
Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst


{kind=link}
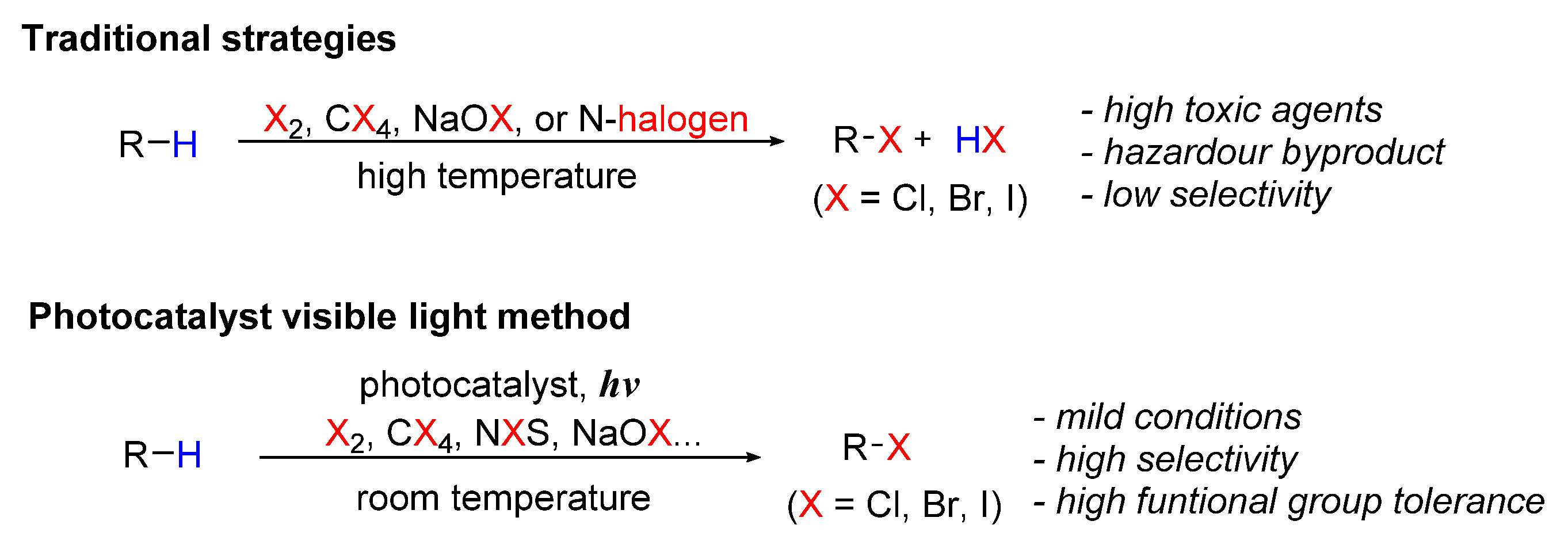{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
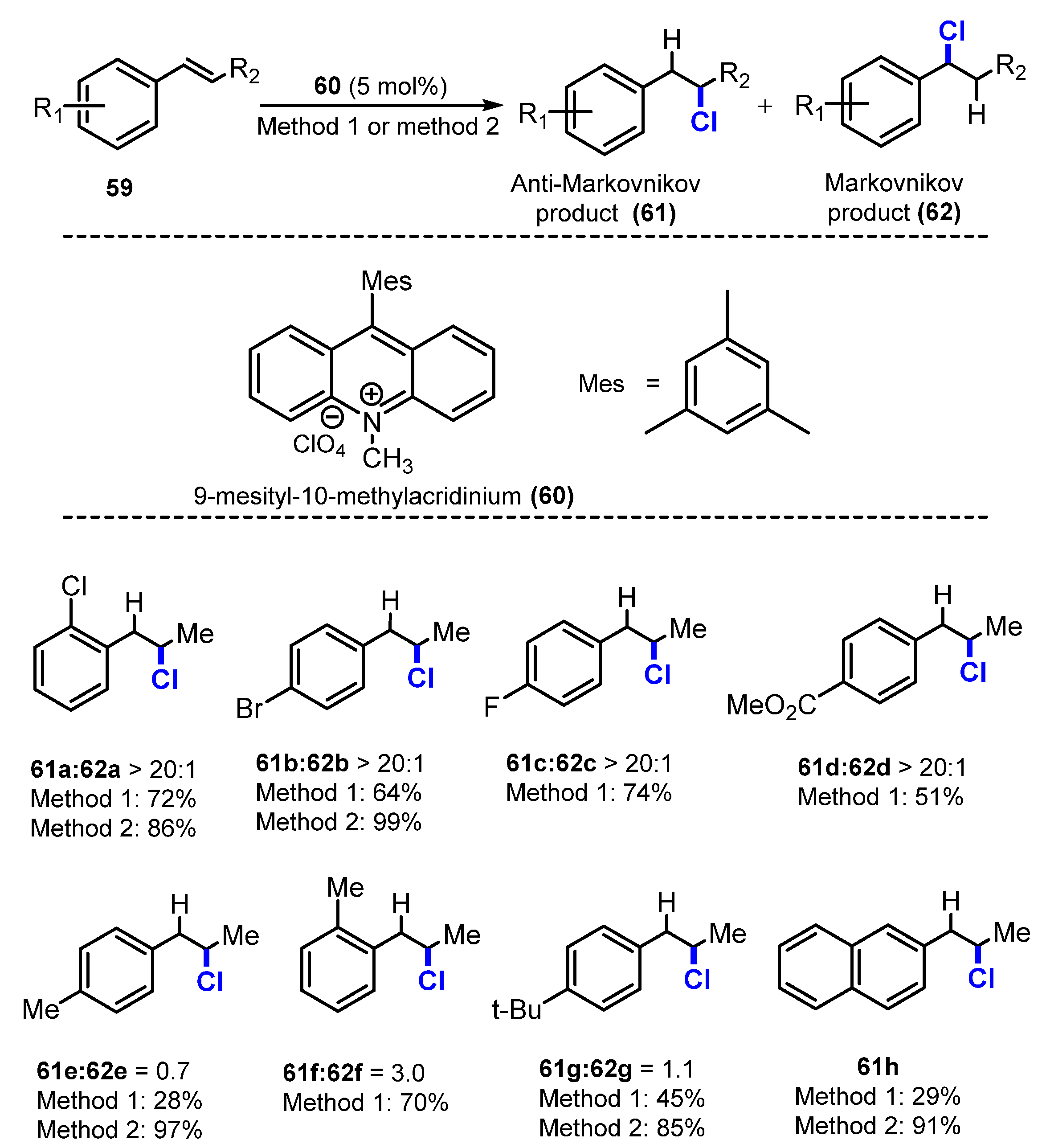{kind=link}
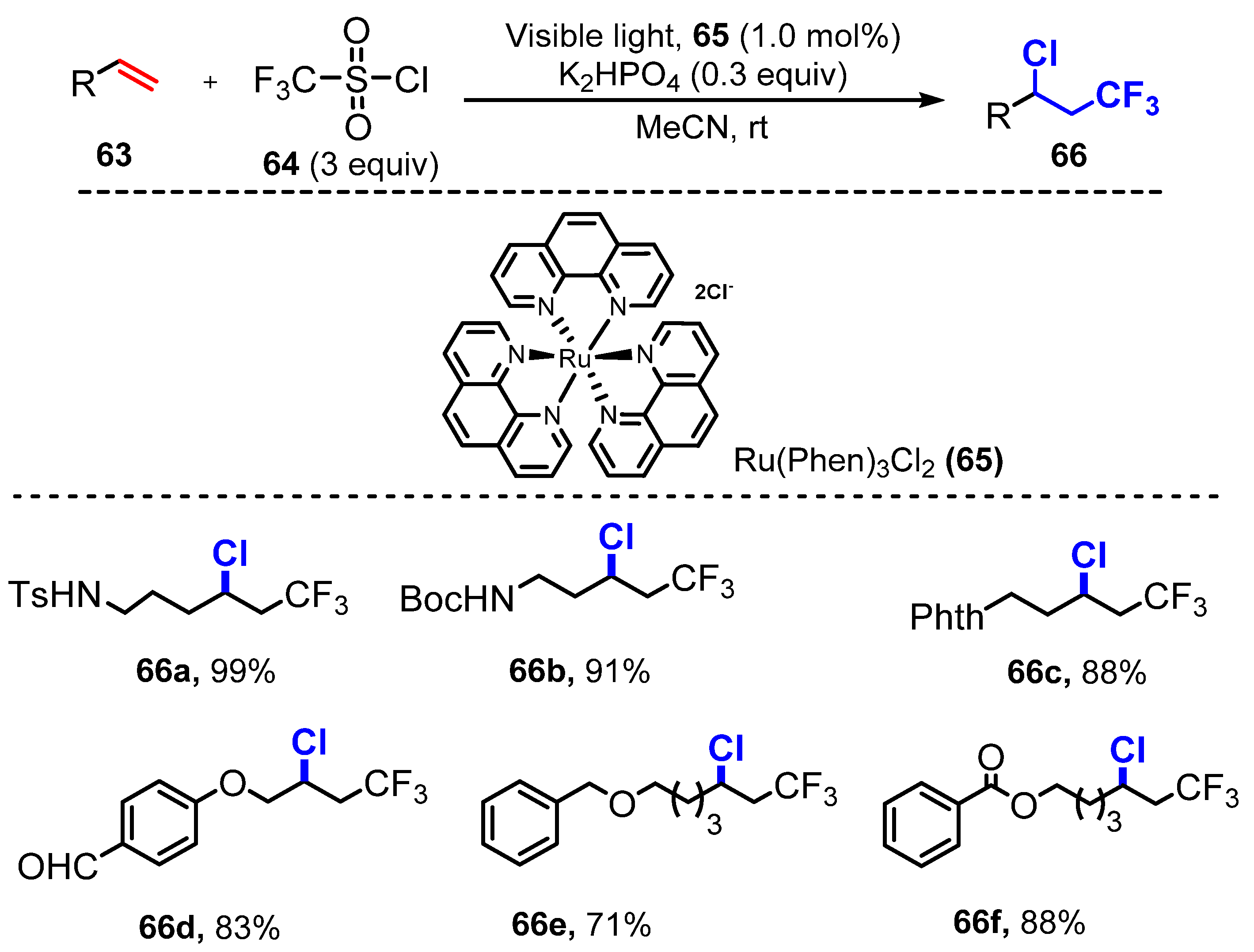{kind=link}
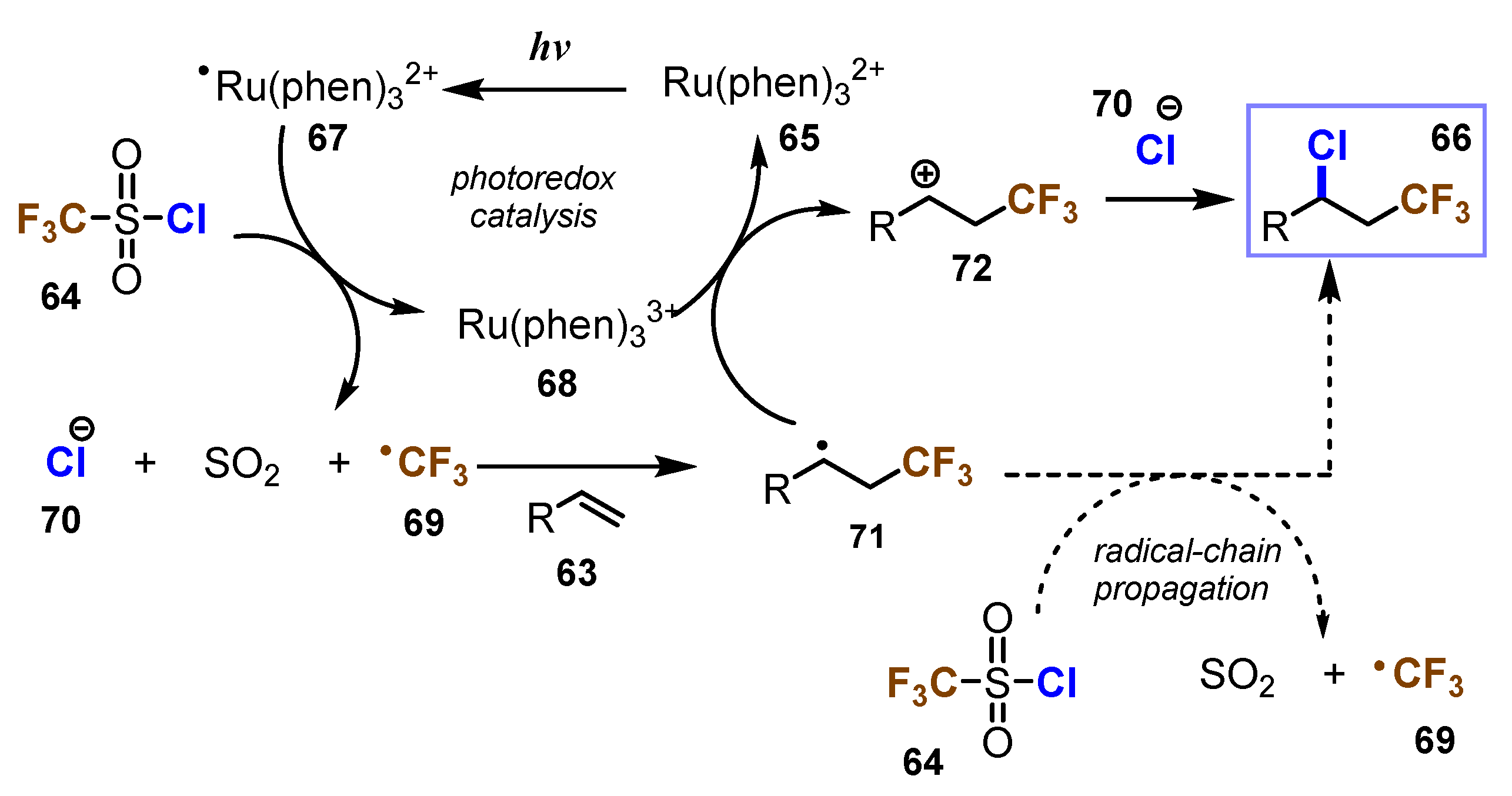{kind=link}
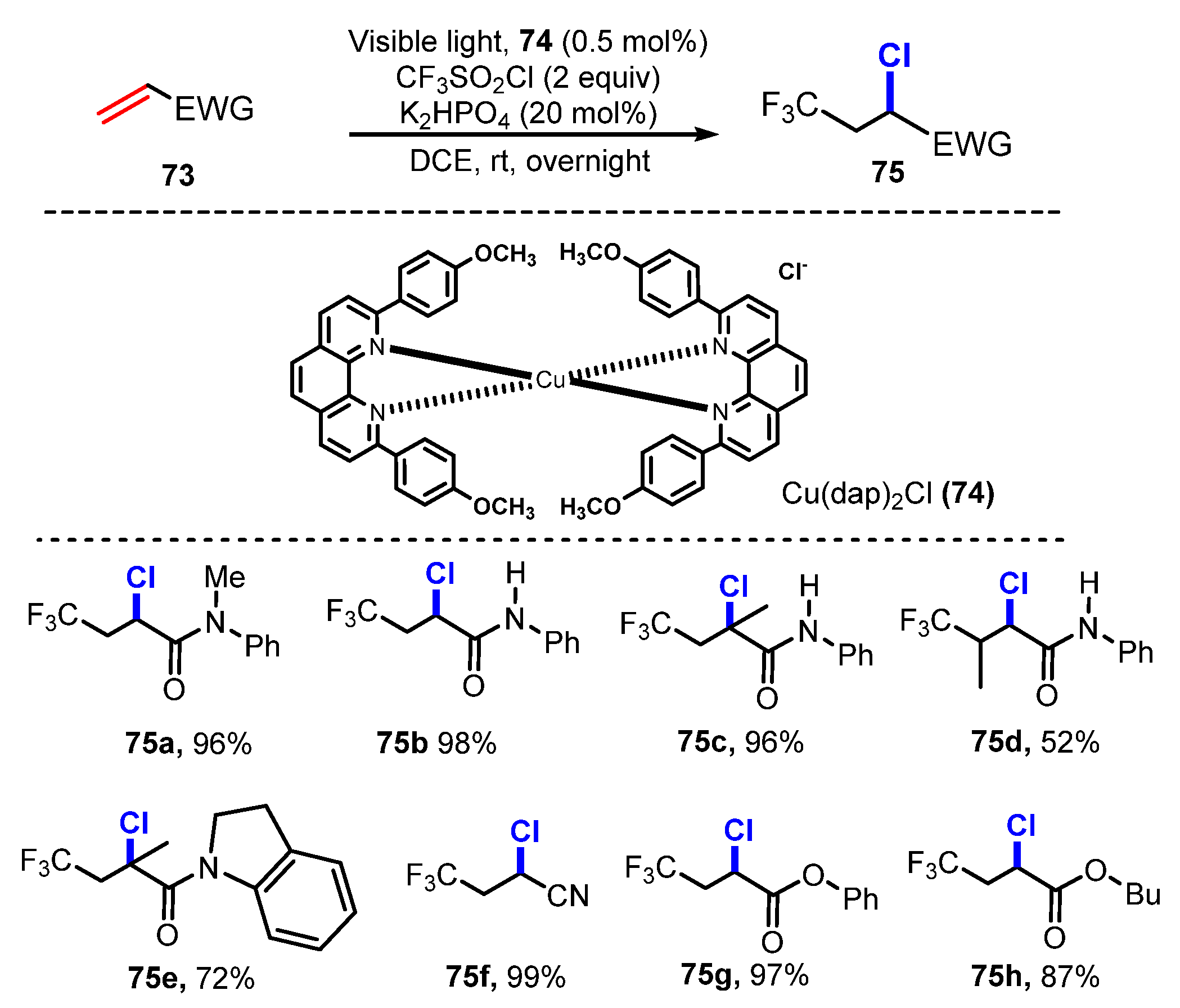{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
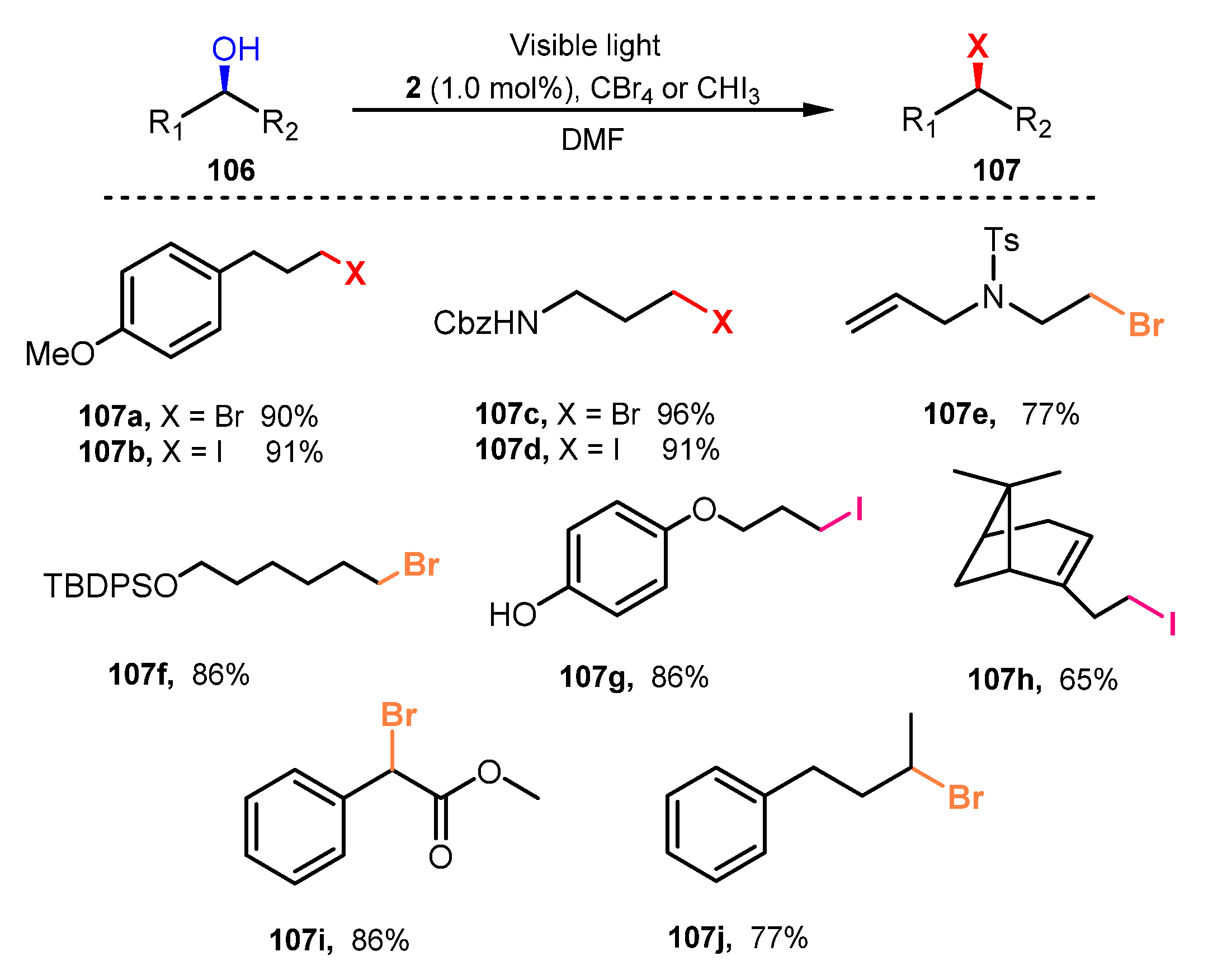{kind=link}
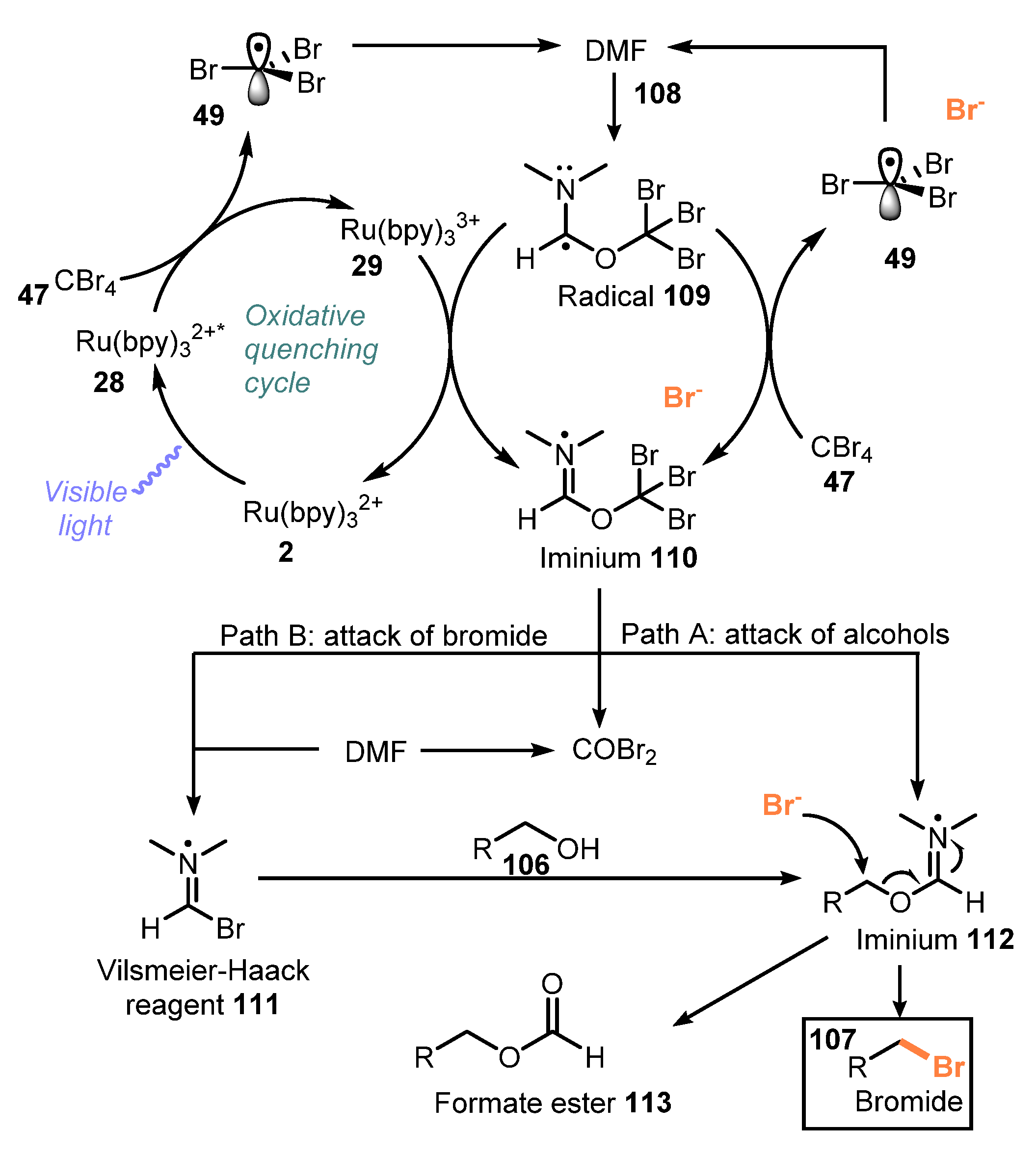{kind=link}
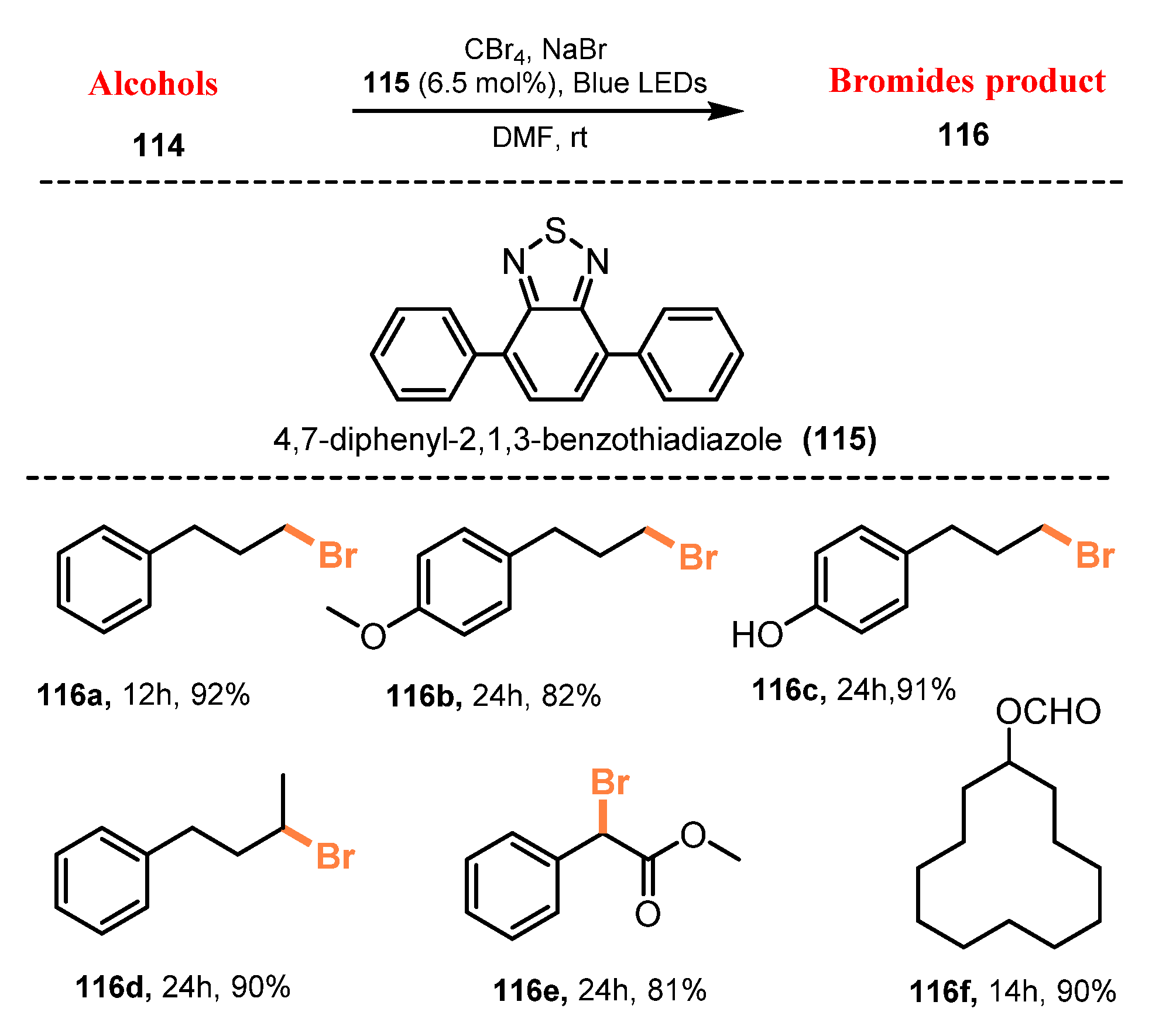{kind=link}
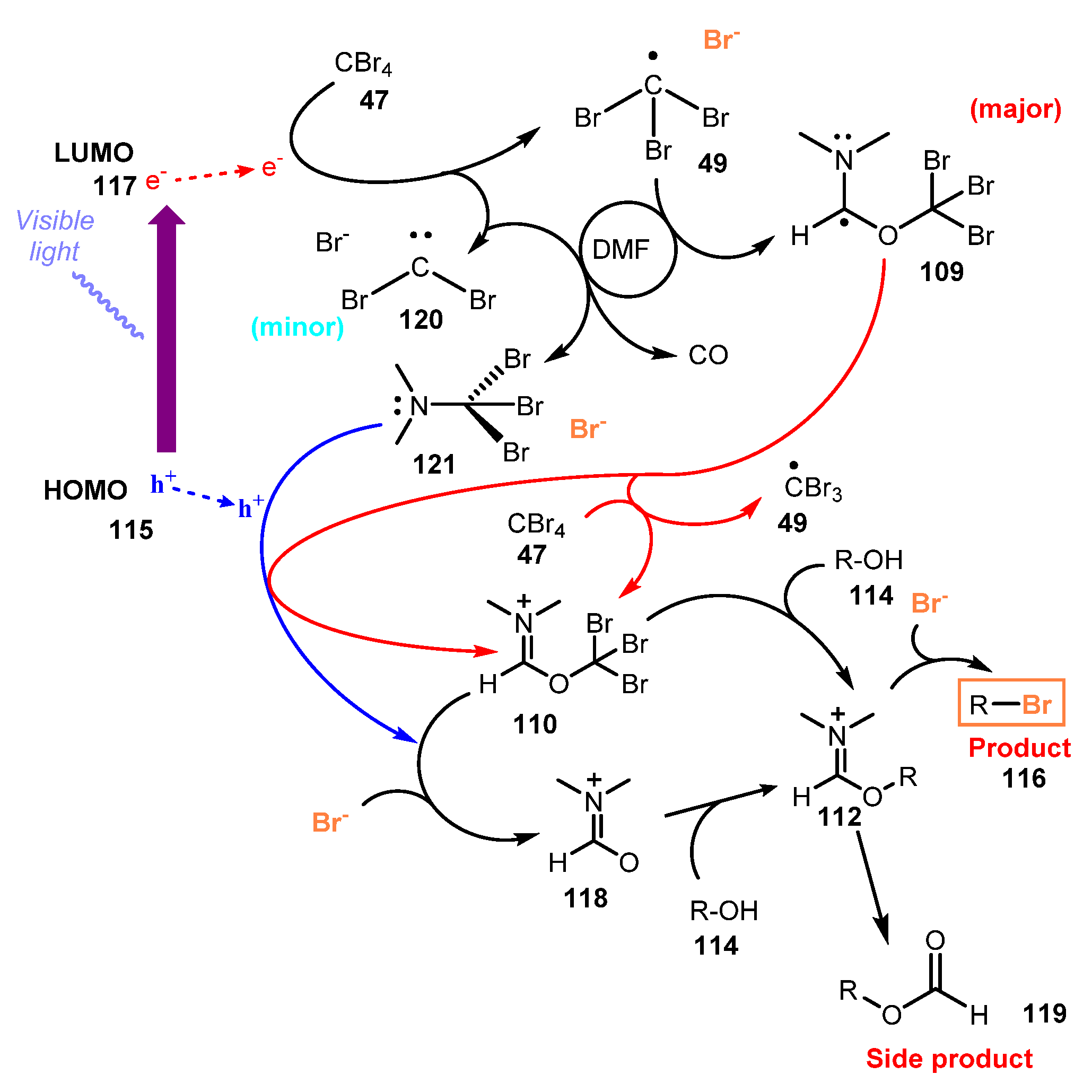{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
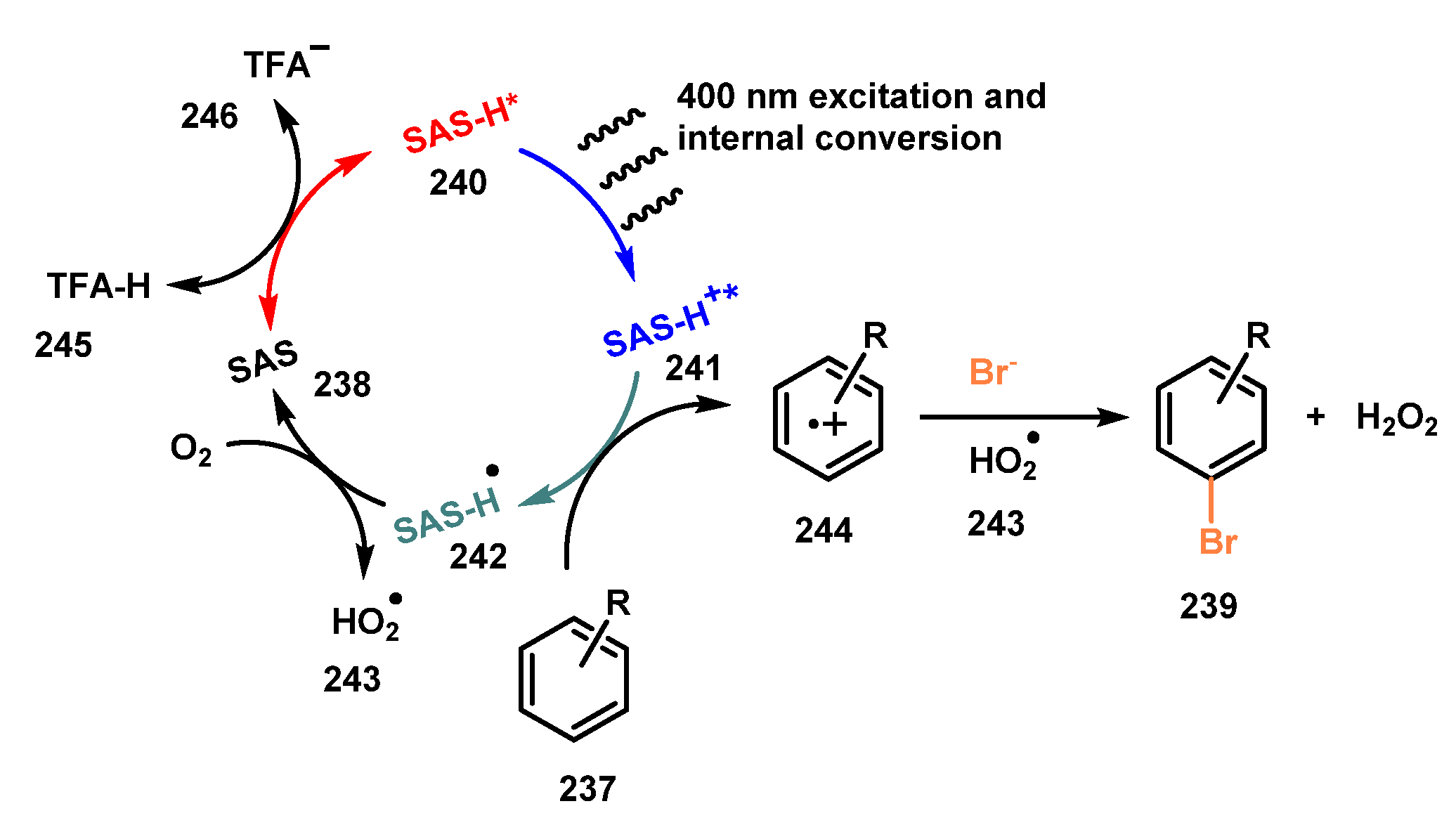{kind=link}
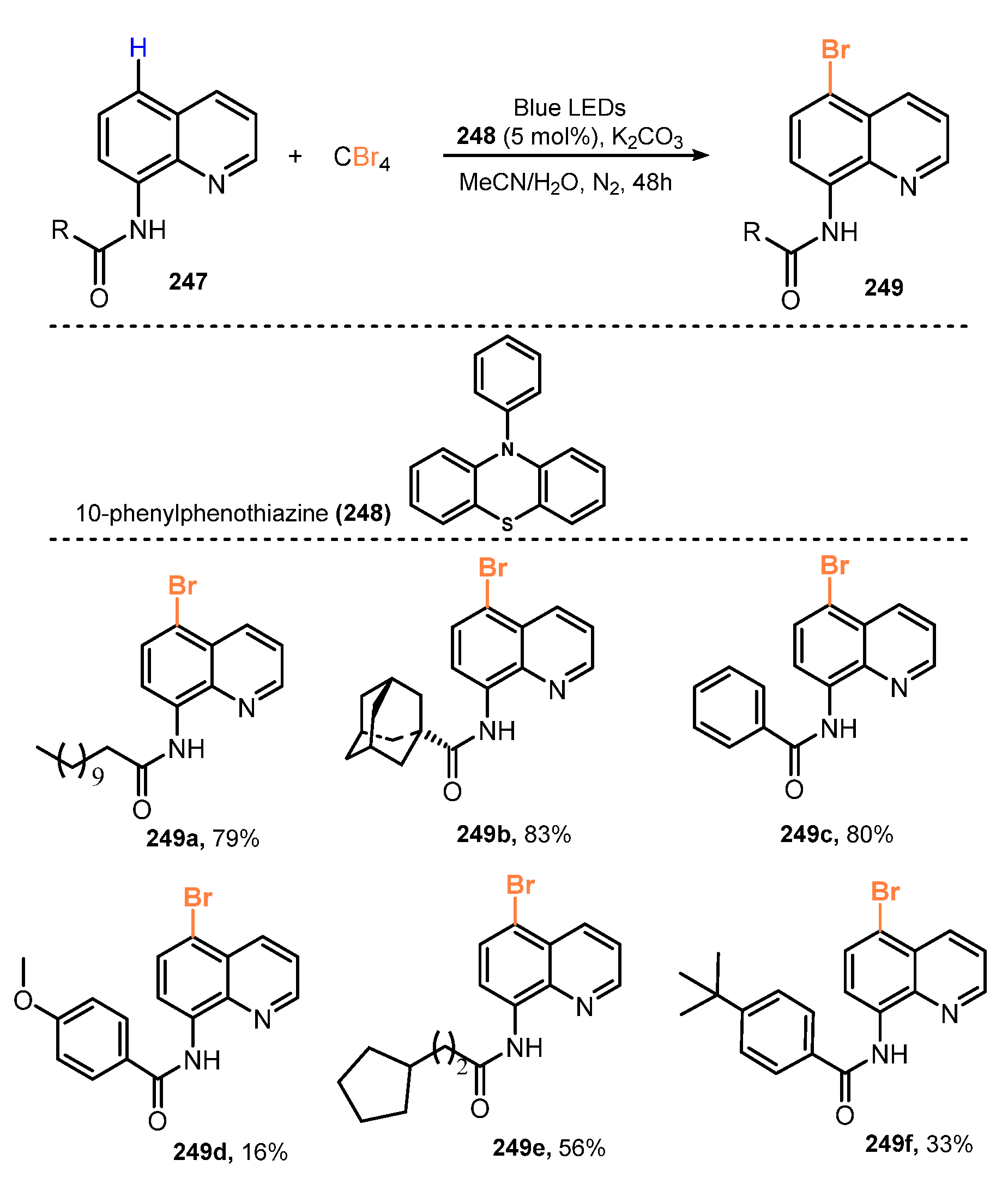{kind=link}
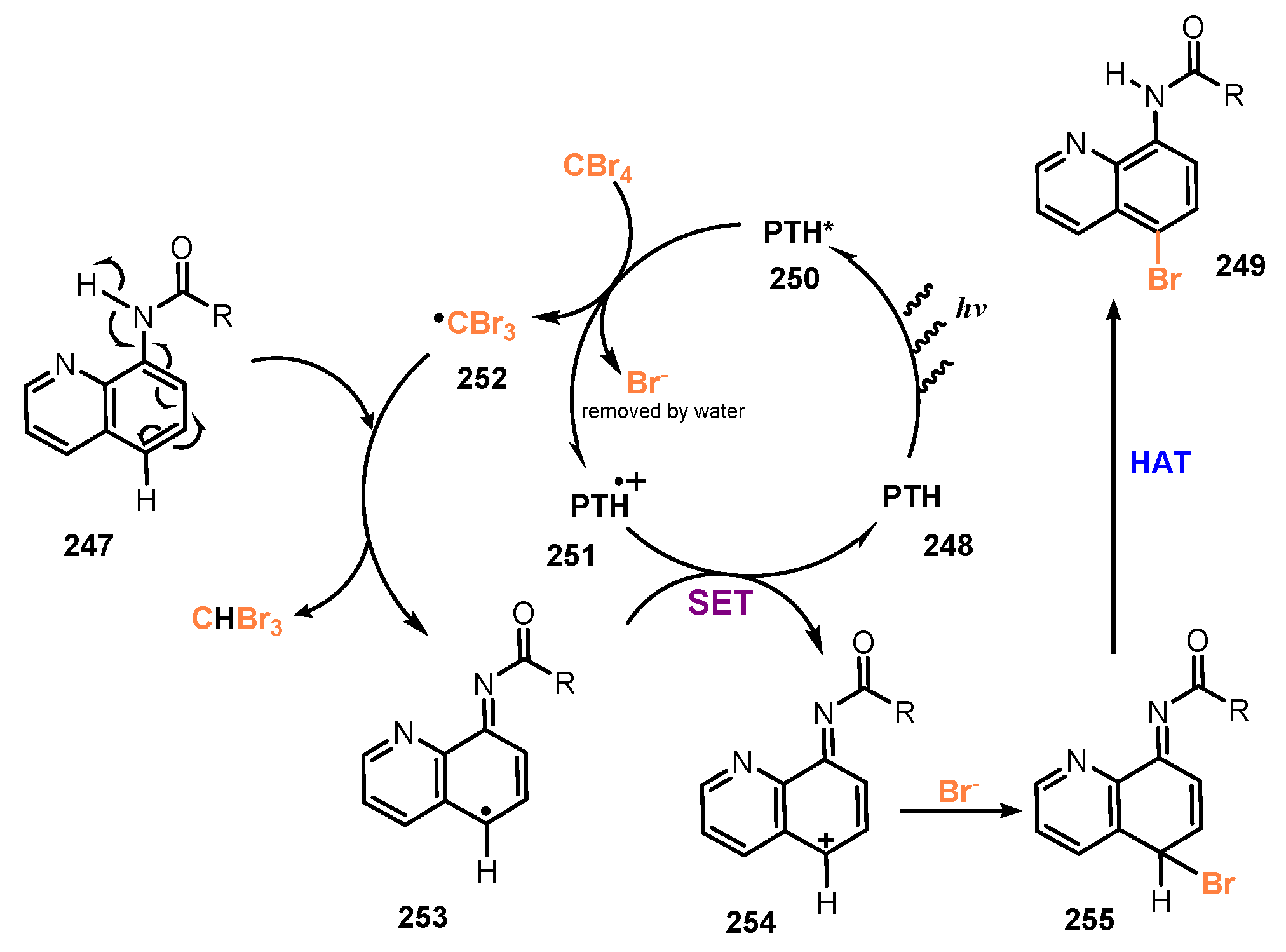{kind=link}
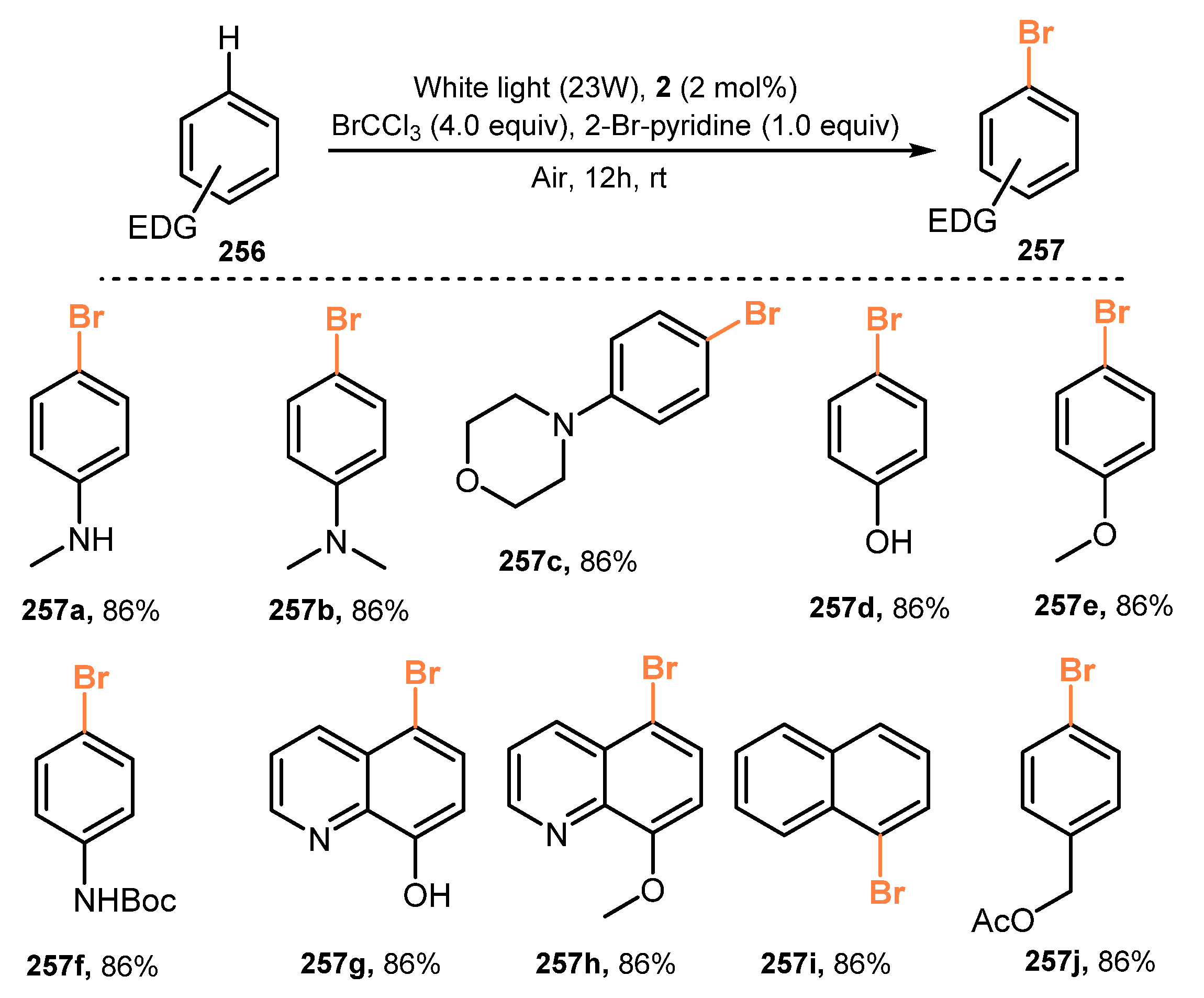{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
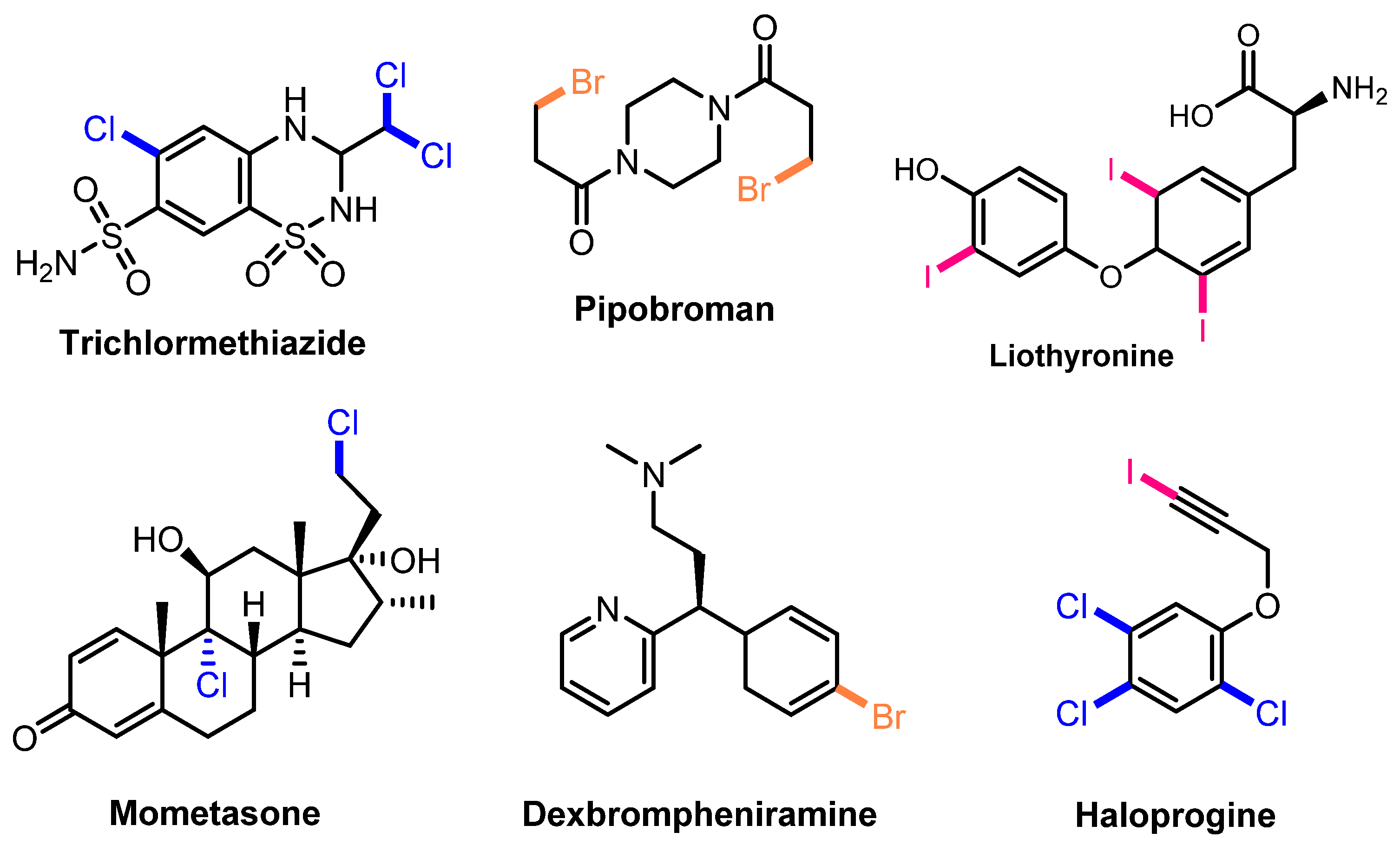
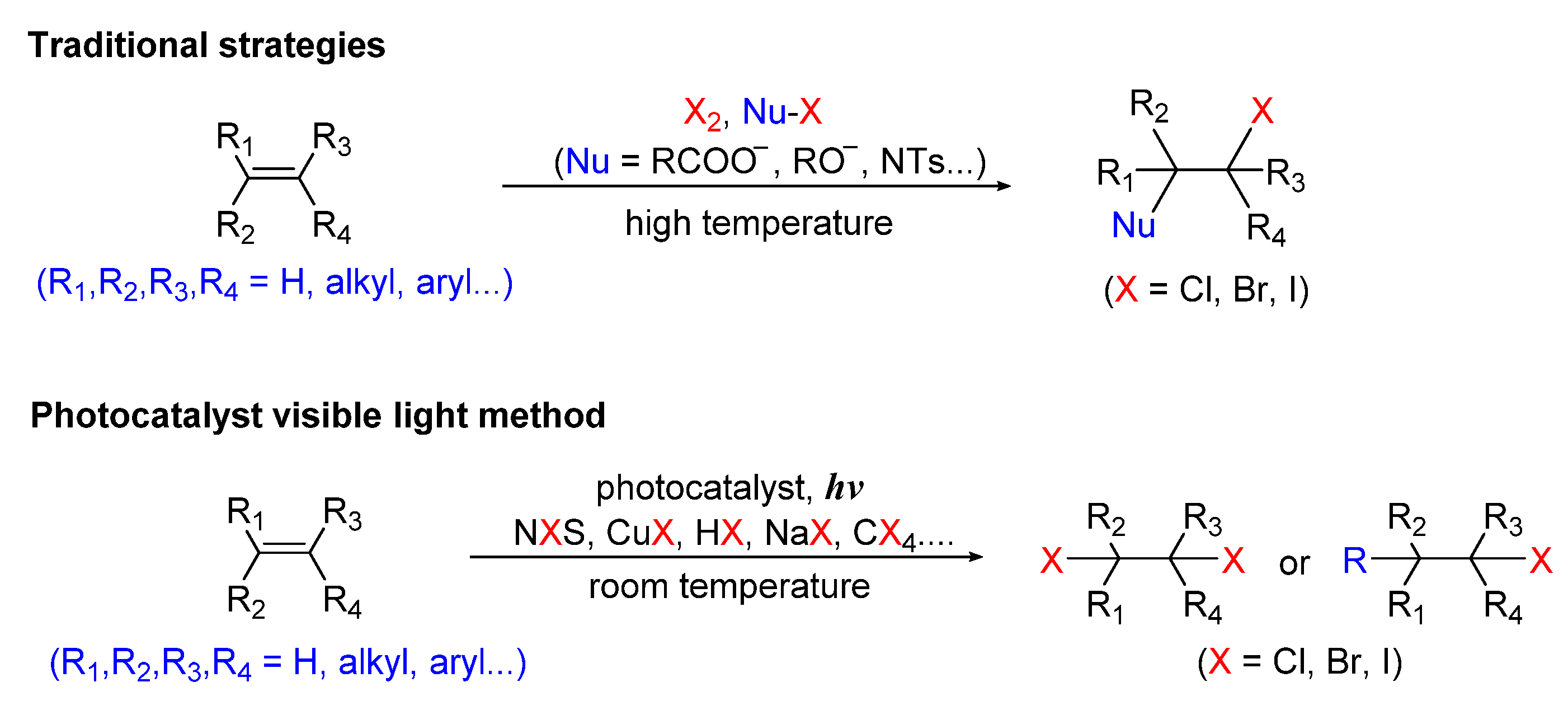
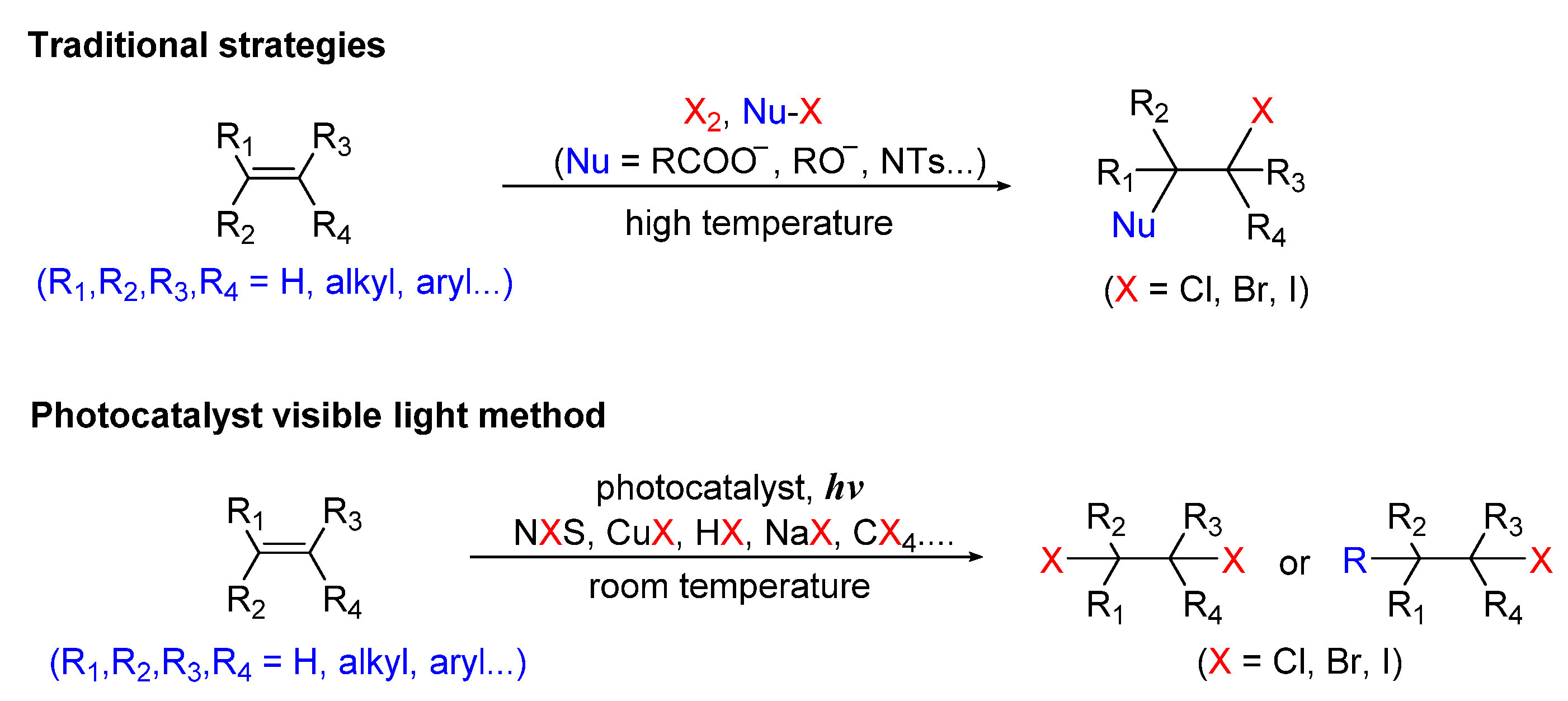
:1. Introduction
2. Photo-Catalyzed Halogenation of Aliphatic C-H Bonds
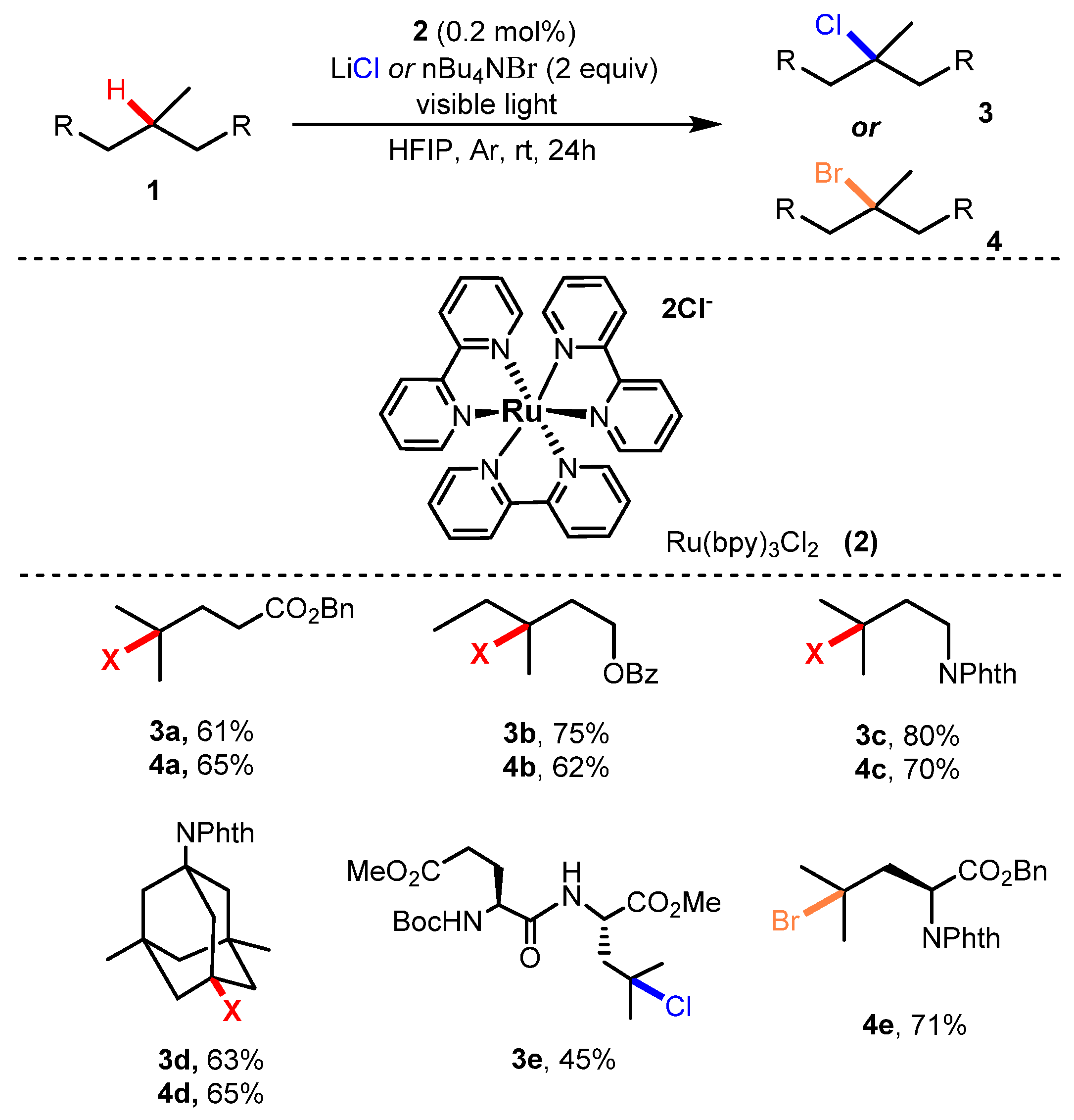
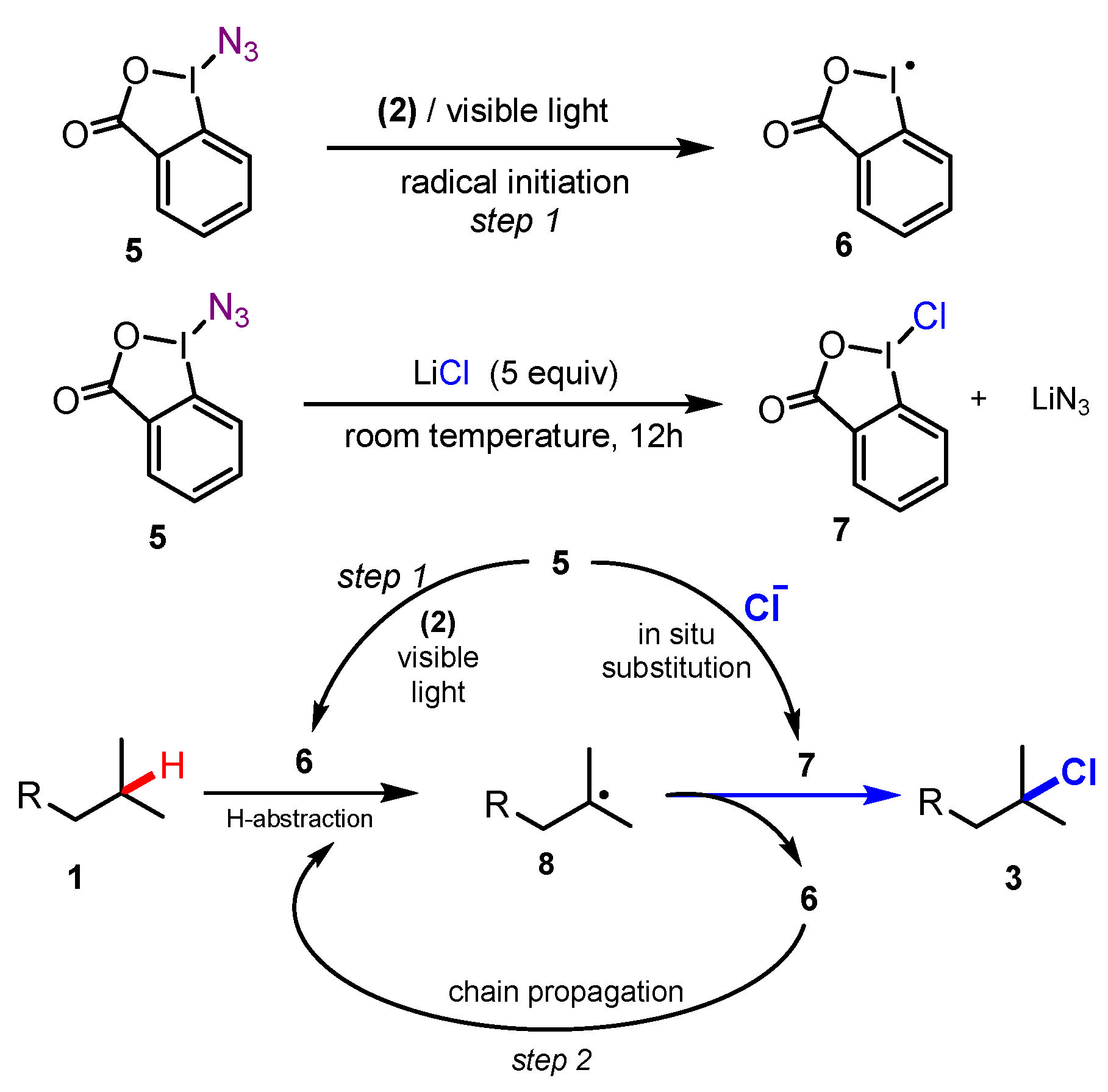
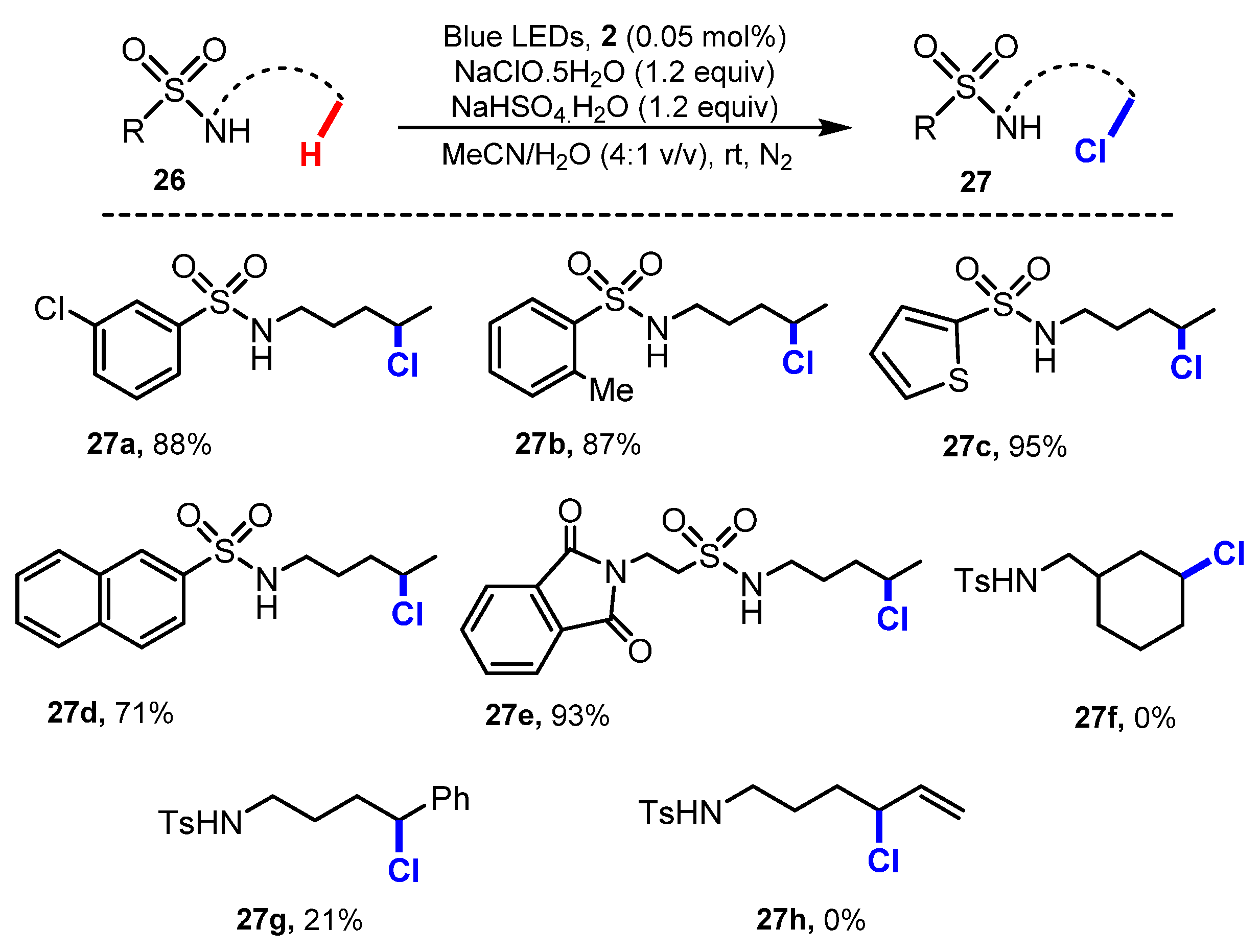
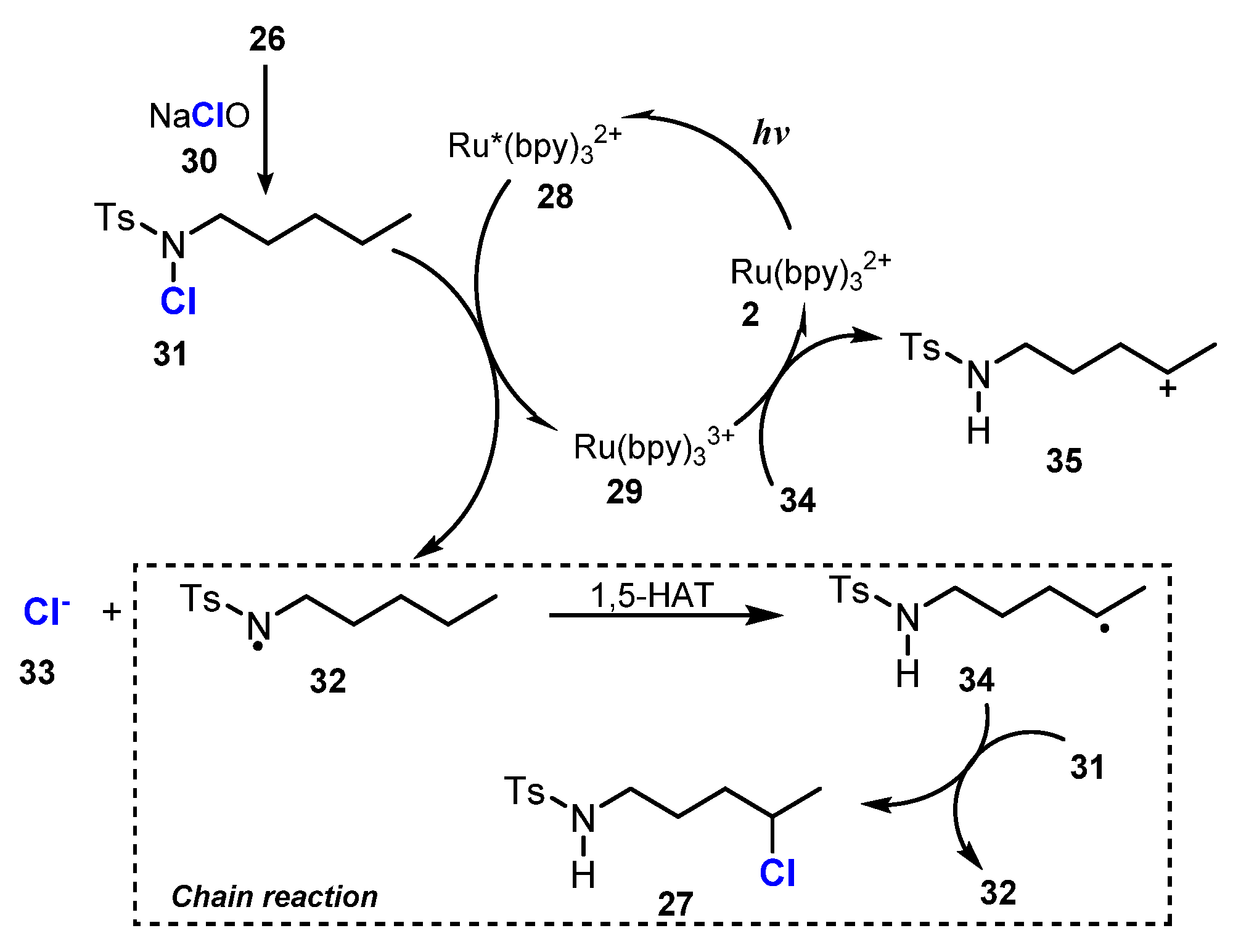
2.1. Chlorination of Aliphatic C-H bBonds
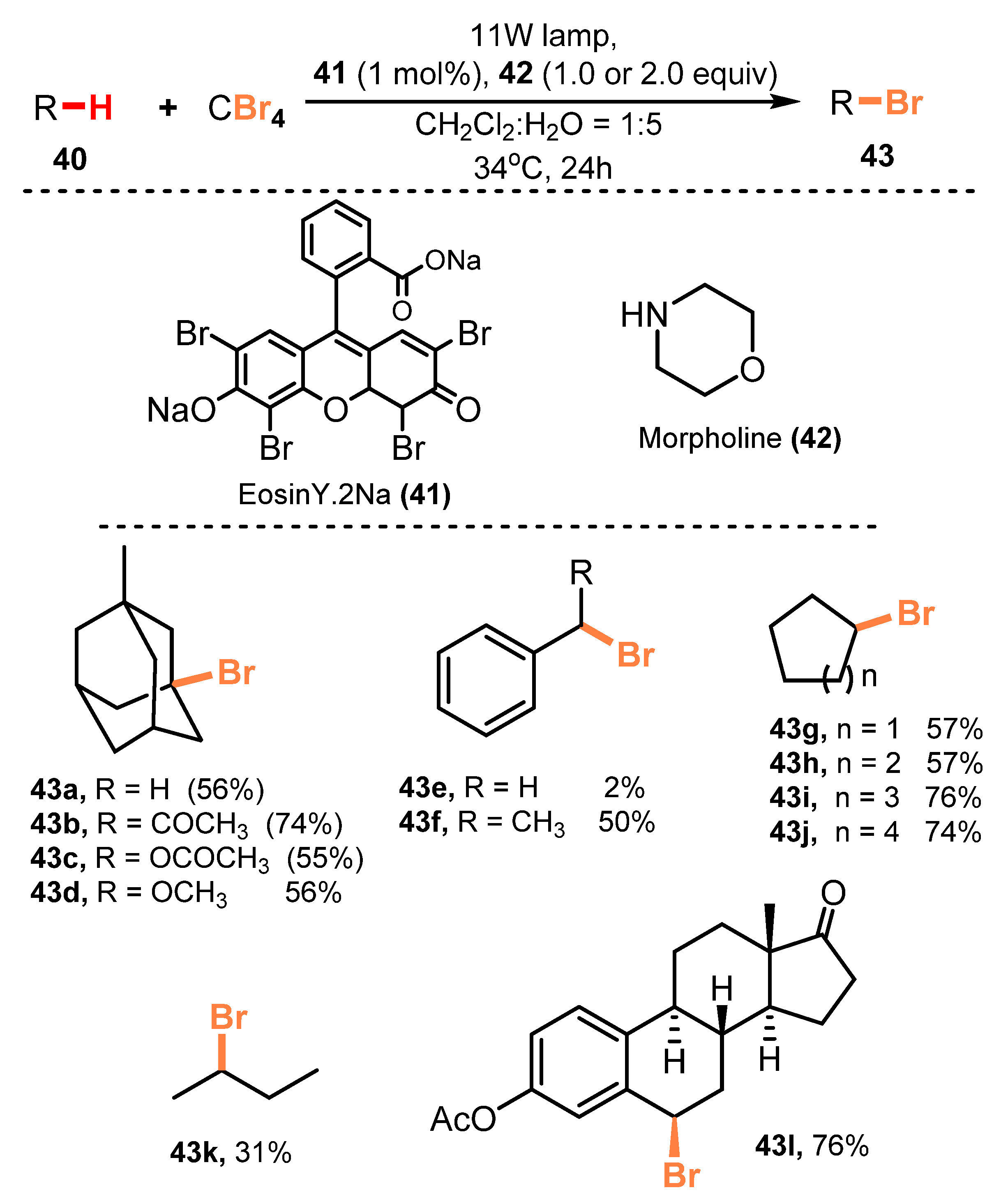
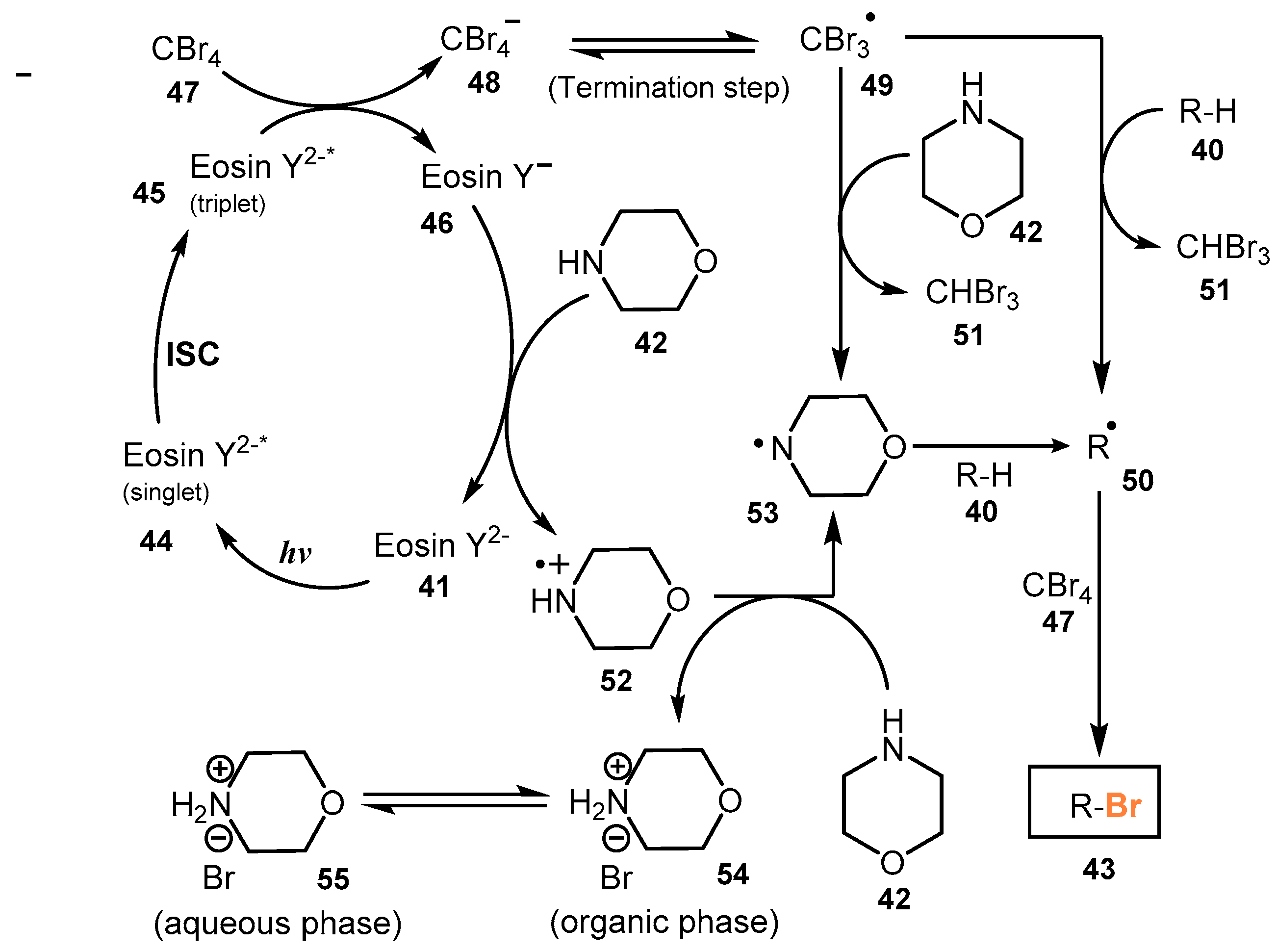
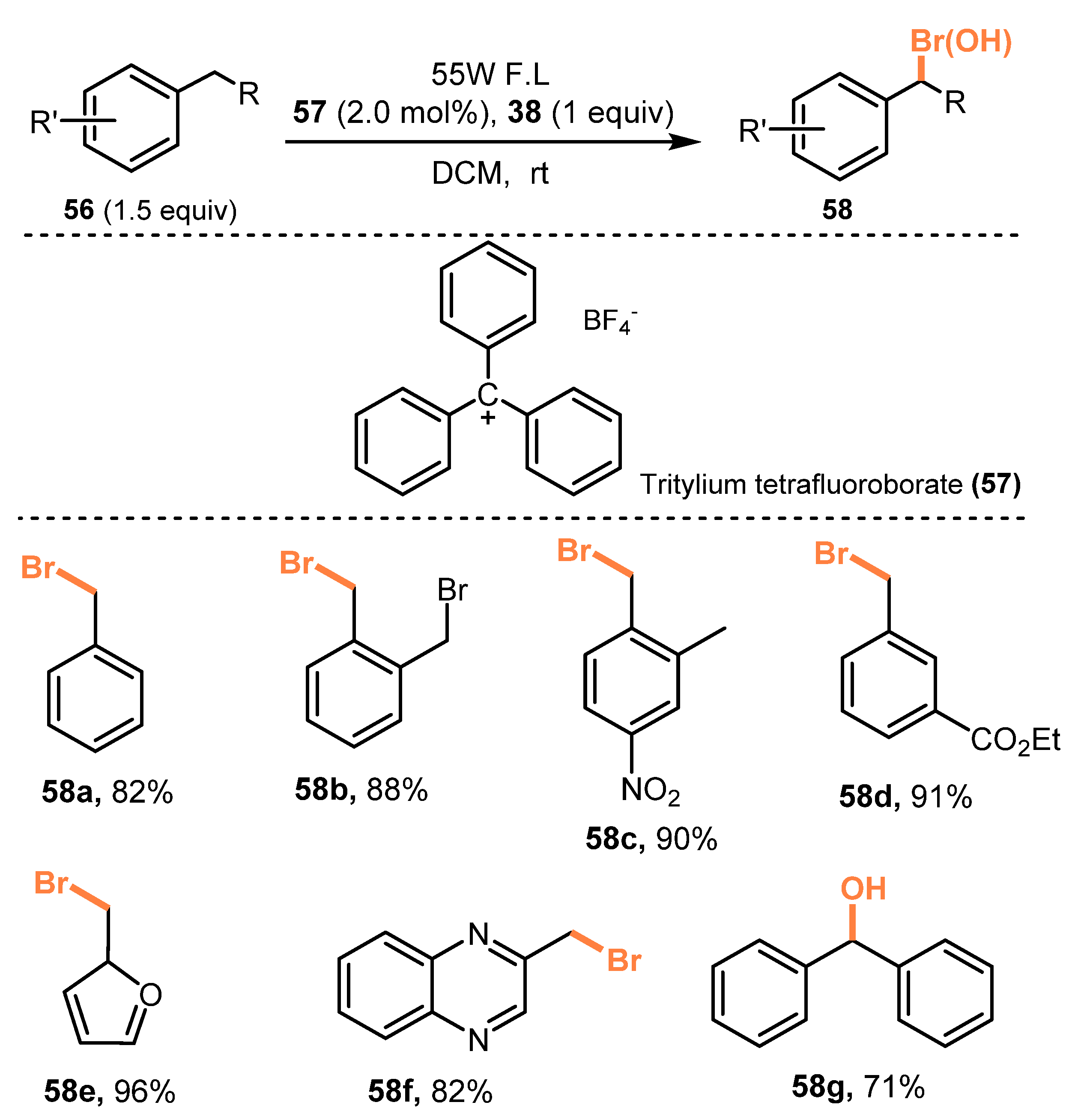
2.2. Bromination of Aliphatic C-H Bonds
3. Photo-Catalyzed Halogenations of Aliphatic Multiple Bonds
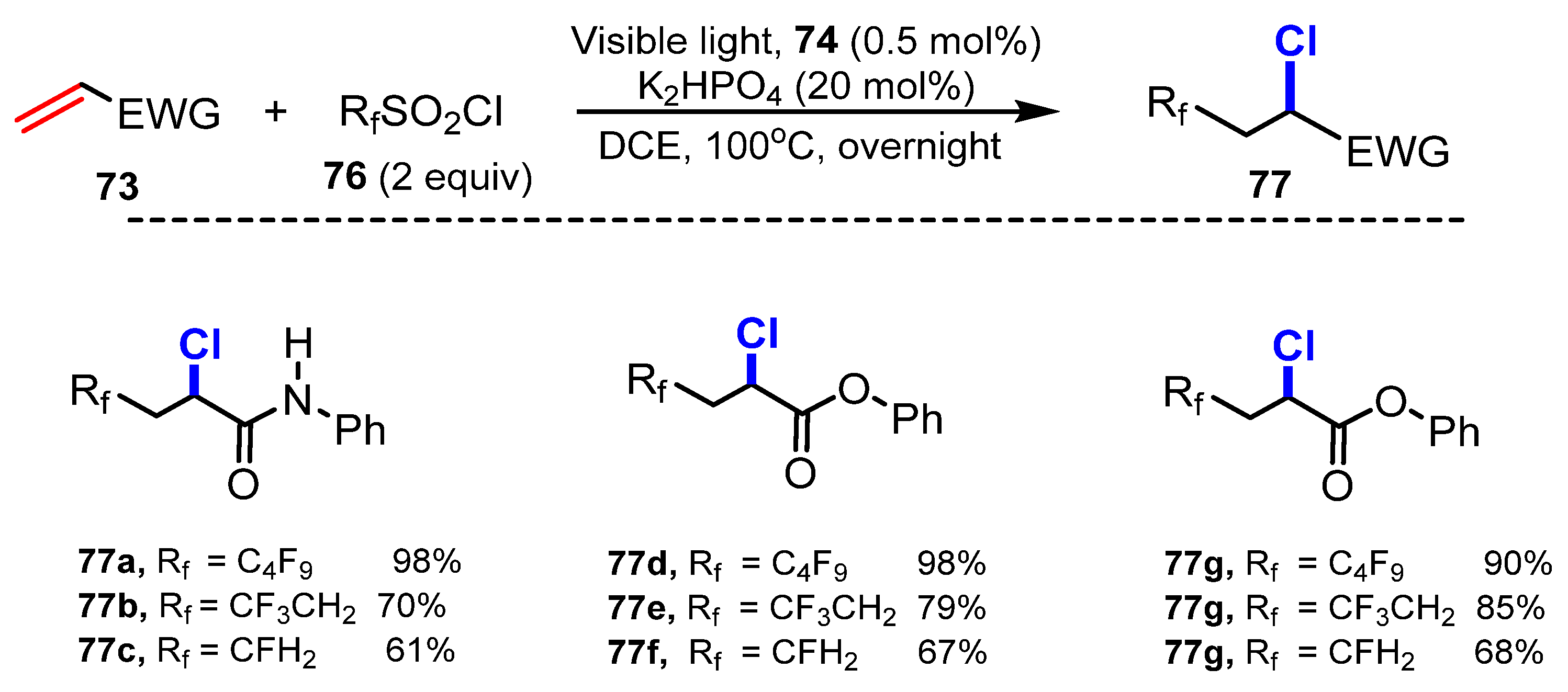
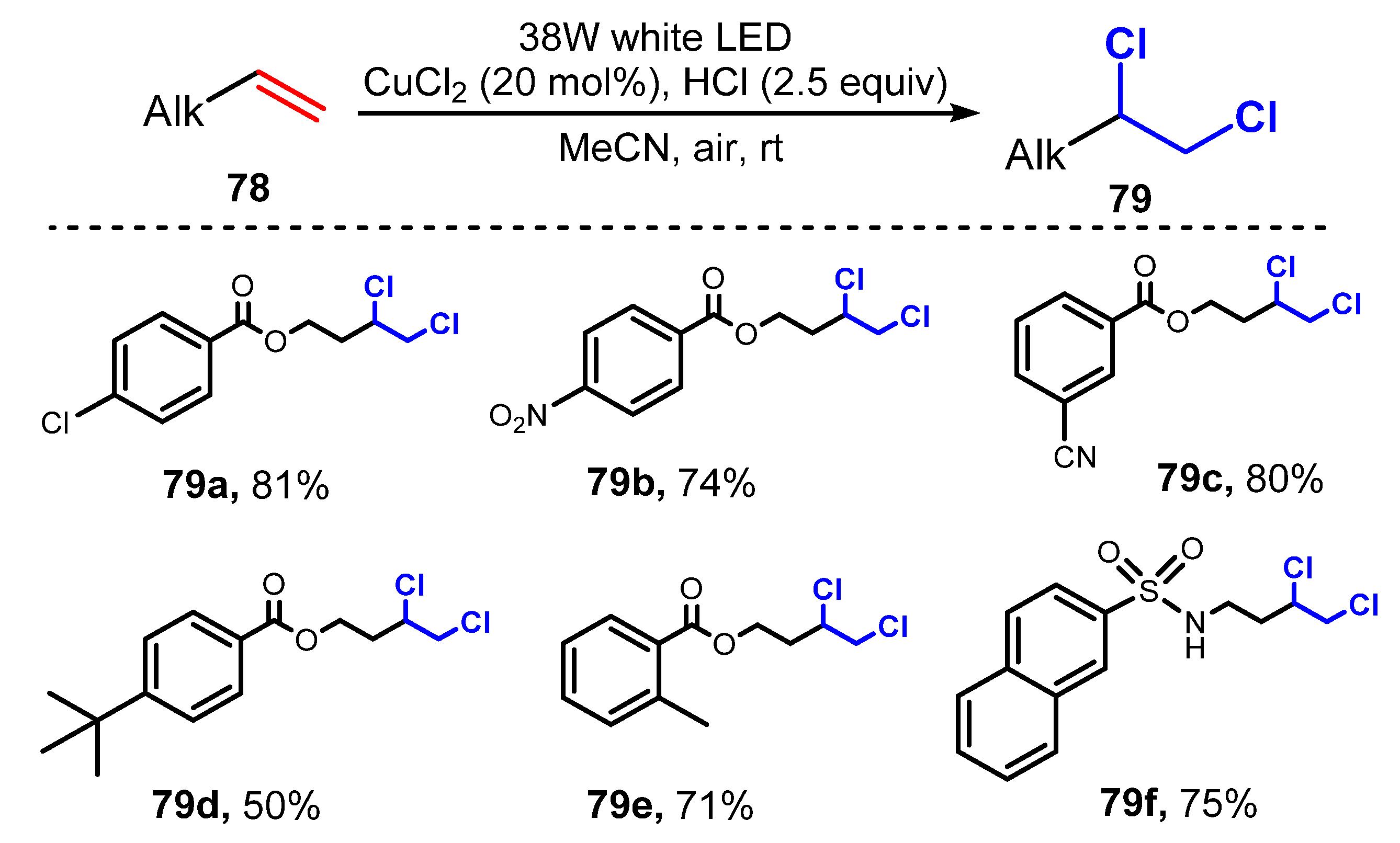
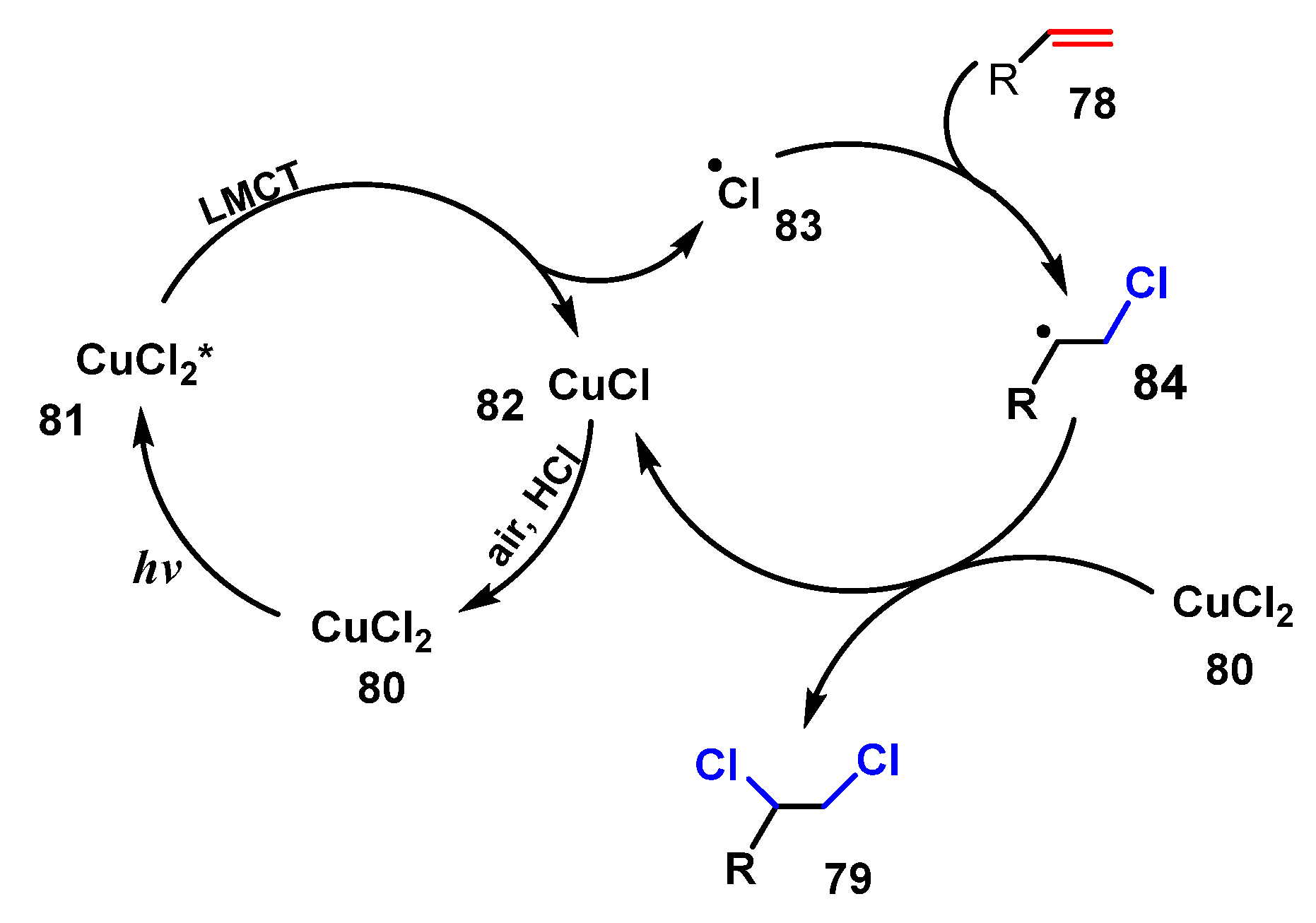
3.1. Chlorination of Aliphatic Multiple Bonds
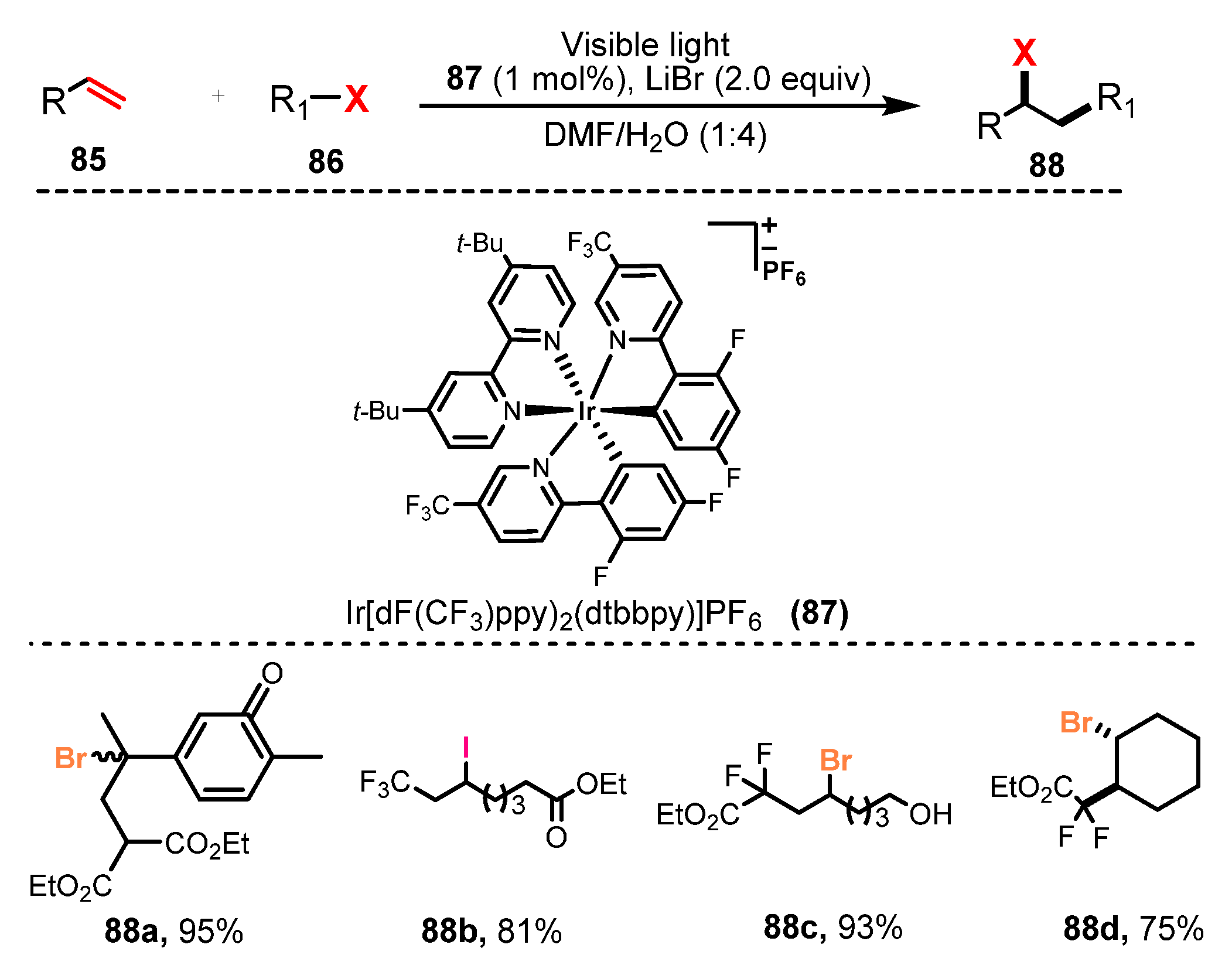
3.2. Bromination of Aliphatic Multiple Bonds
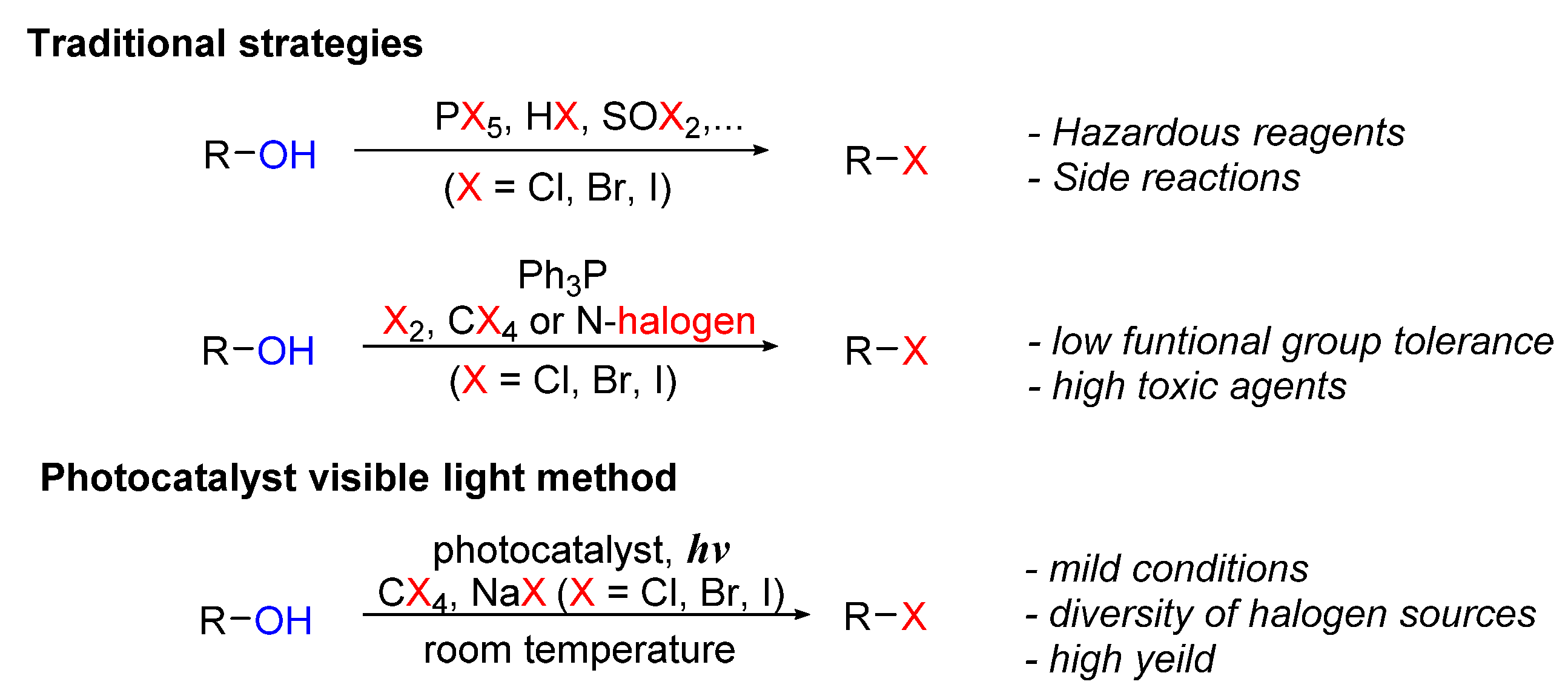
4. Photo-Catalyzed Halogenations of Alcohols
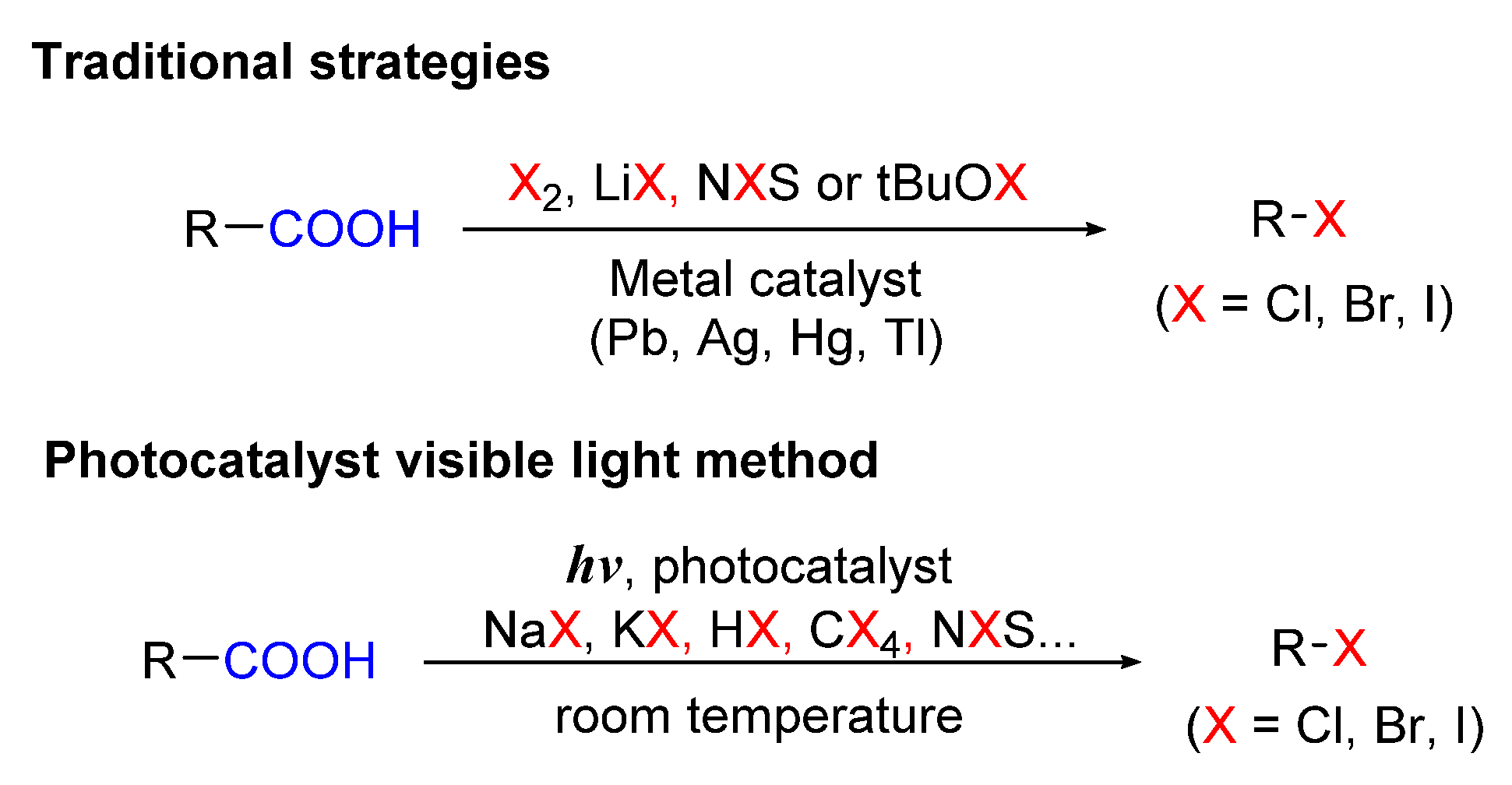
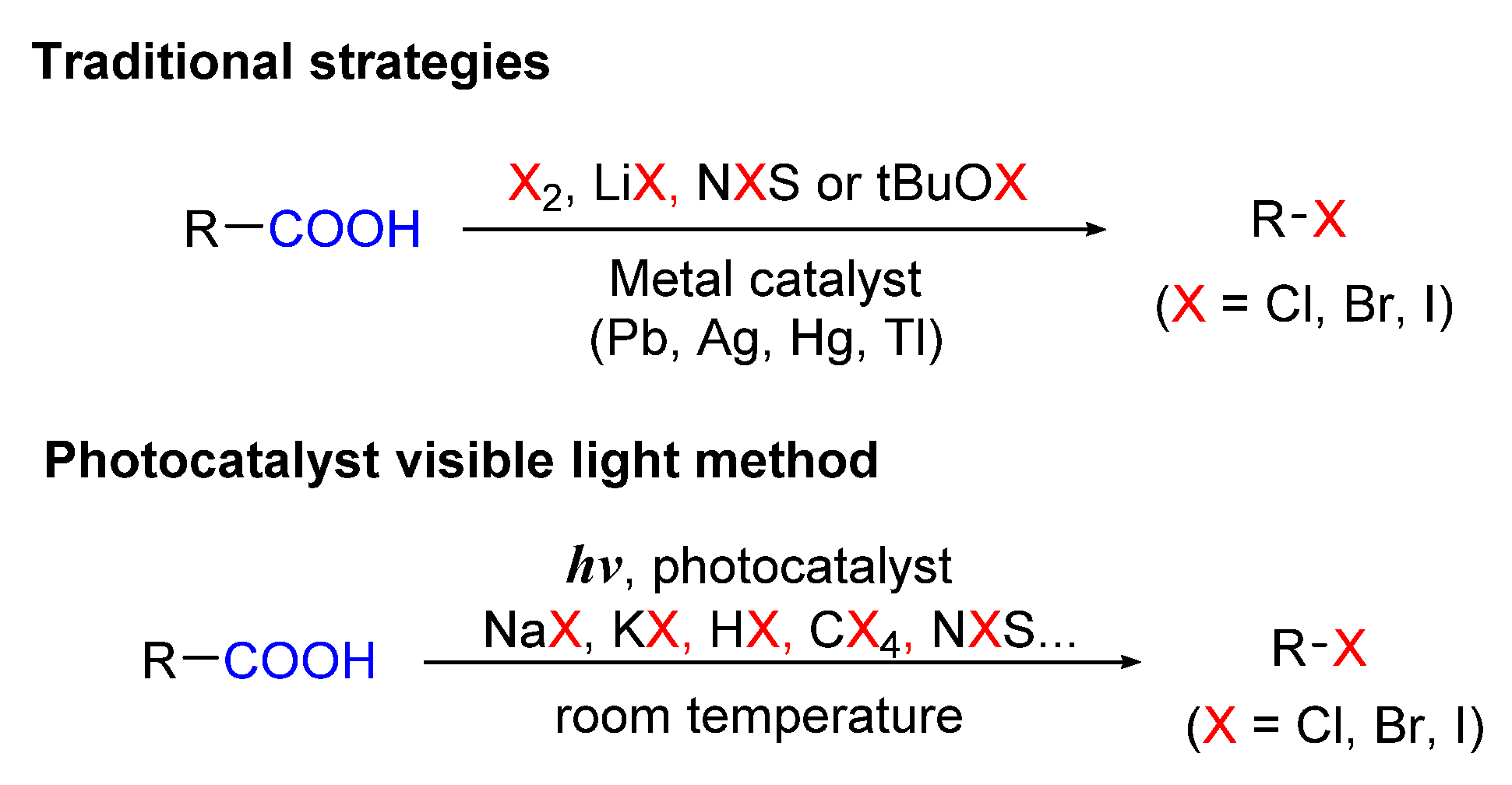
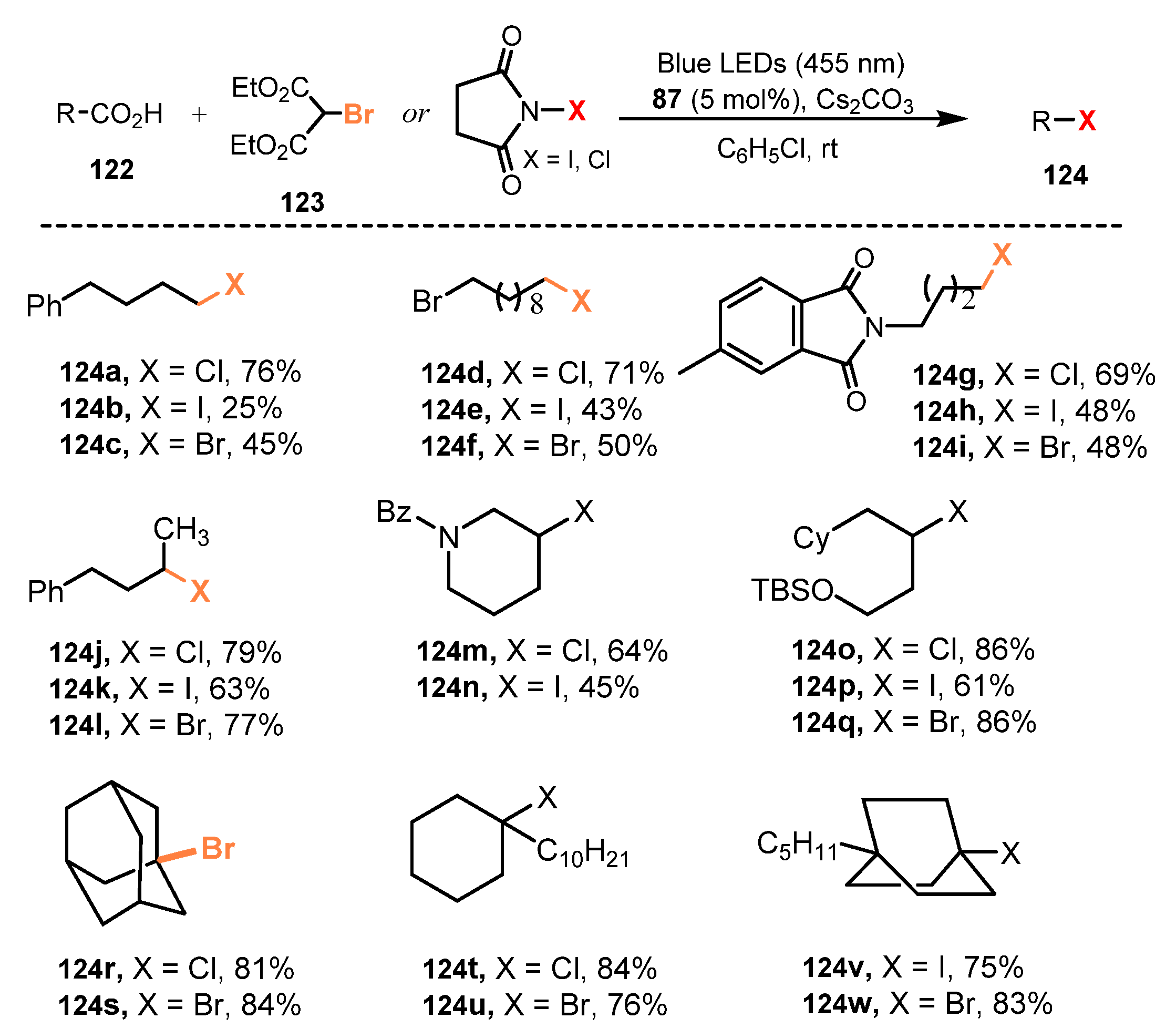
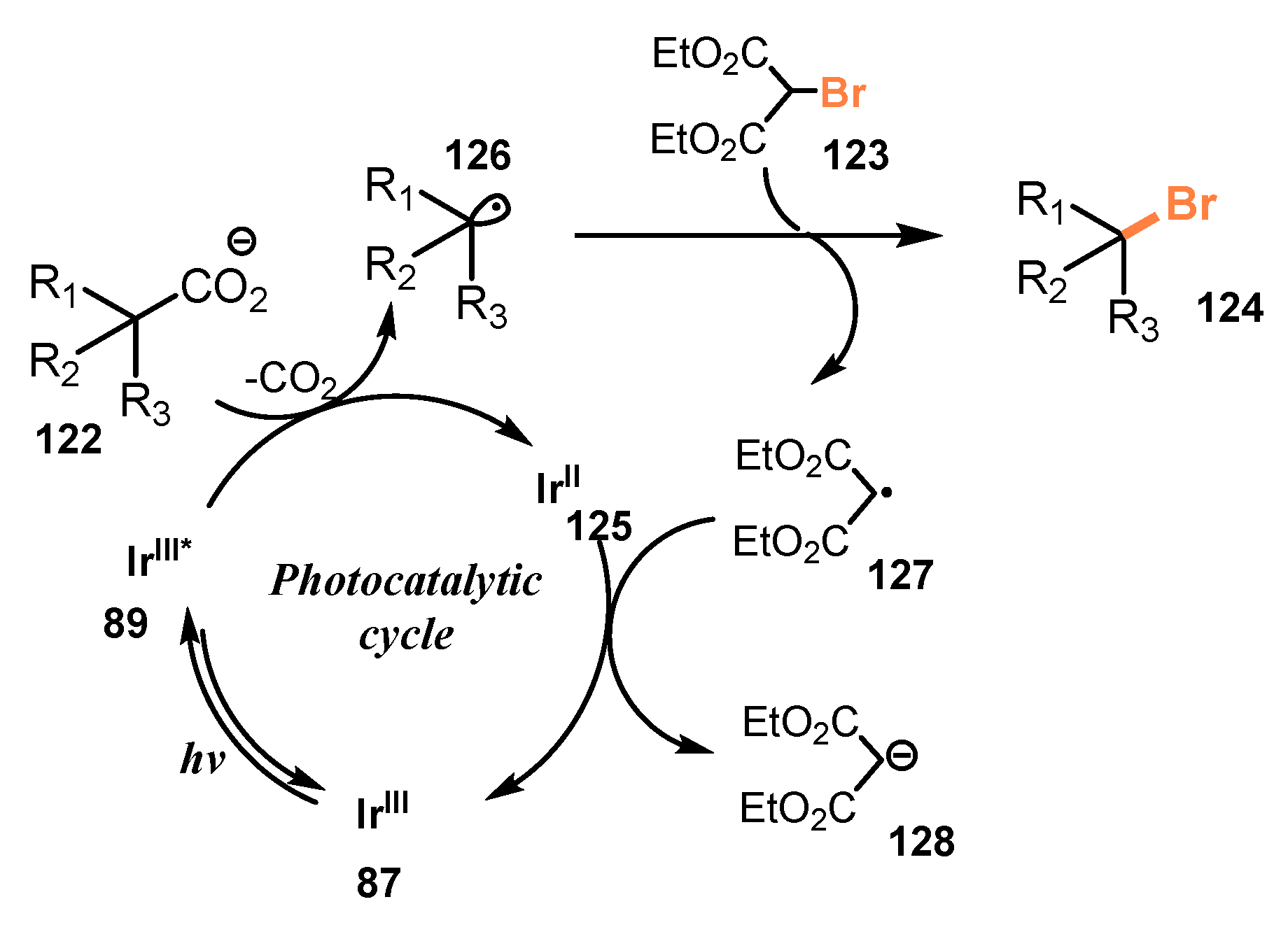
5. Photo-Catalyzed Halogenations of Carboxylic Acids
5.1. Chlorination of Carboxylic Acids
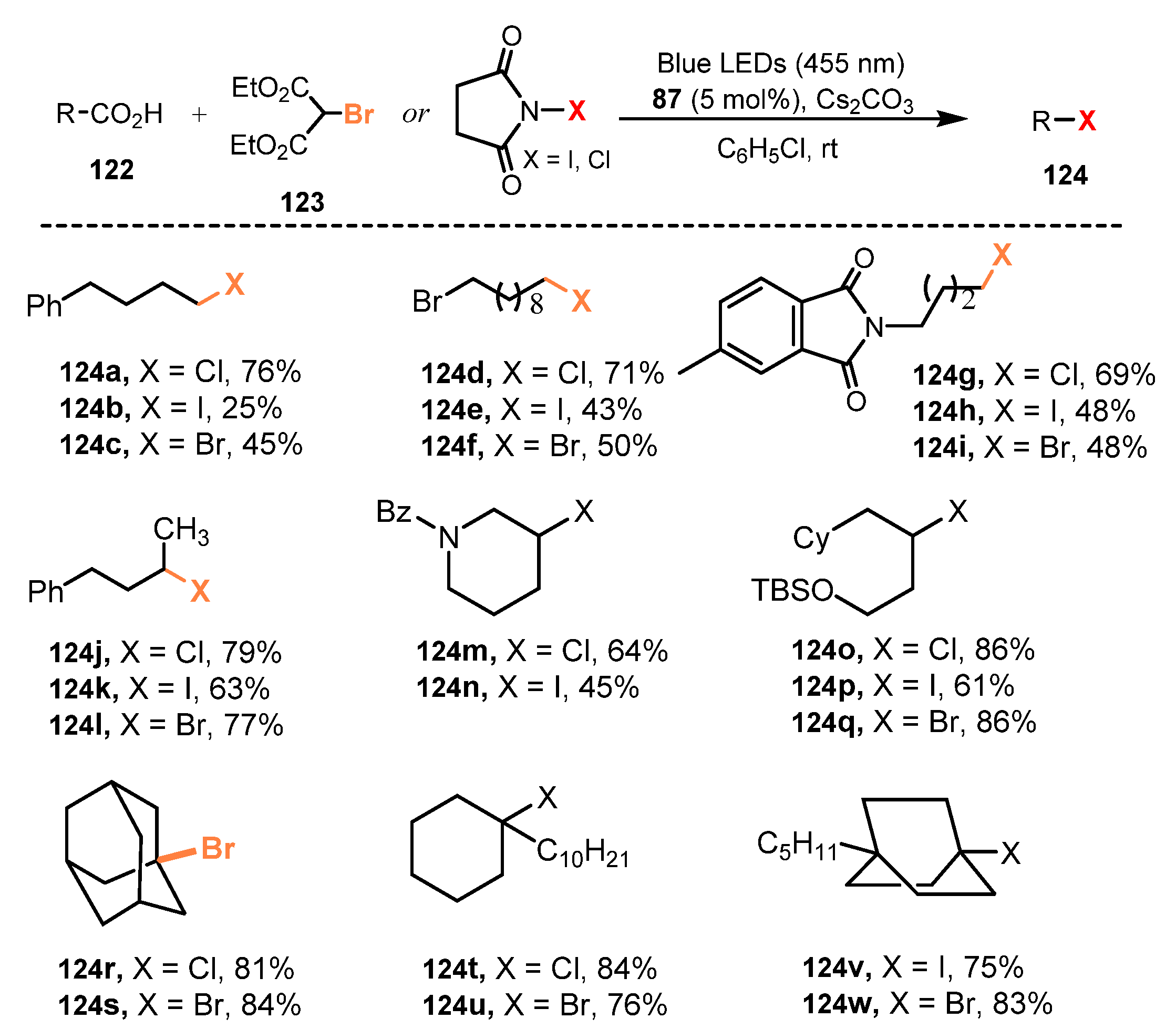
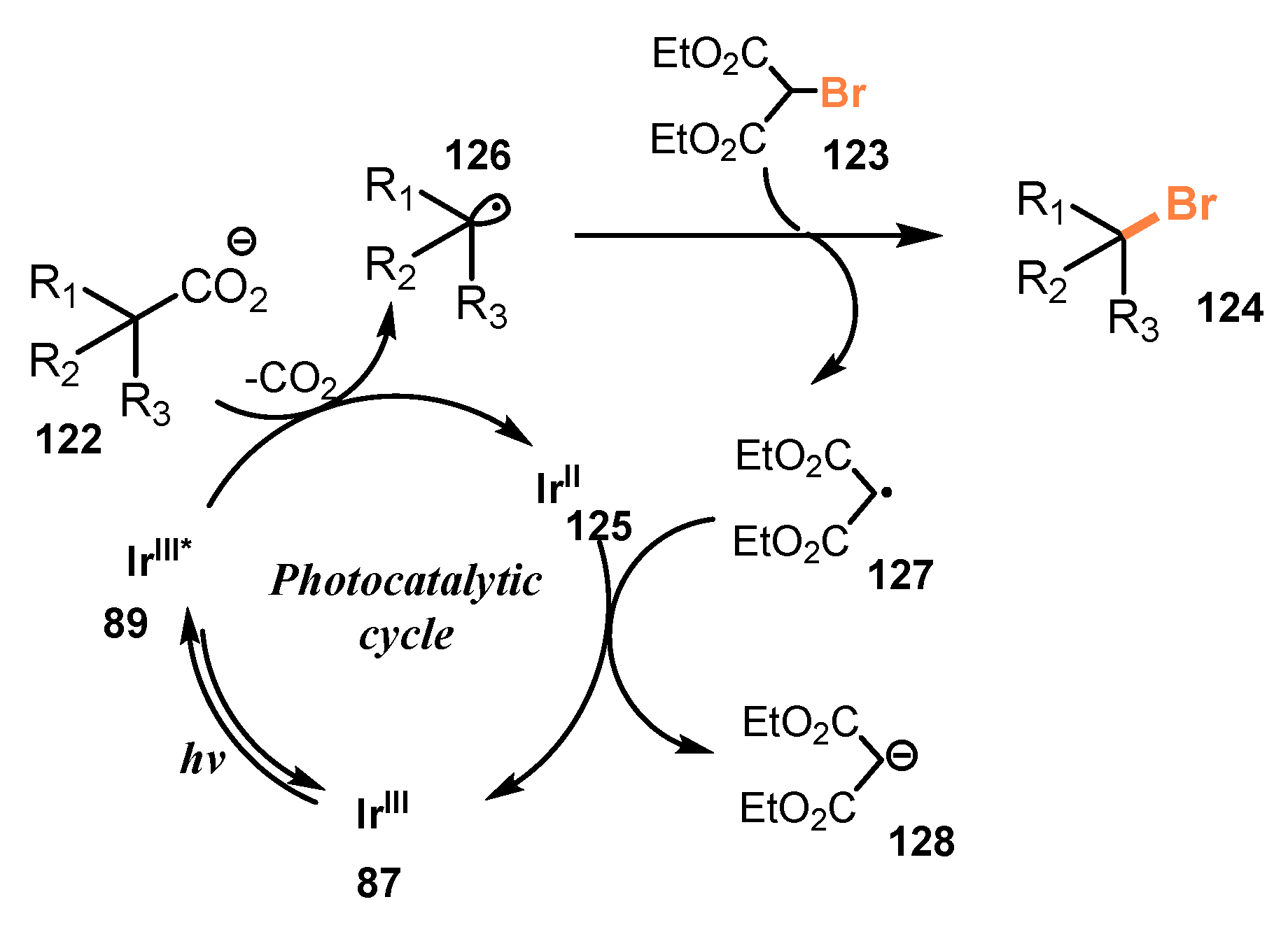
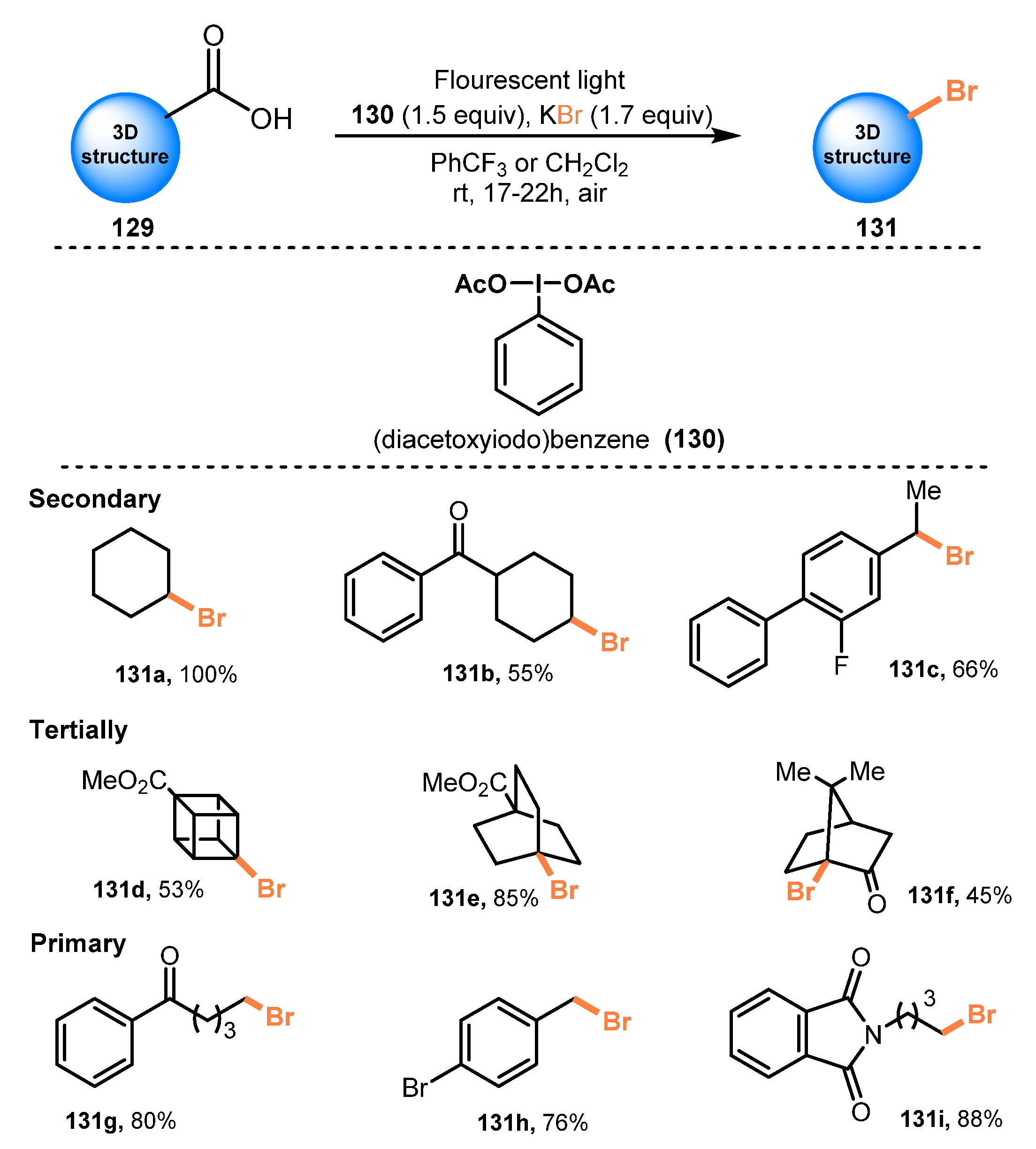
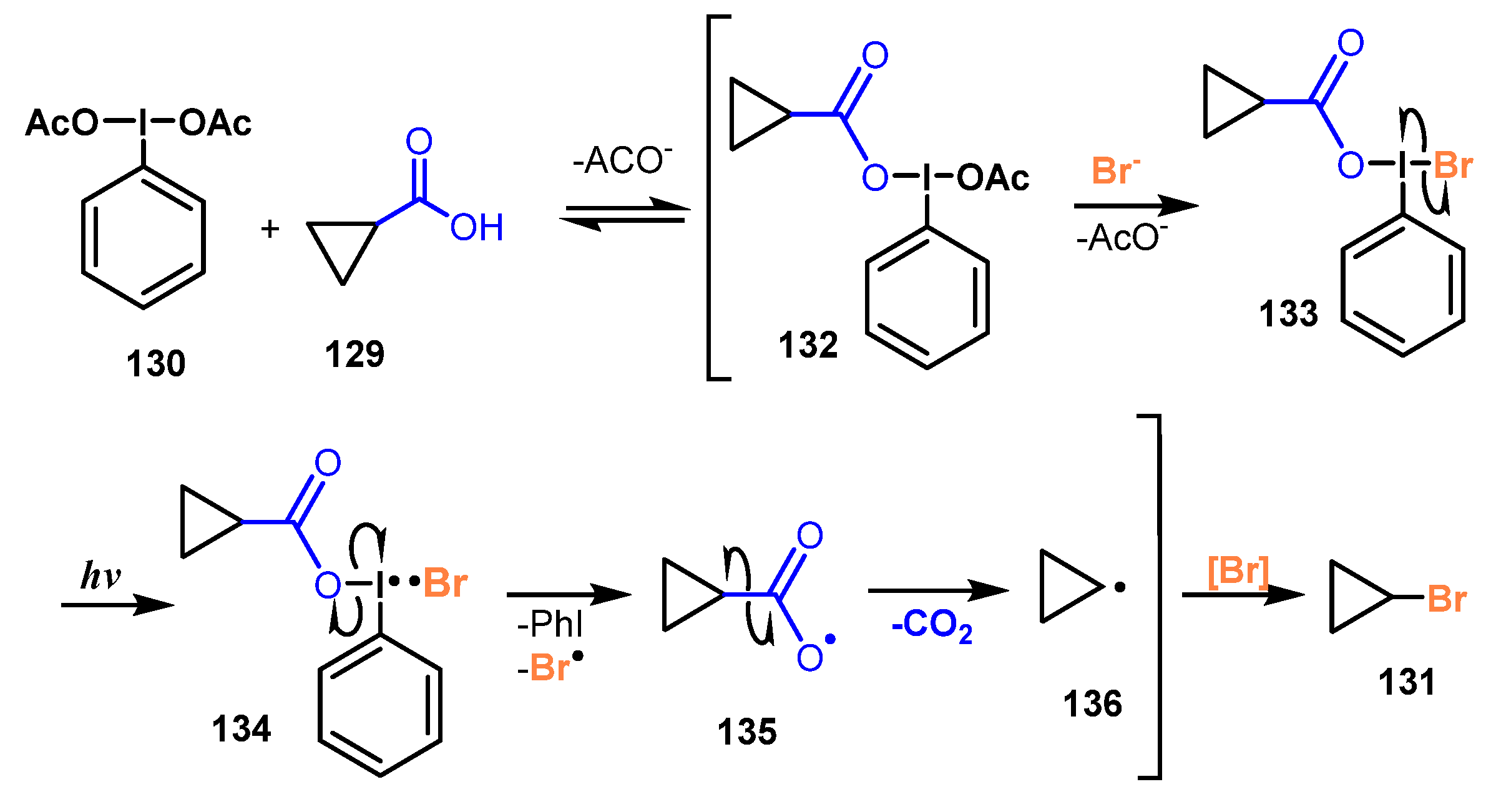
5.2. Bromination of Carboxylic Acids
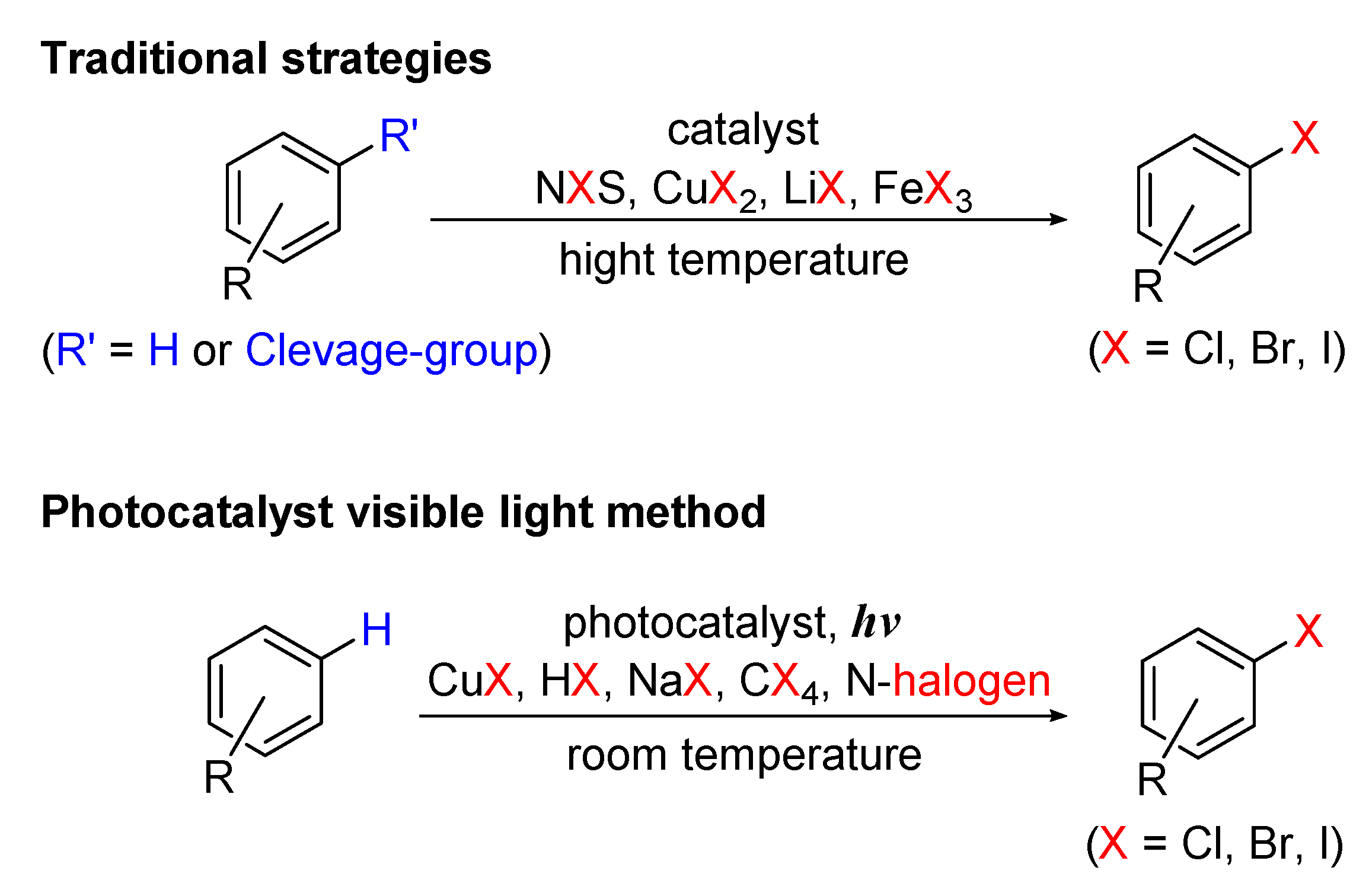
6. Photo-Catalyzed Halogenations of Aromatic C-H Bonds
6.1. Halogenation of Aromatic C-H Bonds
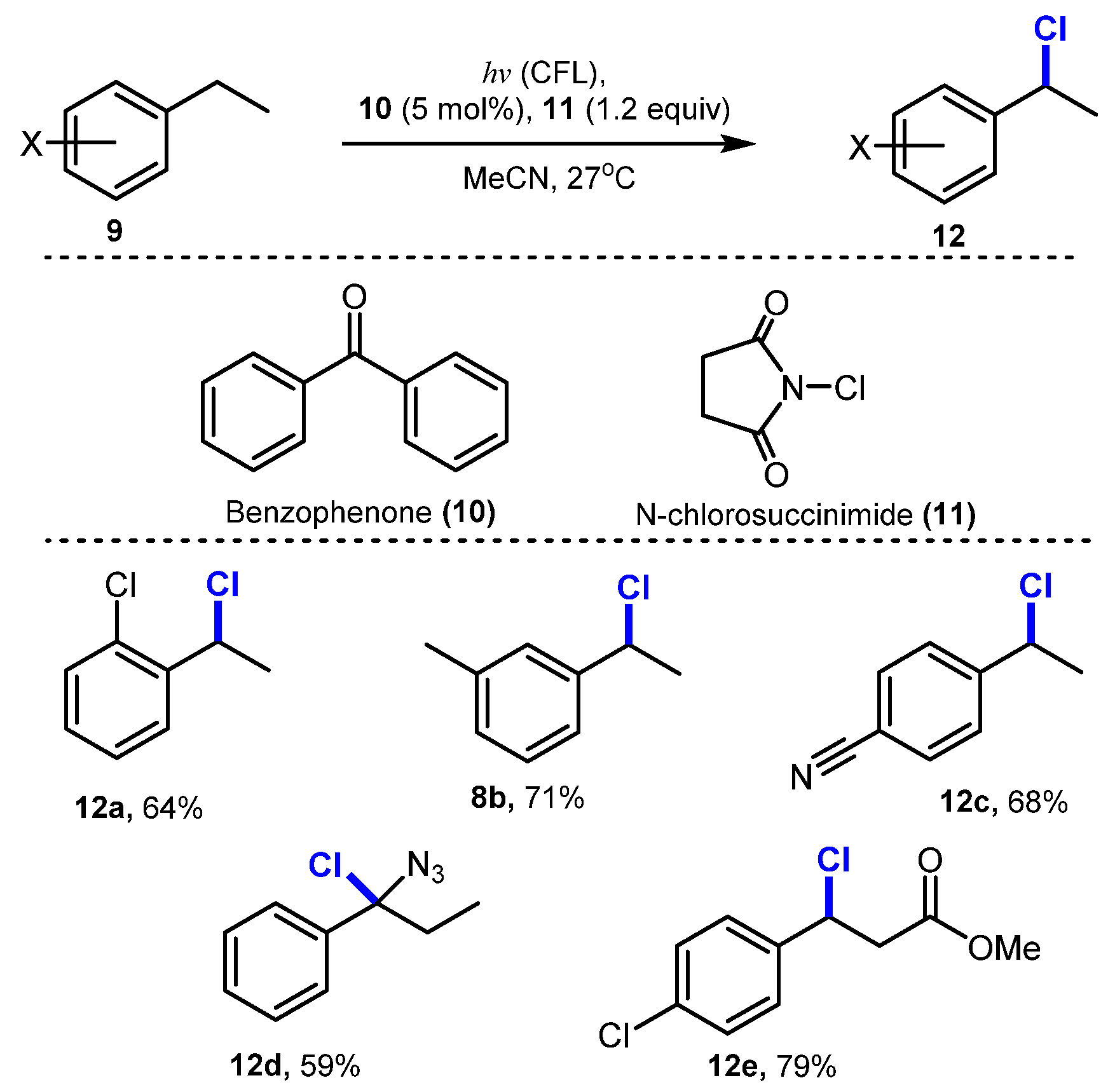
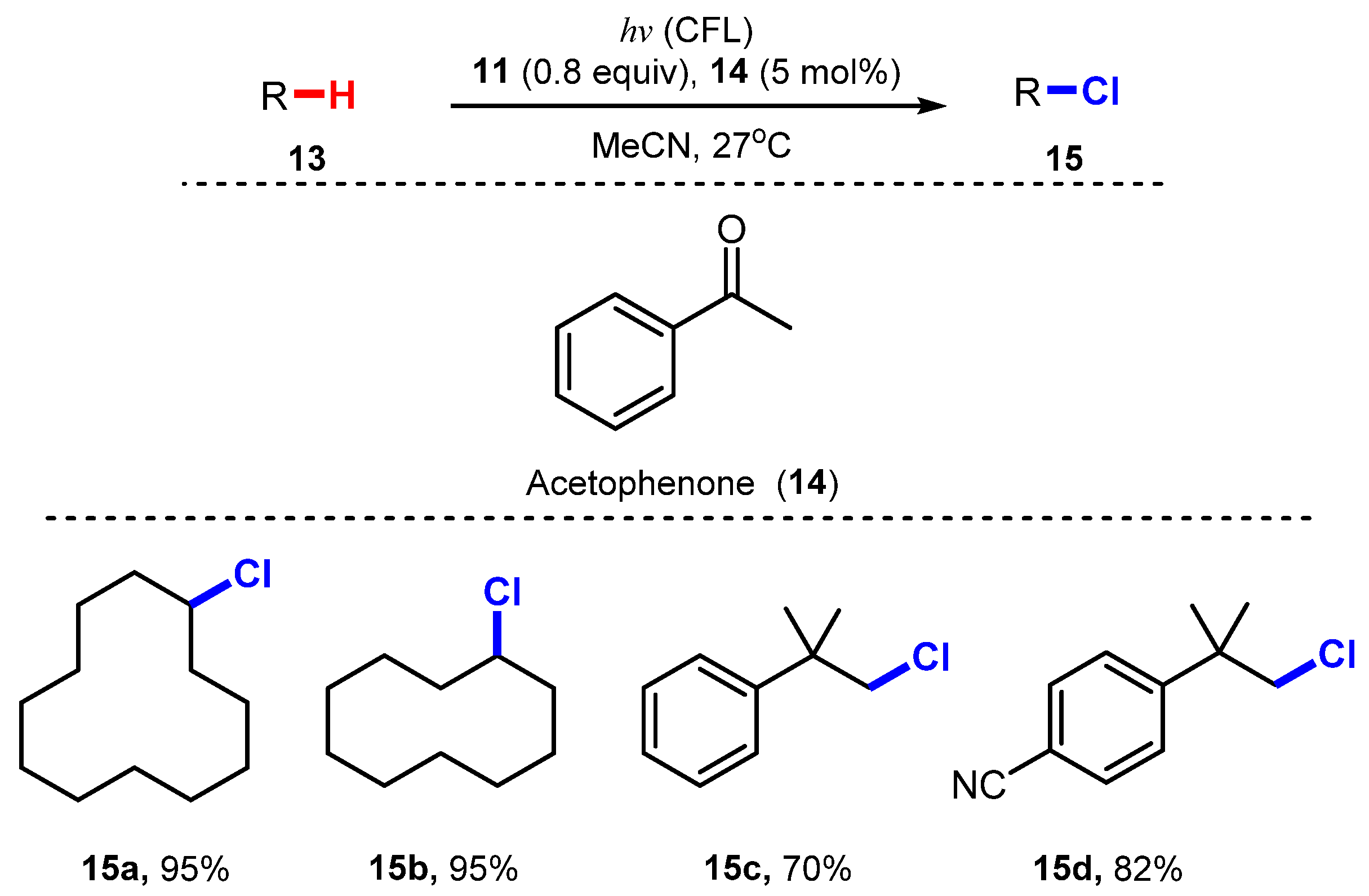
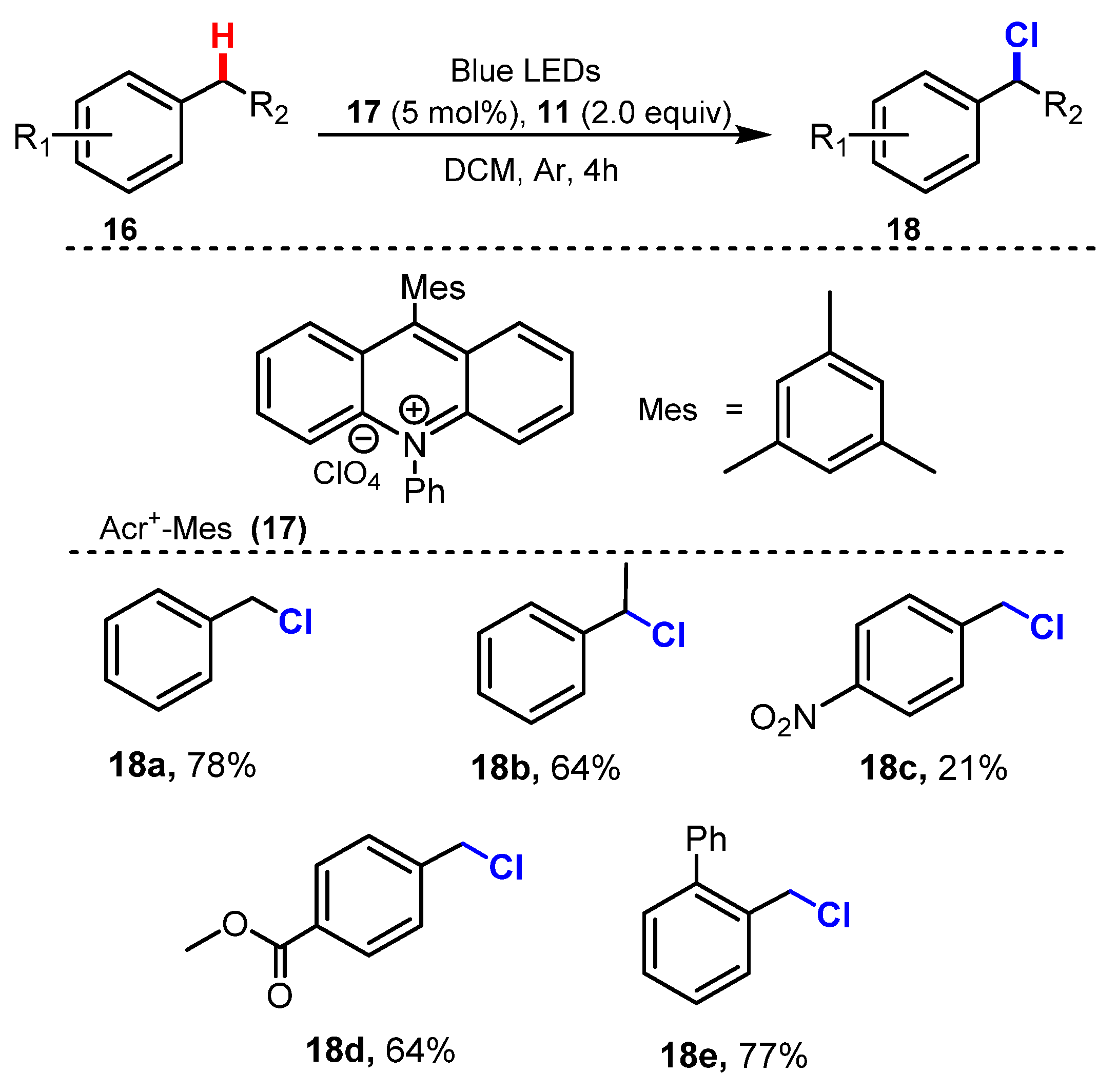
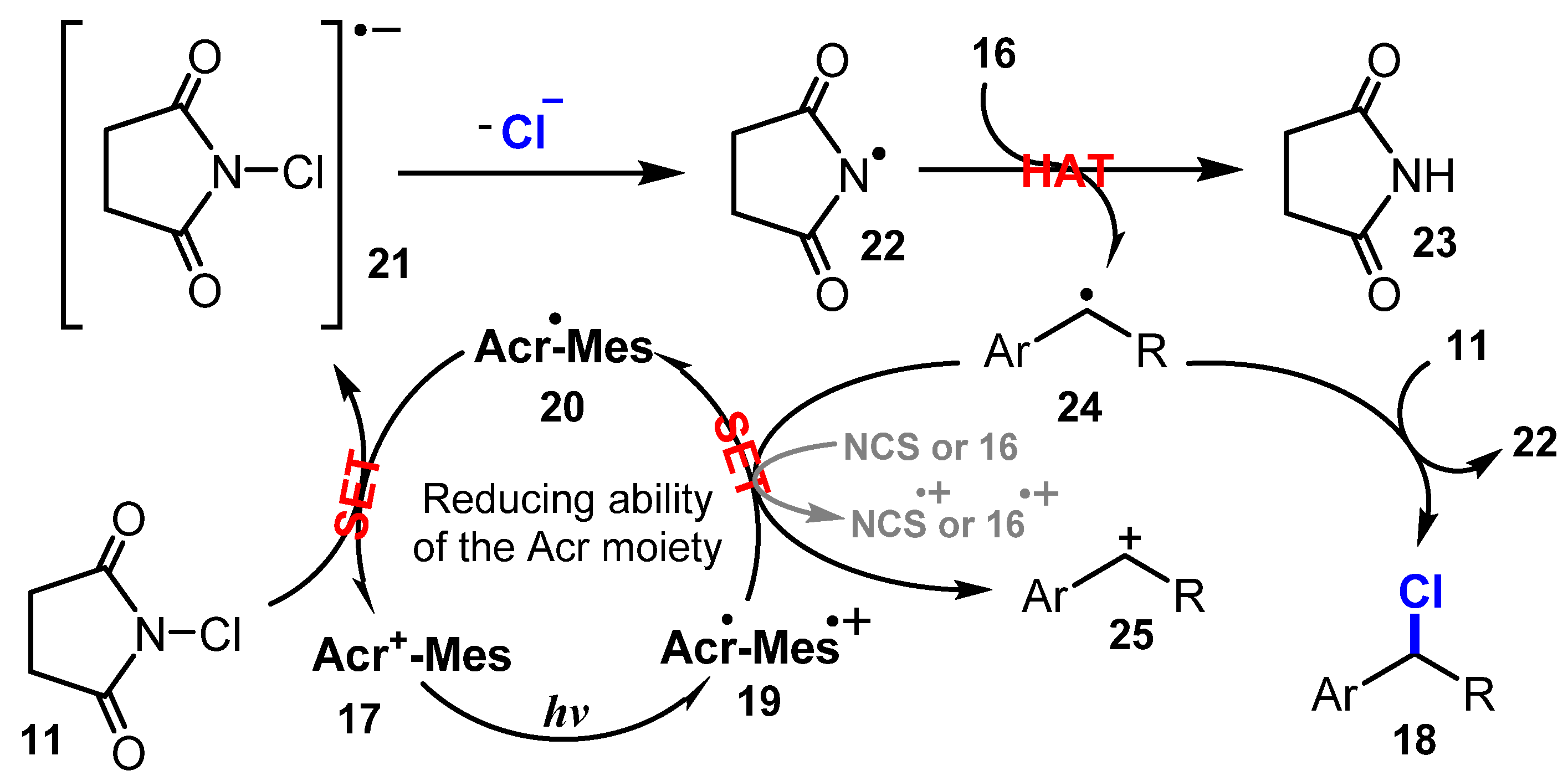
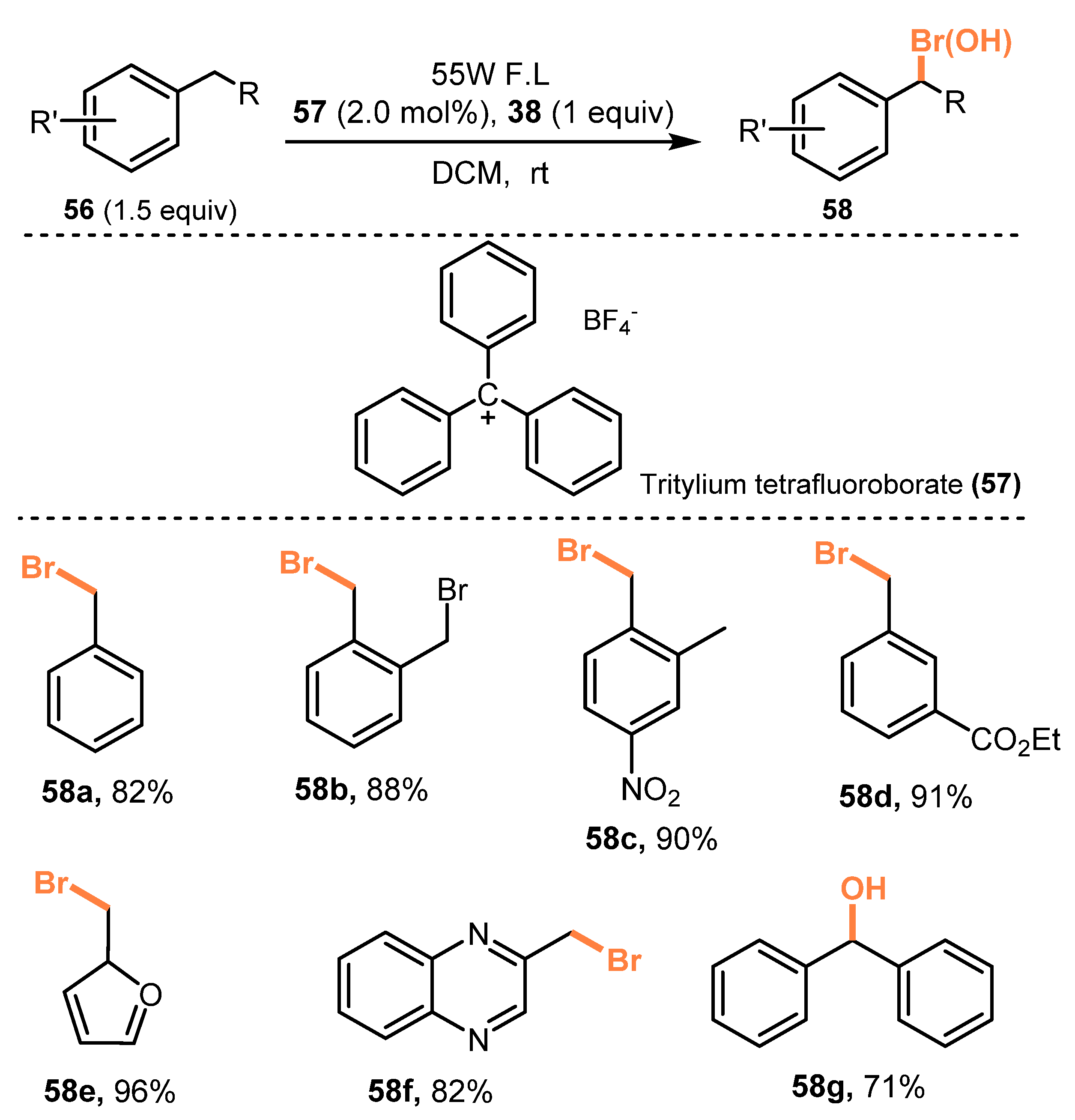
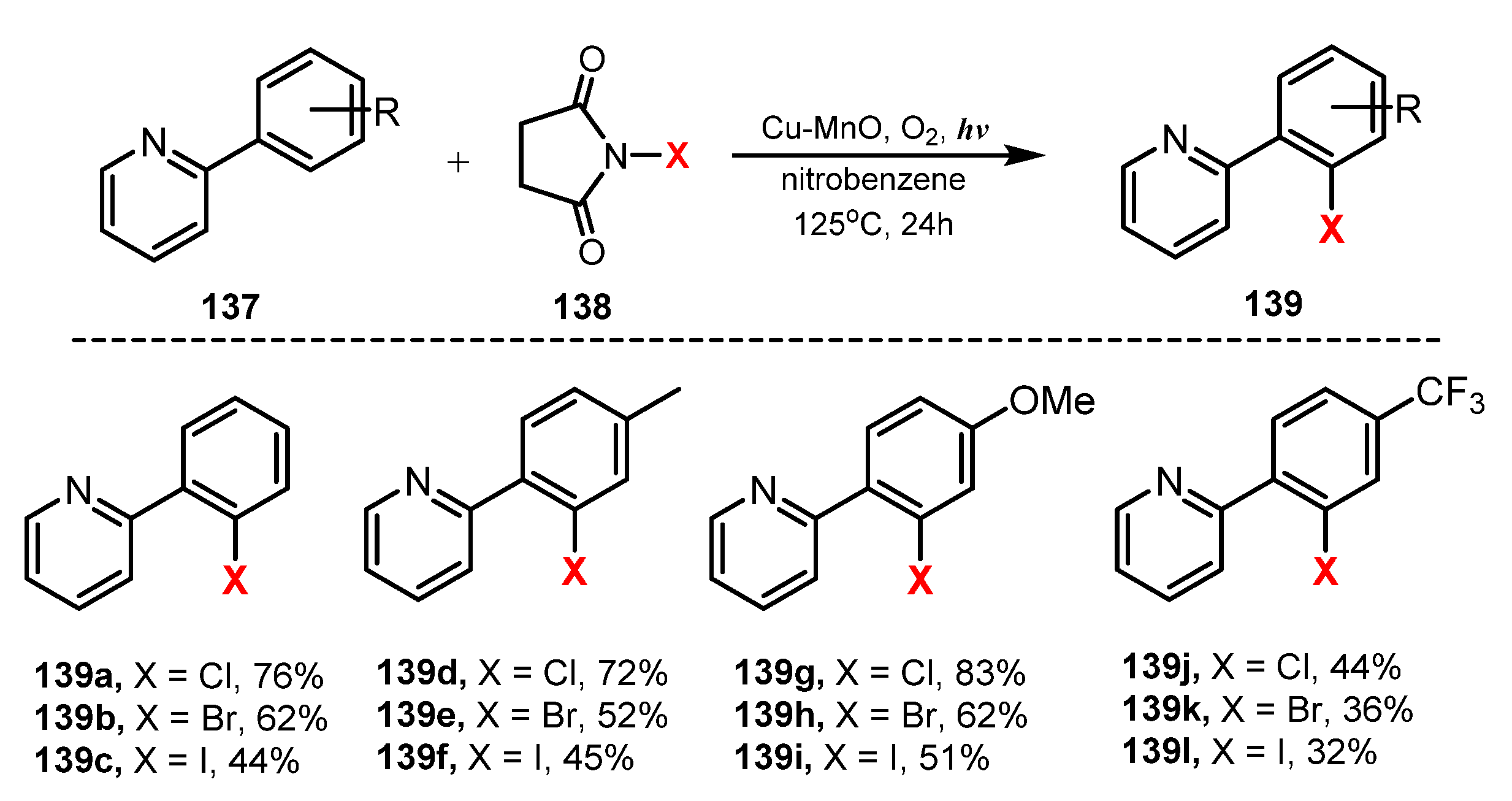
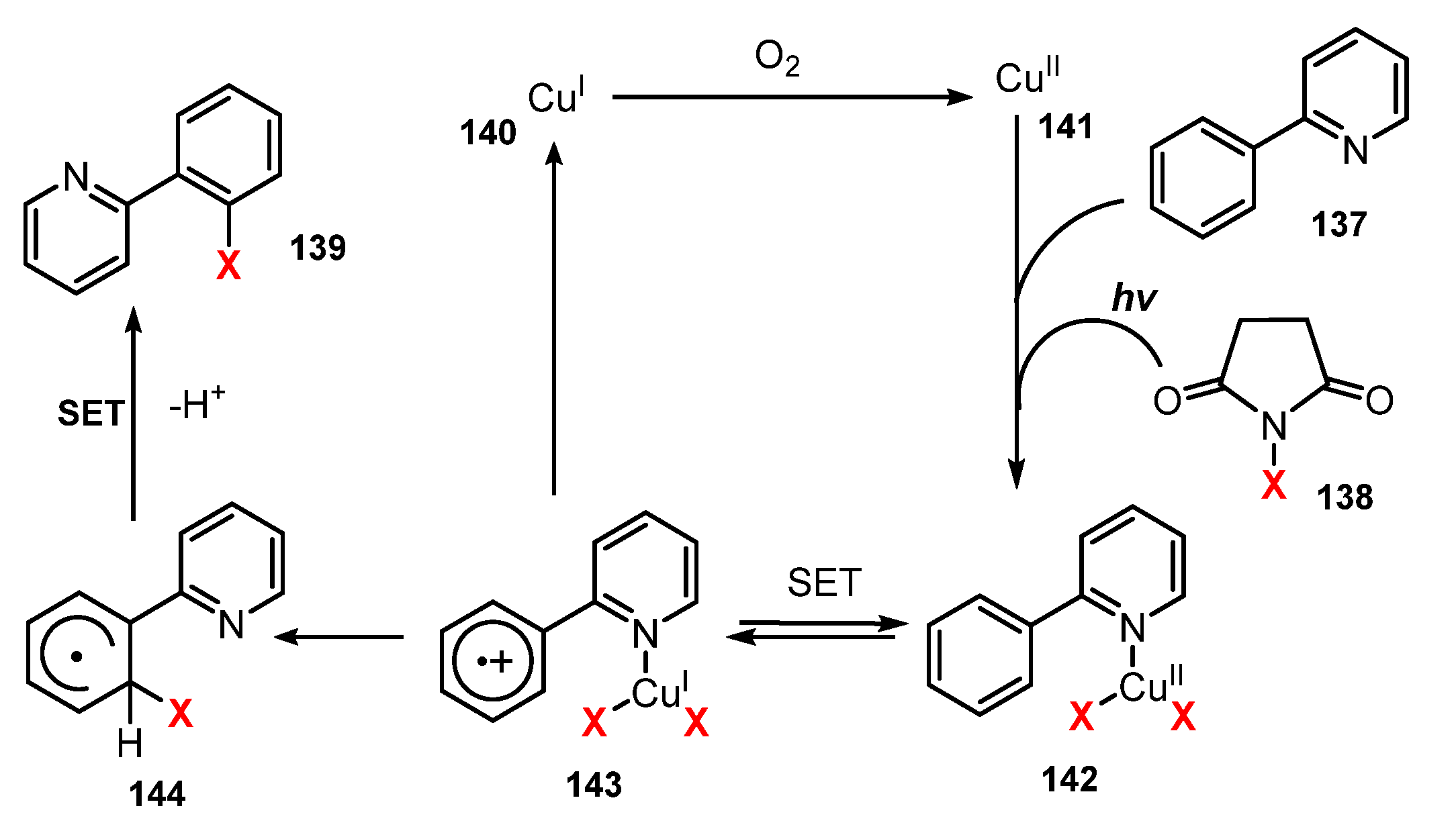
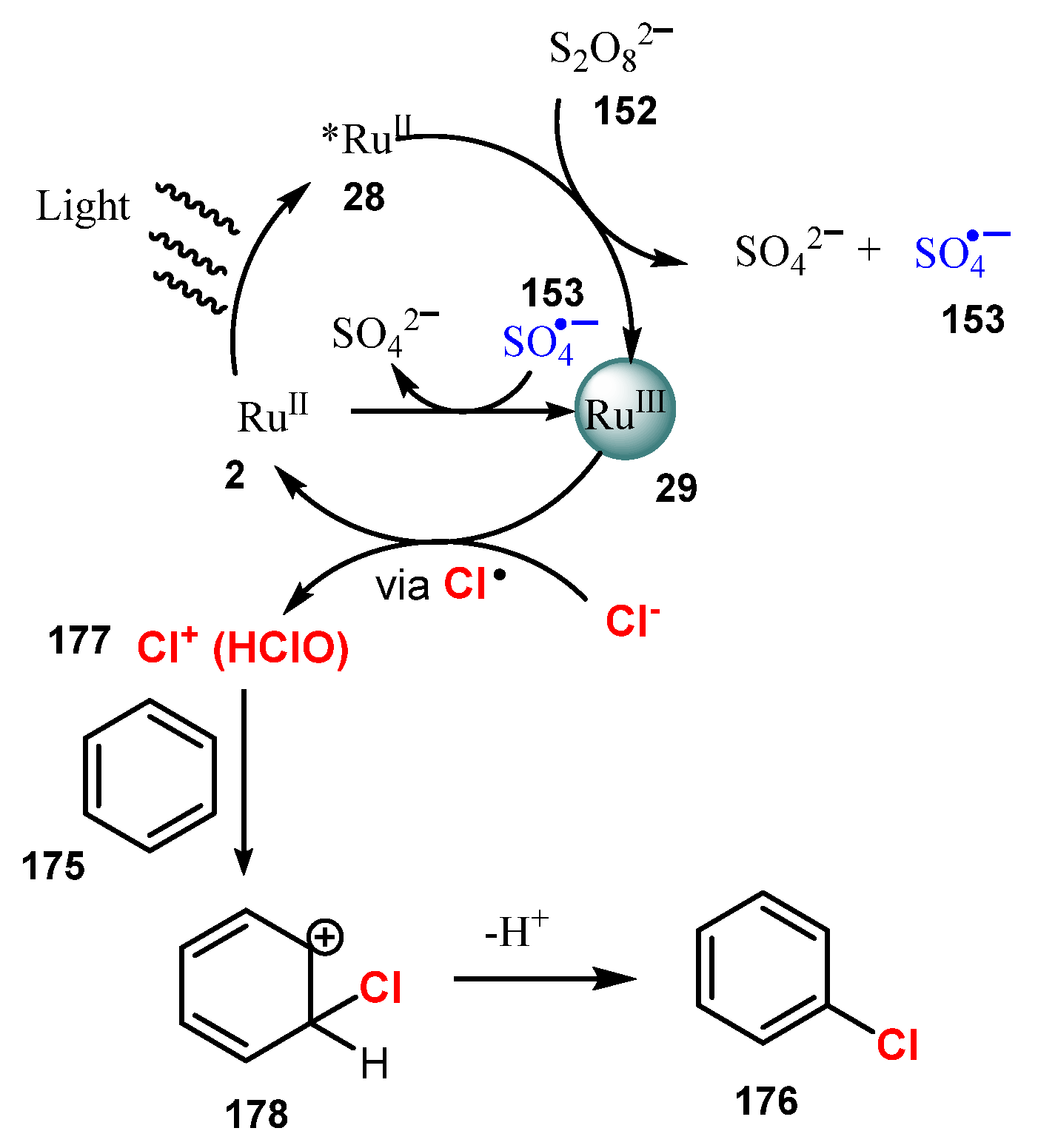
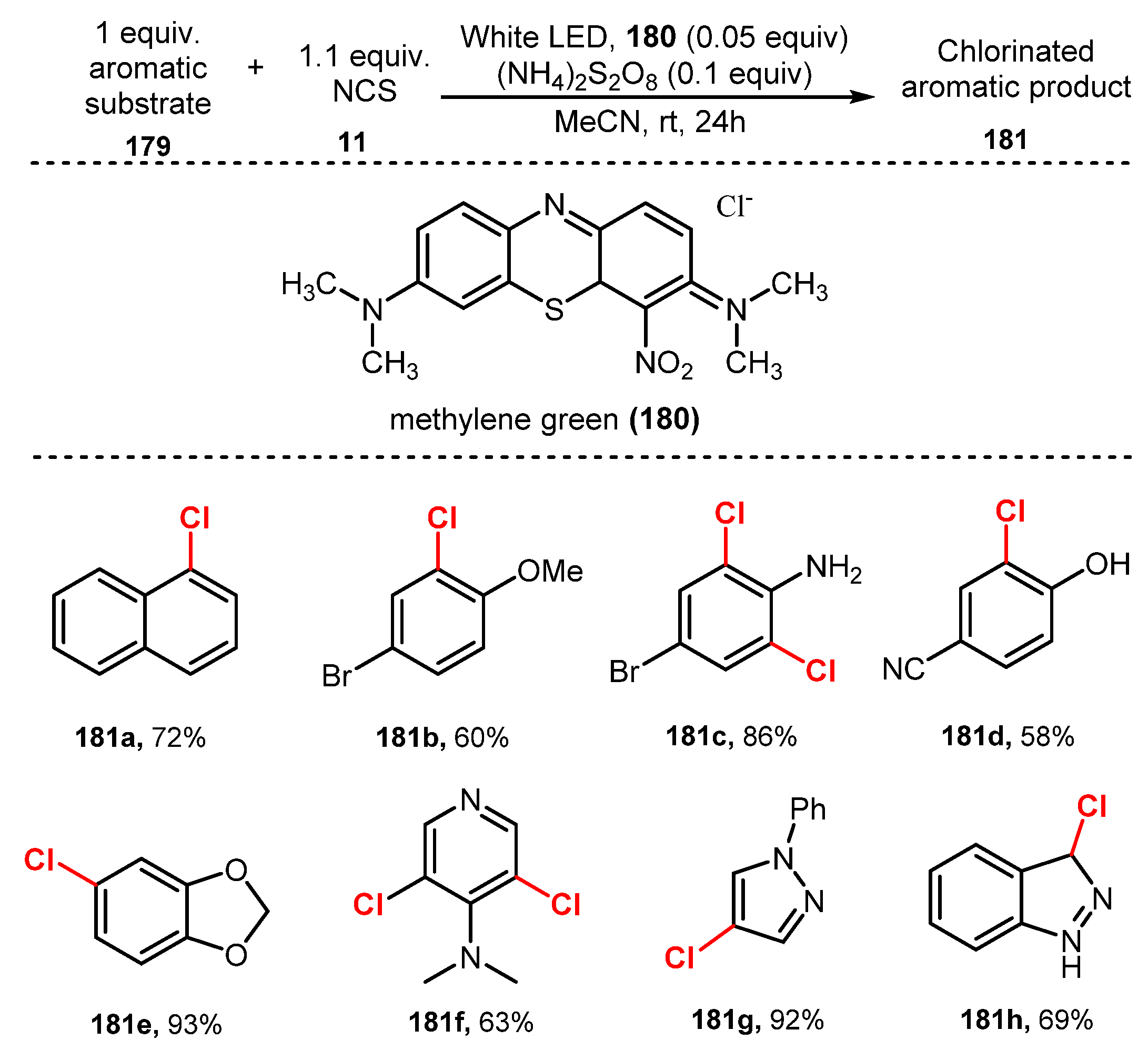
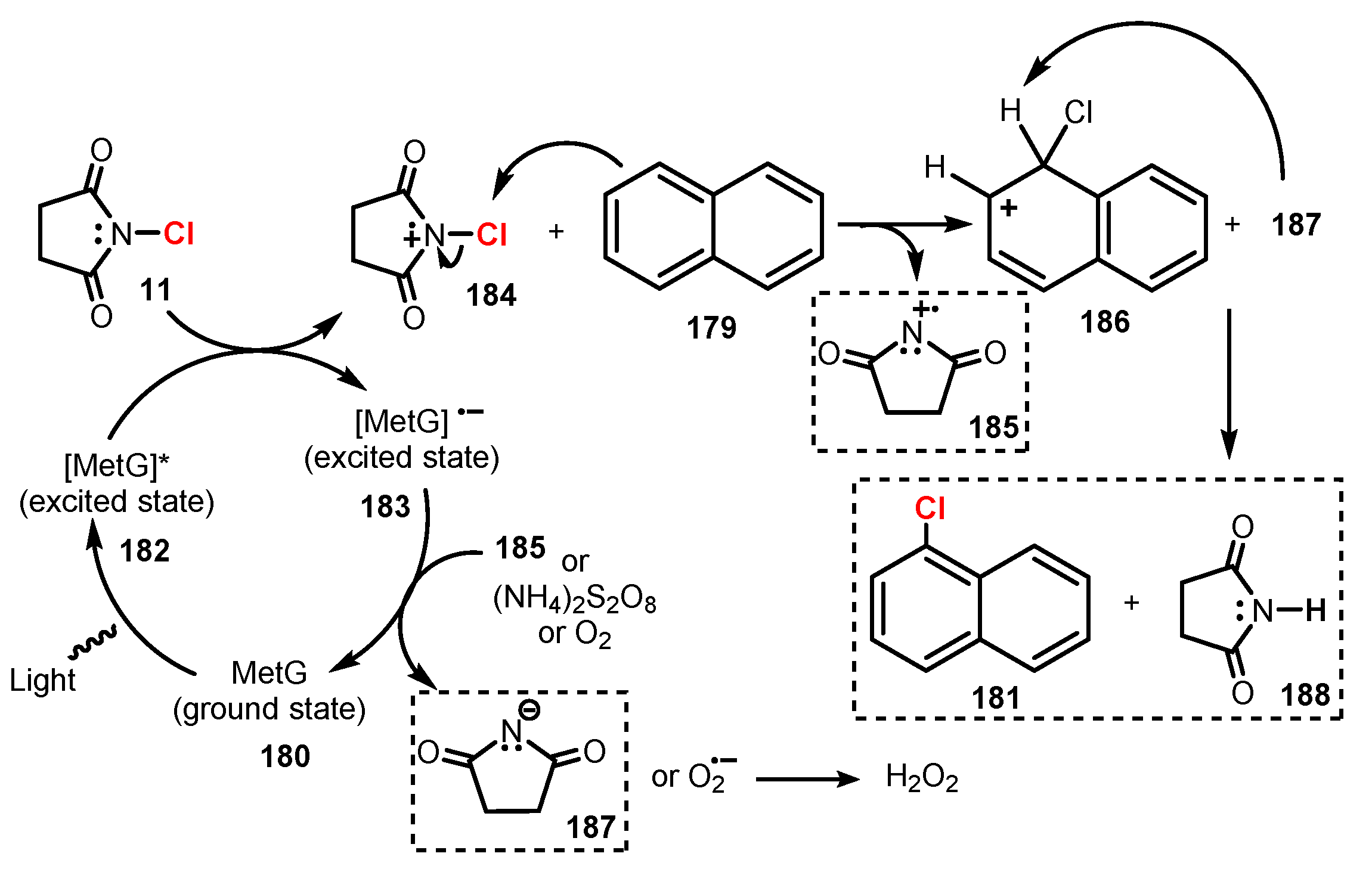
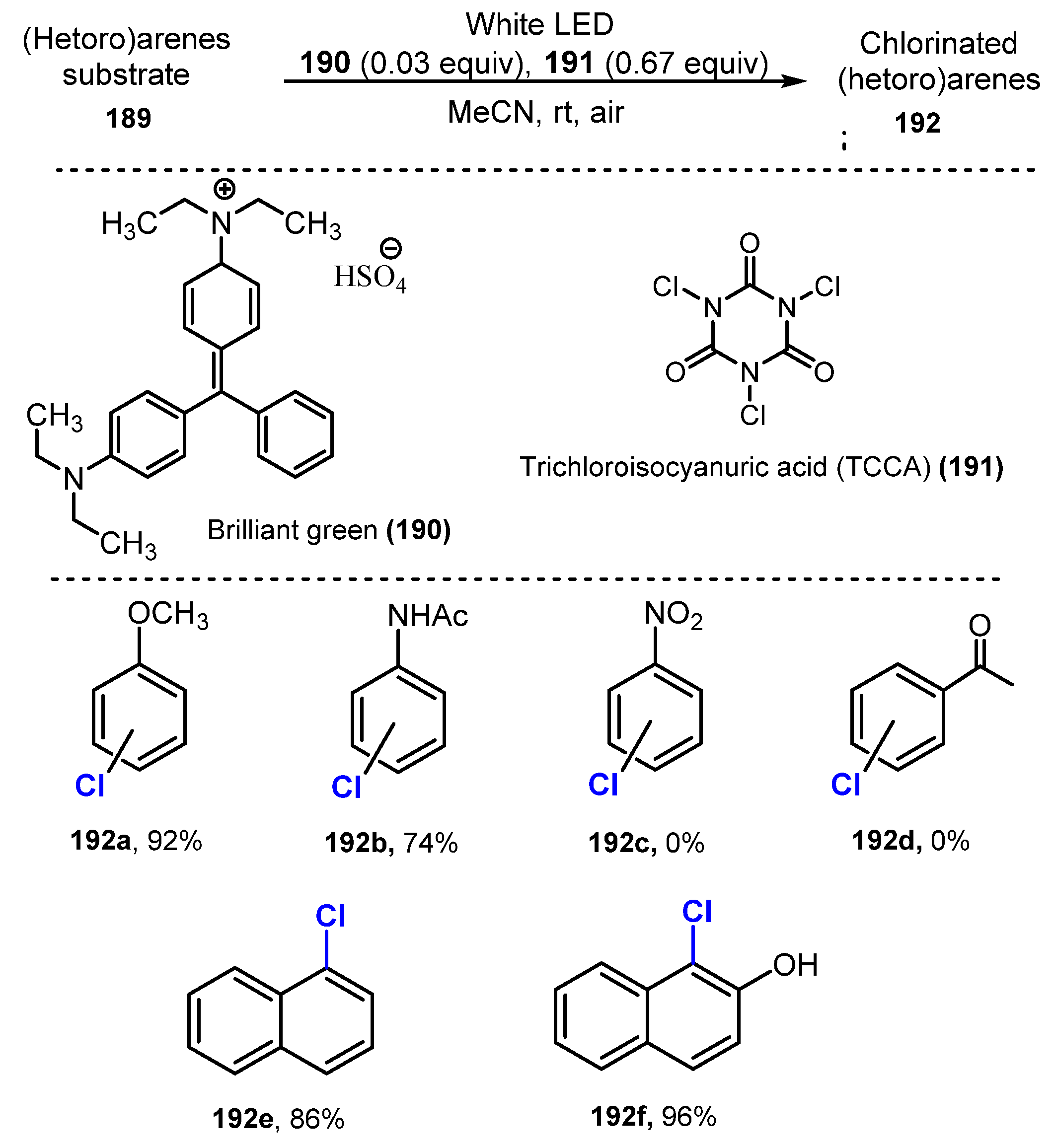
6.2. Chlorination of Aromatic C-H Bonds
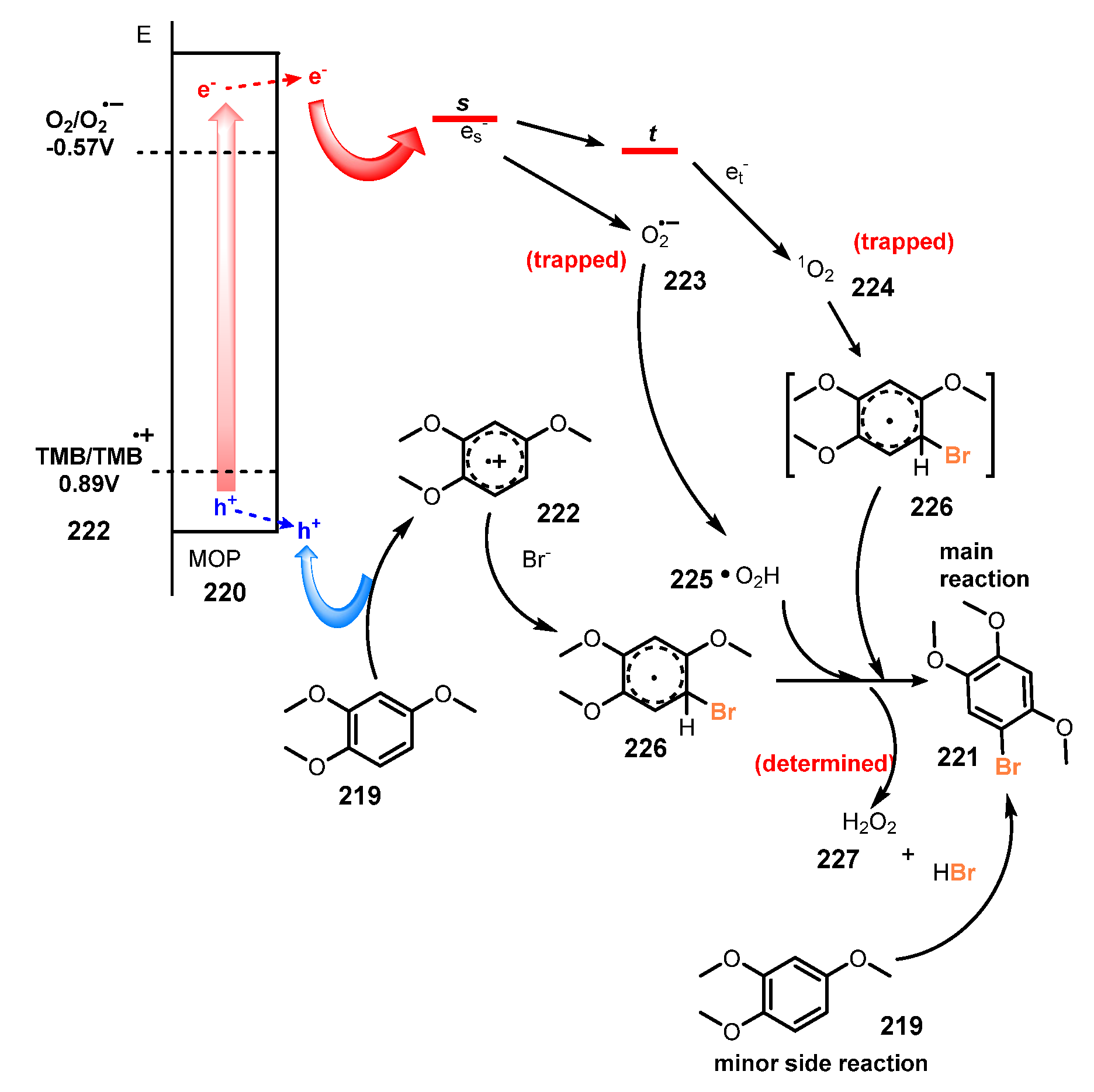
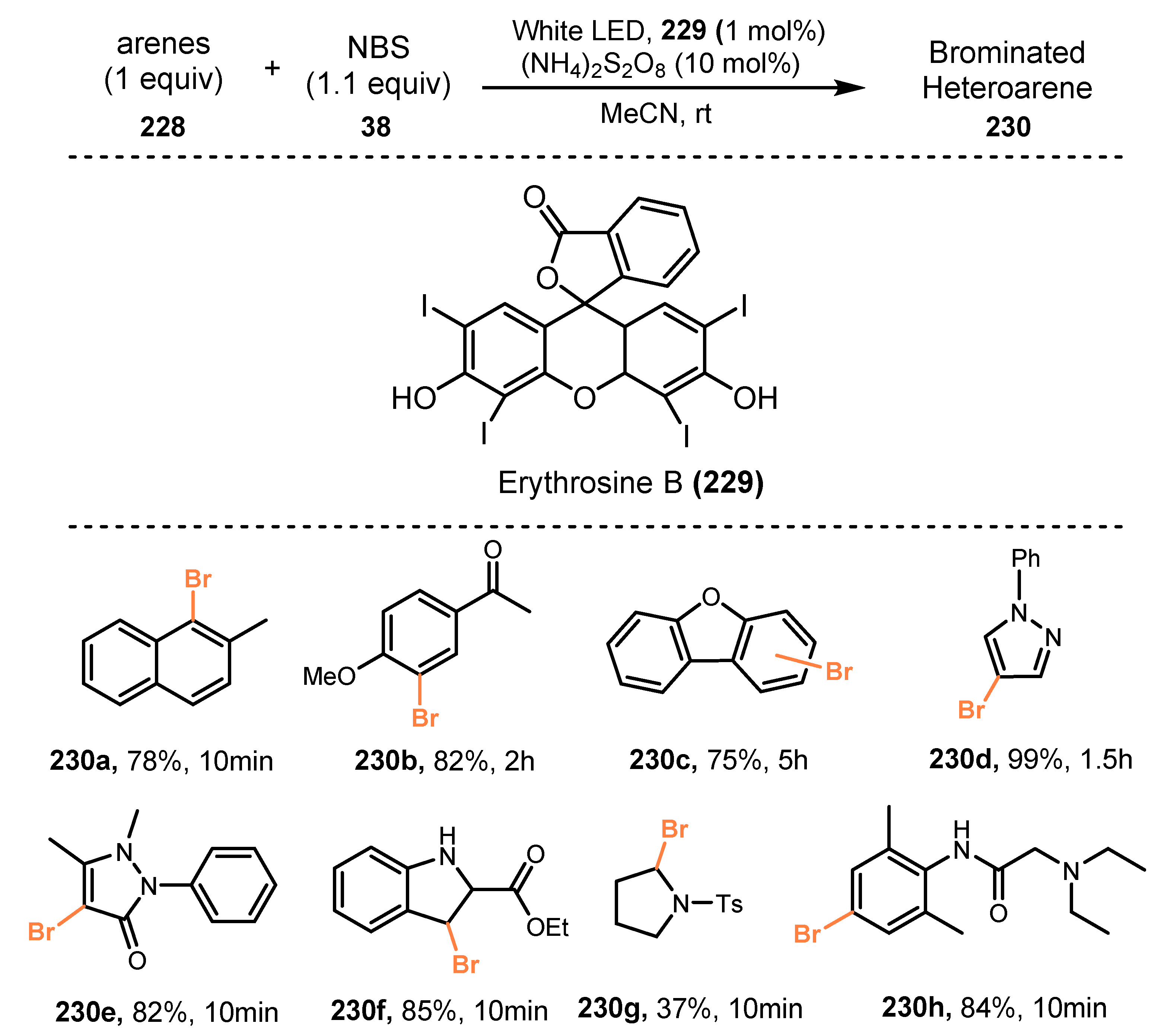
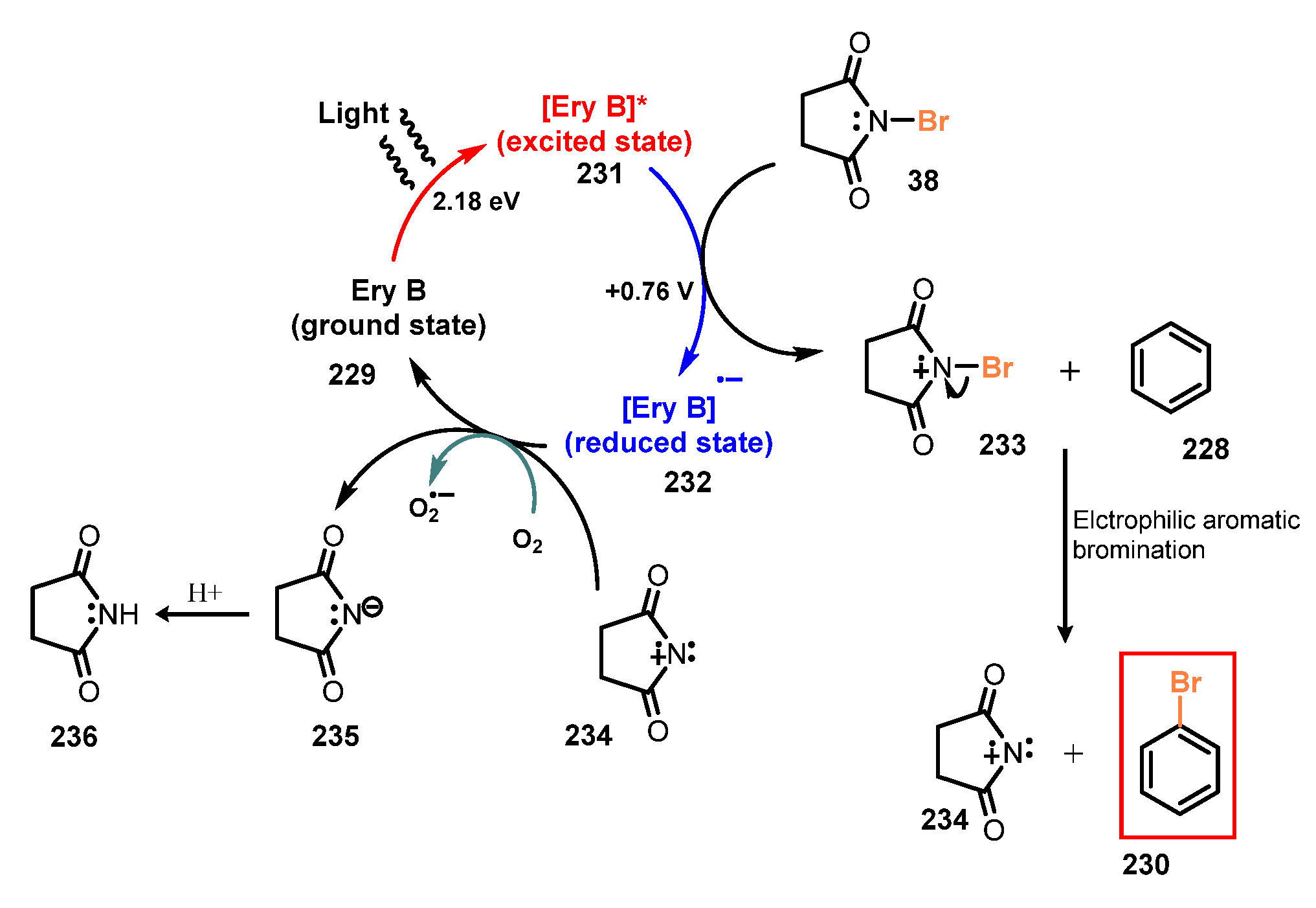
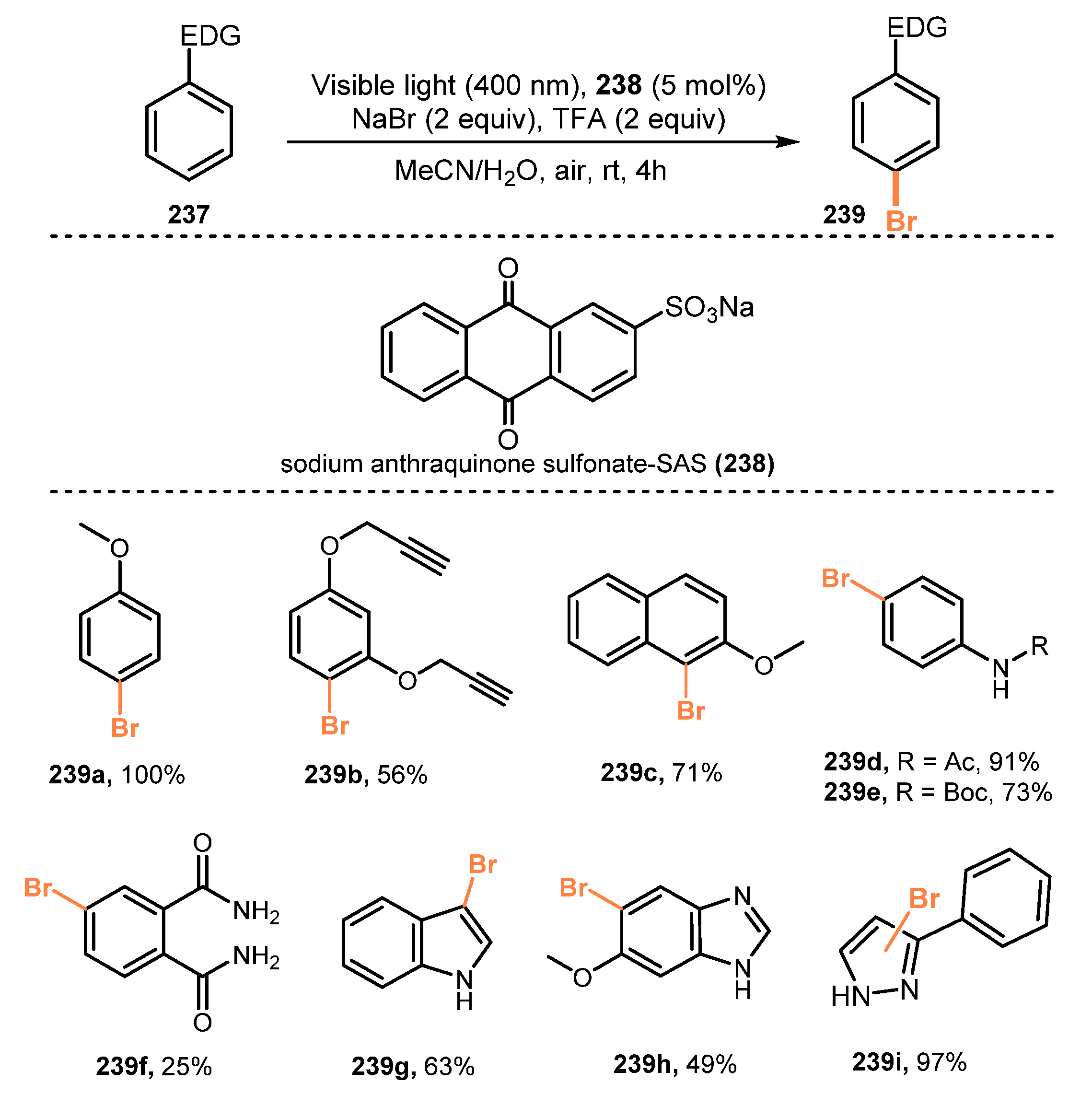
6.3. Bromination of Aromatic C-H Bonds
6.4. Iodination of Aromatic C-H Bonds
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rozhkov, A.V.; Eliseeva, A.A.; Baykov, S.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. One-Pot Route to X-Perfluoroarenes (X = Br, I) Based on FeIII Assisted C−F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding. Cryst. Growth Des. 2020, 20, 5908–5921. [Google Scholar] [CrossRef]
- Nemec, V.; Fotovic, L.; Tomislav, F.; Dominik, C. Large Family of Halogen-Bonded Cocrystals Involving Metal-Organic Building Blocks with Open Coordination Sites. Cryst. Growth Des. 2017, 17, 6169–6173. [Google Scholar] [CrossRef]
- Cotman, A.E.; Guérin, T.; Kovacevic, I.; Tiz, D.B.; Durcik, M.; Fulgheri, F.; Mozina, S.; Secci, D.; Sterle, M.; Ilas, J.; et al. Practical synthesis and application of halogen-doped pyrrole building blocks. ACS Omega 2021, 6, 9723–9730. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Wei, D.; Zhang, J.W.; Li, C.L.; Yu, W.; Han, B. Synthesis of halomethyl isoxazoles/cyclic nitrones via cascade sequence: 1,2-Halogen radical shift as a key link. Org. Lett. 2018, 20, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Constantin, T.; Zanini, M.; Regni, A.; Sheikh, N.S.; Julia, F.; Leonori, D. Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026. [Google Scholar] [CrossRef]
- Kazi, I.; Guha, S.; Sekar, G. Halogen Bond-Assisted Electron-Catalyzed Atom Economic Iodination of Heteroarenes at Room Temperature. J. Org. Chem. 2019, 84, 6642–6654. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking back, Looking forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Latham, J.; Brandenburger, E.; Shepherd, S.A.; Menon, B.R.K.; Micklefield, J. Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev. 2018, 118, 232–269. [Google Scholar] [CrossRef] [Green Version]
- Saccone, M.; Catalano, L. Halogen Bonding beyond Crystals in Materials Science. J. Phys. Chem. B 2019, 123, 9281–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Newly Discovered Naturally Occurring Organohalogens. Arkivoc Part I 2018, 372–410. Available online: http://www.arkat-usa.org/get-file/65071/ (accessed on 1 December 2021). [CrossRef]
- Hennecke, U. New Catalytic Approaches towards the Enantioselective Halogenation of Alkenes. Chem. Asian J. 2012, 7, 456–465. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, M. Visible Light-Mediated Installation of Halogen Functionalities into Multiple Bond Systems. Chem. Sel. 2017, 2, 9136–9146. [Google Scholar] [CrossRef]
- Das, R.; Kapur, M. Transition-Metal-Catalyzed Site-Selective C-H Halogenation Reactions. Asian J. Org. Chem. 2018, 7, 1524–1541. [Google Scholar] [CrossRef]
- Chung, W.J.; Vanderwal, C.D. Stereoselective Halogenation in Natural Product Synthesis. Angew. Chem. Int. Ed. 2016, 55, 4396–4434. [Google Scholar] [CrossRef]
- Tu, H.; Zhu, S.; Qing, F.L.; Chu, L. Visible-Light-Induced Halogenation of Aliphatic C-H Bonds. Tetrahedron Lett. 2018, 59, 173–179. [Google Scholar] [CrossRef]
- Neumann, C.S.; Fujimori, D.G.; Walsh, C.T. Halogenation Strategies in Natural Product Biosynthesis. Chem. Biol. 2008, 15, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podgorsek, A.; Zupan, M.; Iskra, J. Oxidative Halogenation with “Green” Oxidants: Oxygen and Hydrogen Peroxide. Angew. Chem. Int. Ed. 2009, 48, 8424–8450. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Nitelet, A.; Thilmany, P.; Dewez, D.F. Metal-Mediated Halogen Exchange in Aryl and Vinyl Halides: A review. Front. Chem. 2018, 6, 114. [Google Scholar] [CrossRef]
- Fredricks, P.S.; Tedde, J.M. Free-Radical Xubstitution in Aliphatic Compounds. Part II. Halogenation of the N-Butyl Halides. J. Chem. Soc. 1960, 144–150. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; Burkhard, K. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Cavedon, C.; Seeberger, P.H.; Pieber, B. Photochemical Strategies for Carbon–Heteroatom Bond Formation. Eur. J. Org. Chem. 2019, 10, 1379–1392. [Google Scholar] [CrossRef] [Green Version]
- Fukuzumi, S.; Ohkubo, K. Organic Synthetic Transformations Using Organic Dyes as Photoredox Catalysts. Org. Biomol. Chem. 2014, 12, 6059. [Google Scholar] [CrossRef] [Green Version]
- Koike, T. Frontiers in Radical Fluoromethylation by Visible-Light Organic Photocatalysis. Asian J. Org. Chem. 2020, 9, 529–537. [Google Scholar] [CrossRef]
- Bui, T.T.; Hong, W.P.; Kim, H.K. Recent Advances in Visible Light-Mediated Fluorination. J. Fluor. Chem. 2021, 247, 109794. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.X.; Yang, G.; He, G.; Chen, G. A Visible-Light-Promoted Radical Reaction System for Azidation and Halogenation of Tertiary Aliphatic C-H Bonds. Chem. Sci. 2016, 7, 2679–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
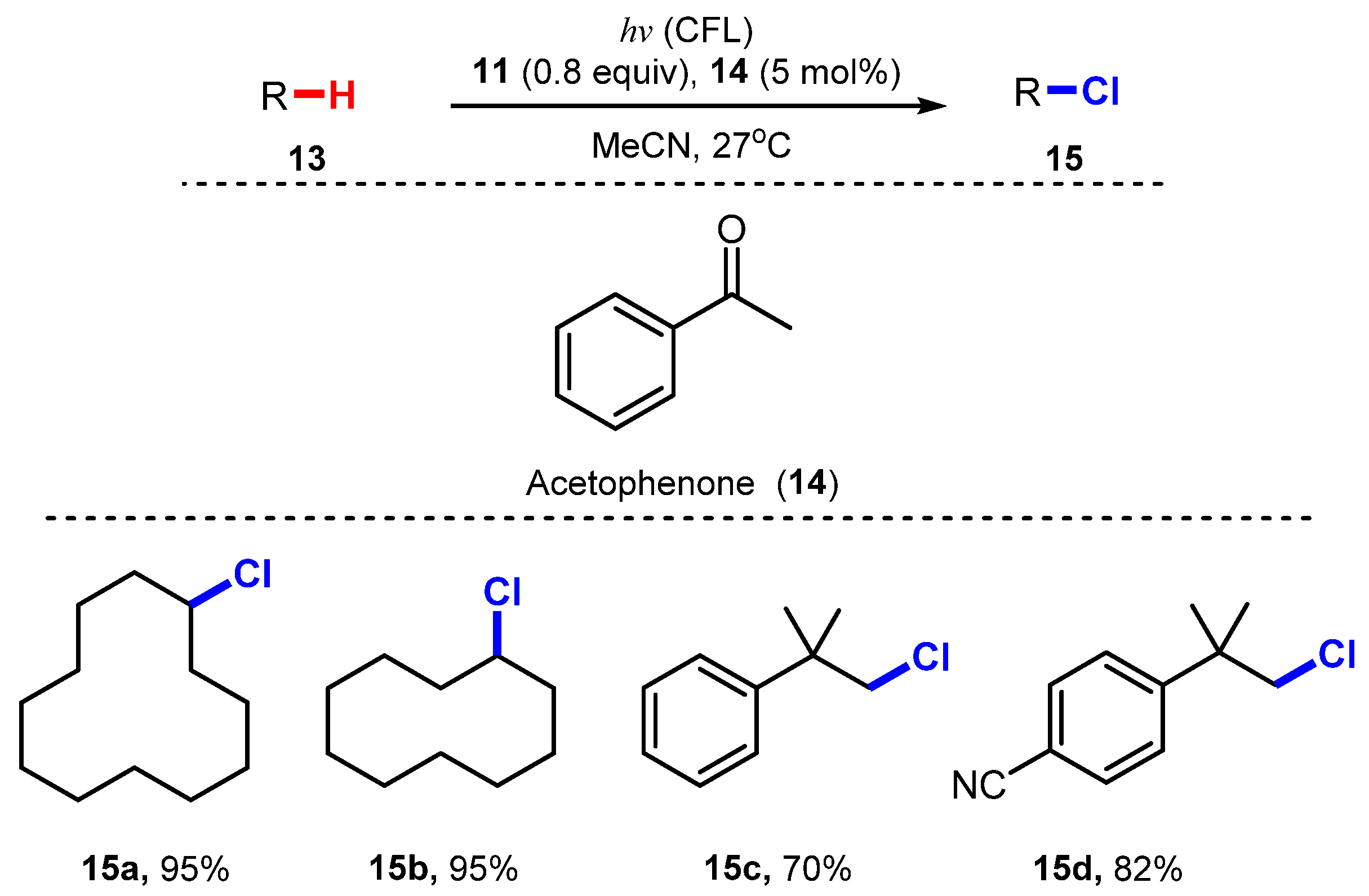
- Han, L.; Xia, J.B.; You, L.; Chen, C. Ketone-Catalyzed Photochemical C(sp3)-H Chlorination. Tetrahedron 2017, 73, 3696–3701. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Zhou, C.; Yang, X.L.; Chen, B.; Tung, C.H.; Wu, L.Z. Visible Light-Catalyzed Benzylic C-H Bond Chlorination by a Combination of Organic Dye (Acr+-Mes) and N-Chlorosuccinimide. J. Org. Chem. 2020, 85, 9080–9087. [Google Scholar] [CrossRef]
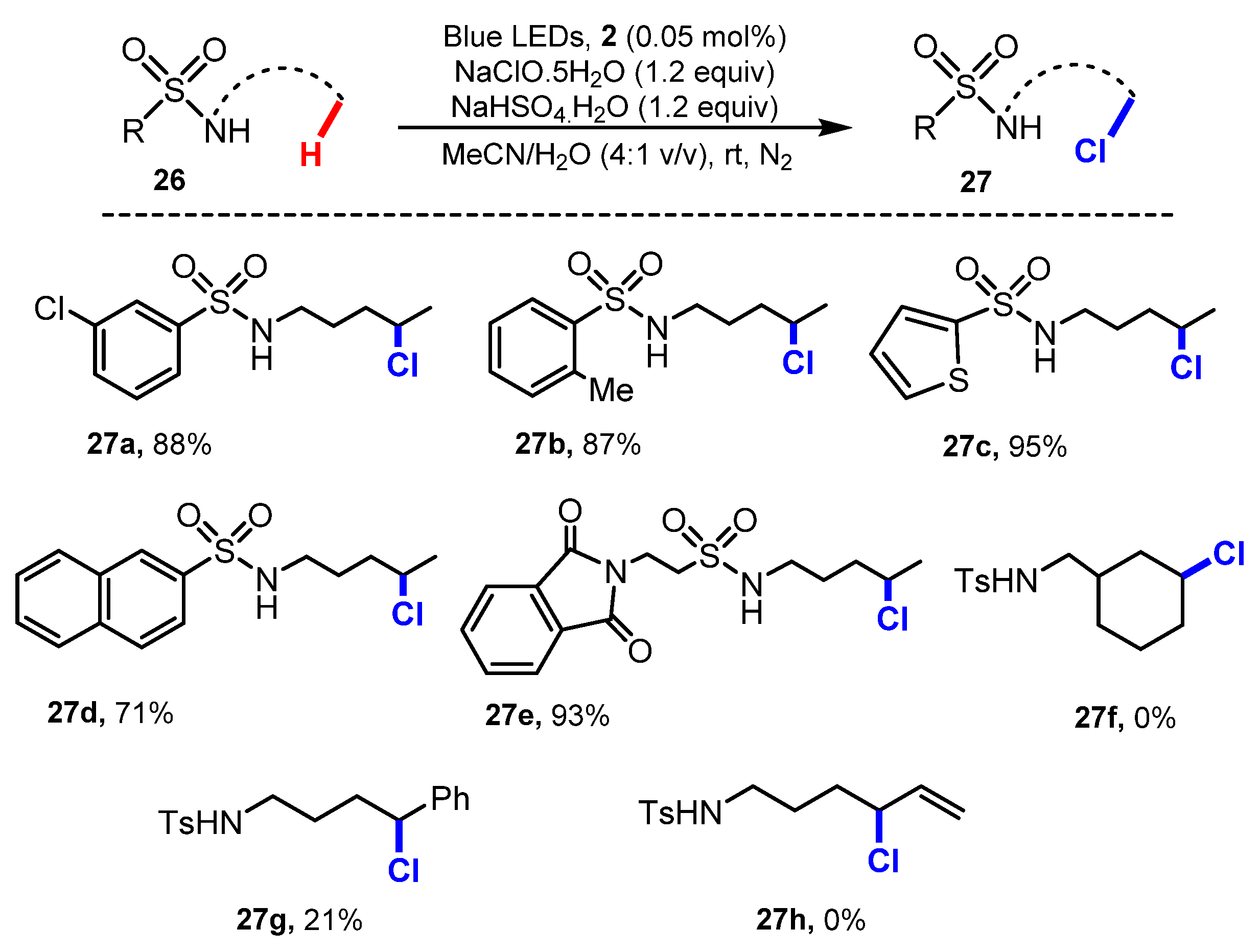
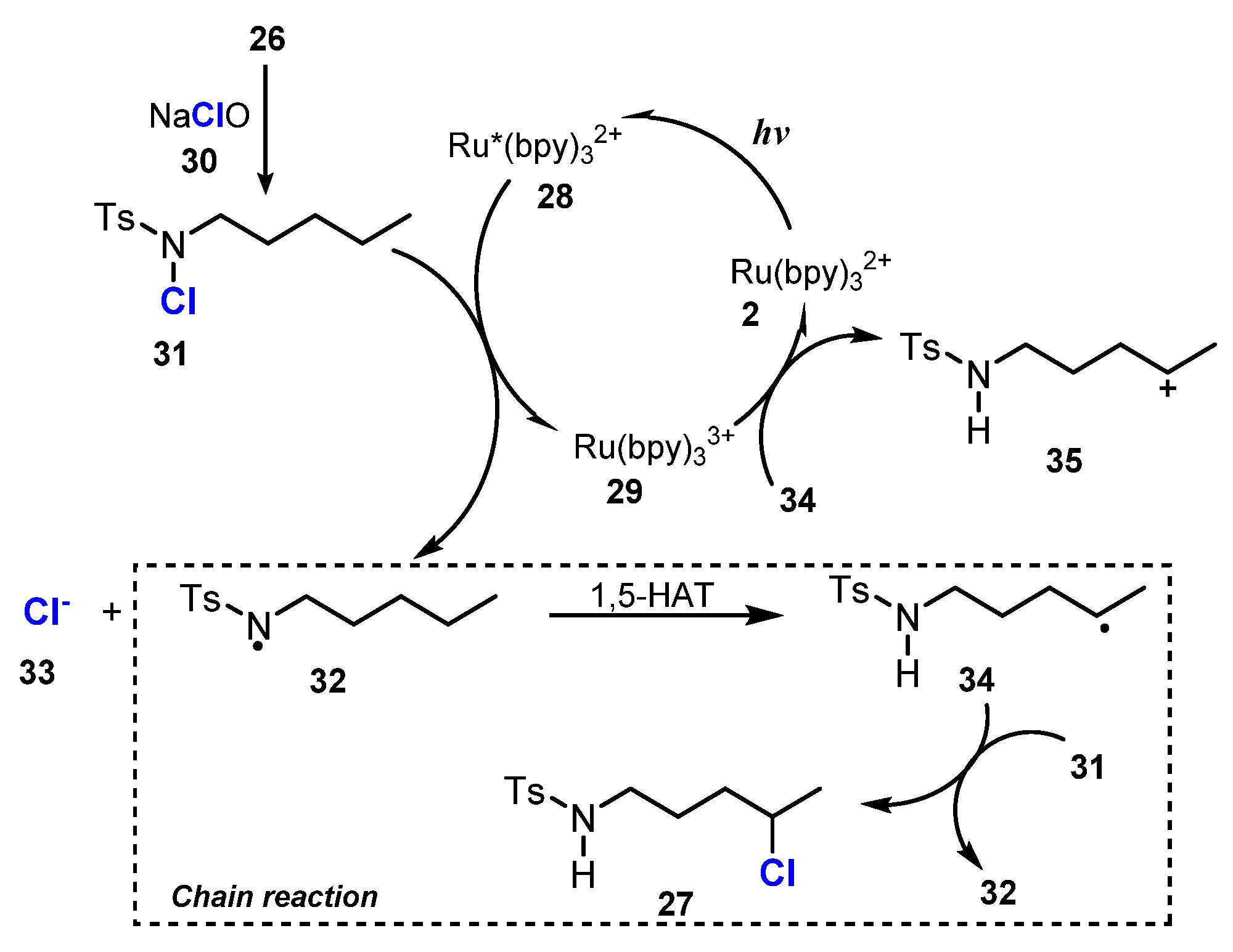
- Zhu, Y.; Wang, J.J.; Wu, D.; Yu, W. Visible-Light-Driven Remote C-H Chlorination of Aliphatic Sulfonamides with Sodium Hypochlorite. Asian J. Org. Chem. 2020, 9, 1650–1654. [Google Scholar] [CrossRef]
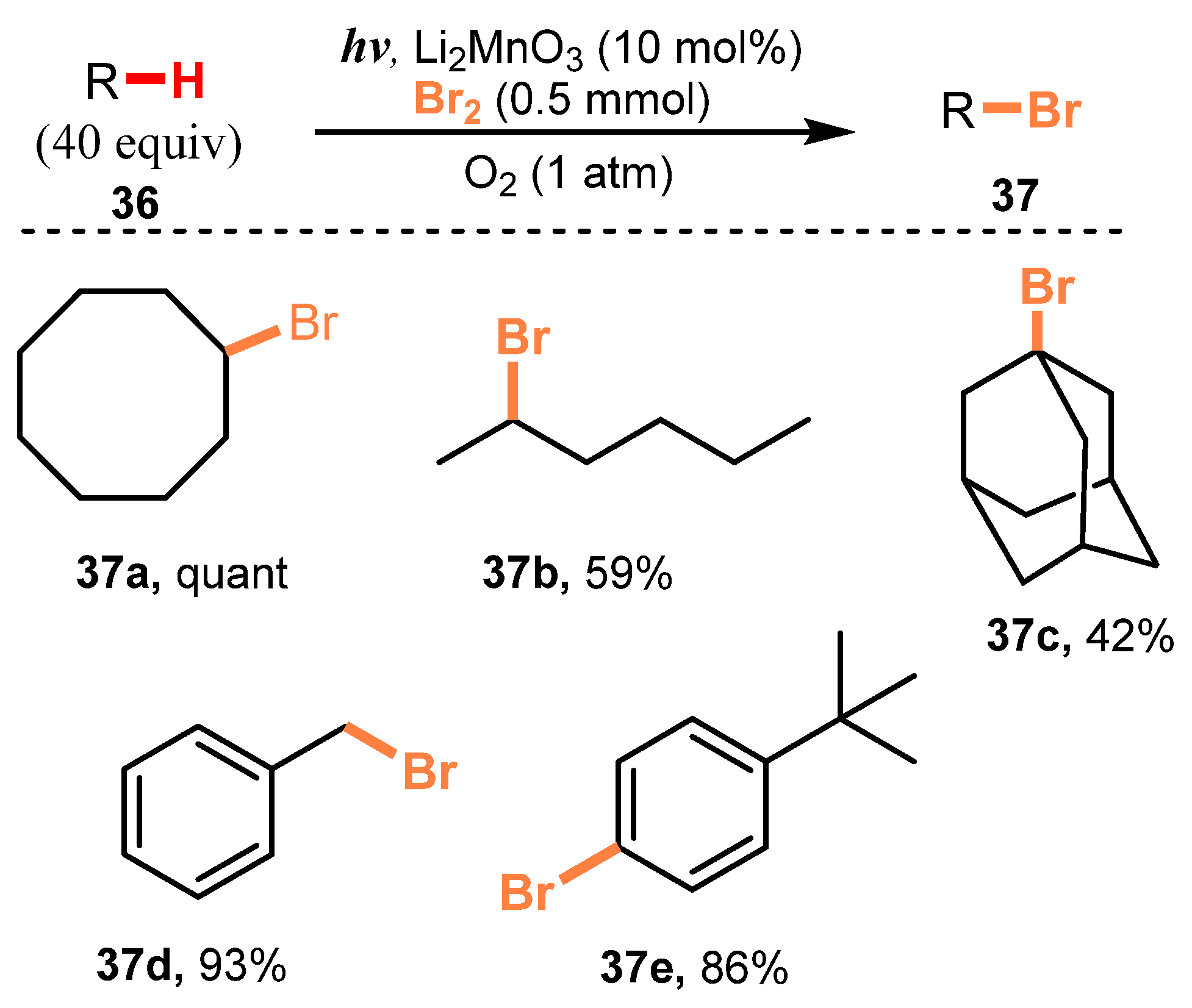
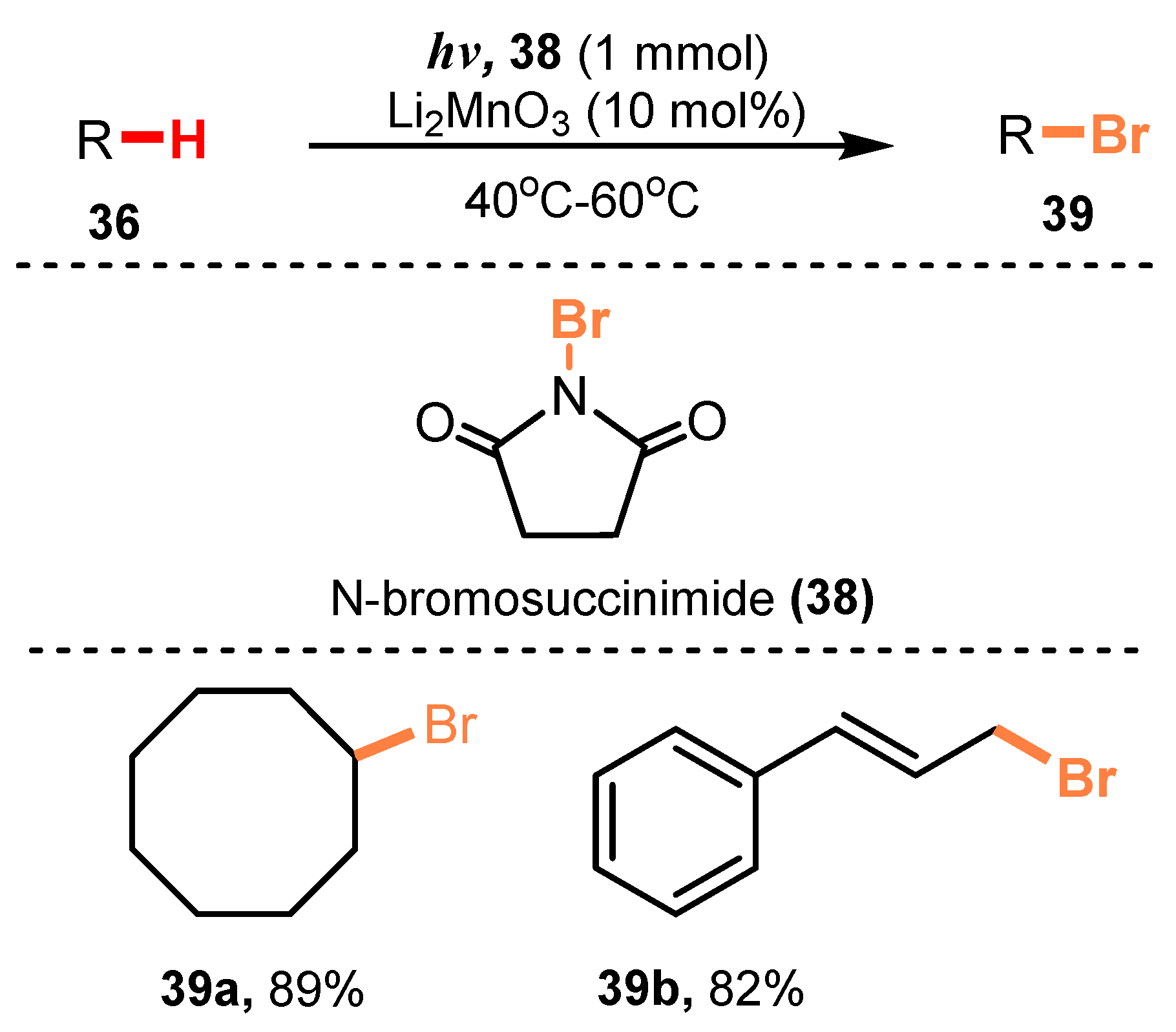
- Nishina, Y.; Morita, J.; Ohtani, B. Direct Bromination of Hydrocarbons Catalyzed by Li2MnO3 under Oxygen and Photo-Irradiation Conditions. RSC Adv. 2013, 3, 2158–2162. [Google Scholar] [CrossRef] [Green Version]
- Kee, C.W.; Chan, K.M.; Wong, M.W.; Tan, C.H. Selective Bromination of sp3 C-H Bonds by Organophotoredox Catalysis. Asian J. Org. Chem. 2014, 3, 536–544. [Google Scholar] [CrossRef]
- Ni, S.; El Remaily, M.A.E.A.A.A.; Franzén, J. Carbocation Catalyzed Bromination of Alkyl Arenes, a Chemoselective sp3 vs. sp2 C-H Functionalization. Adv. Synth. Catal. 2018, 360, 4197–4204. [Google Scholar] [CrossRef]
- Wilger, D.J.; Grandjean, J.M.M.; Lammert, T.R.; Nicewicz, D.A. The Direct Anti-Markovnikov Addition of Mineral Acids to Styrenes. Nat. Chem. 2014, 6, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Malpani, Y.R.; Ha, N.; Jung, Y.S.; Han, S.B. Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis. Org. Lett. 2014, 16, 1310–1313. [Google Scholar] [CrossRef]
- Tang, X.J.; Dolbier, W.R. Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-Deficient Alkenes Inducedby Visible Light. Angew. Chem. Int. Ed. 2015, 54, 4246–4249. [Google Scholar] [CrossRef]
- Lian, P.; Long, W.; Li, J.; Zheng, Y.; Wan, X. Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2. Angew. Chem. Int. Ed. 2020, 59, 23603–23608. [Google Scholar] [CrossRef]
- Nguyen, J.D.; Tucker, J.W.; Konieczynska, M.D.; Stephenson, C.R.J. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, L.; Wang, Z.; Li, P.; Zhang, Y. A Practical Synthesis of α-Bromo/Iodo/Chloroketones from Olefins under Visible-Light Irradiation Conditions. Chin. Chem. Lett. 2021, 32, 429–432. [Google Scholar] [CrossRef]
- Dai, C.; Narayanam, J.M.R.; Stephenson, C.R.J. Visible-Light-Mediated Conversion of Alcohols to Halides. Nat. Chem. 2011, 3, 140–145. [Google Scholar] [CrossRef]
- Li, R.; Gehrig, D.W.; Ramanan, C.; Blom, P.W.M.; Kohl, F.F.; Wagner, M.; Landfester, K.; Zhang, K.A.I. Visible-Light-Mediated Conversion of Alcohols to Bromides by a Benzothiadiazole-Containing Organic Photocatalyst. Adv. Synth. Catal. 2019, 361, 3852–3859. [Google Scholar] [CrossRef]
- Candish, L.; Standley, E.A.; Suárez, A.G.; Mukherjee, S.; Glorius, F. Catalytic Access to Alkyl Bromides, Chlorides and Iodides via Visible Light-Promoted Decarboxylative Halogenation. Chem. Eur. J. 2016, 22, 9971–9974. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Koyamada, K.; Miyamoto, K.; Kanazawa, J.; Uchiyama, M. Decarboxylative Bromination of Sterically Hindered Carboxylic Acids with Hypervalent Iodine(III) Reagents. Org. Process. Res. Dev. 2020, 24, 1328–1334. [Google Scholar] [CrossRef]
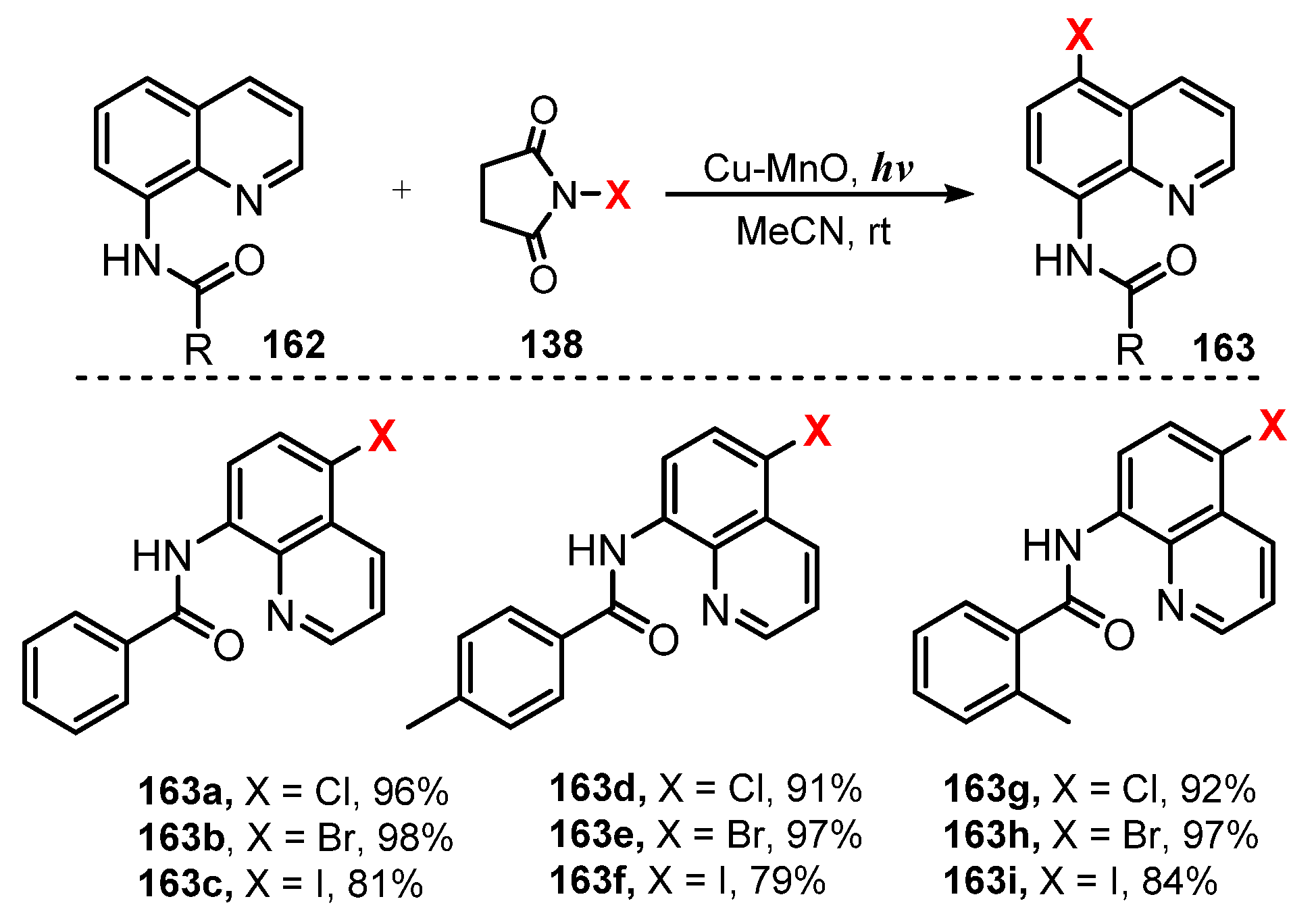
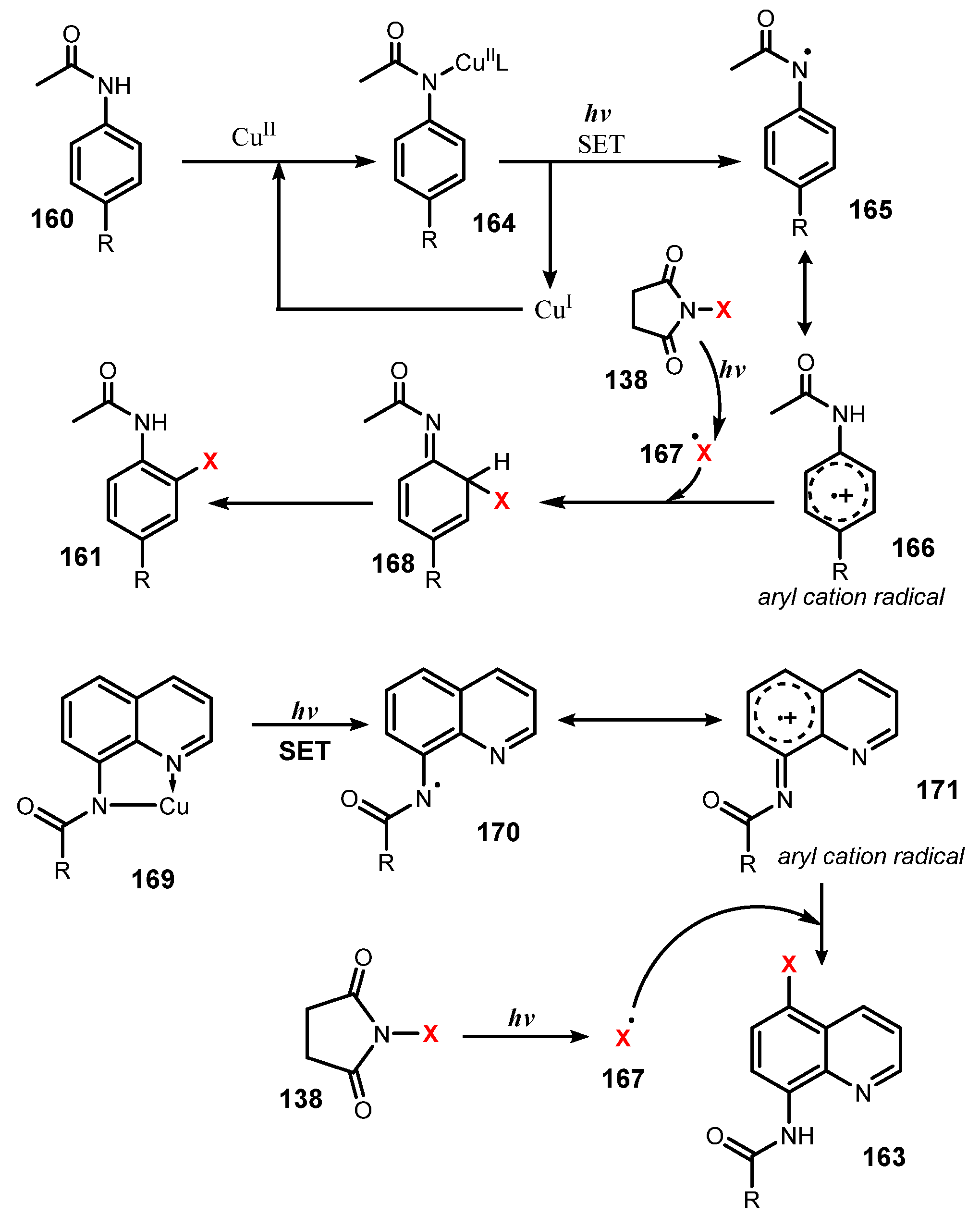
- Pal, P.; Singh, H.; Panda, A.B.; Ghosh, S.C. Heterogeneous Cu-MnO Catalyzed Monoselective Ortho-Halogenation of Aromatic C-H Bonds under Visible Light. Asian J. Org. Chem. 2015, 4, 879–883. [Google Scholar] [CrossRef]
- Wang, X.C.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J.Q. Pd (II)-Catalyzed C-H Iodination Using Molecular I2 as the Sole Oxidant. J. Am. Chem. Soc. 2013, 135, 10326–10329. [Google Scholar] [CrossRef] [Green Version]
- Mo, F.; Yan, J.M.; Qiu, D.; Li, F.; Zhang, Y.; Wang, J. Gold-Catalyzed Halogenation of Aromatics by N-Halosuccinimides. Angew. Chem. Int. Ed. 2010, 49, 2028–2032. [Google Scholar] [CrossRef]
- Wang, L.; Ackermann, L. Ruthenium-Catalyzed Ortho-C-H Halogenations of Benzamides. Chem. Commun. 2014, 50, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.G.; Gensch, T.; de Azambuja, F.; Vasquez-Cespedes, S.; Glorius, F. Co (III)-Catalyzed C-H Activation/Formal SN-type Reactions: Selective and Efficient Cyanation, Halogenation, and Allylation. J. Am. Chem. Soc. 2014, 136, 17722–17725. [Google Scholar] [CrossRef]
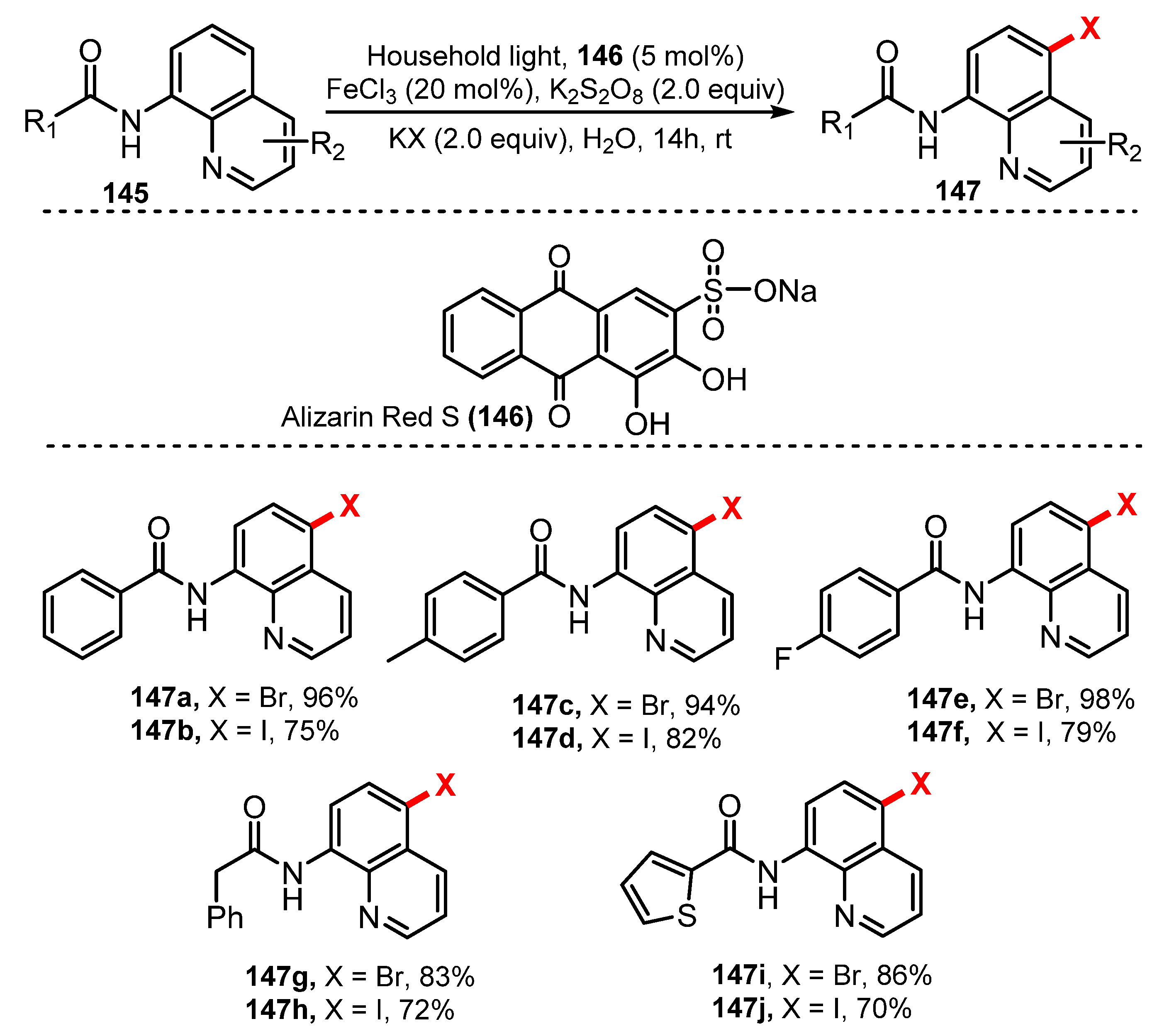
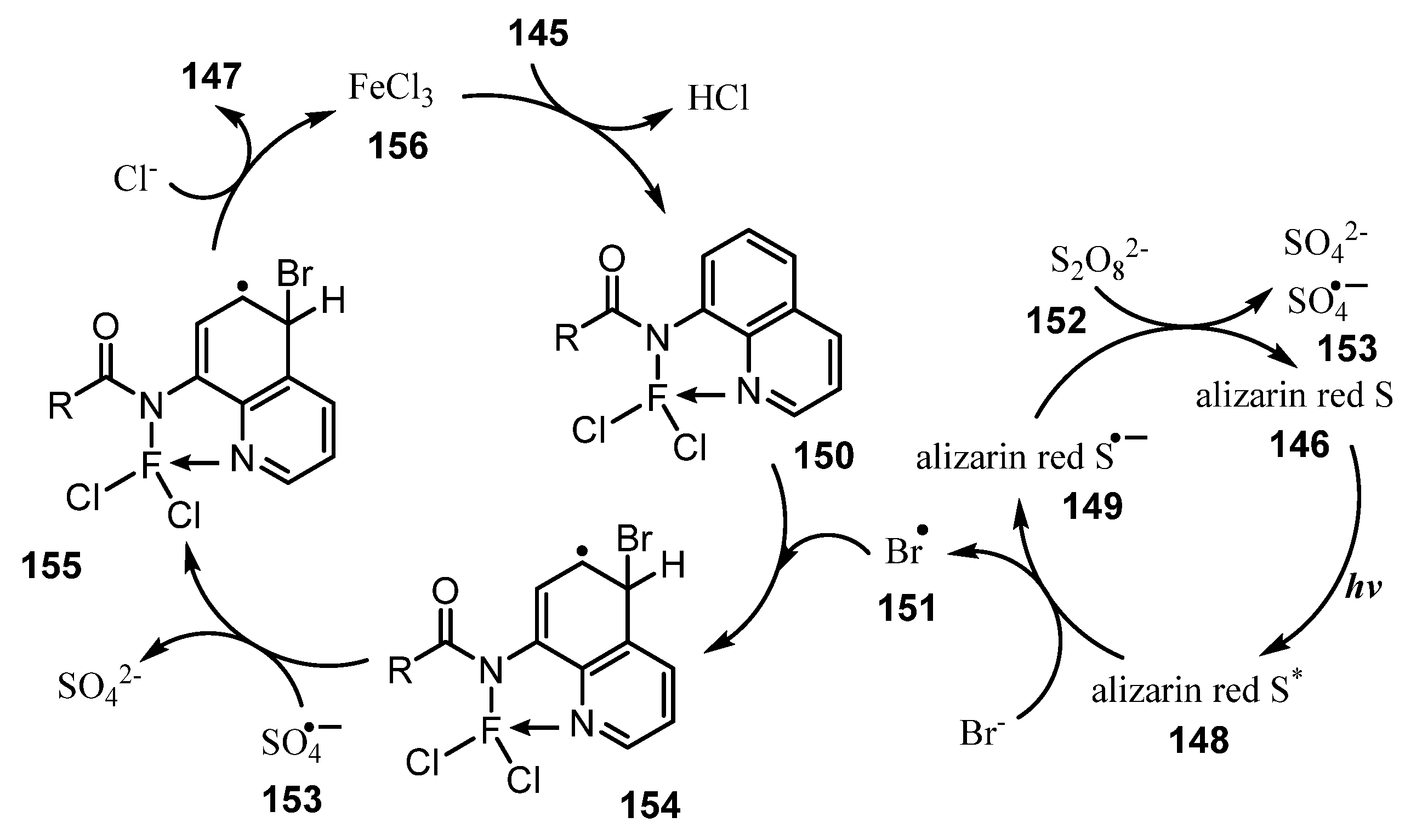
- Qiao, H.; Sun, S.; Yang, F.; Zhu, Y.; Kang, J.; Wu, Y.; Wu, Y. Merging Photoredox Catalysis with Iron (III) Catalysis: C5-H Bromination and Iodination of 8-Aminoquinoline Amides in Water. Adv. Synth. Catal. 2017, 359, 1976–1980. [Google Scholar] [CrossRef]
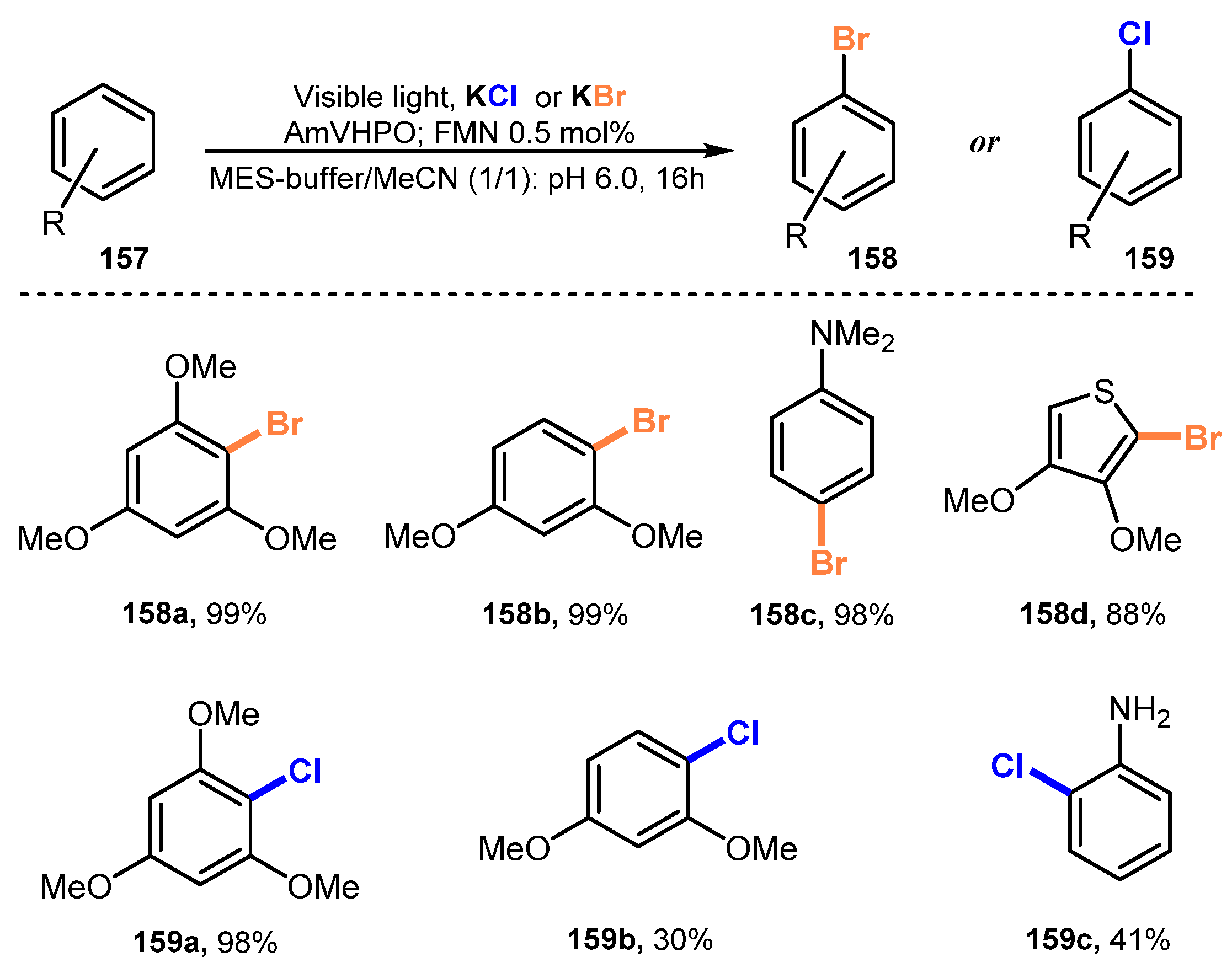
- Seel, C.J.; Králík, A.; Hacker, M.; Frank, A.; König, B.; Gulder, T. Atom-Economic Electron Donors for Photobiocatalytic Halogenations. ChemCatChem 2018, 10, 3960–3963. [Google Scholar] [CrossRef]
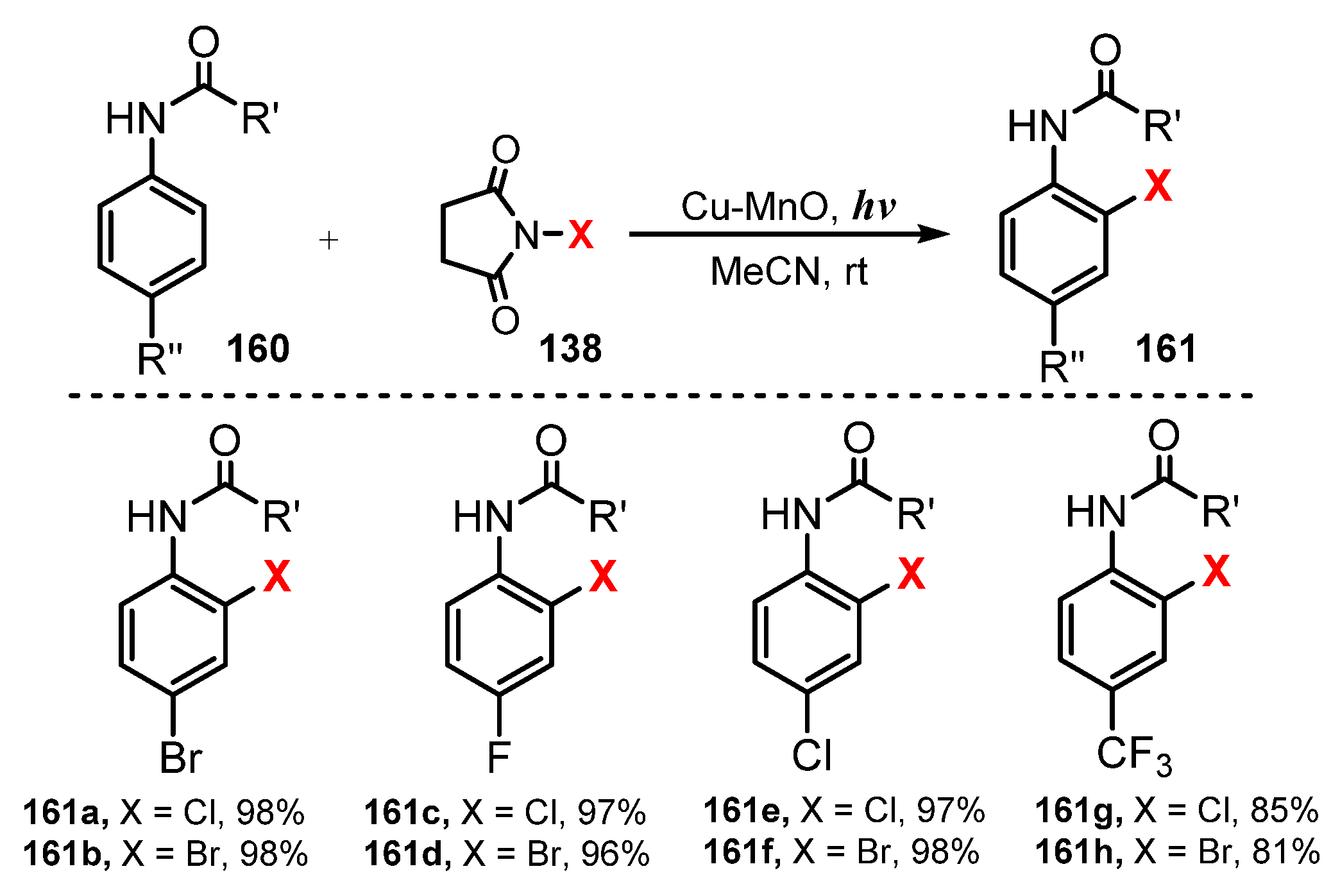
- Singh, H.; Sen, C.; Sahoo, T.; Ghosh, S.C. A Visible Light-Mediated Regioselective Halogenation of Anilides and Quinolines by Using a Heterogeneous Cu-MnO Catalyst. Eur. J. Org. Chem. 2018, 34, 4748–4753. [Google Scholar] [CrossRef]
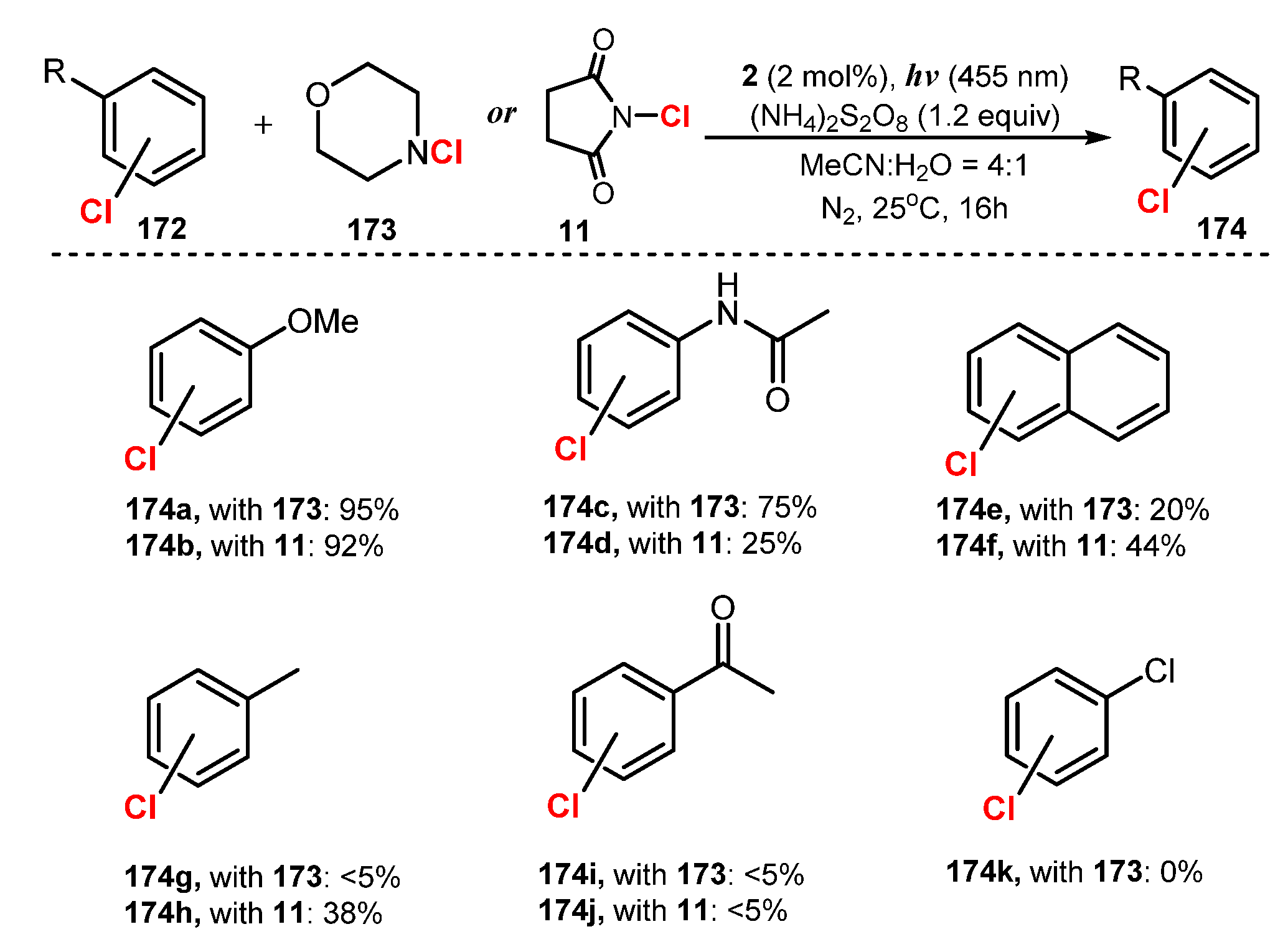
- Hering, T.; König, B. Photocatalytic Activation of N-Chloro Compounds for the Chlorination of Arenes. Tetrahedron 2016, 72, 7821–7825. [Google Scholar] [CrossRef]
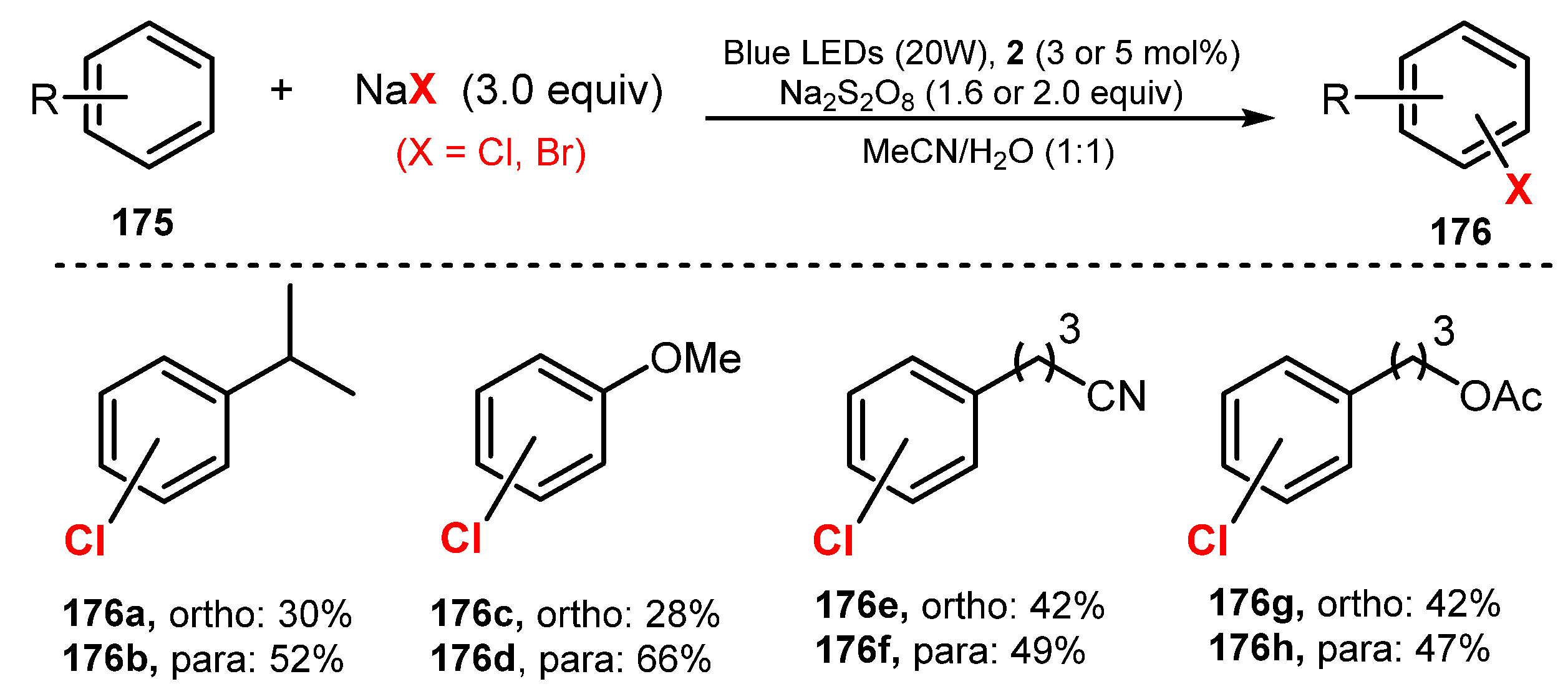
- Zhanga, L.; Hu, X. Room Temperature C(sp2)-H Oxidative Chlorination via Photoredox Catalysis. Chem. Sci. 2017, 8, 7009–7013. [Google Scholar] [CrossRef]
- Rogers, D.A.; Gallegos, J.M.; Hopkins, M.D.; Lignieres, A.A.; Lamar, A.A. Visible-Light Photocatalytic Activation of N-Chlorosuccinimide by Organic Dyes for the Chlorination of Arenes and Heteroarenes. Tetrahedron 2019, 75, 130498. [Google Scholar] [CrossRef]
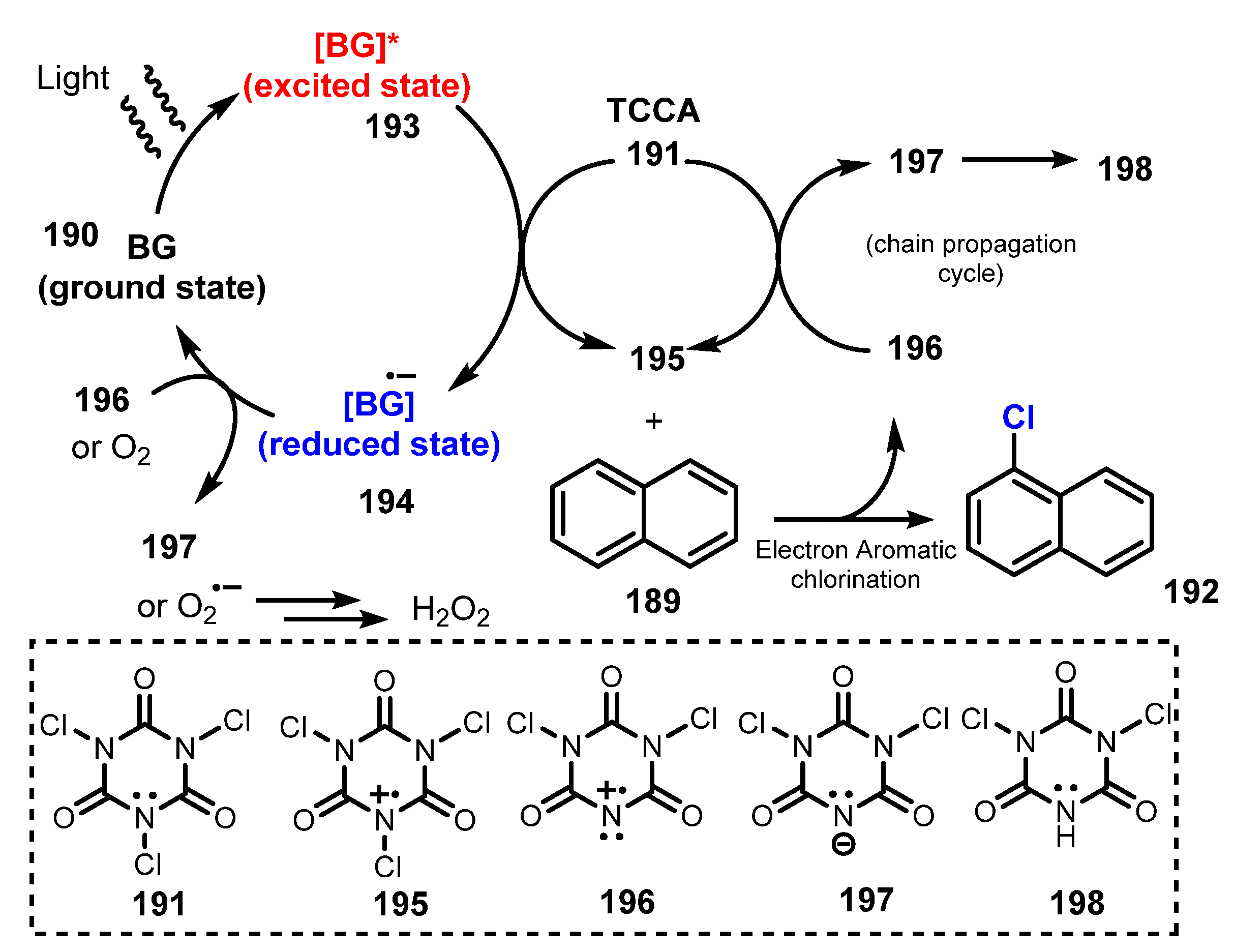
- Rogers, D.A.; Bensalah, A.T.; Espinosa, A.T.; Hoerr, J.L.; Refai, F.H.; Pitzel, A.K.; Alvarado, J.J.; Lamar, A.A. Amplification of Trichloroisocyanuric Acid (TCCA) Reactivity for Chlorination of Arenes and Heteroarenes via Catalytic Organic Dye Activation. Org. Lett. 2019, 21, 4229–4233. [Google Scholar] [CrossRef]
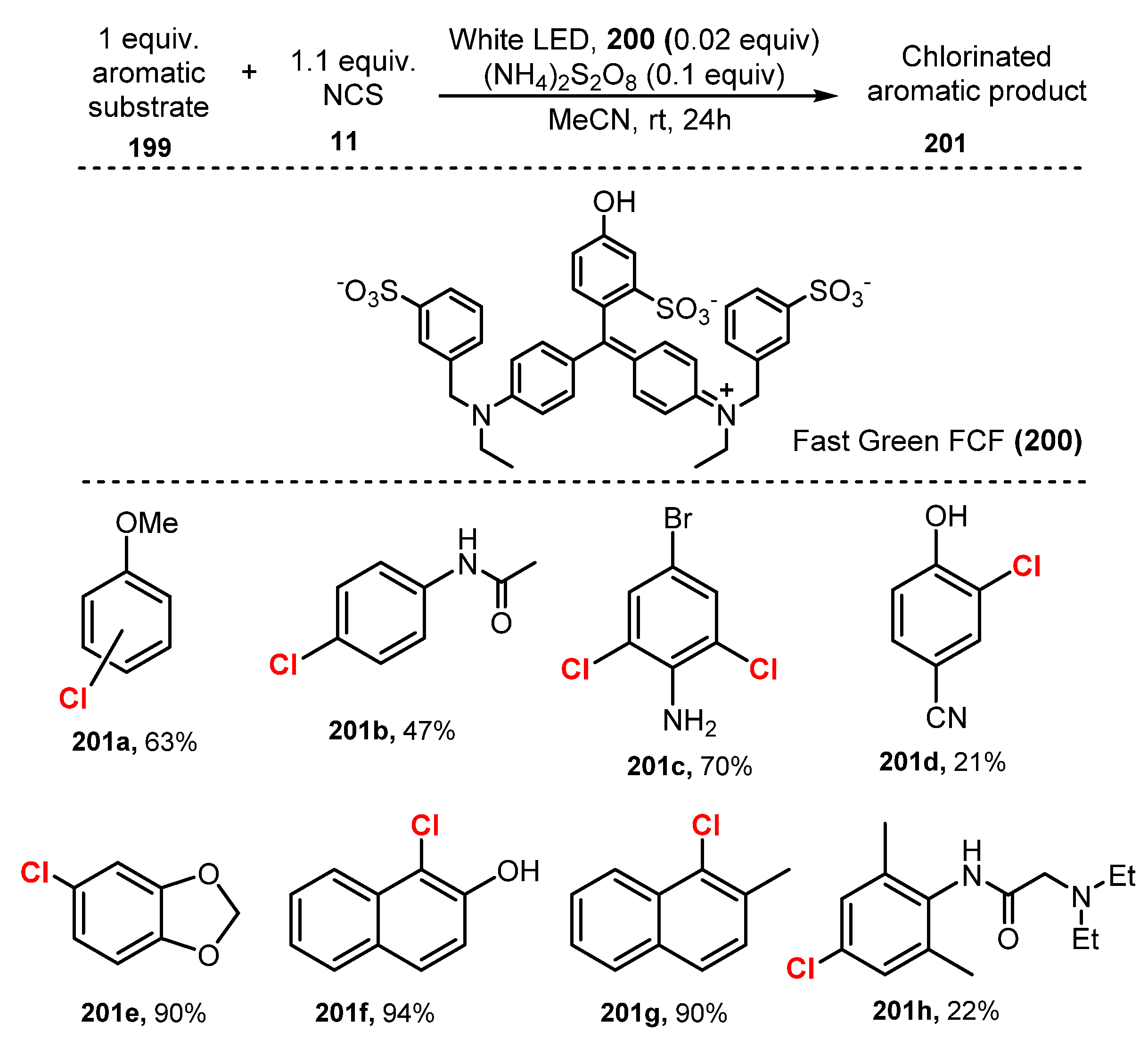
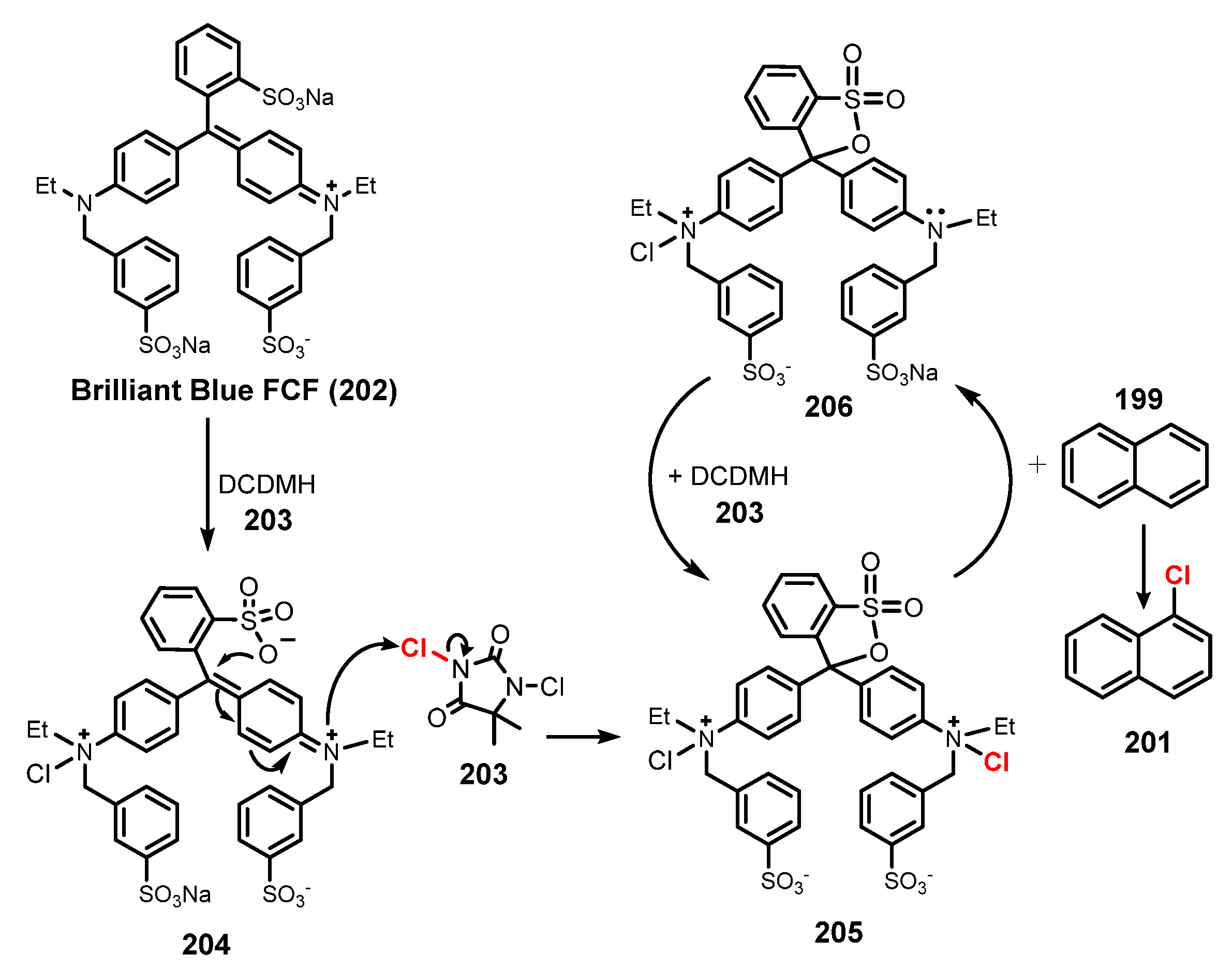
- Rogers, D.A.; Hopkins, M.D.; Rajagopal, N.; Varshney, D.; Howard, H.A.; LeBlanc, G.; Lamar, A.A. U.S. Food and Drug Administration-Certified Food Dyes as Organocatalysts in the Visible Light-Promoted Chlorination of Aromatics and Heteroaromatics. ACS Omega 2020, 5, 7693–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
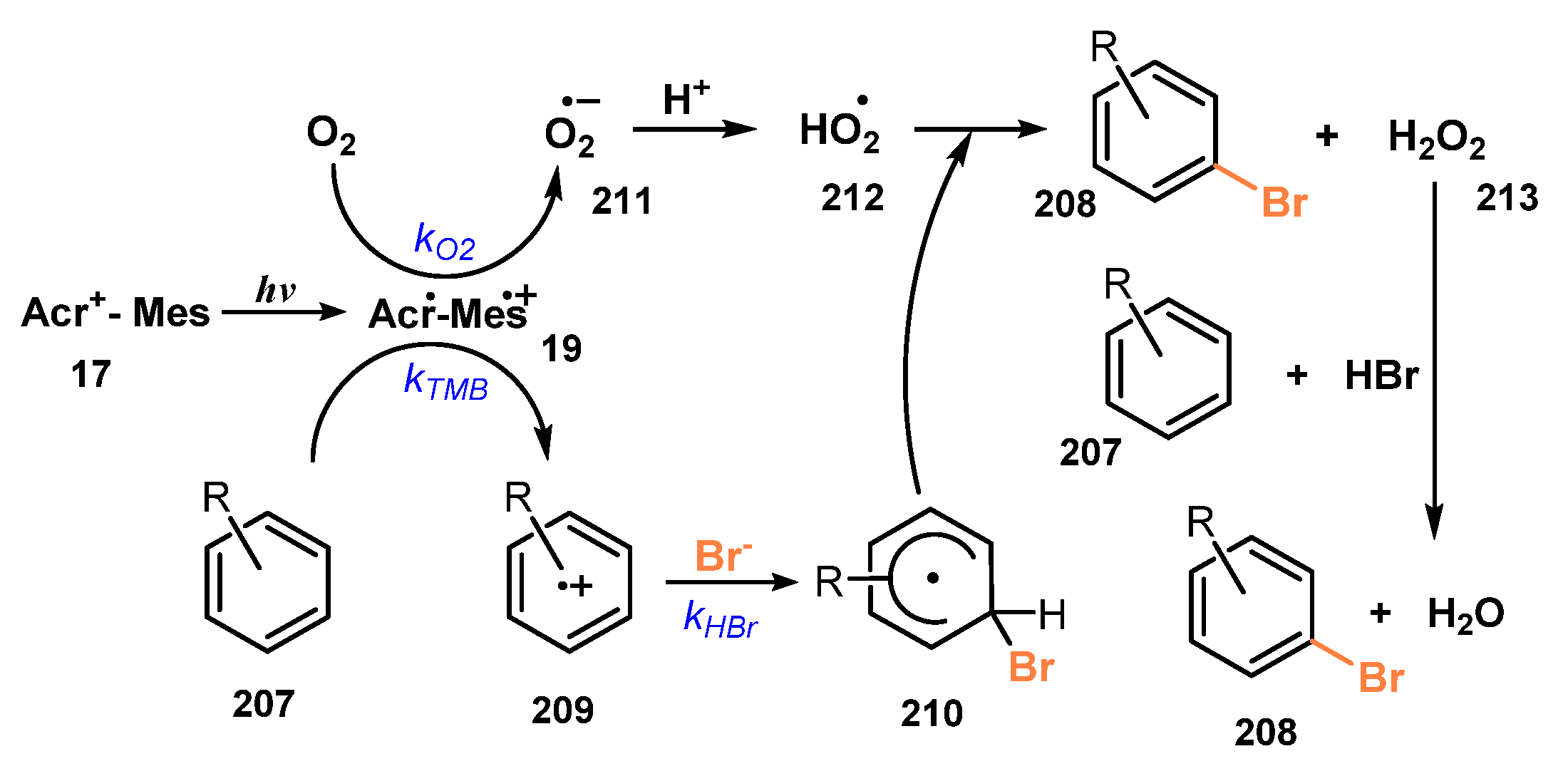
- Ohkubo, K.; Mizushima, K.; Iwata, R.; Fukuzumi, S. Selective Photocatalytic Aerobic Bromination with Hydrogen Bromide via an Electron-Transfer State of 9-mesityl-10-methylacridinium ion. Chem. Sci. 2011, 2, 715–722. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Visible-Light Photoredox Catalysis Enabled Bromination of Phenols and Alkenes. Beilstein J. Org. Chem. 2014, 10, 622–627. [Google Scholar] [CrossRef] [PubMed]
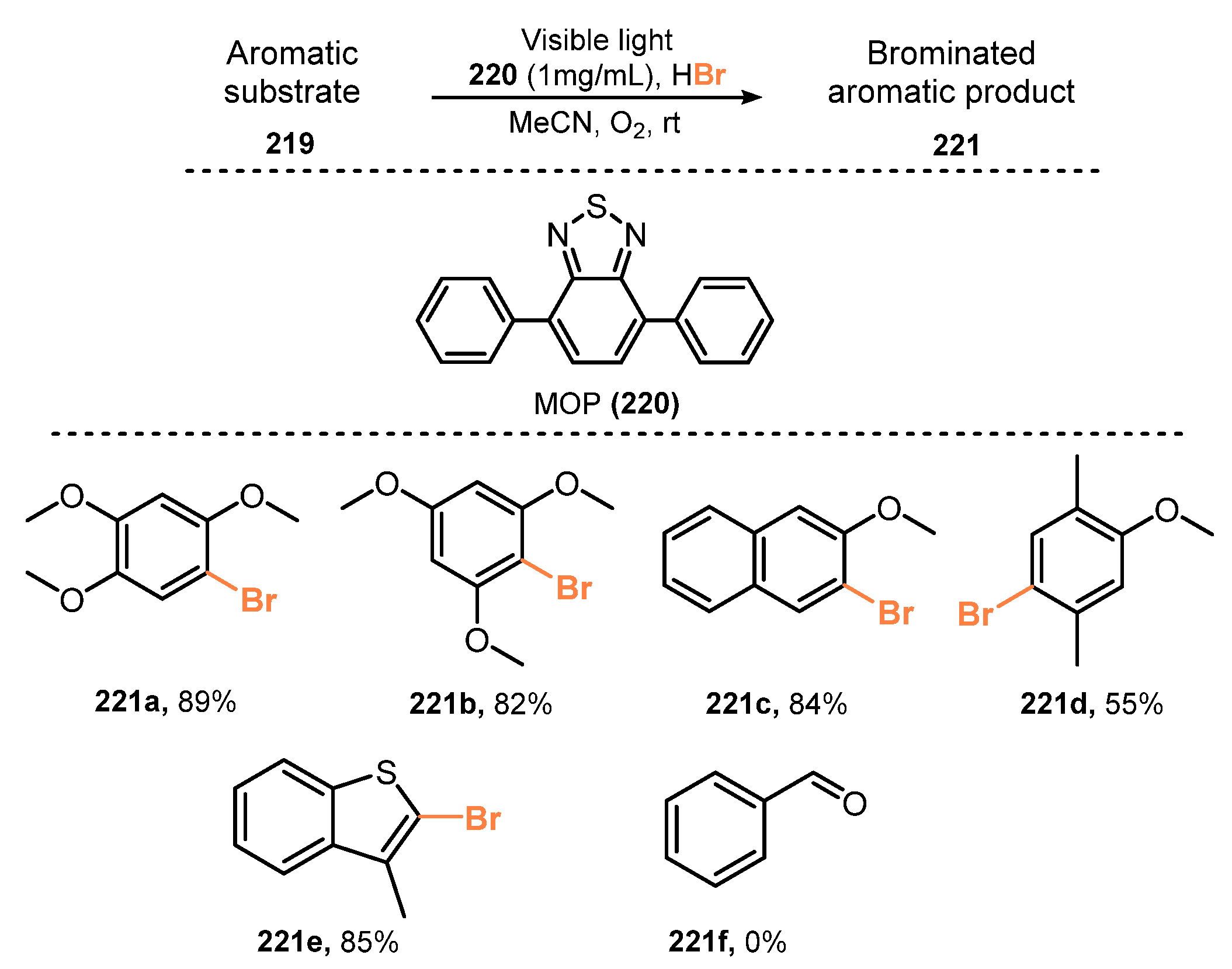
- Li, R.; Wang, Z.J.; Wang, L.; Ma, B.C.; Ghasimi, S.; Lu, H.; Landfester, K.; Zhang, K.A.I. Photocatalytic Selective Bromination of Electron-Rich Aromatic Compounds Using Microporous Organic Polymers with Visible Light. ACS Catal. 2016, 6, 1113–1121. [Google Scholar] [CrossRef]
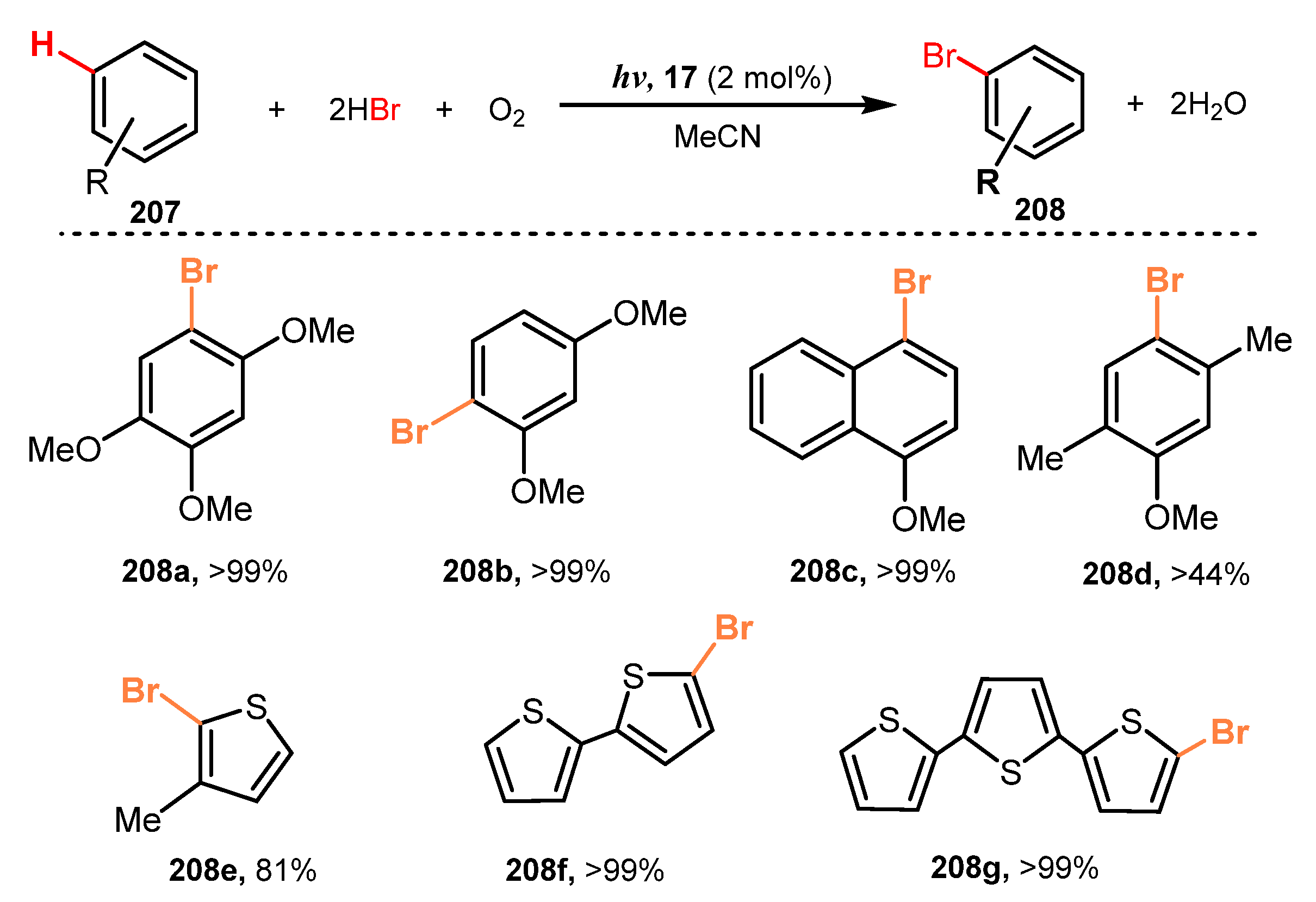
- Rogers, D.A.; Brown, R.G.; Brandeburg, Z.C.; Ko, E.Y.; Hopkins, M.D.; LeBlanc, G.; Lamar, A.A. Organic Dye-Catalyzed, Visible-Light Photoredox Bromination of Arenes and Heteroarenes Using N-Bromosuccinimide. ACS Omega 2018, 3, 12868–12877. [Google Scholar] [CrossRef] [Green Version]
- Petzold, D.; König, B. Photocatalytic Oxidative Bromination of Electron-Rich Arenes and Heteroarenes by Anthraquinone. Adv. Synth. Catal. 2018, 360, 626–630. [Google Scholar] [CrossRef]
- Ma, B.; Lu, F.; Yang, H.; Gu, X.; Li, Z.; Li, R.; Pei, H.; Luo, D.; Zhang, H.; Lei, A. Visible Light Mediated External Oxidant Free Selective C5 Bromination of 8-Aminoquinoline Amides under Ambient Conditions. Asian J. Org. Chem. 2019, 8, 1136–1140. [Google Scholar] [CrossRef]
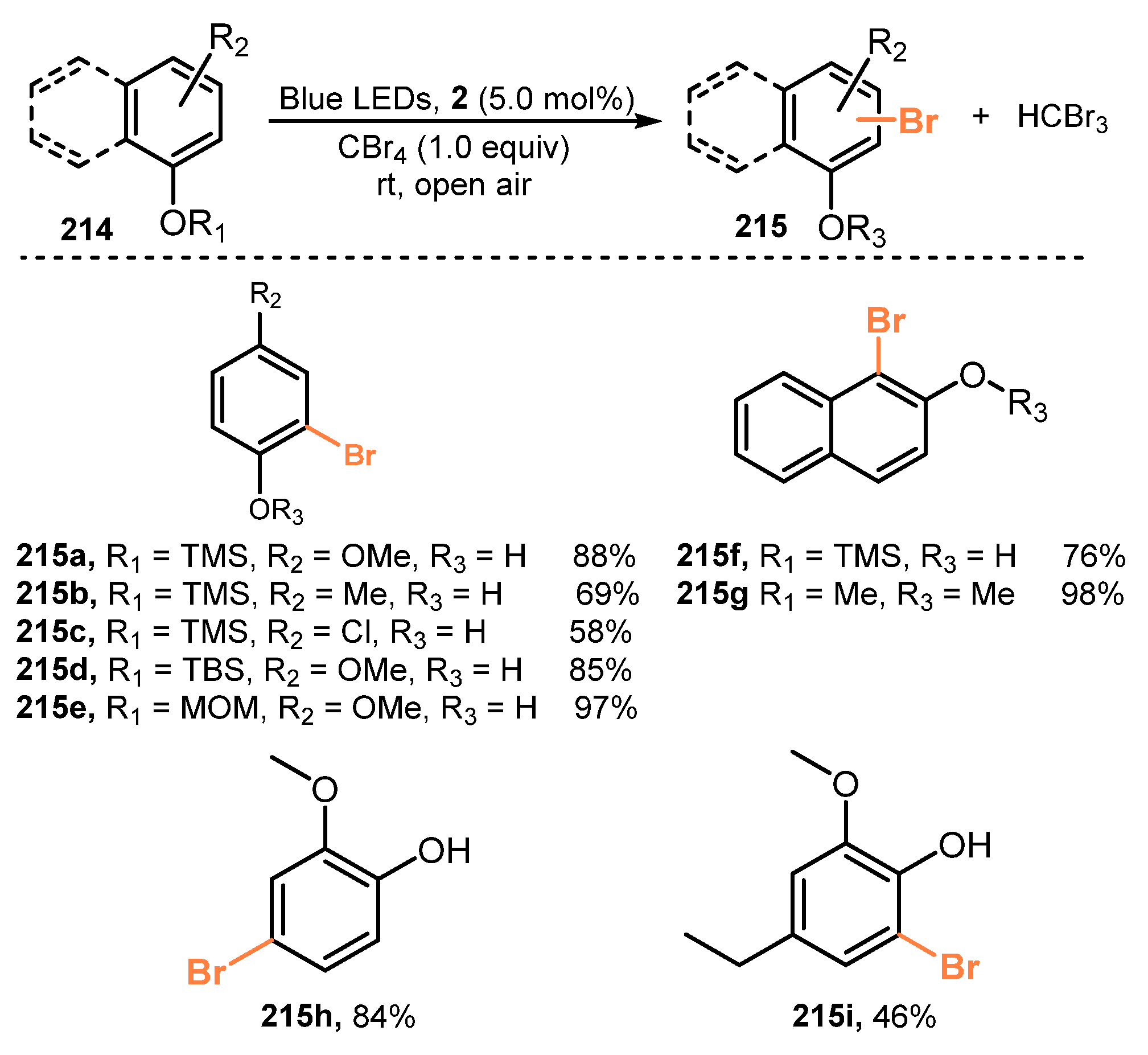
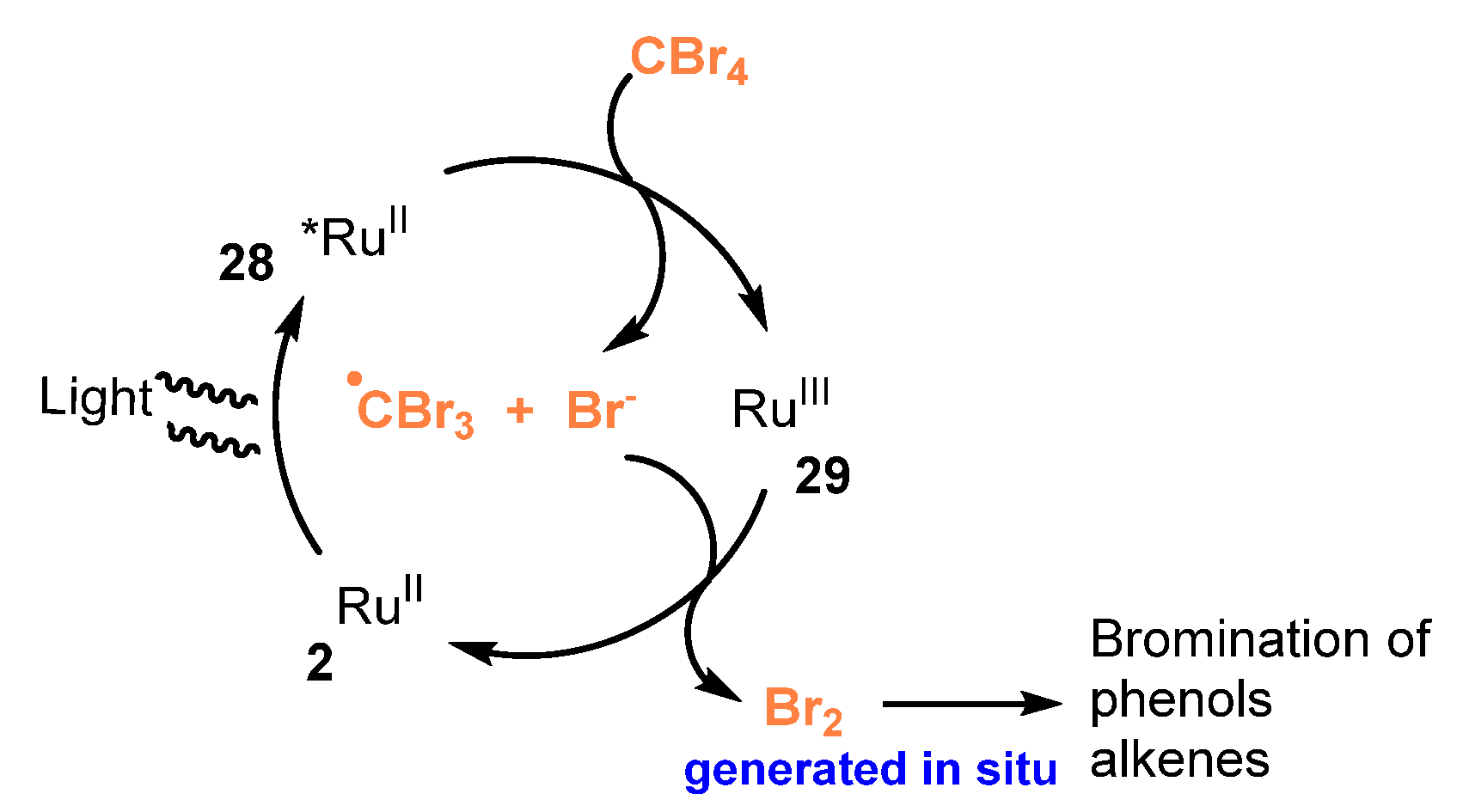
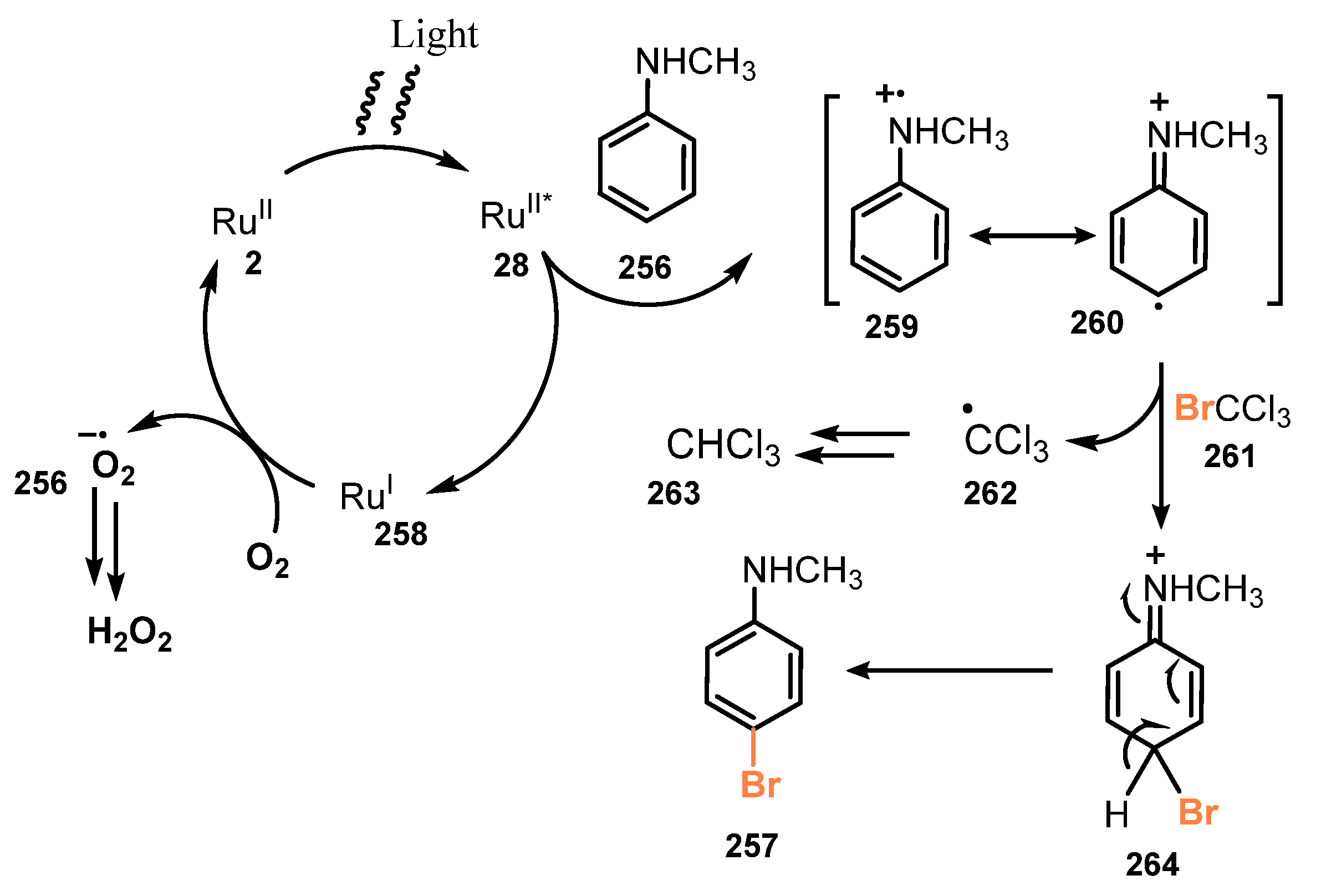
- Fan, J.; Wei, Q.; Zhu, E.; Gao, J.; Cheng, X.; Lu, Y.; Loh, T.P. Visible Light-Induced Mono-Bromination of Arenes with BrCCl3. Chem. Commun. 2021, 57, 5977–5980. [Google Scholar] [CrossRef] [PubMed]
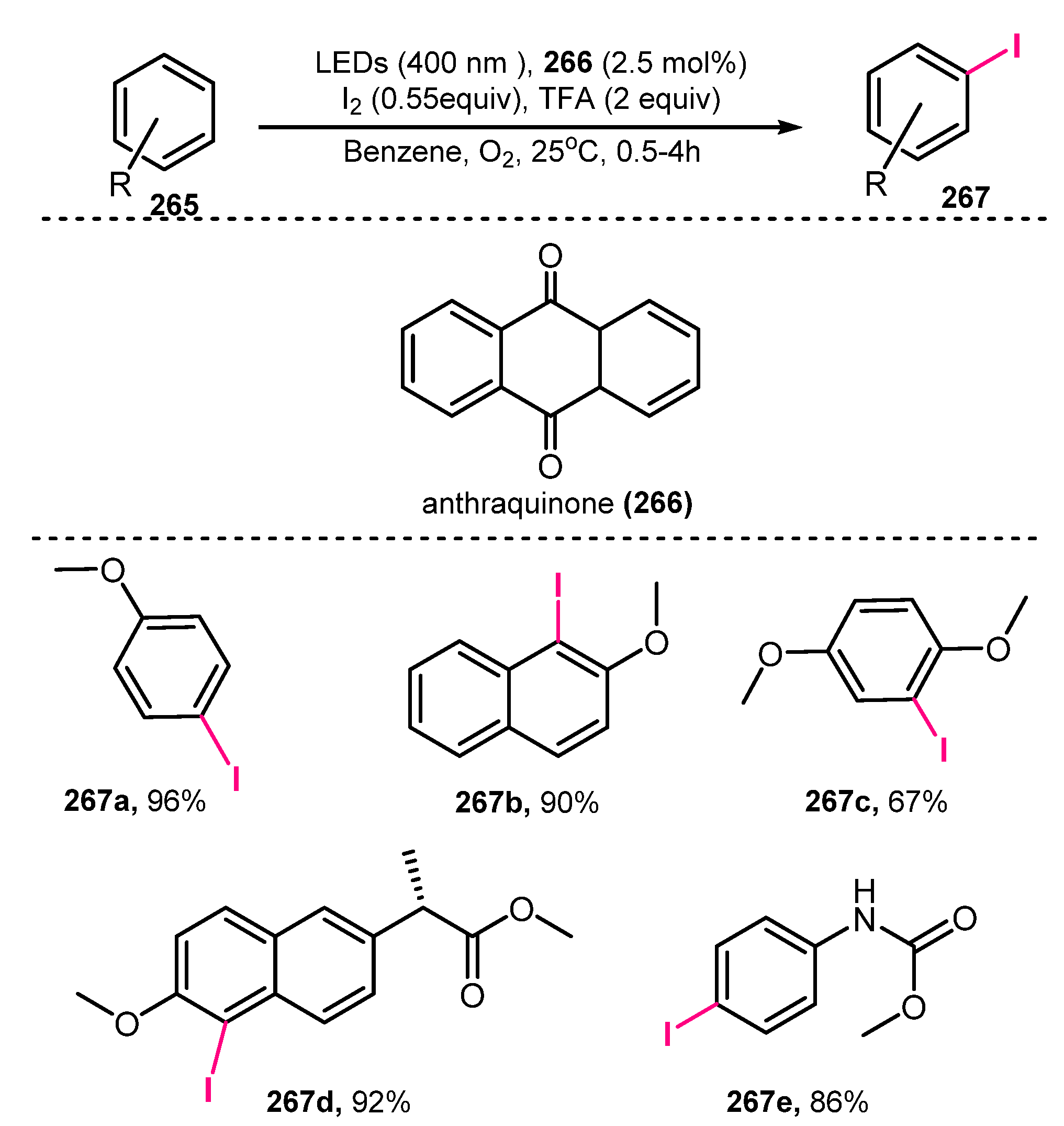
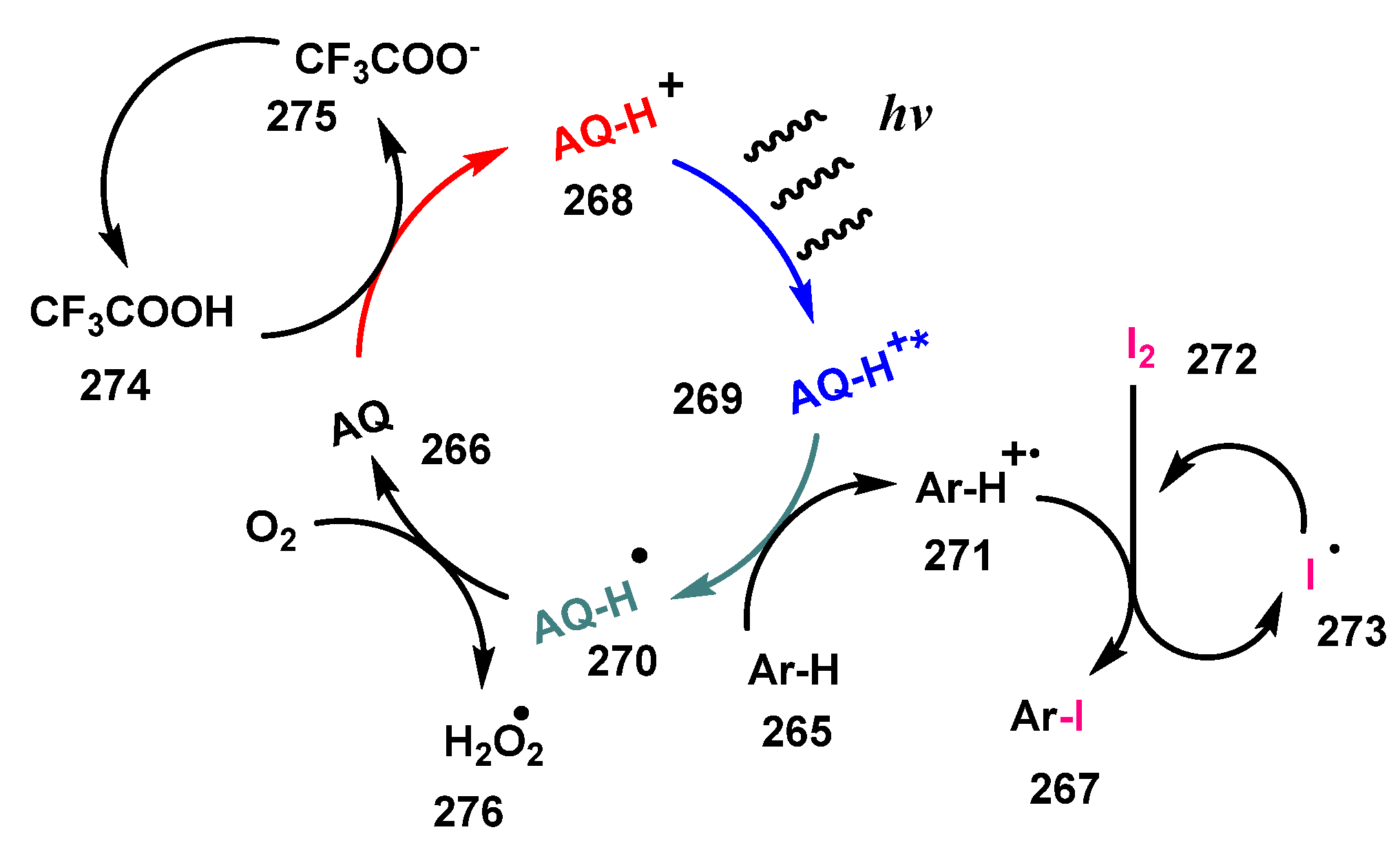
- Narobe, R.; Düsel, S.J.S.; Iskra, J.; König, B. Photocatalytic Oxidative Iodination of Electron-Rich Arenes. Adv. Synth. Catal. 2019, 361, 3998–4004. [Google Scholar] [CrossRef]
































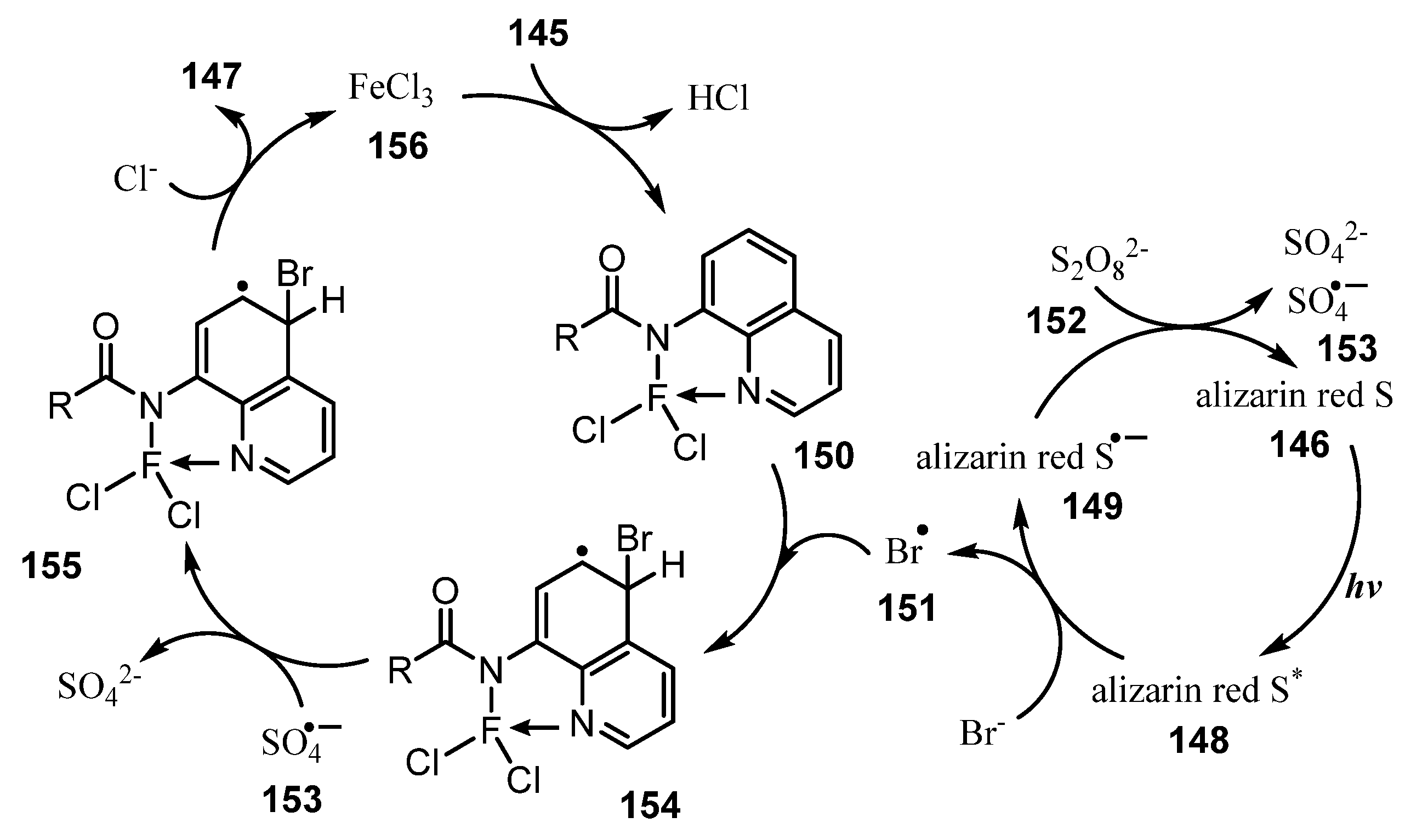






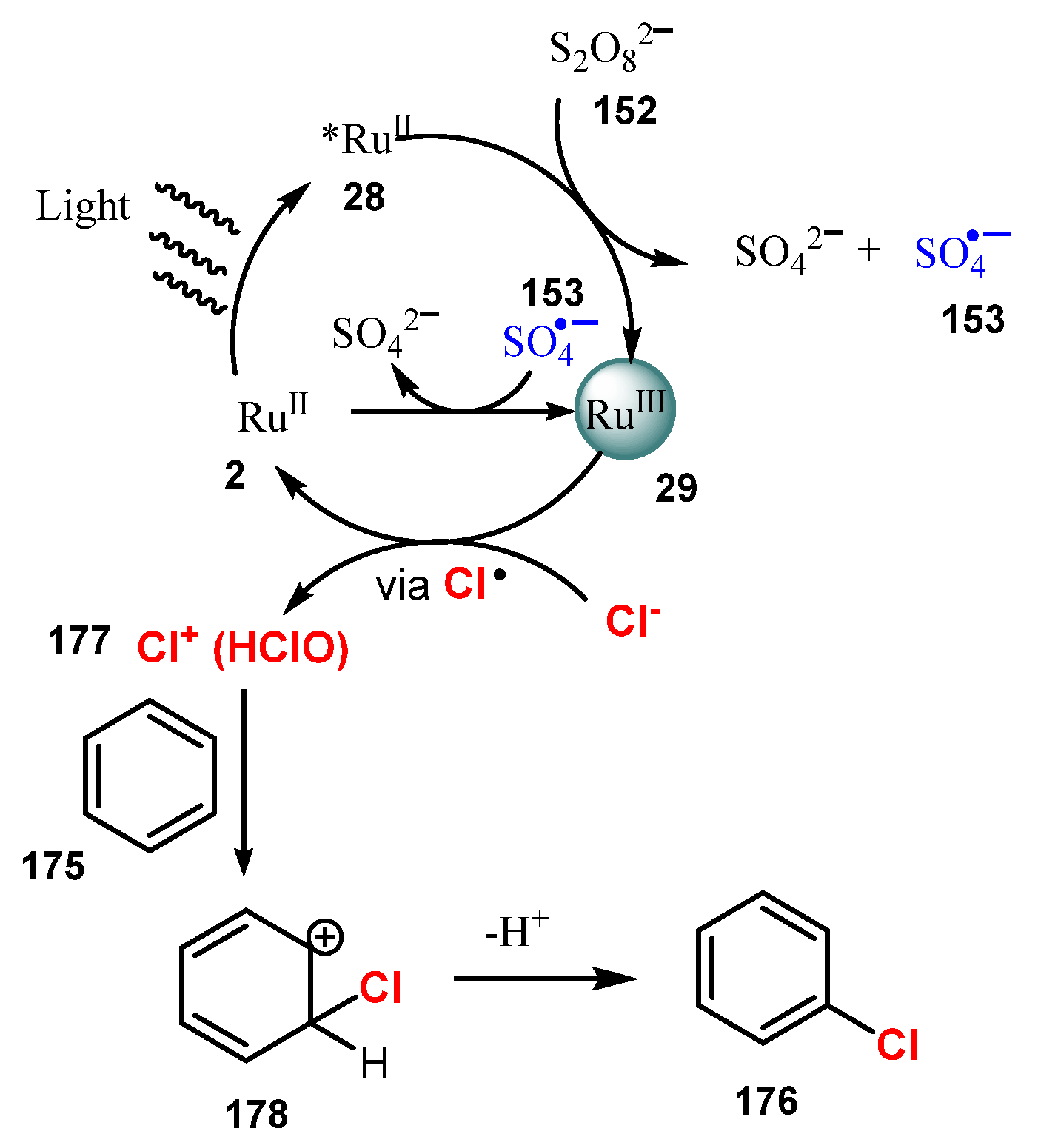





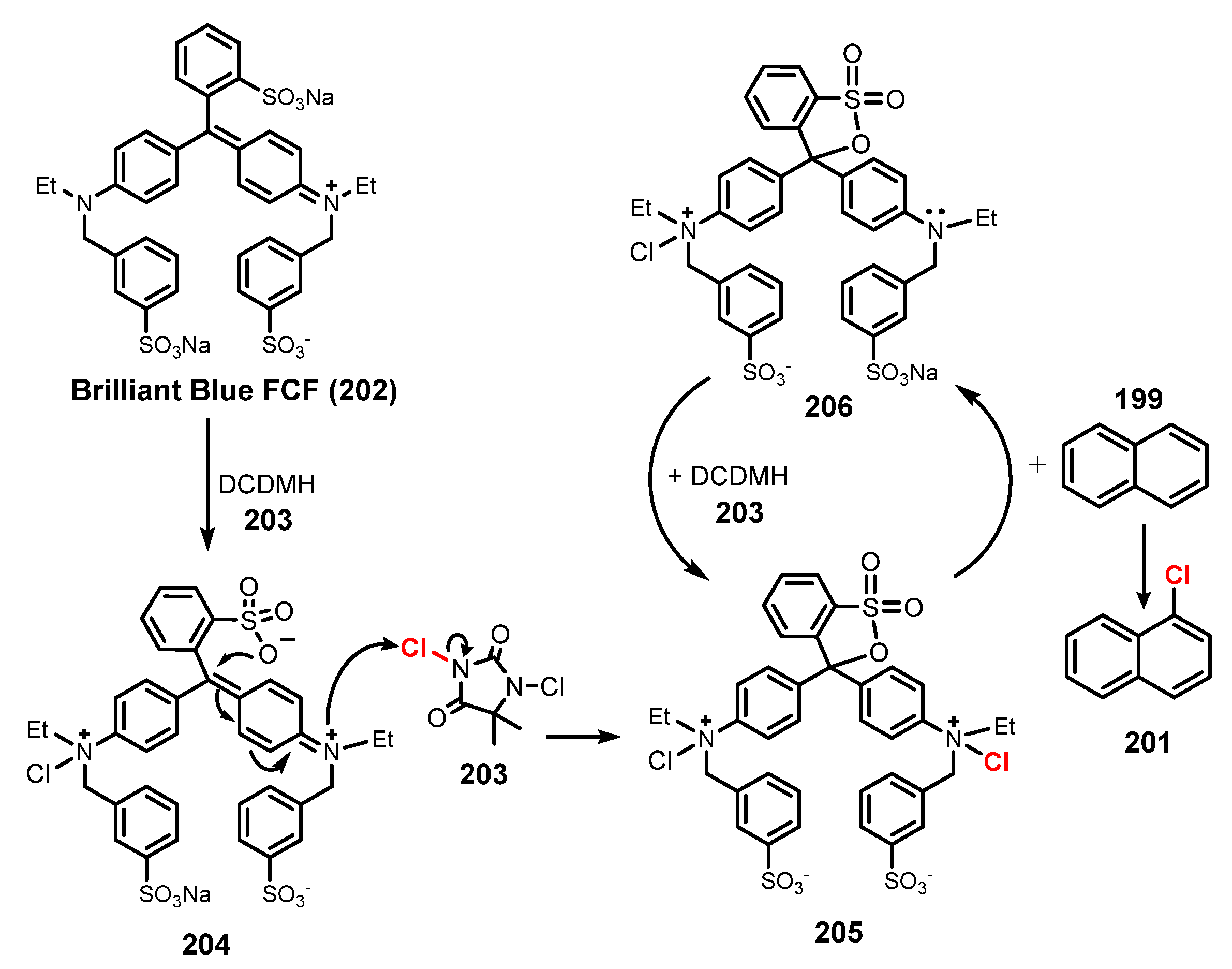





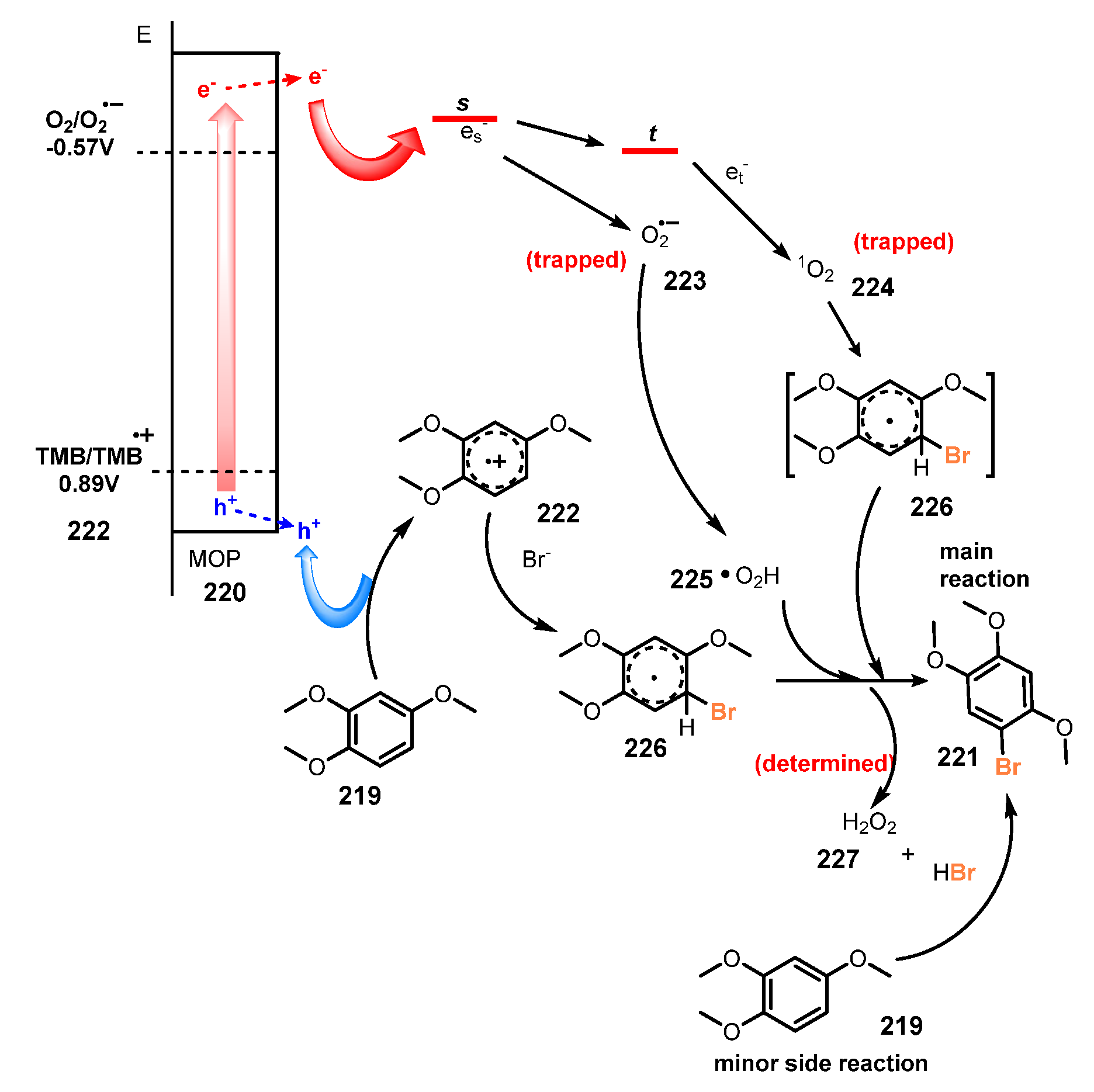









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luu, T.G.; Jung, Y.; Kim, H.-K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules 2021, 26, 7380. https://doi.org/10.3390/molecules26237380
Luu TG, Jung Y, Kim H-K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules. 2021; 26(23):7380. https://doi.org/10.3390/molecules26237380
Chicago/Turabian StyleLuu, Truong Giang, Yongju Jung, and Hee-Kwon Kim. 2021. "Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst" Molecules 26, no. 23: 7380. https://doi.org/10.3390/molecules26237380
APA StyleLuu, T. G., Jung, Y., & Kim, H. -K. (2021). Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules, 26(23), 7380. https://doi.org/10.3390/molecules26237380