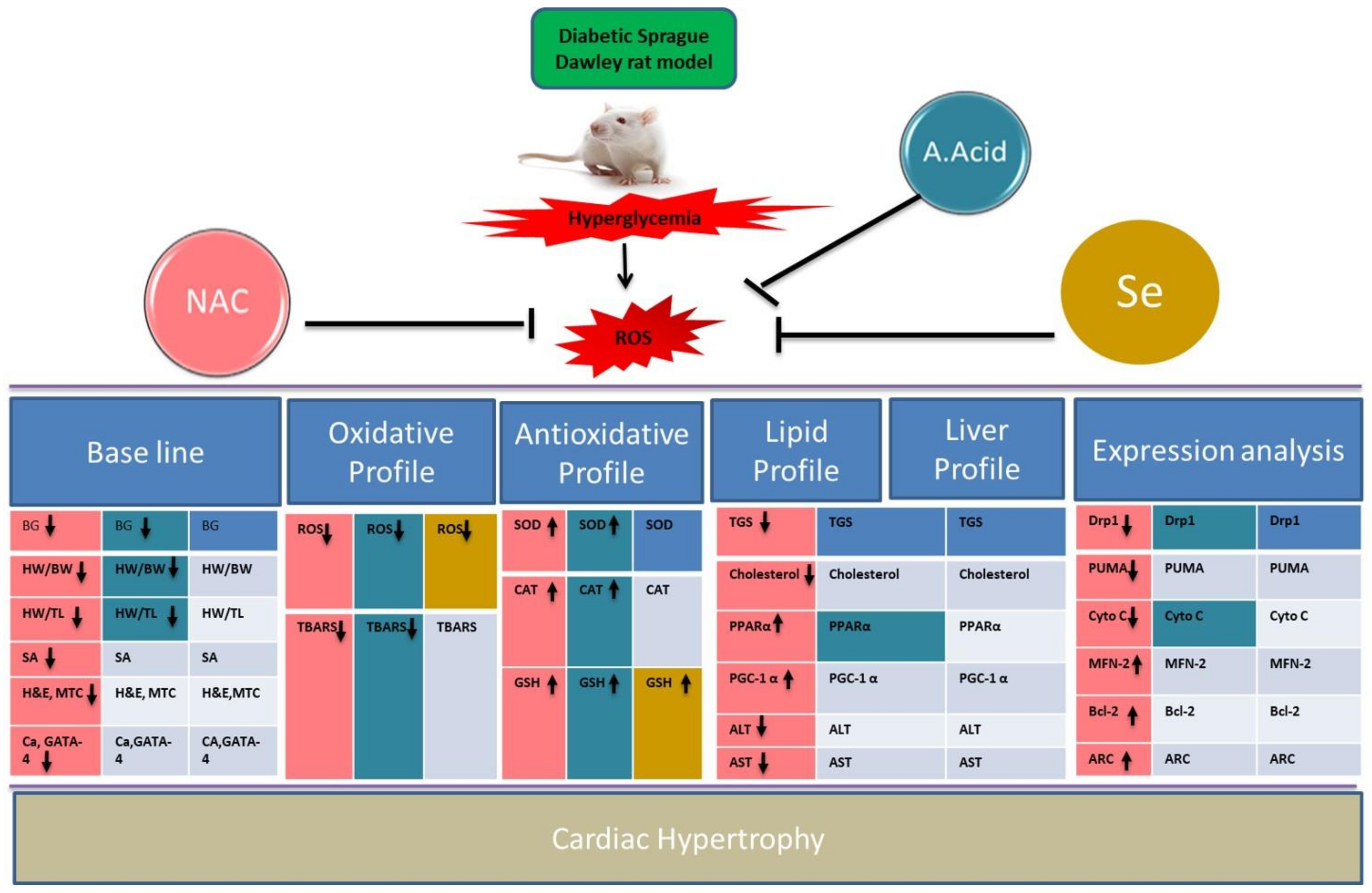
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Study Design
4.3. Measurement of Blood Glucose Level
4.4. Serum Separation from Blood
4.5. Tissue Homogenization
4.6. Reactive Oxygen Species (ROS) Assay
4.7. Thiobarbituric Acid Reactive Substances (TBARS) Assay
4.8. Superoxide Dismutase (SOD) Assay
4.9. Catalase Activity (CAT) Assay
4.10. Reduced Glutathione (GSH) Assay
4.11. Lipid Profiling
4.12. Calcium Test
4.13. Liver Profiling
4.13.1. Alanine aminotransferase (ALT) assay
4.13.2. Aspartate Aminotransferase (AST) Assay
4.14. Western Blot Analysis
4.15. Histological Analysis
4.16. Real-Time PCR Analysis
4.17. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| ROS | Reactive Oxygen Species |
| TBARS | Thiobarbituric Acid Reactive Substances |
| SOD | Superoxide Dismutase assay |
| CAT | Catalase |
| ALT | Alanine Aminotransferase Assay |
| AST | Aspartate Aminotransferase Assay |
| Drp1 | Drp1 (dynamin-related guanosine triphosphatase (GTPase) protein 1) |
| PUMA | p53 upregulated modulator of apoptosis |
| GATA-4 | GATA binding protein 4 |
| MFN-2 | Mitofusin-2 |
| ARC | Apoptosis repressor with caspase recruitment domain |
| NAC | N-acetyl Cysteine |
| PPARα | peroxisome proliferator-activated receptors (PPARs) alpha |
| FIS1 | Mitochondrial fission 1 protein |
| PDK4 | Pyruvate dehydrogenase lipoamide kinase isozyme 4 |
| ACC | acetyl-Co-A carboxylase |
| CPT1 | carnitine palmitoyltransferase I |
| SERCA2a | Sarcoplasmic reticulum calcium pumps |
| AMPK | AMP-dependent protein kinase |
| AGEs | advanced glycation end products |
| BG | Blood glucose |
| SA | surface area |
| TGS | Triglyceride |
References
- Ojiako, O.A.; Chikezie, P.C.; Ogbuji, A.C. Blood glucose level and lipid profile of alloxan-induced hyperglycemic rats treated with single and combinatorial herbal formulations. J. Tradit. Complement. Med. 2015, 6, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Feingold, K.R.; Brinton, E.A.; Grunfeld, C. The effect of endocrine disorders on lipids and lipoproteins. Endotext 2020, 2000. [Google Scholar]
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef]
- Jia, G.; Whaley-Connell, A.; Sowers, J.R. Diabetic cardiomyopathy: A hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia 2017, 61, 21–28. [Google Scholar] [CrossRef]
- Barzilay, J.I.; Kronmal, R.A.; Gottdiener, J.S.; Smith, N.L.; Burke, G.L.; Tracy, R.; Savage, P.J.; Carlson, M. The association of fasting glucose levels with congestive heart failure in diabetic adults ≥65 years: The Cardiovascular Health Study. J. Am. Coll. Cardiol. 2004, 43, 2236–2241. [Google Scholar] [CrossRef] [Green Version]
- Mushtaq, S.; Ali, T.; Altaf, F.; Abdullah, M.; Murtaza, I. Stress-responsive factor regulation in patients suffering from type 2 diabetes and myocardial infarction. Turk. J. Med. Sci. 2015, 45, 148–152. [Google Scholar] [CrossRef]
- Lee, T.-W.; Bai, K.-J.; Chao, T.-F.; Kao, Y.-H.; Chen, Y.-J. PPARs modulate cardiac metabolism and mitochondrial function in diabetes. J. Biomed. Sci. 2017, 24, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Thirupathi, A.; De Souza, C.T. Biochemistry, Multi-regulatory network of ROS: The interconnection of ROS, PGC-1 alpha, and AMPK-SIRT1 during exercise. J. Physiol. Biochem. 2017, 73, 487–494. [Google Scholar] [CrossRef]
- Boudina, S.; Abel, E.D. Diabetic Cardiomyopathy Revisited. Circulation 2007, 115, 3213–3223. [Google Scholar] [CrossRef]
- Mushtaq, S.; Ali, T.; Gul, M.; Javed, Q.; Emanueli, C.; Murtaza, I. Insulin over expression induces heart abnormalities via reactive oxygen species regulation, might be step towards cardiac hypertrophy. Cell. Mol. Boil. 2015, 61, 30–35. [Google Scholar]
- Ali, T.; Waheed, H.; Shaheen, F.; Mahmud, M.; Javed, Q.; Murtaza, I. Increased endogenous serotonin level in diabetic conditions may lead to cardiac valvulopathy via reactive oxygen species regulation. Biologia 2015, 70, 273–278. [Google Scholar] [CrossRef]
- Jan, M.I.; Khan, R.A.; Ali, T.; Bilal, M.; Bo, L.; Sajid, A.; Malik, A.; Urehman, N.; Waseem, N.; Nawab, J.; et al. Interplay of mitochondria apoptosis regulatory factors and microRNAs in valvular heart disease. Arch. Biochem. Biophys. 2017, 633, 50–57. [Google Scholar] [CrossRef]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2019, 17, 170–194. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Dludla, P.V.; Dias, S.; Obonye, N.; Johnson, R.; Louw, J.; Nkambule, B.B. A Systematic Review on the Protective Effect of N-Acetyl Cysteine Against Diabetes-Associated Cardiovascular Complications. Am. J. Cardiovasc. Drugs 2018, 18, 283–298. [Google Scholar] [CrossRef]
- Wang, T.; Qiao, S.; Lei, S.; Liu, Y.; Ng, K.F.J.; Xu, A.; Lam, K.S.L.; Irwin, M.G.; Xia, Z. N-Acetylcysteine and Allopurinol Synergistically Enhance Cardiac Adiponectin Content and Reduce Myocardial Reperfusion Injury in Diabetic Rats. PLoS ONE 2011, 6, e23967. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Otani, H.; Shimazu, T.; Yoshioka, K.; Fujita, M.; Iwasaka, T. Ascorbic acid and N-acetyl cysteine prevent uncoupling of nitric oxide synthase and increase tolerance to ischemia/reperfusion injury in diabetic rat heart. Free. Radic. Res. 2011, 45, 1173–1183. [Google Scholar] [CrossRef]
- Kumar, S.; Sitasawad, S.L. N-acetylcysteine prevents glucose/glucose oxidase-induced oxidative stress, mitochondrial damage and apoptosis in H9c2 cells. Life Sci. 2009, 84, 328–336. [Google Scholar] [CrossRef]
- Xia, Z.; Guo, Z.; Nagareddy, P.; Yuen, V.; Yeung, E.; McNeill, J.H. Antioxidant N-acetylcysteine restores myocardial Mn-SOD activity and attenuates myocardial dysfunction in diabetic rats. Eur. J. Pharmacol. 2006, 544, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Sangweni, N.F.; Mabhida, S.E.; Dludla, P.V.; Mabasa, L.; Riedel, S.; Chapman, C.; Mosa, R.A.; Kappo, A.P.; Louw, J.; et al. An in vitro study on the combination effect of metformin and N-acetyl cysteine against hyperglycae-mia-induced cardiac damage. Nutrients 2019, 11, 2850. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Kuo, K.-H.; Nagareddy, P.; Wang, F.; Guo, Z.; Guo, T.; Jiang, J.; McNeill, J.H. N-acetylcysteine attenuates PKCβ2overexpression and myocardial hypertrophy in streptozotocin-induced diabetic rats. Cardiovasc. Res. 2007, 73, 770–782. [Google Scholar] [CrossRef] [Green Version]
- Fiordaliso, F.; Bianchi, R.; Staszewsky, L.; Cuccovillo, I.; Doni, M.; Laragione, T.; Salio, M.; Savino, C.; Melucci, S.; Santangelo, F. Antioxidant treatment attenuates hyperglycemia-induced cardiomyocyte death in rats. J. Mol. Cell. Cardiol. 2004, 37, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Mushtaq, I.; Maryam, S.; Farhan, A.; Saba, K.; Jan, M.I.; Sultan, A.; Anees, M.; Duygu, B.; Hamera, S.; et al. Interplay of N acetyl cysteine and melatonin in regulating oxidative stress-induced cardiac hypertrophic factors and microRNAs. Arch. Biochem. Biophys. 2018, 661, 56–65. [Google Scholar] [CrossRef]
- Mushtaq, S.; Ali, T.; Javed, Q.; Tabassum, S.; Murtaza, I. N-acetyl cysteine inhibits endothelin-1-induced ROS de-pendent cardiac hypertrophy through superoxide dismutase regulation. Cell J. 2015, 17, 355. [Google Scholar] [PubMed]
- Scalzo, R.L.; Bauer, T.A.; Harrall, K.; Moreau, K.; Ozemek, C.; Herlache, L.; McMillin, S.; Huebschmann, A.G.; Dorosz, J.; Reusch, J.E.B.; et al. Acute vitamin C improves cardiac function, not exercise capacity, in adults with type 2 diabetes. Diabetol. Metab. Syndr. 2018, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aluwong, T.; Ayo, J.O.; Kpukple, A.; Oladipo, O.O. Amelioration of Hyperglycaemia, Oxidative Stress and Dyslipidaemia in Alloxan-Induced Diabetic Wistar Rats Treated with Probiotic and Vitamin C. Nutrients 2016, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdi, Y.; Hornick, J.-L.; Istasse, L.; Dufrasne, I. Selenium in the Environment, Metabolism and Involvement in Body Functions. Molecules 2013, 18, 3292–3311. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Chen, S.; Liu, Z.; Cheng, C.; Li, H.; Wang, M. Toxicity of selenium nanoparticles in male Sprague–Dawley rats at supranutritional and nonlethal levels. Life Sci. 2014, 115, 44–51. [Google Scholar] [CrossRef]
- Żarczyńska, K.; Sobiech, P.; Tobolski, D.; Mee, J.F.; Illek, J. Effect of a single, oral administration of selenitetriglycerides, at two dose rates, on blood selenium status and haematological and biochemical parameters in Holstein-Friesian calves. Ir. Vet. J. 2021, 74, 1–9. [Google Scholar] [CrossRef]
- Takada, H.; Hirooka, T.; Hatano, T.; Hamada, Y.; Yamamoto, M. Inhibition of 7, 12-dimethylbenz [a] anthracene-induced lipid peroxidation and mammary tumor development in rats by vitamin E in conjunction with selenium. Nutr. Cancer 1992, 17, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.R.; Burke, N.; Dongworth, R.K.; Hausenloy, D.J. Mitochondrial fusion and fission proteins: Novel therapeutic targets for combating cardiovascular disease. Br. J. Pharmacol. 2014, 171, 1890–1906. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res. 2010, 88, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Pasula, D.J.; Shi, R.; Shih, A.Z.; Luciani, D.S.; Shi, R. Anti-Apoptotic Bcl-xL Limits Mitochondrial Dysregulation in β-cells during Prolonged Exposure to High Glucose. Can. J. Diabetes 2018, 42, S54. [Google Scholar] [CrossRef]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef]
- Loo, J.F.; Lau, P.; Ho, H.; Kong, S. An aptamer-based bio-barcode assay with isothermal recombinase polymerase amplification for cytochrome-c detection and anti-cancer drug screening. Talanta 2013, 115, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, N.J.; Trapani, J.A. A new quantitative assay for cytochrome c release in apoptotic cells. Cell Death Differ. 2003, 10, 853–855. [Google Scholar] [CrossRef] [Green Version]
- Chandra, D.; Liu, J.-W.; Tang, D.; Canela, N.; Rodriguez-Vilarrupla, A.; Estanyol, J.M.; Dıaz, C.; Pujol, M.J.; Agell, N.; Bachs, O. Early Mitochondrial Activation and Cytochrome c Up-regulation during Apoptosis. J. Biol. Chem. 2002, 277, 50842–50854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giunti, S.; Bruno, G.; Veglio, M.; Gruden, G.; Webb, D.J.; Livingstone, S.; Chaturvedi, N.; Fuller, J.H.; Perin, P.C.; The Eurodiab Iddm Complications Study Group. Electrocardiographic Left Ventricular Hypertrophy in Type 1 Diabetes: Prevalence and relation to coronary heart disease and cardiovascular risk factors: The Eurodiab IDDM Complications Study. Diabetes Care 2005, 28, 2255–2257. [Google Scholar] [CrossRef] [Green Version]
- Novoa, U.; Arauna, D.; Moran, M.; Nuñez, M.; Zagmutt, S.; Saldivia, S.; Valdes, C.; Villaseñor, J.; Zambrano, C.G.; Gonzalez, D.R. High-intensity exercise reduces cardiac fibrosis and hypertrophy but does not restore the nitroso-redox imbalance in diabetic cardiomyopathy. Oxid. Med. Cell. Longev. 2017, 2017, 7921363. [Google Scholar] [CrossRef] [Green Version]
- Whitman, V.; Schuler, H.G.; Neely, J.R. Effect of alloxan-induced diabetes on the hypertrophic response of rat heart. J. Mol. Cell. Cardiol. 1979, 11, 1275–1281. [Google Scholar] [CrossRef]
- Mushtaq, I.; Mushtaq, I.; Akhter, Z.; Murtaza, I.; Qamar, S.; Ayub, S.; Mirza, B.; Butt, T.M.; Janjua, N.K.; Shah, F.U.; et al. Engineering electroactive and biocompatible tetra(aniline)-based terpolymers with tunable intrinsic antioxidant properties in vivo. Mater. Sci. Eng. C 2019, 108, 110456. [Google Scholar] [CrossRef]
- Bisping, E.; Ikeda, S.; Kong, S.W.; Tarnavski, O.; Bodyak, N.; McMullen, J.R.; Rajagopal, S.; Son, J.K.; Ma, Q.; Springer, Z.; et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 14471–14476. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Buja, L.M.; McMillin, J.B. Activation of the cytochrome c gene by electrical stimulation in neonatal rat cardiac myocytes: Role of NRF-1 and c-Jun. J. Biol. Chem. 1998, 273, 12593–12598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, G.; DeMarco, V.; Sowers, J.R. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat. Rev. Endocrinol. 2015, 12, 144–153. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid Metabolism and Toxicity in the Heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian Target of Rapamycin Signaling in Cardiac Physiology and Disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Perino, A.; Ghigo, A.; Hirsch, E.; Shah, A.M. NADPH Oxidases in Heart Failure: Poachers or Gamekeepers? Antioxid. Redox Signal. 2013, 18, 1024–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proud, C.G. Ras, PI3-kinase and mTOR signaling in cardiac hypertrophy. Cardiovasc. Res. 2004, 63, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malhowski, A.J.; Hira, H.; Bashiruddin, S.; Warburton, R.; Goto, J.; Robert, B.; Kwiatkowski, D.J.; Finlay, G.A. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum. Mol. Genet. 2011, 20, 1290–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Cesario, D.A.; Brar, R.; Shivkumar, K. Alterations in Ion Channel Physiology in Diabetic Cardiomyopathy. Endocrinol. Metab. Clin. N. Am. 2006, 35, 601–610. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Adeosun, A.M.; Akinloye, O.A. Alloxan-induced diabetes, a common model for evaluating the glycemic-control potential of therapeutic compounds and plants extracts in experimental studies. Medicina 2017, 53, 365–374. [Google Scholar] [CrossRef]
- Al-Rasheed, N.M.; Al-Rasheed, N.M.; Hasan, I.; Al-Amin, M.A.; Al-Ajmi, H.N.; Mohamad, R.A.; Mahmoud, A.M. Simvastatin Ameliorates Diabetic Cardiomyopathy by Attenuating Oxidative Stress and Inflammation in Rats. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef]
- Zhao, X.-Y.; Hu, S.-J.; Li, J.; Mou, Y.; Chen, B.-P.; Xia, Q. Decreased cardiac sarcoplasmic reticulum Ca2+-ATPase activity contributes to cardiac dysfunction in streptozotocin-induced diabetic rats. J. Physiol. Biochem. 2006, 62, 1–8. [Google Scholar] [CrossRef]
- Endoh, M. Signal Transduction and Ca2+ Signaling in Intact Myocardium. J. Pharmacol. Sci. 2006, 100, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, K.M.; Zhong, Y.; Hoit, B.D.; Grupp, I.L.; Hahn, H.; Dilly, K.W.; Guatimosim, S.; Lederer, W.J.; Matlib, M.A. Defective intracellular Ca2+ signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am. J. Physiol. Circ. Physiol. 2002, 283, H1398–H1408. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gómez, A.M. Mechanisms of [Ca2+] i Transient Decrease in Cardiomyopathy of db/db Type 2 Diabetic Mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, Y.; Matsuda, N.; Kimura, J.; Ishitani, T.; Tamada, A.; Gando, S.; Kemmotsu, O.; Kanno, M. Diminished function and expression of the cardiac Na+-Ca2+exchanger in diabetic rats: Implication in Ca2+overload. J. Physiol. 2000, 527, 85–94. [Google Scholar] [CrossRef]
- Kashihara, H.; Shi, Z.Q.; Yu, J.Z.; McNeill, J.H.; Tibbits, G.F. Effects of diabetes and hyper-tension on myocardial Na+-Ca2+ exchange. Can. J. Physiol. Pharmacol. 1999, 78, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Belke, D.D.; Swanson, E.A.; Dillmann, W.H. Decreased Sarcoplasmic Reticulum Activity and Contractility in Diabetic db/db Mouse Heart. Diabetes 2004, 53, 3201–3208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jweied, E.E.; McKinney, R.D.; Walker, L.A.; Brodsky, I.; Geha, A.S.; Massad, M.G.; Buttrick, P.M.; De Tombe, P.P. Depressed cardiac myofilament function in human diabetes mellitus. Am. J. Physiol. Circ. Physiol. 2005, 289, H2478–H2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trost, S.U.; Belke, D.D.; Bluhm, W.F.; Meyer, M.; Swanson, E.; Dillmann, W.H. Overexpression of the Sarcoplasmic Reticulum Ca2+-ATPase Improves Myocardial Contractility in Diabetic Cardiomyopathy. Diabetes 2002, 51, 1166–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, D.L.; Brooks, C.L. Tailoring the structural integrity process to meet the challenges of aging aircraft. Int. J. Fatigue 1999, 21, 1–14. [Google Scholar] [CrossRef]
- Sen, L.; Cui, G.; Fonarow, G.; Laks, H. Differences in mechanisms of SR dysfunction in ischemic vs. idiopathic dilated cardiomyopathy. Am. J. Physiol. Circ. Physiol. 2000, 279, H709–H718. [Google Scholar] [CrossRef]
- Lopaschuk, G.D. Metabolic Abnormalities in the Diabetic Heart. Heart Fail. Rev. 2002, 7, 149–159. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; McNulty, P.; Young, M.E. Adaptation and maladaptation of the heart in diabetes: Part I: General concepts. Circulation 2002, 105, 1727–1733. [Google Scholar] [CrossRef] [Green Version]
- Stanley, W.C.; Lopaschuk, G.D.; McCormack, J.G. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc. Res. 1997, 34, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Carley, A.; Severson, D.L. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2005, 1734, 112–126. [Google Scholar] [CrossRef]
- Frazier-Wood, A.; Ordovas, J.; Straka, R.; Hixson, J.; Borecki, I.; Tiwari, H.; Arnett, D. The PPAR alpha gene is associated with triglyceride, low-density cholesterol and inflammation marker response to fenofibrate intervention: The GOLDN study. Pharmacogenomics J. 2012, 13, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-I.; Kao, Y.-H.; Chen, Y.-C.; Huang, J.-H.; Hsiao, F.-C.; Chen, Y.-J. Peroxisome prolifera-tor-activated receptors modulate cardiac dysfunction in diabetic cardiomyopathy. Diabetes Res. Clin. Pract. 2013, 100, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.P.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Dobbins, R.L.; Metzger, G.; Sartoni-D’Ambrosia, G.; Arbique, D.; Vongpatanasin, W.; Unger, R.; Victor, R.G. Myocardial triglycerides and systolic function in humans: In vivo evaluation by localized proton spectroscopy and cardiac imaging. Magn. Reson. Med. 2003, 49, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-T.; Grayburn, P.; Karim, A.; Shimabukuro, M.; Higa, M.; Baetens, D.; Orci, L.; Unger, R.H. Lipotoxic heart disease in obese rats: Implications for human obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 1784–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puddu, G.M.; Cravero, E.; Arnone, G.; Muscari, A.; Puddu, P. Molecular aspects of atherogenesis: New insights and unsolved questions. J. Biomed. Sci. 2005, 12, 839–853. [Google Scholar] [CrossRef] [PubMed]
- Finck, B.N.; Chinetti, G.; Staels, B. PPARs/RXRs in Cardiovascular Physiology and Disease. PPAR Res. 2008, 2008, 1. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef]
- Boye, A.; Barku, V.Y.A.; Acheampong, D.O.; Ofori, E.G. Abrus precatorius Leaf Extract Reverses Allox-an/Nicotinamide-Induced Diabetes Mellitus in Rats through Hormonal (Insulin, GLP-1, and Glucagon) and Enzymatic (α-Amylase/α-Glucosidase) Modulation. BioMed Res. Int. 2021, 2021. [Google Scholar] [CrossRef]
- Naz, I.; Khan, M.R.; Zai, J.A.; Batool, R.; Maryam, S.; Majid, M. Indigofera linifolia ameliorated CCl4 induced endoplasmic reticulum stress in liver of rat. J. Ethnopharmacol. 2021, 114826. [Google Scholar] [CrossRef]
- Ali, T.; Ishtiaq, A.; Mushtaq, I.; Ayaz, N.; Jan, M.I.; Khan, W.; Khan, U.; Murtaza, I. Mentha longifolia Alleviates Exog-enous Serotonin-Induced Diabetic Hypoglycemia and Relieves Renal Toxicity via ROS Regulation. Plant Foods Hum. Nutr. 2021, 76, 1–6. [Google Scholar] [CrossRef]
- Ebuehi, O.; Ajuluchukwu, A.; Afolabi, O.; Akinwande, A. Oxidative stress in alloxan-induced diabetes in female and male rats. Adv. Med Dent. Sci. 2010, 3, 71–75. [Google Scholar]
- Surya, D.; Vijayakumar, R.S.; Nalini, N. Oxidative stress and the role of cumin (Cuminum cyminum Linn.) in allox-an-induced diabetic rats. J. Herbs Spices Med. Plants 2005, 11, 127–139. [Google Scholar] [CrossRef]
- Ma, H.; Shieh, K.J.J. Western blotting method. J. Am. Sci. 2006, 2, 23–27. [Google Scholar]
- Hayashi, I.; Morishita, Y.; Imai, K.; Nakamura, M.; Nakachi, K.; Hayashi, T. High-throughput spectrophotometric assay of reactive oxygen species in serum. Mutat. Res. Toxicol. Environ. Mutagen. 2007, 631, 55–61. [Google Scholar] [CrossRef]
- Buege, J.A.; Aust, S.D. Microsomal Lipid Peroxidation. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 1978; Volume 52, pp. 302–310. [Google Scholar]
- Jevremović, S.; Petrić, M.; Živković, S.; Trifunović, M.; Subotić, A. Superoxide dismutase activity and isoen-zyme profiles in bulbs of snake’s head fritillary in response to cold treatment. Arch. Biol. Sci. 2010, 62, 553–558. [Google Scholar] [CrossRef]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef]
- Schipke, J.; Brandenberger, C.; Rajces, A.; Manninger, M.; Alogna, A.; Post, H.; Mühlfeld, C. Assessment of cardiac fibrosis: A morphometric method comparison for collagen quantification. J. Appl. Physiol. 2017, 122, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Samra, G.; Ko, K.A.; Vu, H.T.; Thomas, T.N.; Luo, W.; Chang, J.; Reddy, A.K.; Fujiwara, K.; Abe, J.-I. Topoisomerase 2B Decrease Results in Diastolic Dysfunction via p53 and Akt: A Novel Pathway. Front. Cardiovasc. Med. 2020, 7, 594123. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Gene ID | Primer | Sequence | |
|---|---|---|---|---|
| Rattus norvegicus | PPARα | 25747 | FP | 5′-ATGAACAAAGACGGGATGC-3′ |
| RP | 5′-TCAAACTTGGGTTCCATGAT-3′ | |||
| GATA-4 | 54254 | FP | 5′-CCCATGGGTCCTCCATC-3′ | |
| RP | 5′-GGGGGTGCTGATTACG-3′ | |||
| DRP1 | 114114 | FP | 5′-GATGCCATAGTTGAAGTGGTGAC-3′ | |
| RP | 5′-CCACAAGCATCAGCAAAGTCTGG-3′ | |||
| PUMA | 317673 | FP | 5′-ACGACCTCAACGCACAGTACGA-3′ | |
| RP | 5′-CCTAATTGGGCTCCATCTCGGG-3′ | |||
| MFN-2 | 64476 | FP | 5-′TCAAGACCGTGAACCAGC-3′ | |
| RP | 5′-AGAAGTGGACACTTGGAGTTG-3′ | |||
| GAPDH | 24383 | FP | 5′-TTCAACAGCAACTCCCATT -3′ | |
| RP | 5′-CACCACCCTGTTGCTGTA-3′ | |||
| ARC | 85383 | FP | 5-TGCCAGGAGCTGCTACGCTGT-3 | |
| RP | 5-TGGGCATGGAGGGTCATAGCTG-3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mushtaq, I.; Bashir, Z.; Sarwar, M.; Arshad, M.; Ishtiaq, A.; Khan, W.; Khan, U.; Tabassum, S.; Ali, T.; Fatima, T.; et al. N-Acetyl Cysteine, Selenium, and Ascorbic Acid Rescue Diabetic Cardiac Hypertrophy via Mitochondrial-Associated Redox Regulators. Molecules 2021, 26, 7285. https://doi.org/10.3390/molecules26237285
Mushtaq I, Bashir Z, Sarwar M, Arshad M, Ishtiaq A, Khan W, Khan U, Tabassum S, Ali T, Fatima T, et al. N-Acetyl Cysteine, Selenium, and Ascorbic Acid Rescue Diabetic Cardiac Hypertrophy via Mitochondrial-Associated Redox Regulators. Molecules. 2021; 26(23):7285. https://doi.org/10.3390/molecules26237285
Chicago/Turabian StyleMushtaq, Iram, Zainab Bashir, Mehvish Sarwar, Maria Arshad, Ayesha Ishtiaq, Wajiha Khan, Uzma Khan, Sobia Tabassum, Tahir Ali, Tahzeeb Fatima, and et al. 2021. "N-Acetyl Cysteine, Selenium, and Ascorbic Acid Rescue Diabetic Cardiac Hypertrophy via Mitochondrial-Associated Redox Regulators" Molecules 26, no. 23: 7285. https://doi.org/10.3390/molecules26237285
APA StyleMushtaq, I., Bashir, Z., Sarwar, M., Arshad, M., Ishtiaq, A., Khan, W., Khan, U., Tabassum, S., Ali, T., Fatima, T., Valadi, H., Nawaz, M., & Murtaza, I. (2021). N-Acetyl Cysteine, Selenium, and Ascorbic Acid Rescue Diabetic Cardiac Hypertrophy via Mitochondrial-Associated Redox Regulators. Molecules, 26(23), 7285. https://doi.org/10.3390/molecules26237285