
A Dispersion Corrected DFT Investigation of the Inclusion Complexation of Dexamethasone with β-Cyclodextrin and Molecular Docking Study of Its Potential Activity against COVID-19

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
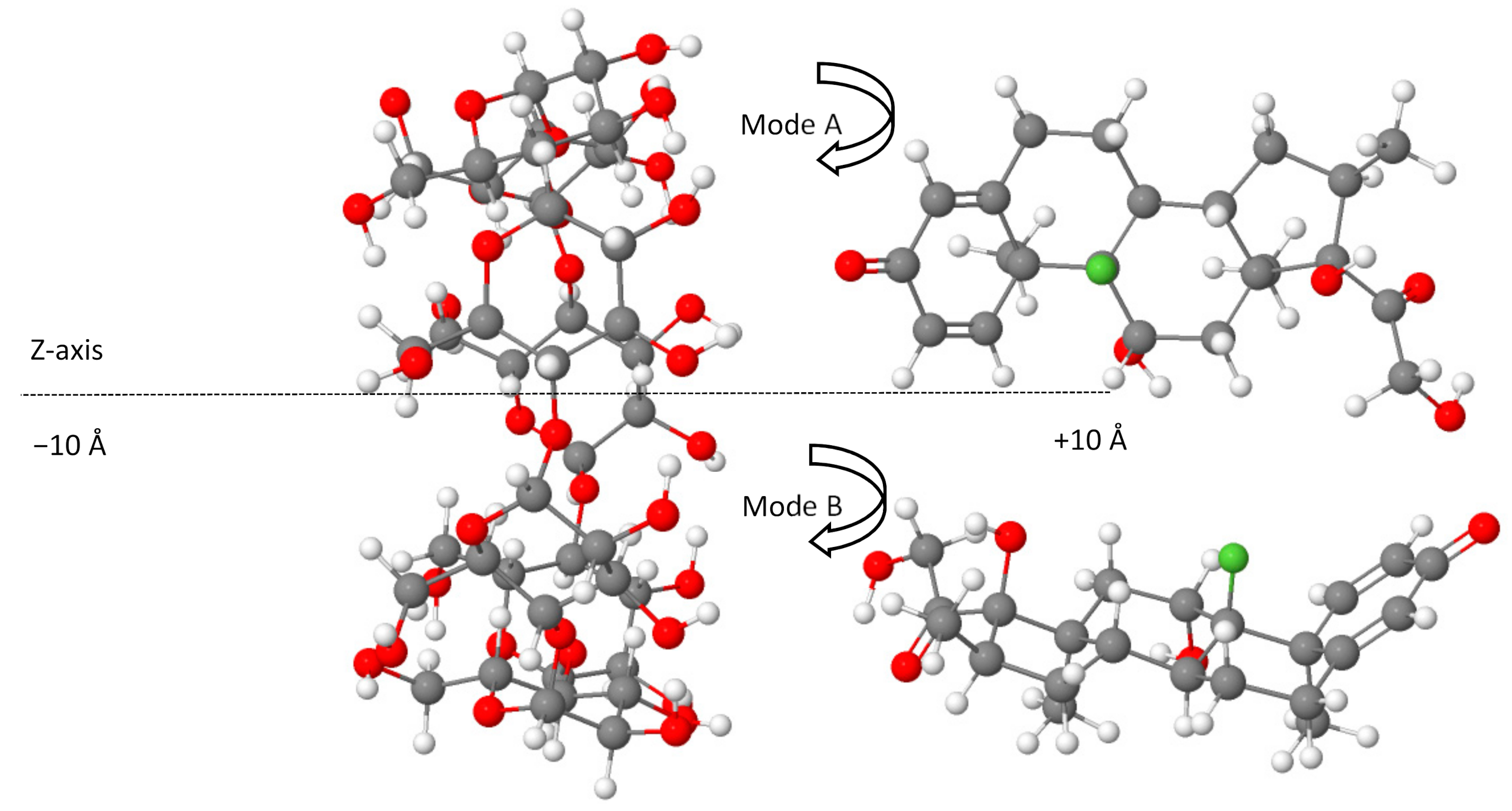
2.1. DFT-D4 Calculations of Complexation Energies
2.2. Analysis of the Non-Covalent Intermolecular Interactions
2.3. Contribution of Intermolecular Hydrogen Bonds
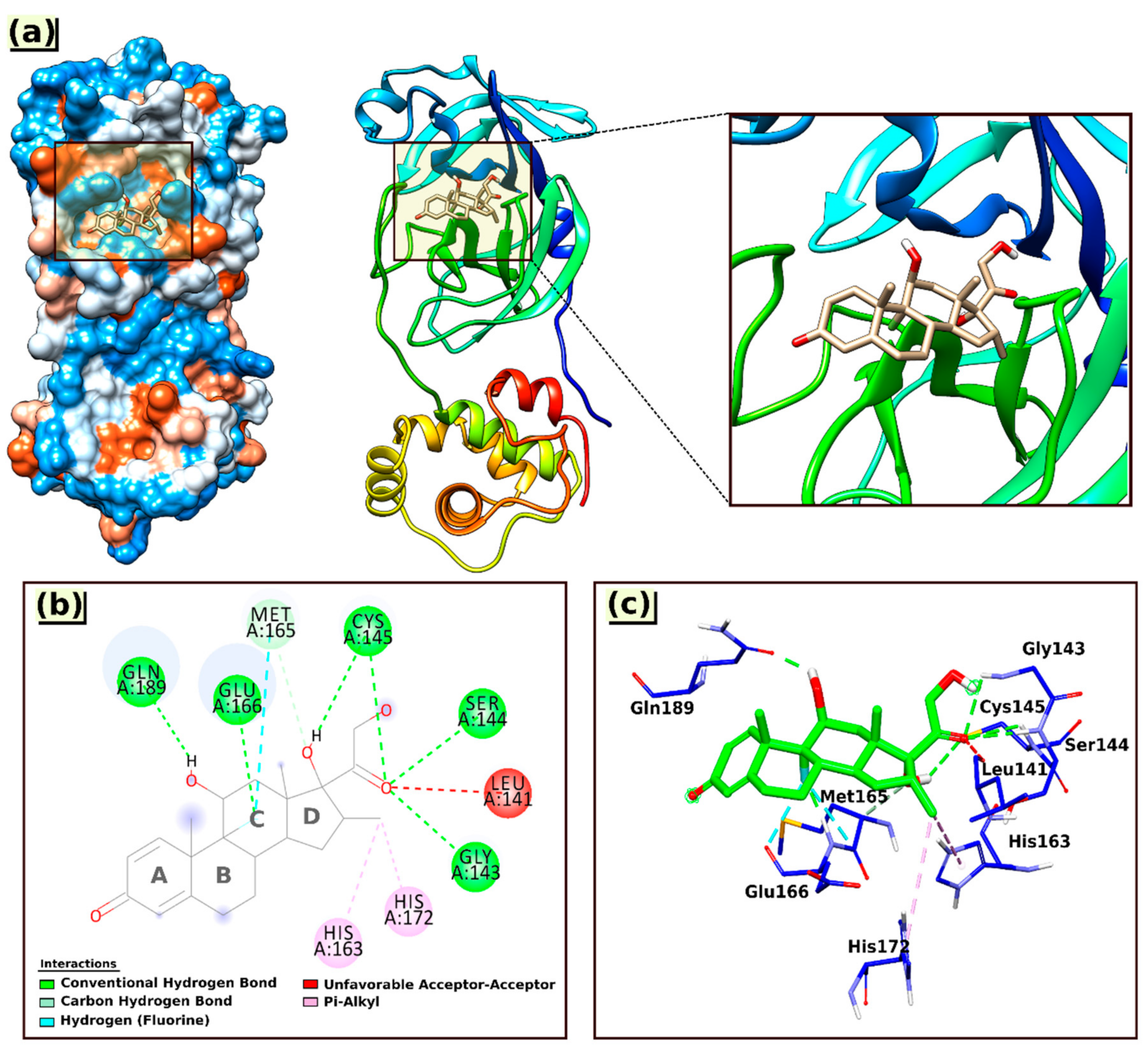
2.4. AutoDock Docking Result Analysis
3. Computational Procedure
3.1. DFT Calculations
3.2. Molecular Docking Study Using AutoDock
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Theoharides, T.; Conti, P. Dexamethasone for COVID-19? Not so fast. J. Biol. Regul. Homeost. Agents 2020, 34, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Villar, J.; Ferrando, C.; Martínez, D.; Ambrós, A.; Muñoz, T.; Soler, J.A.; Aguilar, G.; Alba, F.; González-Higueras, E.; Conesa, L.A. Dexamethasone treatment for the acute respiratory distress syndrome: A multicentre, randomised controlled trial. Lancet Respir. Med. 2020, 8, 267–276. [Google Scholar] [CrossRef]
- Hajjo, R.; Sabbah, D.A.; Bardaweel, S.K. Chemocentric informatics analysis: Dexamethasone versus combination therapy for COVID-19. ACS Omega 2020, 5, 29765–29779. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, V.; Dapaah-Afriyie, K.; Finn, A.; Flanigan, T.P. Short-term dexamethasone in Sars-CoV-2 patients. RI Med. J. 2020, 103, 39–43. [Google Scholar]
- Girgis, N.I.; Farid, Z.; Mikhall, I.; Farrag, I.; Sultan, Y.; Kilpatrick, M.E. Dexamethasone treatment for bacterial meningitis in children and adults. Pediatric Infect. Dis. J. 1989, 8, 848–851. [Google Scholar] [CrossRef]
- Alexanian, R.; Dimopoulos, M.A.; Delasalle, K.; Barlogie, B. Primary dexamethasone treatment of multiple myeloma. Blood 1992, 80, 887–890. [Google Scholar] [CrossRef] [Green Version]
- Thwaites, G.E.; Bang, N.D.; Dung, N.H.; Quy, H.T.; Oanh, D.T.T.; Thoa, N.T.C.; Hien, N.Q.; Thuc, N.T.; Hai, N.N.; Lan, N.T.N. Dexamethasone for the treatment of tuberculous meningitis in adolescents and adults. N. Engl. J. Med. 2004, 351, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Lester, M.; Sahin, A.; Pasyar, A. The use of dexamethasone in the treatment of COVID-19. Ann. Med. Surg. 2020, 56, 218–219. [Google Scholar] [CrossRef]
- Lammers, T.; Sofias, A.M.; van der Meel, R.; Schiffelers, R.; Storm, G.; Tacke, F.; Koschmieder, S.; Brümmendorf, T.H.; Kiessling, F.; Metselaar, J.M. Dexamethasone nanomedicines for COVID-19. Nat. Nanotechnol. 2020, 15, 622–624. [Google Scholar] [CrossRef]
- Horby, P.; Landrain, M. Low-cost dexamethasone reduces death by up to one third in hospitalised patients with severe respiratory complications of COVID-19. Nuffield Dep. Popul. Health 2020, 56, 218–219. [Google Scholar]
- Tomazini, B.M.; Maia, I.S.; Cavalcanti, A.B.; Berwanger, O.; Rosa, R.G.; Veiga, V.C.; Avezum, A.; Lopes, R.D.; Bueno, F.R.; Silva, M.V.A. Effect of dexamethasone on days alive and ventilator-free in patients with moderate or severe acute respiratory distress syndrome and COVID-19: The CoDEX randomized clinical trial. JAMA 2020, 324, 1307–1316. [Google Scholar] [CrossRef]
- Carvallo, H.E.; Hirsch, R.R.; Farinella, M.E. Safety and Efficacy of the combined use of ivermectin, dexamethasone, enoxaparin and aspirin against COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Sharun, K.; Tiwari, R.; Dhama, J.; Dhama, K. Dexamethasone to combat cytokine storm in COVID-19: Clinical trials and preliminary evidence. Int. J. Surg. 2020, 82, 179–181. [Google Scholar] [CrossRef]
- Stauffer, W.M.; Alpern, J.D.; Walker, P.F. COVID-19 and dexamethasone: A potential strategy to avoid steroid-related strongyloides hyperinfection. JAMA 2020, 324, 623–624. [Google Scholar] [CrossRef]
- Ortolani, C.; Pastorello, E.A. Hydroxychloroquine and dexamethasone in COVID-19: Who won and who lost? Clin. Mol. Allergy 2020, 18, 1–7. [Google Scholar] [CrossRef]
- Sterne, J.A.; Murthy, S.; Diaz, J.V.; Slutsky, A.S.; Villar, J.; Angus, D.C.; Annane, D.; Azevedo, L.C.P.; Berwanger, O.; Cavalcanti, A.B. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID-19: A meta-analysis. JAMA 2020, 324, 1330–1341. [Google Scholar] [CrossRef]
- Vianna, R.F.; Bentley, M.V.L.; Ribeiro, G.; Carvalho, F.S.; Neto, A.F.; de Oliveira, D.C.; Collett, J.H. Formation of cyclodextrin inclusion complexes with corticosteroids: Their characterization and stability. Int. J. Pharm. 1998, 167, 205–213. [Google Scholar] [CrossRef]
- Piras, A.M.; Zambito, Y.; Burgalassi, S.; Monti, D.; Tampucci, S.; Terreni, E.; Fabiano, A.; Balzano, F.; Uccello-Barretta, G.; Chetoni, P. A water-soluble, mucoadhesive quaternary ammonium chitosan-methyl-β-cyclodextrin conjugate forming inclusion complexes with dexamethasone. J. Mater. Sci. Mater. Med. 2018, 29, 42. [Google Scholar] [CrossRef]
- Li, J.; Loh, X.J. Cyclodextrin-based supramolecular architectures: Syntheses, structures, and applications for drug and gene delivery. Adv. Drug Deliv. Rev. 2008, 60, 1000–1017. [Google Scholar] [CrossRef]
- Cal, K.; Centkowska, K. Use of cyclodextrins in topical formulations: Practical aspects. Eur. J. Pharm. Biopharm. 2008, 68, 467–478. [Google Scholar] [CrossRef]
- Bonnet, V.; Gervaise, C.; Djedaïni-Pilard, F.; Furlan, A.; Sarazin, C. Cyclodextrin nanoassemblies: A promising tool for drug delivery. Drug Discov. Today 2015, 20, 1120–1126. [Google Scholar] [CrossRef]
- Astray, G.; Mejuto, J.C.; Morales, J.; Rial-Otero, R.; Simal-Gandara, J. Factors controlling flavors binding constants to cyclodextrins and their applications in foods. Food Res. Int. 2010, 43, 1212–1218. [Google Scholar] [CrossRef]
- Harata, K.; Kawano, K.; Fukunaga, K.; Ohtani, Y. Structure of β-Cyclodextrin Inclusion Complex with Nicotinamide. Chem. Pharm. Bull. 1983, 31, 1428–1430. [Google Scholar] [CrossRef] [Green Version]
- Kurkov, S.V.; Loftsson, T. Cyclodextrins. Int. J. Pharm. 2013, 453, 167–180. [Google Scholar] [CrossRef]
- Stella, V.J.; He, Q. Cyclodextrins. Toxicol. Pathol. 2008, 36, 30–42. [Google Scholar] [CrossRef]
- Daghrery, A.; Aytac, Z.; Dubey, N.; Mei, L.; Schwendeman, A.; Bottino, M.C. Electrospinning of dexamethasone/cyclodextrin inclusion complex polymer fibers for dental pulp therapy. Colloids Surf. B Biointerfaces 2020, 191, 111011. [Google Scholar] [CrossRef]
- Allal, H.; Belhocine, Y.; Rahali, S.; Damous, M.; Ammouchi, N. Structural, electronic, and energetic investigations of acrolein adsorption on B 36 borophene nanosheet: A dispersion-corrected DFT insight. J. Mol. Model. 2020, 26, 128. [Google Scholar] [CrossRef]
- Athar, M.; Behzadi, H.; Makki, S. Understanding non-covalent interactions by NMR in urea-and thiourea-substituted calixarene complexes. Mon. Für Chem. Chem. Mon. 2020, 151, 743–749. [Google Scholar] [CrossRef]
- Kasprzak, A.; Borys, K.M.; Molchanov, S.; Adamczyk-Woźniak, A. Spectroscopic insight into supramolecular assemblies of boric acid derivatives and β-cyclodextrin. Carbohydr. Polym. 2018, 198, 294–301. [Google Scholar] [CrossRef]
- Lee, J.-u.; Lee, S.-S.; Lee, S.; Oh, H.B. Noncovalent Complexes of Cyclodextrin with Small Organic Molecules: Applications and Insights into Host–Guest Interactions in the Gas Phase and Condensed Phase. Molecules 2020, 25, 4048. [Google Scholar] [CrossRef]
- Gropp, C.; Quigley, B.L.; Diederich, F. Molecular recognition with resorcin [4] arene cavitands: Switching, halogen-bonded capsules, and enantioselective complexation. J. Am. Chem. Soc. 2018, 140, 2705–2717. [Google Scholar] [CrossRef] [PubMed]
- Gassoumi, B.; Chaabene, M.; Ghalla, H.; Chaabane, R.B. Role of hydrogen bonding interactions within of the conformational preferences of calix [n= 4, 6, 8] arene: DFT and QTAIM analysis. J. Mol. Model. 2020, 26, 12. [Google Scholar] [CrossRef] [PubMed]
- Assaba, I.M.; Rahali, S.; Belhocine, Y.; Allal, H. Inclusion complexation of chloroquine with α and β-cyclodextrin: Theoretical insights from the new B97-3c composite method. J. Mol. Struct. 2021, 1227, 129696. [Google Scholar] [CrossRef]
- Oqmhula, K.; Hongo, K.; Maezono, R.; Ichibha, T. Ab Initio Evaluation of Complexation Energies for Cyclodextrin-Drug Inclusion Complexes. ACS Omega 2020, 5, 19371–19376. [Google Scholar] [CrossRef]
- Aree, T. Inclusion complex of β-cyclodextrin with coffee chlorogenic acid: New insights from a combined crystallographic and theoretical study. Acta Crystallogr. Sect. C Struct. Chem. 2019, 75, 15–21. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Rahali, S.; Belhocine, Y.; Allal, H.; Bouhadiba, A.; Assaba, I.M.; Seydou, M. A DFT investigation of the host–guest interactions between boron-based aromatic systems and β-cyclodextrin. Struct. Chem. 2021, 1–12. [Google Scholar] [CrossRef]
- Chhetri, A.; Chettri, S.; Rai, P.; Mishra, D.K.; Sinha, B.; Brahman, D. Synthesis, characterization and computational study on potential inhibitory action of novel azo imidazole derivatives against COVID-19 main protease (Mpro: 6LU7). J. Mol. Struct. 2021, 1225, 129230. [Google Scholar] [CrossRef]
- Garg, S.; Roy, A. In silico analysis of selected alkaloids against main protease (Mpro) of SARS-CoV-2. Chem. Biol. Interact. 2020, 332, 109309. [Google Scholar] [CrossRef]
- Baildya, N.; Ghosh, N.N.; Chattopadhyay, A.P. Inhibitory activity of hydroxychloroquine on COVID-19 main protease: An insight from MD-simulation studies. J. Mol. Struct. 2020, 1219, 128595. [Google Scholar] [CrossRef]
- Irfan, A.; Imran, M.; Khalid, M.; Ullah, M.S.; Khalid, N.; Assiri, M.A.; Thomas, R.; Muthu, S.; Basra, M.A.R.; Hussein, M. Phenolic and flavonoid contents in Malva sylvestris and exploration of active drugs as antioxidant and anti-COVID19 by quantum chemical and molecular docking studies. J. Saudi Chem. Soc. 2021, 25, 101277. [Google Scholar] [CrossRef]
- Rangsinth, P.; Sillapachaiyaporn, C.; Nilkhet, S.; Tencomnao, T.; Ung, A.T.; Chuchawankul, S. Mushroom-derived bioactive compounds potentially serve as the inhibitors of SARS-CoV-2 main protease: An in silico approach. J. Tradit. Complement. Med. 2021, 11, 158–172. [Google Scholar] [CrossRef]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Caldeweyher, E.; Ehlert, S.; Hansen, A.; Neugebauer, H.; Spicher, S.; Bannwarth, C.; Grimme, S. A generally applicable atomic-charge dependent London dispersion correction. J. Chem. Phys. 2019, 150, 154122. [Google Scholar] [CrossRef]
- Eichkorn, K.; Treutler, O.; Öhm, H.; Häser, M.; Ahlrichs, R. Auxiliary basis sets to approximate Coulomb potentials. Chem. Phys. Lett. 1995, 240, 283–290. [Google Scholar] [CrossRef]
- Brandenburg, J.G.; Zen, A.; Alfè, D.; Michaelides, A. Interaction between water and carbon nanostructures: How good are current density functional approximations? J. Chem. Phys. 2019, 151, 164702. [Google Scholar] [CrossRef] [Green Version]
- Rězáč, J. Non-covalent interactions atlas benchmark data sets: Hydrogen bonding. J. Chem. Theory Comput. 2020, 16, 2355–2368. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.-X. Use of quantum chemical methods to study cyclodextrin chemistry. J. Incl. Phenom. Macrocycl. Chem. 2004, 50, 95–103. [Google Scholar] [CrossRef]
- Jmol: An Open-Source Java Viewer for Chemical Structures in 3D. Available online: http://www.jmol.org/ (accessed on 30 March 2021).
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Olson, A.J. Using autodock for ligand-receptor docking. Curr. Protoc. Bioinform. 2008, 24. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biovia, D.S. Discovery studio modeling environment. Release. 2017. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Configurations | Mode A | Mode B |
|---|---|---|
| −10 | −101.30 | −100.70 |
| −8 | −104.96 | −91.19 |
| −6 | −94.04 | −134.17 |
| −4 | −115.24 | −115.77 |
| −2 | −162.29 | −142.11 |
| 0 | −161.26 | −174.67 |
| 2 | −137.76 | −147.21 |
| 4 | −153.71 | −107.88 |
| 6 | −179.50 | −164.56 |
| 8 | −175.09 | −137.95 |
| 10 | −175.42 | −116.61 |
| Complex | Donor | Acceptor | H-bond (Å) | E(2) (kJ/mol) |
|---|---|---|---|---|
| Dex@β-CD | β-CD (Donor) | Dex (Acceptor) | ||
| LP(2) O132 | BD*(1) O164–H176 | 1.80 | 63.39 | |
| Dex (Donor) | β-CD (Acceptor) | |||
| LP(3) F148 | BD*(1) O45–H59 | 2.03 | 10.67 | |
| LP(1) O187 | BD*(1) O87–H101 | 1.79 | 19.87 | |
| LP(2) O187 | BD*(1) O87–H101 | 1.79 | 37.66 | |
| LP(1) O194 | BD*(1) O20–H27 | 1.90 | 19.41 | |
| LP(2) O194 | BD*(1) O20–H27 | 1.90 | 12.30 |
| BE a | KiC b | TIE c | FIE d | EE e | |
|---|---|---|---|---|---|
| AutoDock4 | AutoDock Vina | ||||
| −29.97 | −32.19 | 5.59 | −36.20 | −3.55 | −0.59 |
| Amino Acids Involved in the Interactions (Interaction Site) | Distances (Å) | |
|---|---|---|
| 6LU7@Dex | Gln189(A), Glu166(A), Cys145(A), Ser144(A), Gly143(A), Met165(A), His172(A), His163(A), and Leu141(A). | Lig−Glu166(A) (1.77, 3.08) Lig−Gln189(A) (1.80) Lig−Cys145(A) (2.91, 2.91) Lig−Ser144(A) (2.33) Lig−Gly143(A) (2.70) Lig−Met165(A) (3.36) Lig−His172(A) (5.38) Lig−His163(A) (4.40) Lig−Leu141(A) (2.55) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belhocine, Y.; Rahali, S.; Allal, H.; Assaba, I.M.; Ghoniem, M.G.; Ali, F.A.M. A Dispersion Corrected DFT Investigation of the Inclusion Complexation of Dexamethasone with β-Cyclodextrin and Molecular Docking Study of Its Potential Activity against COVID-19. Molecules 2021, 26, 7622. https://doi.org/10.3390/molecules26247622
Belhocine Y, Rahali S, Allal H, Assaba IM, Ghoniem MG, Ali FAM. A Dispersion Corrected DFT Investigation of the Inclusion Complexation of Dexamethasone with β-Cyclodextrin and Molecular Docking Study of Its Potential Activity against COVID-19. Molecules. 2021; 26(24):7622. https://doi.org/10.3390/molecules26247622
Chicago/Turabian StyleBelhocine, Youghourta, Seyfeddine Rahali, Hamza Allal, Ibtissem Meriem Assaba, Monira Galal Ghoniem, and Fatima Adam Mohamed Ali. 2021. "A Dispersion Corrected DFT Investigation of the Inclusion Complexation of Dexamethasone with β-Cyclodextrin and Molecular Docking Study of Its Potential Activity against COVID-19" Molecules 26, no. 24: 7622. https://doi.org/10.3390/molecules26247622