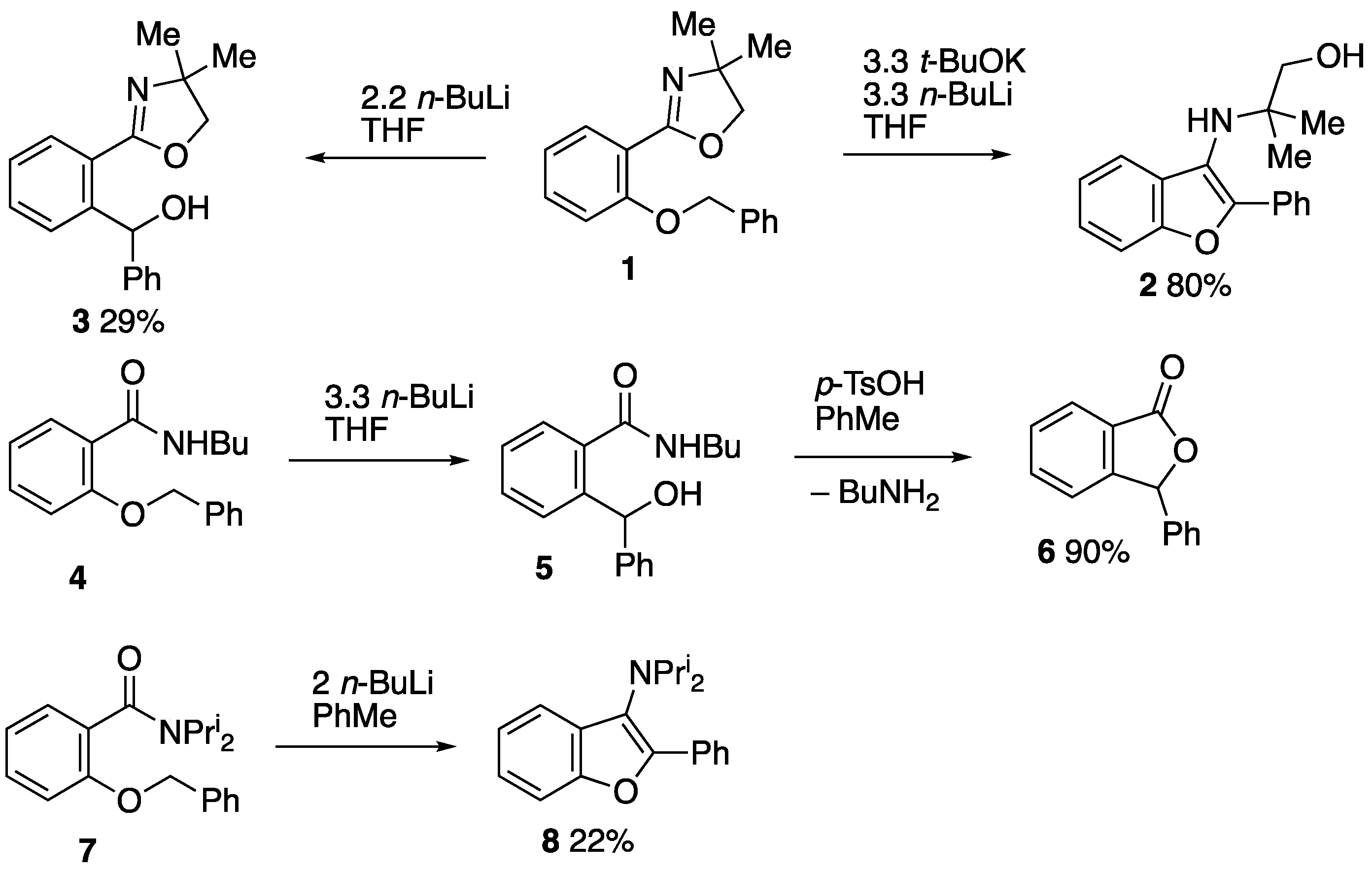
3.2. Preparation and Reactions of 3-Benzyloxy-2-thienyl Systems
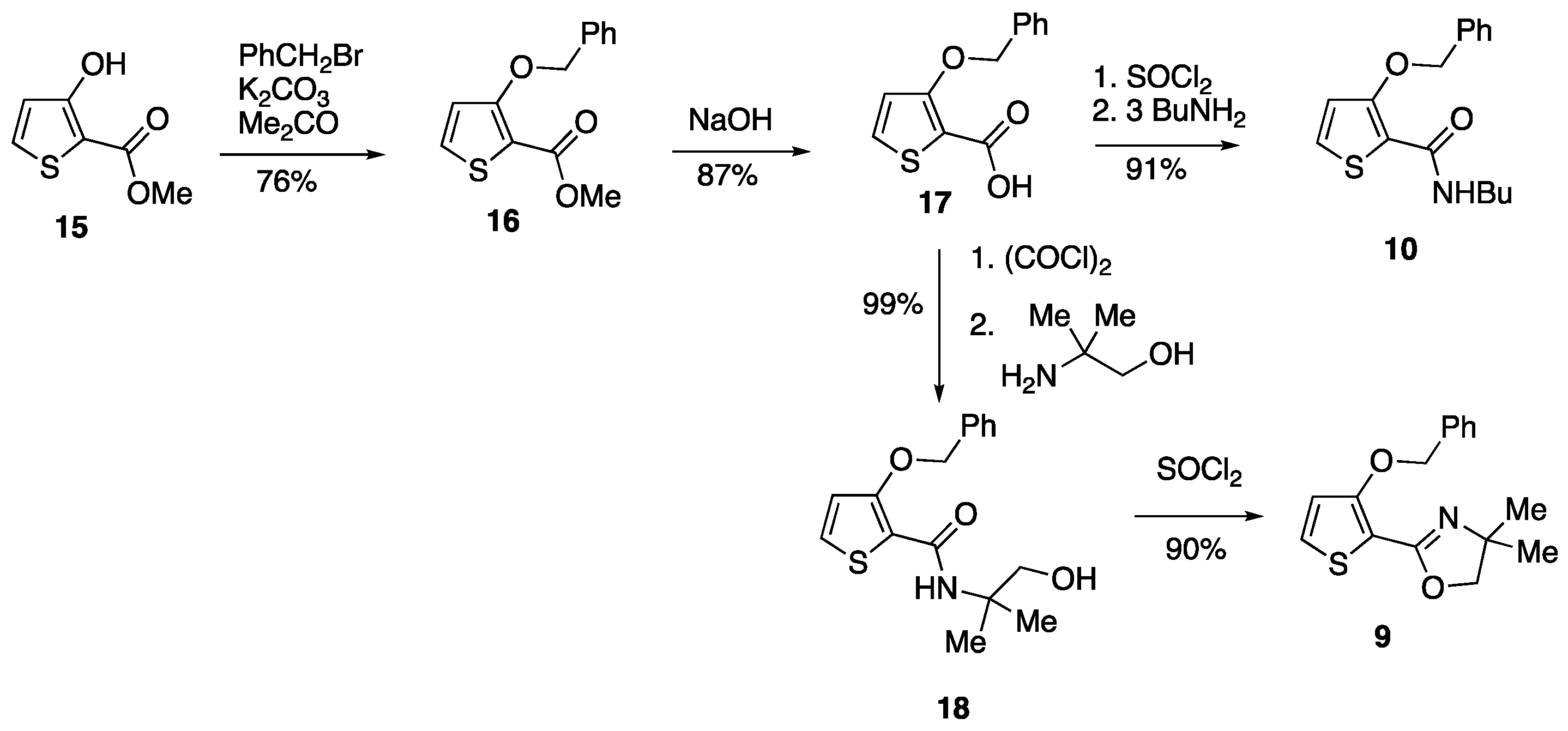
3.2.1. Methyl 3-(Benzyloxy)thiophene-2-carboxylate 16
A literature procedure [
17] was modified as follows: benzyl bromide (11.9 cm
3, 17.11 g, 0.100 mol) was added to a stirred mixture of methyl 3-hydroxythiophene-2-carboxylate
15 (15.85 g, 0.100 mol) and potassium carbonate (27.60 g, 0.200 mol) in acetone (50 cm
3) and the reaction mixture was heated at reflux for 18 h. After cooling to rt, the inorganic salts were removed by filtration and the filtrate was concentrated
in vacuo. The residue was dissolved in CH
2Cl
2 (150 cm
3) and washed with water (100 cm
3) before being dried and evaporated. The crude residue was recrystallised (aq. MeOH) to give
16 (18.94 g, 76%) as pale yellow crystals; mp 69–72 °C; (lit. [
17] 66–67 °C); δ
H (500 MHz) 7.46–7.44 (2H, m, Ph), 7.39–7.35 (2H, m, Ph), 7.35 (1H, d,
J 5.5, 5-H), 7.32–7.29 (1H, m, Ph), 6.82 (1H, d,
J 5.5, 4-H), 5.24 (2H, s, CH
2) and 3.85 (3H, s, CH
3); δ
C (125 MHz) 162.1 (C), 160.8 (C), 136.4 (C), 130.4 (CH), 128.6 (2CH), 127.9 (CH), 126.8 (2CH), 117.5 (CH), 110.4 (C), 73.2 (CH
2) and 51.6 (CH
3). The
1H NMR spectral data were in accordance with those previously reported [
17].
13C NMR data are reported for the first time.
3.2.2. 3-(Benzyloxy)thiophene-2-carboxylic Acid 17
Following a literature procedure [
17], a mixture of methyl 3-(benzyloxy)thiophene-2-carboxylate
16 (18.40 g, 74.1 mmol) and sodium hydroxide (5.99 g, 0.150 mol) in water (150 cm
3) was heated at reflux for 2.5 h. After cooling to rt, the aqueous layer was washed with CH
2Cl
2 (2 × 50 cm
3) before being acidified to pH 1 by the addition of 2 M HCl. The resultant suspension was extracted with CH
2Cl
2 (2 × 100 cm
3) and the combined organic layers were dried and evaporated. The crude residue was recrystallised (aq. MeOH) to give
17 (15.07 g, 87%) as tan-coloured crystals; mp 122–125 °C; (lit. [
17] 125–126 °C); δ
H (400 MHz, CD
3SOCD
3) 12.49 (1H, br s, CO
2H), 7.74 (1H, d,
J 5.6, 5-H), 7.48–7.45 (2H, m, Ph), 7.41–7.37 (2H, m, Ph), 7.35–7.30 (1H, m, Ph), 7.14 (1H, d,
J 5.6, 4-H) and 5.25 (2H, s, CH
2); δ
C (100 MHz, CD
3SOCD
3) 162.4 (C), 160.1 (C), 136.8 (C), 131.2 (CH), 128.4 (2CH), 127.9 (CH), 127.3 (2CH), 118.5 (Ar CH), 110.5 (C) and 72.4 (CH
2). The
1H NMR spectral data were in accordance with those previously reported [
17].
13C NMR data are reported for the first time.
3.2.3. 3-(Benzyloxy)-N-(1-hydroxy-2-methylpropan-2-yl)thiophene-2-carboxamide 18
Oxalyl chloride (3.0 cm3, 4.50 g, 35.5 mmol) was added to a suspension of 3-(benzyloxy)thiophene-2-carboxylic acid 17 (4.00 g, 17.1 mmol) and in Et2O (25 cm3) and the mixture was stirred for 18 h. Evaporation gave 3-(benzyloxy)thiophene-2-carbonyl chloride as a brown oil which was used without further purification.
A solution of 3-(benzyloxy)thiophene-2-carbonyl chloride (assuming 17.1 mmol) in CH2Cl2 (40 cm3) was added dropwise to a solution of 2-amino-2-methylpropan-1-ol (3.10 g, 34.8 mmol) in CH2Cl2 (40 cm3) stirred at 0 °C. After the addition, the mixture was allowed to warm to rt and stirred for 18 h before being poured into water. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (2 × 20 cm3). The combined organic layers were washed successively with 2M HCl, 2M NaOH and water before being dried and evaporated to give 18 (5.14 g, 99%) as a pale yellow solid which was used without further purification; mp 112–115 °C; νmax/cm–1 3358, 3065, 2968, 1626, 1531, 1425, 1240, 1057, 777, 704 and 600; δH (500 MHz) 7.43–7.40 (6H, m, NH and Ph), 7.41 (1H, d, J 5.5, 5-H), 6.93 (1H, d, J 5.5, 4-H), 5.18 (2H, s, OCH2Ph), 3.58 (2H, s, CH2OH) and 1.19 (6H, s, CH3); δC (125 MHz) 162.5 (C), 155.3 (C), 135.2 (C), 129.2 (CH), 129.0 (CH), 128.8 (2CH), 128.0 (2CH), 117.6 (C), 116.2 (CH), 74.1 (CH2), 70.9 (CH2), 56.3 (CMe2) and 24.8 (2CH3); HRMS (NSI+): found 306.1150. C16H20NO3S (M + H) requires 306.1158.
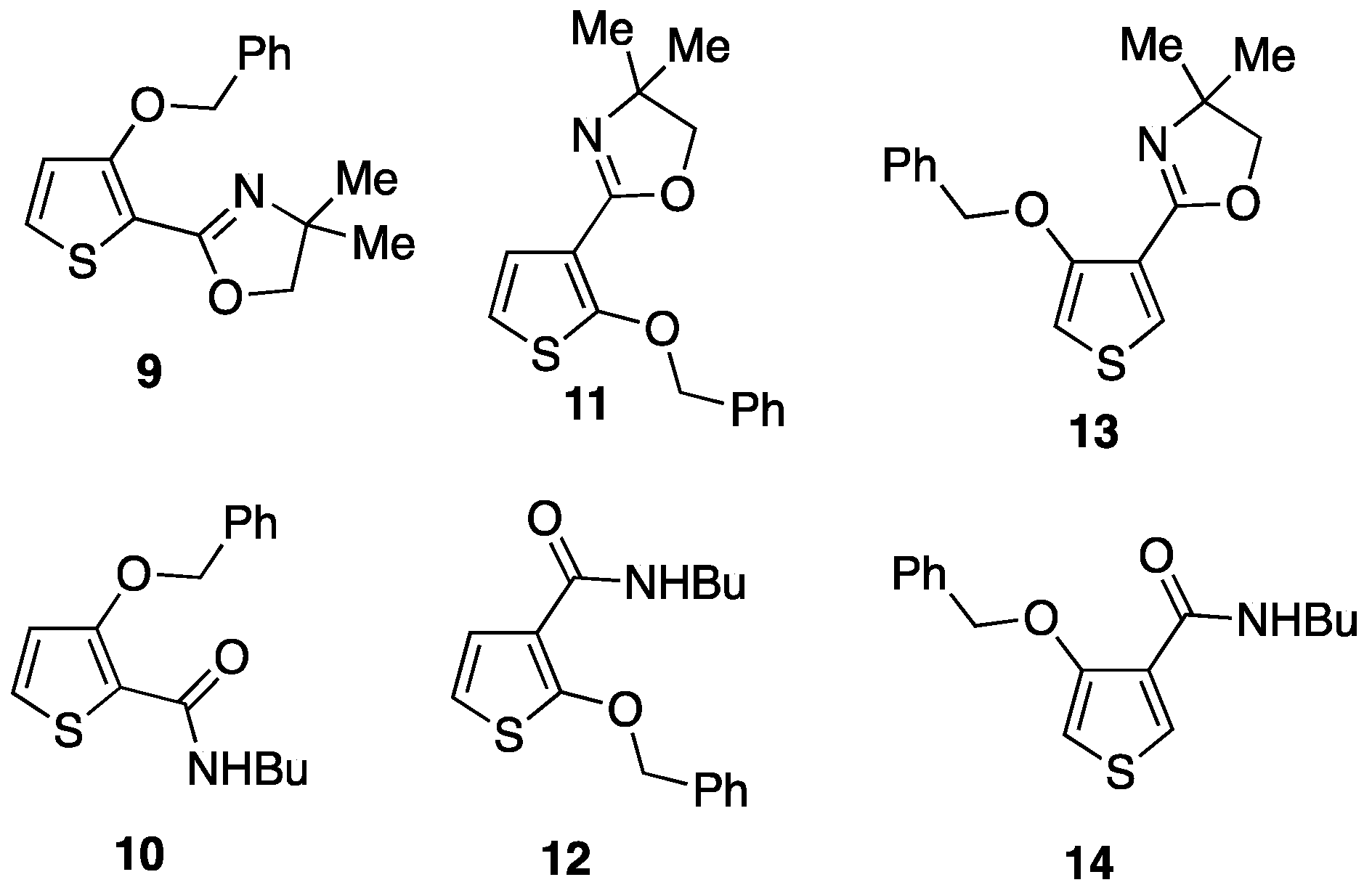
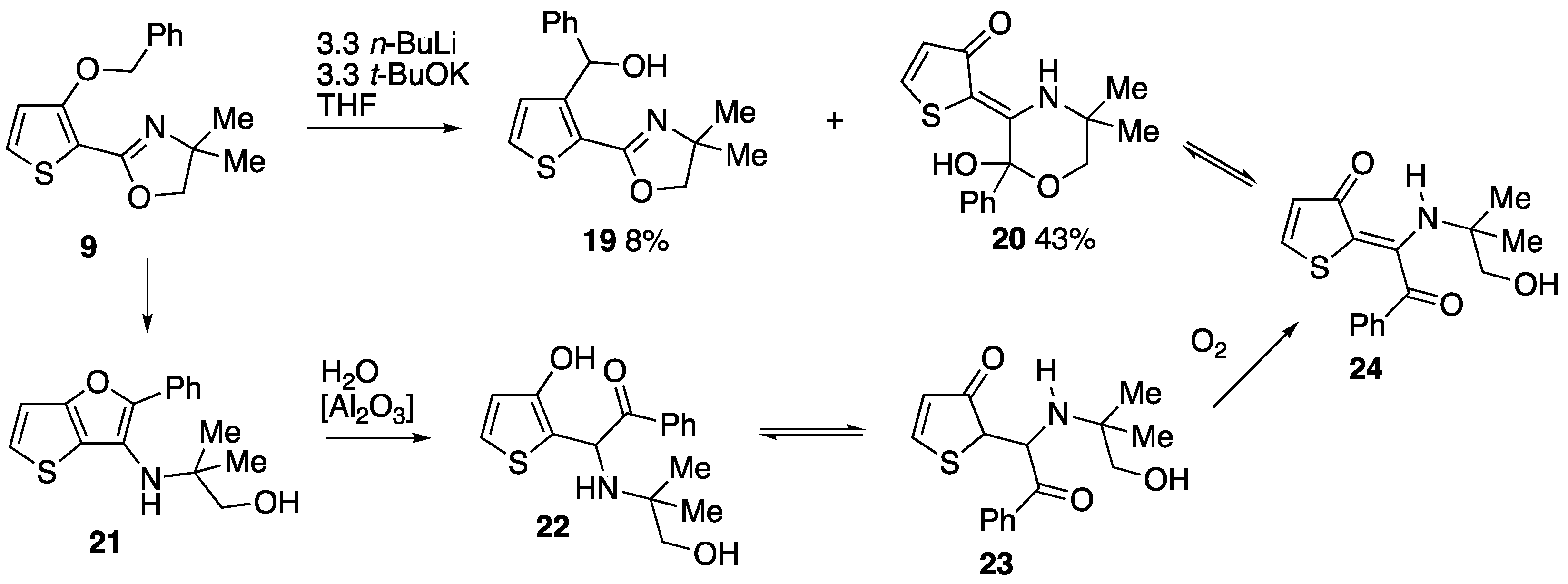
3.2.4. 2-(3-(Benzyloxy)thiophen-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 9
Thionyl chloride (1.4 cm3, 2.28 g, 19.2 mmol) was added to a solution of 3-(benzyloxy)-N-(1-hydroxy-2-methylpropan-2-yl)thiophene-2-carboxamide 18 (4.71 g, 15.4 mmol) in CH2Cl2 (50 cm3) and the mixture was stirred at room temperature for 18 h. The mixture was washed with 2M NaOH and water before being dried and evaporated to give 9 (4.01 g, 90%) as a pale brown oil which solidified on standing as a tan-coloured solid; mp 65–68 °C; νmax/cm–1 3080, 2965, 1632, 1545, 1260, 1231, 1200, 1069, 1026, 766 and 745; δH (500 MHz) 7.45–7.42 (2H, m, Ph), 7.37–7.33 (2H, m, Ph), 7.31–7.27 (1H, m, Ph), 7.24 (1H, d, J 5.5, 5-H), 6.78 (1H, d, J 5.5, 4-H), 5.24 (2H, s, OCH2Ph), 4.09 (2H, s, OCH2) and 1.38 (6H, s, CH3) δC (125 MHz) 157.6 (C), 157.3 (C), 136.8 (C), 128.4 (2CH), 127.8 (2CH), 126.9 (2CH), 118.1 (CH), 108.9 (C), 79.2 (CH2), 73.4 (CH2), 67.1 (CMe2) and 28.3 (2CH3); HRMS (ESI+): found 288.1047. C16H18NO2S (M + H) requires 288.1053.
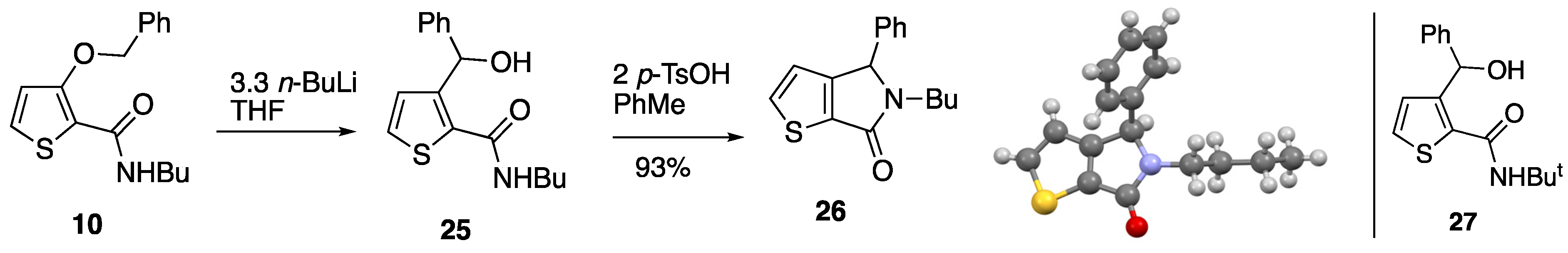
3.2.5. 3-(Benzyloxy)-N-butylthiophene-2-carboxamide 10
Thionyl chloride (2.5 cm3, 4.08 g, 34.3 mmol) was added to a suspension of 3-(benzyloxy)thiophene-2-carboxylic acid 17 (4.00 g, 17.1 mmol) in toluene (30 cm3) and the mixture was heated under reflux for 3 h. After cooling to room temperature, the mixture was evaporated to give 3-(benzyloxy)thiophene-2-carbonyl chloride as a brown oil which was used without further purification.
A solution of 3-(benzyloxy)thiophene-2-carbonyl chloride (assuming 17.1 mmol) in toluene (30 cm3) was added dropwise to a solution of n-butylamine (5.1 cm3, 3.77 g, 51.6 mmol) in toluene (10 cm3) stirred at 0 °C. Once the addition was complete, the reaction mixture was allowed to warm to room temperature over 1 h before being poured into water. The organic layer was separated and washed with 2M NaOH and brine, dried and evaporated to give, after purification by column chromatography (SiO2, Et2O/hexane 7:3), at Rf 0.65, 10 (4.49 g, 91%) as an orange oil; νmax/cm–1 3364, 2961, 1628, 1558, 1435, 1364, 1310, 1074, 976, 773 and 606; δH (500 MHz) 7.43–7.38 (5H, m, Ph), 7.36 (1H, d, J 5.5, 5-H), 7.19 (1H, br s, NH), 6.89 (1H, d, J 5.5, 4-H), 5.19 (2H, s, OCH2), 3.36 (2H, td, J 7.0, 5.5, NCH2), 1.47–1.41 (2H, m, NCH2CH2), 1.28–1.20 (2H, m, CH2CH3) and 0.85 (3H, t, J 7.5, CH3); δC (125 MHz) 161.7 (C), 154.9 (C), 135.6 (C), 128.84 (2CH), 128.76 (CH), 128.5 (CH), 127.7 (2CH), 118.1 (C), 116.2 (CH), 73.9 (OCH2), 38.9 (NCH2), 31.5 (CH2), 20.0 (CH2) and 13.7 (CH3); HRMS (ESI+): found 312.1017. C16H19NaNO2S (M + Na) requires 312.1029.
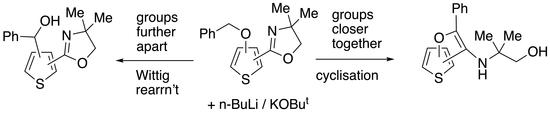
3.2.6. (2-(4,4-Dimethyl-4,5-dihydrooxazol-2-yl)thiophen-3-yl)(phenyl)methanol 19 and (E)-2-(2-Hydroxy-5,5-dimethyl-2-phenylmorpholin-3-ylidene)thiophen-3(2H)-one 20
Under a nitrogen atmosphere,
n-butyllithium (2.5 M in hexanes, 0.66 cm
3, 1.65 mmol) was added to a stirred mixture of 2-(3-(benzyloxy)thiophen-2-yl)-4,4-dimethyl-4,5-dihydrooxazole
9 (0.1440 g, 0.50 mmol) and potassium
tert-butoxide (0.1850 g, 1.65 mmol) in dry THF (5 cm
3). The mixture was stirred at rt for 2 h before being quenched by the addition of saturated aq. NH
4Cl and extracted with Et
2O (3 × 10 cm
3). The combined extracts were dried and evaporated to give, after purification by preparative TLC (Al
2O
3, Et
2O/hexane 7:3), at R
f 0.65,
19 (12 mg, 8%) as an orange oil; δ
H (400 MHz) 7.42–7.38 (2H, m, Ph), 7.34–7.30 (3H, m, ArH and Ph), 7.28–7.23 (1H, m, Ph), 6.71 (1H, d,
J 5.2, ArH), 6.02 (1H, s, C
HOH), 4.11 and 4.09 (2H, AB pattern,
J 8.2, CH
2), 1.40 (3H, s, CH
3) and 1.27 (3H, s, CH
3); δ
C (125 MHz) 158.6 (C), 150.4 (C), 142.8 (C), 130.1 (CH), 128.2 (CH), 128.0 (2CH), 127.1 (CH), 126.5 (2CH), 124.7 (C), 79.5 (CH
2), 70.9 (CHOH), 68.1 (
CMe
2), 28.3 (CH
3) and 28.1 (CH
3). The
1H NMR spectral data were consistent with those previously reported [
18].
13C NMR data are reported for the first time.
This was followed by a second fraction, at Rf 0.15, to give 20 (64.5 mg, 42%) in slightly impure form as brown crystals; mp 103–105 °C; νmax/cm–1 1582, 1537, 1449, 1317, 1260, 1221, 1067, 768, 698 and 669; δH (400 MHz, CD3COCD3) 7.65–7.62 (2H, m, Ph), 7.52 (1H, d, J 5.6, 5-H), 7.40–7.35 (3H, m, Ph), 6.32 (1H, d, J 5.6, 4-H), 4.12 and 3.69 (2H, AB pattern, J 11.6, CH2), 3.06 (2H, br s, OH and NH), 1.53 (3H, s, CH3) and 1.39 (3H, s, CH3); δC (125 MHz, CD3COCD3) 182.1 (C=O), 162.7 (C)), 141.5 (C), 138.5 (CH), 129.5 (CH), 128.6 (2CH), 127.6 (2CH), 122.1 (CH), 103.5 (C), 95.0 (C), 68.3 (CH2), 51.4 (CMe2), 26.8 (CH3) and 26.5 (CH3); HRMS (NSI+): found 304.1004. C16H18NO3S (M + H) requires 304.1002.
3.2.7. N-Butyl-3-(hydroxy(phenyl)methyl)thiophene-2-carboxamide 25 and 5-Butyl-4-phenyl-4,5-dihydro-6H-thieno[2,3-c]pyrrol-6-one 26
Under a nitrogen atmosphere, n-butyllithium (2.5 M in hexanes, 6.6 cm3, 16.5 mmol) was added dropwise to a stirred solution of 3-(benzyloxy)-N-butylthiophene-2-carboxamide 10 (1.45 g, 5.01 mmol) in dry THF (50 cm3). After stirring at room temperature for 2 h, the reaction mixture was quenched by the addition of saturated aq. NH4Cl and extracted with Et2O (3 × 30 cm3). The combined organic extracts were washed with NaOH and water before being dried and evaporated to give 25 as a pale brown oil which was used without further purification; νmax/cm–1 3256, 3086, 2957, 2930, 1612, 1545, 1450, 1302, 1026, 698 and 669; δH (400 MHz) 7.37–7.29 (4H, m, Ph), 7.27–7.23 (1H, m, Ph), 7.21 (1H, d, J 5.0, 5-H), 6.96 (1H, t, J 5.6, NH), 6.71 (1H, d, J 5.0, 4-H), 6.02 (1H, s, CHOH), 5.87 (1H, br s, OH), 3.30 (2H, td, J 7.2, 5.6, NCH2), 1.52–1.45 (2H, m, NCH2CH2), 1.35–1.26 (2H, m, CH2CH3) and 0.89 (3H, t, J 7.2, CH3); δC (75 MHz) 163.1 (C=O), 147.8 (C), 142.3 (C), 133.4 (C), 130.4 (CH), 128.2 (2CH), 127.3 (CH), 126.7 (CH), 126.2 (2CH), 70.9 (CHOH), 39.9 (NCH2), 31.3 (CH2), 20.0 (CH2) and 13.7 (CH3); HRMS (ESI+): found 312.1023. C16H19NaNO2S (M + Na) requires 312.1029.
A mixture of N-butyl-3-(hydroxy(phenyl)methyl)thiophene-2-carboxamide 25 (assuming 5.01 mmol) and p-toluenesulfonic acid monohydrate (1.90 g, 9.99 mmol) in toluene (50 cm3) was heated at reflux for 1 h. After cooling to room temperature, the reaction mixture was washed with water (50 cm3), 2 M NaOH (50 cm3) and brine (50 cm3) before being dried and evaporated. The crude residue was purified by filtration through a silica plug (Et2O) to give 26 (1.26 g, 93%) as a tan-coloured solid; mp 90–93 °C; νmax/cm–1 2955, 1668, 1441, 1398, 1310, 1069, 781, 743, 698 and 637; δH (400 MHz) 7.55 (1H, d, J 4.8, 5-H), 7.38–7.32 (3H, m, Ph), 7.16–7.13 (2H, m, Ph), 6.79 (1H, d, J 4.8, 4-H), 5.39 (1H, s, CHPh), 3.84 (1H, dt, J 14.4, 7.8, NCH), 2.86–2.79 (1H, m, NCH), 1.54–1.46 (2H, m, NCH2CH2), 1.34–1.24 (2H, m, CH2CH3) and 0.88 (3H, t, J 7.2, CH3); δC (125 MHz) 164.4 (C=O), 155.7 (C), 136.0 (C), 134.8 (C), 134.5 (CH), 129.0 (2CH), 128.6 (CH), 127.3 (2CH), 120.7 (CH), 62.9 (CHPh), 40.3 (NCH2), 30.6 (CH2), 19.9 (CH2) and 13.7 (CH3); HRMS (NSI+): found 272.1103. C16H18NOS (M + H) requires 272.1104.
3.3. Preparation and Reactions of 2-Benzyloxy-3-thienyl Systems
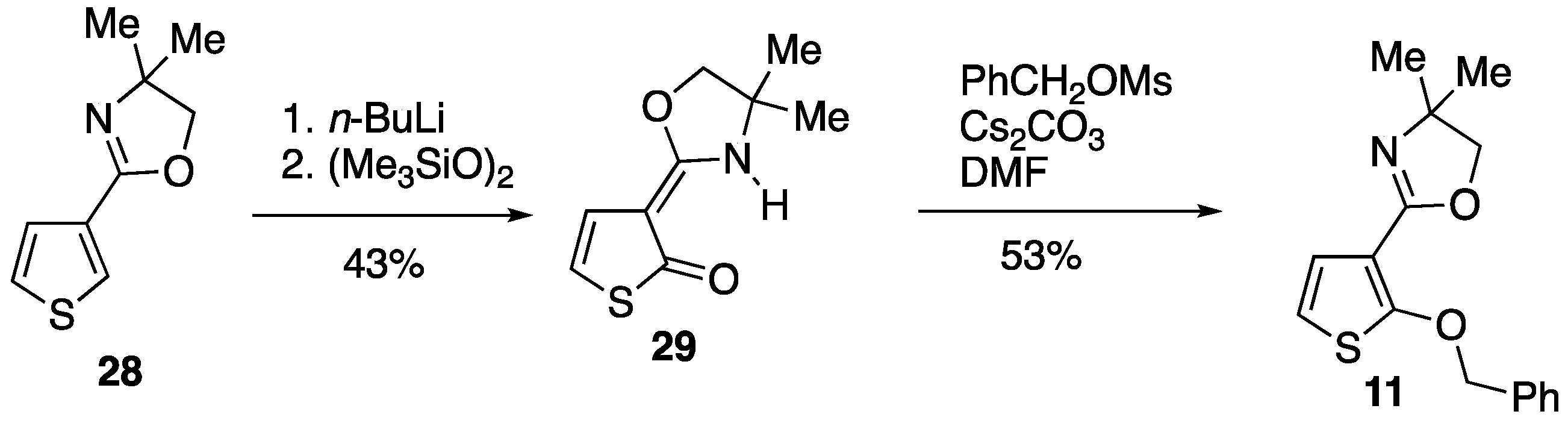
3.3.1. Attempted Cyclisation of 2-(2-(Benzyloxy)thiophen-3-yl)-4,4-dimethyl-4,5-dihydrooxazole 11
Under a nitrogen atmosphere,
n-butyllithium (2.5 M in hexanes, 0.66 cm
3, 1.65 mmol) was added to a stirred mixture of 2-(2-(benzyloxy)thiophen-3-yl)-4,4-dimethyl-4,5-dihydrooxazole
11 [
11] (0.1437 g, 0.50 mmol) and potassium
tert-butoxide (0.1875 g, 1.67 mmol) in dry THF (5 cm
3). The mixture was stirred at rt for 2 h before being quenched by the addition of saturated aq. NH
4Cl and extracted with Et
2O (3 × 10 cm
3). The combined extracts were dried and evaporated to give, after purification by preparative TLC (Al
2O
3, Et
2O/hexane 1:1), at R
f 0.50, (3-(4,4-Dimethyl-4,5-dihydrooxazol-2-yl)thiophen-2-yl)(phenyl)methanol
30 (96.7 mg, 67%) as an orange oil; ν
max/cm
–1 3177, 2965, 1636, 1535, 1452, 1288, 1194, 1148, 974 and 698; δ
H (500 MHz) 8.04 (1H, br s, OH), 7.49 (2H, d,
J 7.0, Ph), 7.36–7.28 (4H, m, ArH and Ph), 7.06 (1H, d,
J 5.0, ArH), 6.10 (1H, s, C
HOH), 4.09 and 4.06 (2H, AB pattern,
J 6.8, CH
2), 1.38 (3H, s, CH
3) and 1.28 (3H, s, CH
3); δ
C (125 MHz) 159.6 (C), 153.7 (C), 141.8 (C), 128.6 (CH), 127.9 (2CH), 127.7 (CH), 126.8 (2CH), 125.5 (C), 123.2 (CH), 79.0 (CH
2), 69.5 (CHOH), 67.3 (4ry,
CMe
2), 28.5 (CH
3) and 28.1 (CH
3); HRMS (NSI
+): found 288.1052. C
16H
18NO
2S (M + H) requires 288.1053.
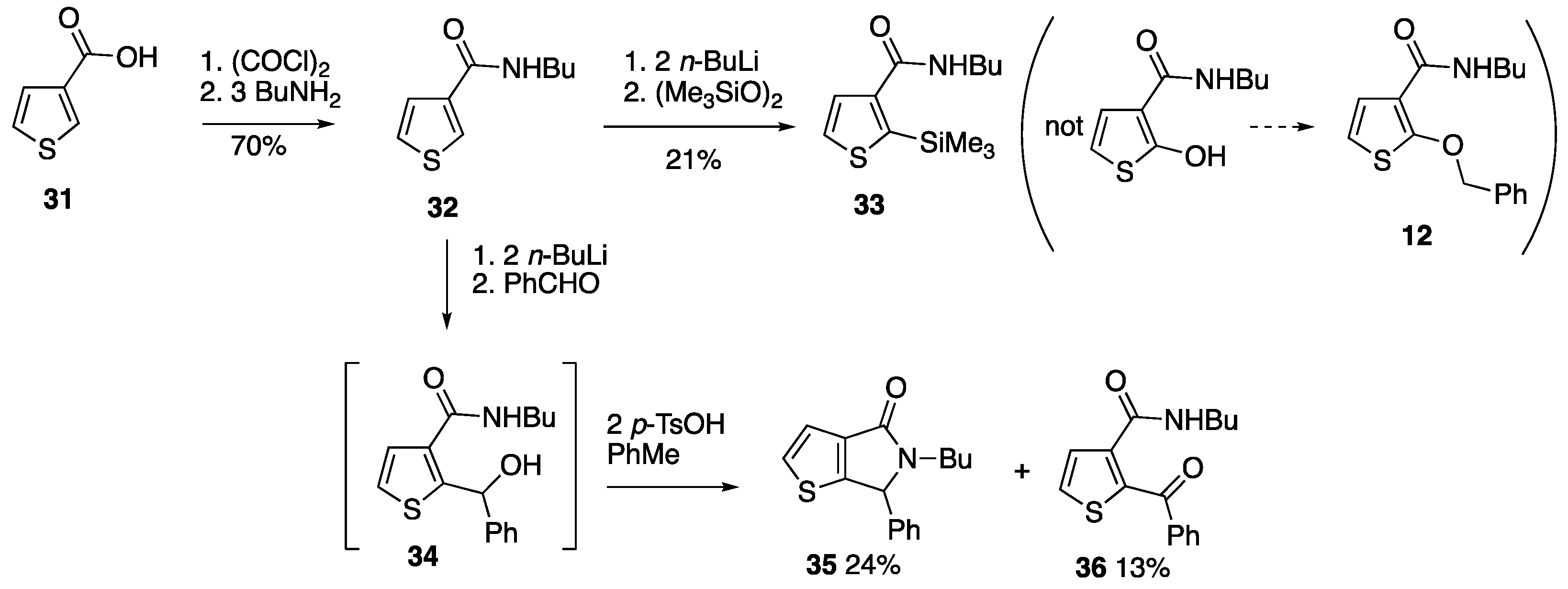
3.3.2. N-Butylthiophene-3-carboxamide 32
Oxalyl chloride (2.0 cm3, 3.00 g, 23.6 mmol) was added to a solution of thiophene-3-carboxylic acid 31 (2.52 g, 19.7 mmol) in CH2Cl2 (30 cm3) and the mixture was stirred for 18 h. Evaporation gave thiophene-3-carbonyl chloride as a pale-yellow solid which was used immediately without further purification.
A solution of thiophene-3-carbonyl chloride (assuming 19.7 mmol) in toluene (30 cm
3) was added dropwise to a solution of
n-butylamine (5.8 cm
3, 4.29 g, 58.7 mmol) in toluene (30 cm
3) stirred at 0 °C. Once the addition was complete, the reaction mixture was allowed to warm to rt over 1 h before being poured into water. The organic layer was separated and washed with 2M NaOH and brine, dried and evaporated to give, after recrystallisation (EtOAc/hexane),
32 (2.54 g, 70%) as colourless crystals; mp 66–68 °C; (lit. [
19] 53–55 °C); ν
max/cm
–1 3253, 3085, 2921, 1617, 1555, 1301, 1220, 1127, 881, 831, 741 and 707; δ
H (500 MHz) 7.84 (1H, dd,
J 3.0, 1.5, ArH), 7.37 (1H, dd,
J 5.0, 1.5, ArH), 7.33 (1H, dd,
J 5.0, 3.0, ArH), 6.02 (1H, br s, NH), 3.43 (2H, td,
J 7.0, 6.0, NCH
2), 1.62–1.56 (2H, m, NCH
2C
H2), 1.44–1.37 (2H, m, C
H2CH
3) and 0.95 (3H, t,
J 7.5, CH
3). The
1H NMR spectral data were in accordance with those previously reported [
19]. IR data are reported for the first time.
3.3.3. N-Butyl-2-(trimethylsilyl)thiophene-3-carboxamide 33
Under a nitrogen atmosphere, n-butyllithium (2.5 M in hexane, 5.2 cm3, 13.0 mmol) was added dropwise to a stirred −78 °C solution of N-butylthiophene-3-carboxamide 32 (1.10 g, 6.00 mmol) in dry THF (30 cm3). After stirring at −78 °C for 5 min, the reaction mixture was allowed to warm to rt for 1 h, before being cooled to −78 °C and treated with bis(trimethylsilyl) peroxide (1.28 g, 7.18 mmol). The reaction mixture was allowed to warm to rt over 18 h before being poured into sat. aq. NH4Cl (100 cm3) and extracted with Et2O (3 × 50 cm3). The combined organic extracts were dried and evaporated and the crude residue was purified by column chromatography (SiO2, Et2O/hexane 3:2) to give, at Rf 0.90, 33 (0.32 g, 21%) as tan-coloured crystals; mp 86–89 °C; νmax/cm–1 3285, 2957, 1620, 1558, 1402, 1296, 1240, 1005, 833, 746, 704 and 604; δH (500 MHz) 7.50 (1H, d, J 5.0, ArH), 7.29 (1H, d, J 5.0, ArH), 5.94 (1H, br s, NH), 3.41 (2H, td, J 7.0, 6.0, NCH2), 1.61–1.55 (2H, m, NCH2CH2), 1.43–1.36 (2H, m, CH2CH3), 0.94 (3H, t, J 7.5, CH2CH3) and 0.39 (9H, s, SiMe3); δC (125 MHz) 164.6 (C=O), 145.7 (C), 143.0 (C), 130.2 (CH), 126.9 (CH), 39.6 (NCH2), 31.7 (CH2), 20.1 (CH2), 13.8 (CH3) and 0.0 (SiMe3); HRMS (NSI+): found 256.1184. C12H22NOSSi (M + H) requires 256.1186.
3.3.4. 5-Butyl-6-phenyl-5,6-dihydro-4H-thieno[2,3-c]pyrrol-4-one 35 and 2-Benzoyl-N-butylthiophene-3-carboxamide 36
Under a nitrogen atmosphere, n-butyllithium (2.5 M in hexane, 4.2 cm3, 10.5 mmol) was added dropwise to a stirred −78 °C solution of N-butylthiophene-3-carboxamide 32 (0.9158 g, 5.00 mmol) in dry THF (50 cm3). After stirring at −78 °C for 5 min, the reaction mixture was allowed to warm to rt for 1 h before benzaldehyde (0.57 cm3, 0.60 g, 5.61 mmol) was added and stirring was continued for 18 h. The reaction mixture was poured into sat. aq. NH4Cl (100 cm3) and extracted with Et2O (3 × 50 cm3) and the combined organic layers were dried and evaporated.
The residue was dissolved in toluene (100 cm3) and treated with p-toluenesulfonic acid monohydrate (1.90 g, 9.99 mmol) before being heated at reflux for 1 h. After cooling to rt, the reaction mixture was washed with water (50 cm3), 2 M NaOH (50 cm3) and brine (50 cm3) before being dried and evaporated. The crude residue was purified by column chromatography (SiO2, Et2O/hexane 3:2) to give, at Rf 0.50, 35 (0.3223 g, 24%) as a pale yellow solid; mp 96–98 °C; νmax/cm–1 2953, 1668, 1454, 1412, 1375, 1308, 1267, 1070, 760, 700 and 575; δH (400 MHz) 7.40–7.34 (4H, m, ArH), 7.27 (1H, d, J 4.8, ArH), 7.17–7.13 (2H, m, ArH), 5.51 (1H, s, CHPh), 3.84 (1H, dt, J 14.0, 8.0, NCH), 2.81 (1H, ddd, J 14.0, 7.6, 6.4, NC), 1.53–1.45 (2H, m, NCH2CH2), 1.35–1.24 (2H, m, CH2CH3) and 0.88 (3H, t, J 7.4, CH3); δC (125 MHz) 165.1 (C=O), 155.0 (C), 139.8 (C), 136.7 (C), 130.2 (CH), 129.2 (2CH), 129.0 (CH), 127.3 (2CH), 120.1 (CH), 62.9 (CHPh), 40.3 (NCH2), 30.7 (CH2), 20.0 (CH2) and 13.7 (CH3); HRMS (ASAP+): found 272.1115. C16H18NOS (M + H) requires 272.1104.
This was followed by a second fraction to give, at Rf 0.30, 36 (0.1817 g, 13%) as an orange oil; νmax/cm–1 3277, 2957, 2870, 1630, 1549, 1449, 1406, 1279, 847 and 691; δH (400 MHz) 8.65 (1H, br s, NH), 7.84–7.81 (2H, m, Ph), 7.75 (1H, d, J 5.0, ArH), 7.64–7.60 (1H, m, Ph), 7.55 (1H, d, J 5.0, ArH), 7.51–7.46 (2H, m, Ph), 3.33 (2H, td, J 7.2, 5.6, NCH2), 1.56–1.49 (2H, m, NCH2CH2), 1.42–1.33 (2H, m, CH2CH3) and 0.92 (3H, t, J 7.4, CH3); δC (100 MHz, signals for major amide rotamer only) 190.6 (COPh), 162.5 (CONHBu), 142.6 (C), 138.8 (C), 138.1 (C), 133.3 (CH), 132.8 (CH), 130.4 (CH), 129.8 (2CH), 128.3 (2CH), 39.7 (NCH2), 31.2 (CH2), 20.2 (CH2) and 13.7 (CH3); HRMS (NSI+): found 288.1052. C16H18NO2S (M + H) requires 288.1053.
3.4. Preparation and Reactions of 4-Benzyloxy-3-thienyl Systems
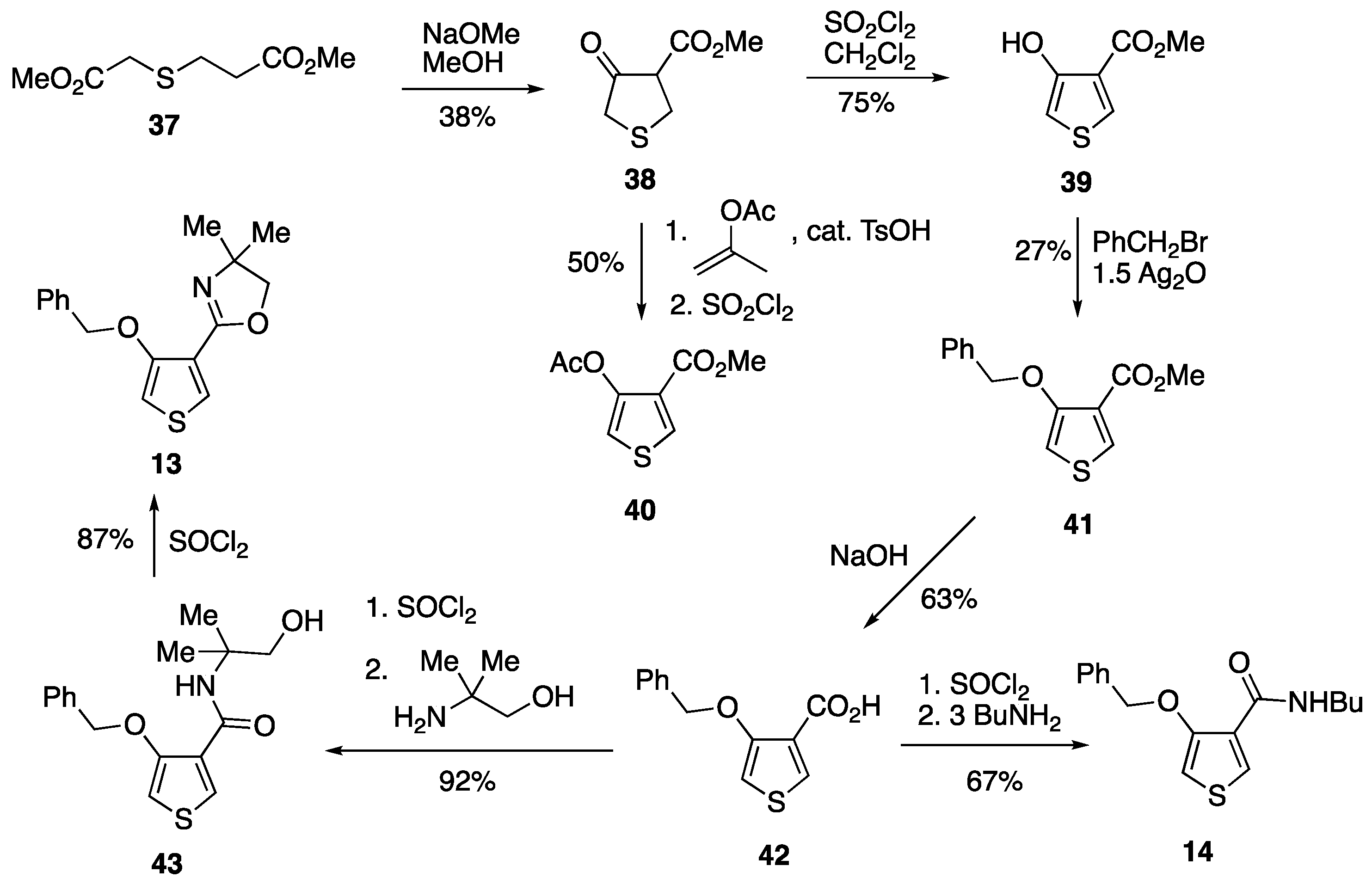
3.4.1. Methyl 3-((2-Methoxy-2-oxoethyl)thio)propanoate 37
Following a literature procedure [
12], methyl acrylate (18.37 g, 0.213 mol) was added dropwise to a stirred mixture of methyl thioglycolate (21.18 g, 0.200 mol) and piperidine (0.2 cm
3, 0.17 g, 2.02 mmol). Once approximately half of the methyl acrylate had been added, further piperidine (0.2 cm
3, 0.17 g, 2.02 mmol) was added. Once the addition was complete, the reaction mixture was heated at 80 °C for 30 min. After cooling to rt, the reaction mixture was diluted with Et
2O (150 cm
3) and washed with water (5 × 50 cm
3) before being dried and evaporated to give
37 (37.12 g, 97%) as a pale yellow oil which was used without further purification; δ
H (400 MHz) 3.75 (3H, s, CH
3), 3.71 (3H, s, CH
3), 3.26 (2H, s, MeO
2CCH
2S), 2.92 (2H, t,
J 7.2, SC
H2CH
2CO
2Me) and 2.66 (2H, t,
J 7.2, SCH
2C
H2CO
2Me); δ
C (100 MHz) 172.0 (C=O), 170.7 (C=O), 52.4 (CH
3), 51.8 (CH
3), 34.1 (CH
2), 33.4 (CH
2) and 27.5 (CH
2). The
1H NMR spectral data were in accordance with those previously reported [
11].
13C NMR data are reported for the first time.
3.4.2. Methyl 4-Oxotetrahydrothiophene-3-carboxylate 38
Following a literature procedure [
13], sodium methoxide was prepared by the addition of sodium (12.51 g, 0.544 mol) in small portions to methanol (90 cm
3). Once the sodium had fully dissolved, a solution of methyl 3-((2-methoxy-2-oxoethyl)thio)propanoate
37 (37.12 g, 0.193 mol) in methanol (30 cm
3) was added dropwise and the reaction mixture was heated at reflux for 1 h. After cooling to rt, the reaction mixture was poured into a mixture of crushed ice (400 g) and conc. HCl (100 cm
3) before being extracted with CH
2Cl
2 (2 × 300 cm
3). The combined organic layers were washed with sat. aq. NaHCO
3 (250 cm
3) before being dried and evaporated. The crude residue was purified by distillation to give
38 (11.69 g, 38%) as a colourless oil which partially crystallised on standing; bp 103 °C/4.9 Torr; (lit. [
20] 109 °C/4 Torr).
3.4.3. Methyl 4-Hydroxythiophene-3-carboxylate 39
Following a literature procedure [
14], sulfuryl chloride (9.7 cm
3, 16.15 g, 0.120 mol) was added dropwise to a stirred 0 °C solution of methyl 4-oxotetrahydrothiophene-3-carboxylate
973 (17.41 g, 0.109 mol) in CH
2Cl
2 (110 cm
3) over a period of 1 h. The reaction mixture was stirred at 0 °C for 30 min before being washed with sat. aq. NaHCO
3 (150 cm
3) and water (3 × 50 cm
3). The organic layer was dried and evaporated to give after filtration through a silica plug (Et
2O),
39 (12.93 g, 75%) as a light brown low-melting solid; δ
H (300 MHz) 8.72 (1H, s, OH), 7.90 (1H, d,
J 3.6, ArH), 6.39 (1H, d,
J 3.6, ArH) and 3.92 (3H, s, CH
3); δ
C (125 MHz) 165.4 (C=O), 155.3 (C–O), 131.0 (CH), 119.0 (C), 100.0 (CH) and 51.8 (CH
3). The
1H NMR spectral data were in accordance with those previously reported [
15].
13C NMR data are reported for the first time.
3.4.4. Methyl 4-Acetoxythiophene-3-carboxylate 40
Following a literature procedure [
15], a mixture of methyl 4-oxotetrahydrothiophene-3-carboxylate
38 (30.80 g, 0.192 mol) and
p-toluenesulfonic acid monohydrate (0.19 g, 1.00 mmol) in isopropenyl acetate (70 cm
3) was heated at reflux for 18 h. After cooling to rt, the reaction mixture was concentrated
in vacuo to give methyl 4-acetoxy-2,5-dihydrothiophene-3-carboxylate as a dark brown oil which was used without further purification.
Following a literature procedure [
15], sulfuryl chloride (19.5 cm
3, 32.47 g, 0.241 mol) was added dropwise to a stirred −25 °C solution of methyl 4-acetoxy-2,5-dihydrothiophene-3-carboxylate (assuming 0.192 mol) in CH
2Cl
2 (80 cm
3) over a period of 1 h. The reaction mixture was allowed to warm to rt for 18 h before being evaporated to give
40 (19.10 g, 50%) as a dark brown oil which was used without further purification; δ
H (500 MHz) 8.07 (1H, d,
J 3.8, ArH), 6.98 (1H, d,
J 3.8, ArH), 3.83 (3H, s, OCH
3) and 2.33 (3H, s, COCH
3). The
1H NMR spectral data were in accordance with those previously reported [
15].
3.4.5. Silver(I) Oxide
A solution of sodium hydroxide (14.61 g, 0.365 mol) in water (440 cm3) was added dropwise to a stirred solution of silver nitrate (60.00 g, 0.353 mol) in water (110 cm3). Once the addition was complete, the precipitate was collected by filtration and washed with water until the washings were neutral before being dried in vacuo to give the title compound (39.60 g, 97%) as a brown solid which was stored in the dark and used without further purification.
3.4.6. Methyl 4-(Benzyloxy)thiophene-3-carboxylate 41
A mixture of methyl 4-hydroxythiophene-3-carboxylate 39 (12.91 g, 81.6 mmol), silver(I) oxide (28.40 g, 0.123 mol) and benzyl bromide (10.7 cm3, 15.39 g, 90.0 mmol) in CH2Cl2 (500 cm3) was heated at reflux for 3 d. After cooling to rt, the reaction mixture was filtered and evaporated and the crude residue was purified by column chromatography (SiO2, Et2O/hexane 1:1) to give, at Rf 0.90, 983 (5.47 g, 27%) as a red oil; νmax/cm–1 3113, 2949, 1726, 1541, 1449, 1265, 1082, 770 and 698; δH (500 MHz) 8.01 (1H, d, J 3.5, ArH), 7.48 (2H, d, J 7.5, Ph), 7.38 (2H, t, J 7.5, Ph), 7.31 (1H, t, J 7.5, Ph), 6.31 (1H, d, J 3.5, ArH), 5.13 (2H, s, CH2) and 3.86 (3H, s, CH3); δC (125 MHz) 162.2 (C=O), 156.1 (C–O), 136.5 (C), 133.2 (CH), 128.5 (2CH), 127.8 (CH), 126.8 (2CH), 123.7 (C), 99.9 (CH), 72.3 (CH2) and 51.6 (CH3); HRMS (ESI+): found 271.0393. C13H12NaO3S (M + Na) requires 271.0399.
3.4.7. 4-(Benzyloxy)thiophene-3-carboxylic Acid 42
A mixture of methyl 4-(benzyloxy)thiophene-3-carboxylate 41 (5.17 g, 20.8 mmol) and sodium hydroxide (1.74 g, 43.5 mmol) in water (45 cm3) was heated at reflux for 18 h. After cooling to rt, the reaction mixture was washed with CH2Cl2 (30 cm3) before being adjusted to pH 1 by the addition of 2 M HCl and extracted with CH2Cl2 (2 × 50 cm3). The combined organic extracts were dried and evaporated to give, after recrystallisation (PhMe), 42 (3.08 g, 63%) as brown crystals; mp 101–105 °C; νmax/cm–1 3123, 1667, 1537, 1447, 1285, 1196, 1088, 880 and 764; δH (500 MHz) 9.96 (1H, br s, CO2H), 8.21 (1H, d, J 3.5, ArH), 7.45–7.37 (5H, m, Ph), 6.48 (1H, d, J 3.5, ArH) and 5.20 (2H, s, CH2); δC (125 MHz) 164.7 (C=O), 155.0 (C–O), 135.5 (C), 135.2 (CH), 128.6 (2CH), 128.3 CH), 127.2 (2CH), 122.7 (C), 100.4 (CH) and 72.9 (CH2); HRMS (ESI+): found 257.0239. C12H10NaO3S (M + Na) requires 257.0243.
3.4.8. 4-(Benzyloxy)-N-(1-hydroxy-2-methylpropan-2-yl)thiophene-3-carboxamide 43
Thionyl chloride (0.31 cm3, 0.51 g, 4.25 mmol) was added to a suspension of 4-(benzyloxy)thiophene-3-carboxylic acid 42 (0.50 g, 2.13 mmol) in toluene (5 cm3) and the mixture was heated under reflux for 3 h. After cooling to rt, the mixture was evaporated to give 4-(benzyloxy)thiophene-3-carbonyl chloride as a red oil which was used immediately without further purification.
A solution of 4-(benzyloxy)thiophene-3-carbonyl chloride (assuming 2.13 mmol) in CH2Cl2 (30 cm3) was added dropwise to a solution of 2-amino-2-methylpropan-1-ol (0.41 g, 4.60 mmol) in CH2Cl2 (10 cm3) stirred at 0 °C. After the addition, the mixture was allowed to warm to rt and stirred for 18 h before being poured into water. The organic layer was separated and the aqueous layer extracted with CH2Cl2 (2 × 20 cm3). The combined organic layers were washed successively with 2M HCl, 2M NaOH and water before being dried and evaporated to give 43 (0.60 g, 92%) as a pale-yellow solid which was used without further purification; mp 90–92 °C; νmax/cm–1 3366, 2970, 1628, 1560, 1435, 1364, 1310, 1074, 976 and 606; δH (400 MHz) 8.07 (1H, d, J 3.6, ArH), 7.65 (1H, br s, NH), 7.46–7.37 (5H, m, Ph), 6.46 (1H, d, J 3.6, ArH), 5.22 (1H, br s, OH), 5.08 (2H, s, OCH2Ph), 3.57 (2H, s, CH2OH) and 1.16 (6H, s, CH3); δC (100 MHz) 162.1 (C=O), 153.1 (C–O), 135.1 (C), 131.9 (CH), 128.8 (CH), 128.7 (2CH), 128.1 (2CH), 126.6 (C), 99.7 (CH), 73.2 (CH2), 70.7 (CH2), 56.0 (CMe2) and 24.5 (2CH3); HRMS (NSI+): found 306.1150. C16H20NO3S (M + H) requires 306.1158.
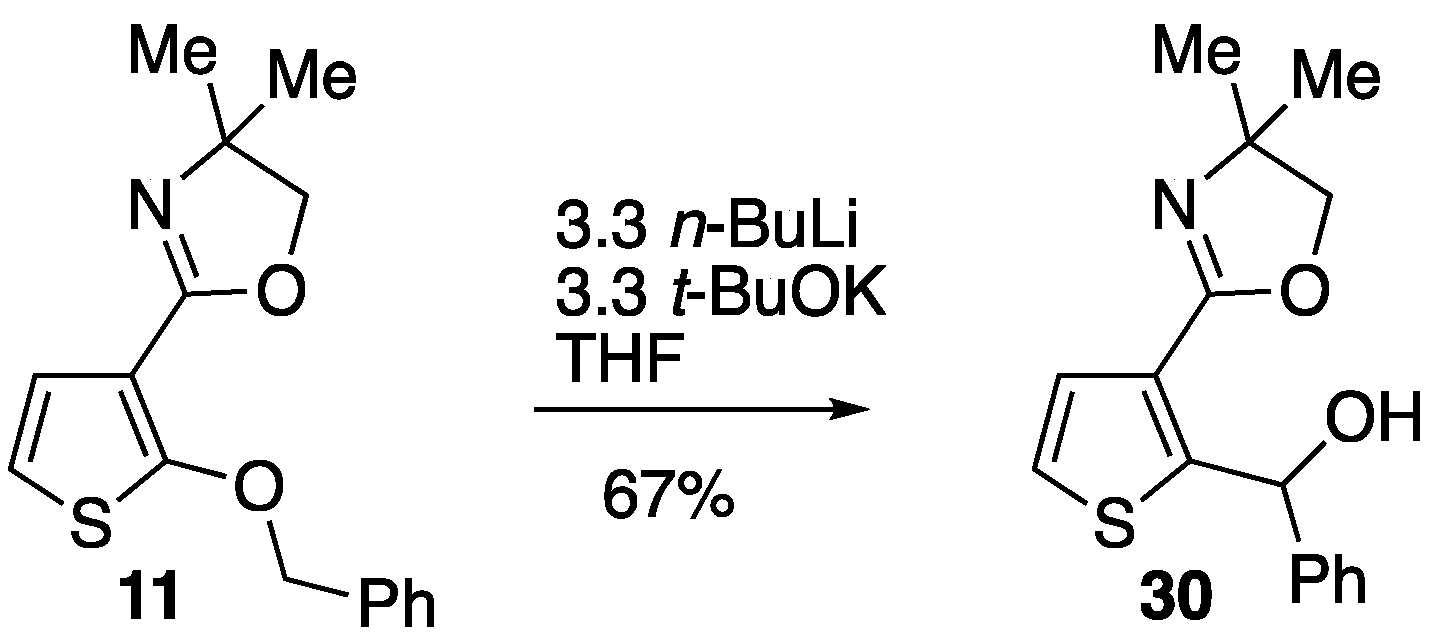
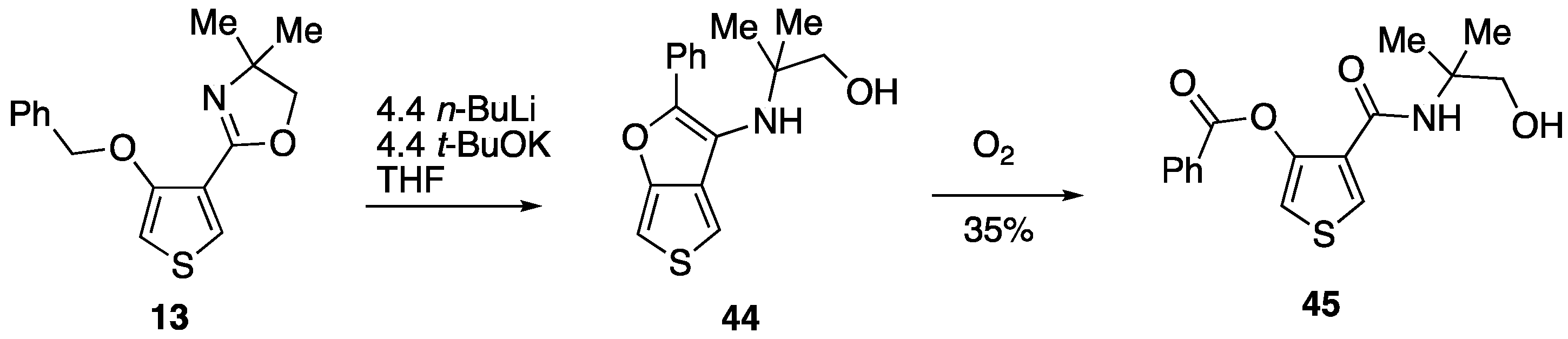
3.4.9. 2-(4-(Benzyloxy)thiophen-3-yl)-4,4-dimethyl-4,5-dihydrooxazole 13
Thionyl chloride (0.15 cm3, 0.24 g, 2.06 mmol) was added to a solution of 4-(benzyloxy)-N-(1-hydroxy-2-methylpropan-2-yl)thiophene-3-carboxamide 43 (0.50 g, 1.64 mmol) in CH2Cl2 (20 cm3) and the mixture was stirred at rt for 18 h. The mixture was washed with 2M NaOH and water before being dried and evaporated to give, after purification by column chromatography (SiO2, Et2O/hexane 3:2), at Rf 0.40, 13 (0.41 g, 87%) as a pale yellow solid; mp 77–80 °C; νmax/cm–1 2965, 1651, 1535, 1449, 1371, 1211, 1194, 1042, 733 and 698; δH (400 MHz) 7.79 (1H, d, J 3.6, ArH), 7.47 (2H, d, J 7.6, Ph), 7.36 (2H, t, J 7.4, Ph), 7.29 (1H, t, J 7.2, Ph), 6.30 (1H, d, J 3.6, ArH), 5.16 (2H, s, OCH2Ph), 4.05 (2H, s, OCH2) and 1.39 (6H, s, CH3); δC (100 MHz) 157.2 (C), 155.3 (C), 136.9 (C), 129.1 (CH), 128.4 (2CH), 127.6 CH), 126.7 (2CH), 121.6 (C), 100.3 (CH), 78.5 (OCH2), 72.4 (OCH2), 67.5 (CMe2) and 28.4 (2CH3); HRMS (ESI+): found 288.1047. C16H18NO2S (M + H) requires 288.1053.
3.4.10. 4-((1-Hydroxy-2-methylpropan-2-yl)carbamoyl)thiophen-3-yl Benzoate 45
Under a nitrogen atmosphere, n-butyllithium (2.5 M in hexanes, 0.88 cm3, 2.20 mmol) was added to a stirred mixture of 2-(4-(benzyloxy)thiophen-3-yl)-4,4-dimethyl-4,5-dihydrooxazole 13 (0.1443 g, 0.50 mmol) and potassium tert-butoxide (0.2485 g, 2.21 mmol) in dry THF (5 cm3). The mixture was stirred at rt for 2 h before being quenched by the addition of saturated aq. NH4Cl and extracted with Et2O (3 × 10 cm3). The combined extracts were dried and evaporated to give, after purification by preparative TLC (Al2O3, Et2O/hexane 1:1), at Rf 0.35, 45 (55.8 mg, 35%) in slightly impure form as a brown oil; νmax/cm–1 3335, 2972, 1744, 1638, 1545, 1450, 1246, 1177, 1047, 908, 766 and 700; δH (400 MHz) 8.19–8.16 (2H, m, Ph), 7.99 (1H, d, J 3.6, ArH), 7.71–7.67 (1H, m, Ph), 7.55 (2H, t, J 7.8, Ph), 7.34 (1H, d, J 3.6, ArH), 6.61 (1H, br s, NH), 4.58 (1H, br s, OH), 3.59 (2H, s, CH2) and 1.24 (6H, s, CH3); δC (125 MHz) 163.7 (CO), 162.2 (CO), 143.2 (C), 134.4 (CH), 130.0 (2CH), 129.9 (CH), 129.3 (C), 128.9 (2CH), 128.4 (C), 113.7 (CH), 69.9 (OCH2), 56.3 (CMe2) and 24.6 (2CH3); HRMS (NSI+): found 320.0953. C16H18NO4S (M + H) requires 320.0951.
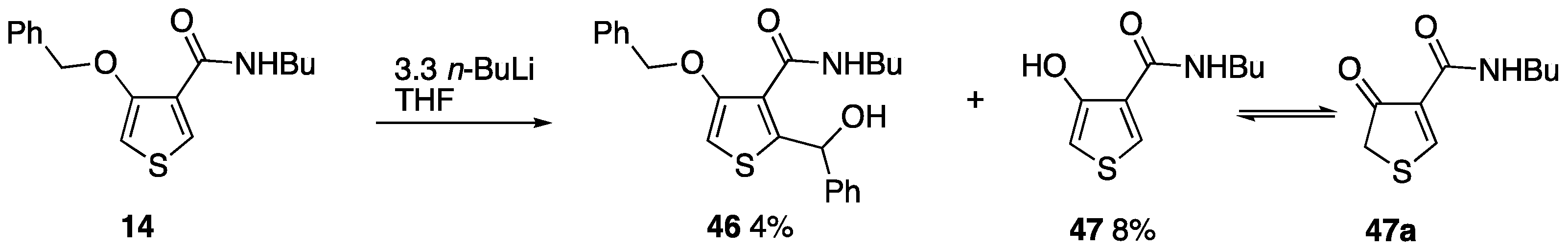
3.4.11. 4-(Benzyloxy)-N-butylthiophene-3-carboxamide 14
Thionyl chloride (1.0 cm3, 1.63 g, 13.7 mmol) was added to a suspension of 4-(benzyloxy)thiophene-3-carboxylic acid 42 (1.50 g, 6.40 mmol) in toluene (15 cm3) and the mixture was heated under reflux for 3 h. After cooling to rt, the mixture was evaporated to give 4-(benzyloxy)thiophene-3-carbonyl chloride as a red oil which was used immediately without further purification.
A solution of 4-(benzyloxy)thiophene-3-carbonyl chloride (assuming 6.40 mmol) in toluene (30 cm3) was added dropwise to a solution of n-butylamine (1.9 cm3, 1.41 g, 19.2 mmol) in toluene (10 cm3) stirred at 0 °C. Once the addition was complete, the reaction mixture was allowed to warm to rt over 1 h before being poured into water. The organic layer was separated and washed with 2M NaOH and brine, dried and evaporated to give, after purification by column chromatography (SiO2, Et2O/hexane 3:2), at Rf 0.50, 14 (1.24 g, 67%) as a tan-coloured solid; mp 51–53 °C; νmax/cm–1 3385, 3111, 2970, 1630, 1557, 1435, 1364, 1265, 1184, 1074, 988, 714 and 579; δH (400 MHz) 8.10 (1H, d, J 3.6, ArH), 7.45–7.37 (6H, m, NH and Ph), 6.43 (1H, d, J 3.6, ArH), 5.10 (2H, s, OCH2), 3.34 (2H, td, J 6.8, 5.6, NCH2), 1.45–1.38 (2H, m, NCH2CH2), 1.25–1.15 (2H, m, CH2CH3) and 0.81 (3H, t, J 7.2, CH3); δC (100 MHz) 161.5 (C=O), 153.5 (C–O), 135.5 (C), 131.5 (CH), 128.8 (2CH), 128.7 (CH), 127.9 (2CH), 127.1 (C), 99.5 (CH), 73.1 (OCH2), 38.7 (NCH2), 31.3 (CH2), 20.0 (CH2) and 13.7 (CH3); HRMS (NSI+): found 290.1207. C16H20NO2S (M + H) requires 290.1209.
3.4.12. Attempted [1,2]-Wittig Rearrangement of 4-(Benzyloxy)-N-butylthiophene-3-carboxamide 14
Under a nitrogen atmosphere, n-butyllithium (2.5 M, 2.7 cm3, 6.75 mmol) was added dropwise to a stirred solution of 4-(benzyloxy)-N-butylthiophene-3-carboxamide 14 (0.5787 g, 2.00 mmol) in dry THF (20 cm3). After stirring at rt for 2 h, the reaction mixture was quenched by the addition of saturated aq. NH4Cl and extracted with Et2O (3 × 30 cm3). The combined organic extracts were washed with NaOH and water before being dried and evaporated to give, after purification by column chromatography (SiO2, Et2O/hexane 3:2), at Rf 0.65, 4-(benzyloxy)-N-butyl-2-(hydroxy(phenyl)methyl)thiophene-3-carboxamide 46 (27.8 mg, 4%) in slightly impure form as a tan-coloured solid; mp 74–76 °C; νmax/cm–1 3393, 3084, 2930, 2870, 1624, 1557, 1443, 1217, 1173, 1016 and 696; δH (400 MHz) 7.73 (1H, t, J 5.0, NH), 7.52–7.48 (2H, m, Ph), 7.44–7.30 (8H, m, Ph), 6.77 (1H, br s, OH), 6.26 (1H, s, ArH), 6.21 (1H, s, CHOH), 5.04 (2H, s, OCH2), 3.41–3.25 (2H, m, NCH2), 1.42–1.35 (2H, m, NCH2CH2), 1.21–1.12 (2H, m, CH2CH3) and 0.80 (3H, t, J 7.2, CH3); δC (100 MHz) 163.6 (C=O), 155.9 (C), 154.3 (C), 140.9 (C), 135.2 (C), 128.8 (3CH), 128.03 (2CH), 128.01 (2CH), 127.97 (CH), 127.3 (2CH), 121.9 (C), 97.5 (CH), 73.0 (OCH2), 70.8 (CHOH), 39.0 (NCH2), 31.0 (CH2), 19.9 (CH2) and 13.7 (CH3); HRMS (ESI+): found 418.1439. C23H25NaNO3S (M + Na) requires 418.1447.
This was followed by a second fraction at Rf 0.50 which was further purified by preparative TLC (SiO2, CH2Cl2) to give, at Rf 0.35 N-butyl-4-hydroxythiophene-3-carboxamide 47 (30.9 mg, 8%) in slightly impure form as a brown oil; νmax/cm–1 3327, 2957, 2930, 1634, 1557, 1441, 1273, 739 and 698; 1H NMR revealed a 3:2 mixture of enol and keto tautomers; δH (500 MHz, enol tautomer 47) 10.36 (1H, br s, OH), 7.54 (1H, d, J 3.3, ArH), 6.36 (1H, d, J 3.3, ArH), 6.35 (1H, br s, NH), 3.41 (2H, td, J 7.3, 6.0, NCH2), 1.62–1.51 (2H, m, NCH2CH2), 1.44–1.33 (2H, m, CH2CH3) and 0.95 (3H, t, J 7.5, CH3); δC (125 MHz, enol tautomer 47) 165.1 (C=O), 156.1 (C–O), 124.6 (CH), 121.7 (C), 100.1 (CH), 39.1 (NCH2), 31.6 (CH2), 20.08 (CH2) and 13.7 (CH3); δH (500 MHz, keto tautomer 47a) 9.33 (1H, s, CH), 8.06 (1H, br s, NH), 3.89 (2H, s, SCH2), 3.36 (2H, td, J 7.0, 6.0, NCH2), 1.62–1.51 (2H, m, NCH2CH2), 1.44–1.33 (2H, m, CH2CH3) and 0.93 (3H, t, J 7.3, CH3); δC (125 MHz, keto tautomer 47a) 199.9 (C=O), 174.7 (CH), 159.9 (CONH), 128.2 (C), 42.7 (SCH2), 38.7 (NCH2), 31.5 (CH2), 20.11 (CH2) and 13.7 (CH3); HRMS (NSI+): found 200.0740. C9H14NO2S (M + H) requires 200.0740.
3.5. X-ray Structure Determination
Data have been deposited at the Cambridge Crystallographic Data Centre as CCDC 2111424 (
9), 2111425 (
20), 2111426 (
11), 2111427 (
26) and 2111428 (
13). The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via
http://www.ccdc.cam.ac.uk/getstructures. In all cases, data were collected on a Rigaku XtaLAB 200 diffractometer using graphite monochromated Mo-Kα radiation, λ = 0.71075 Å and the structures were solved by direct methods and refined by full-matrix least-squares against F2 (SHELXL Version 2014/7 [
21]).
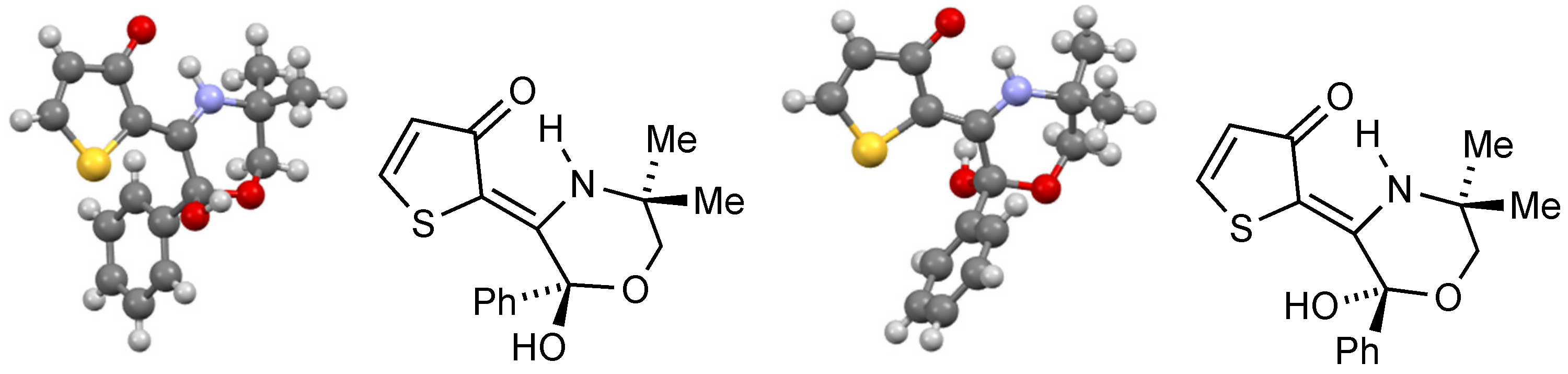
Compound 20
Slow evaporation of an acetone/CH2Cl2 solution gave tan-coloured crystals suitable for X-ray structure determination. Crystal data for 20: 2C16H17NO3S•CH2Cl2•Me2CO, M = 749.76, yellow prism, crystal dimensions 0.10 × 0.10 × 0.10 mm, monoclinic, space group P21/n, a = 17.4520, b = 9.9840, c = 21.1050 Å, β = 92.3760°, V = 3674.1899 Å3, Z = 4, Dc = 1.355 Mg m–3, T = 93 K, R = 0.0929, RW = 0.2464 for 4473 reflections with I > 2σ(I) and 458 variables.
Compound 26
Slow evaporation of a CH2Cl2 solution gave crystals suitable for X-ray structure determination. Crystal data for 26: C16H17NOS, M = 271.38, colourless needle, crystal dimensions 0.12 × 0.02 × 0.02 mm, monoclinic, space group P21, a = 8.237(3), b = 5.717(2), c = 15.097(6) Å, β = 90.539(10)°, V = 710.9(5) Å3, Z = 2, Dc = 1.268 Mg m–3, T = 93 K, R = 0.0481, RW = 0.1086 for 2072 reflections with I > 2σ(I) and 172 variables.
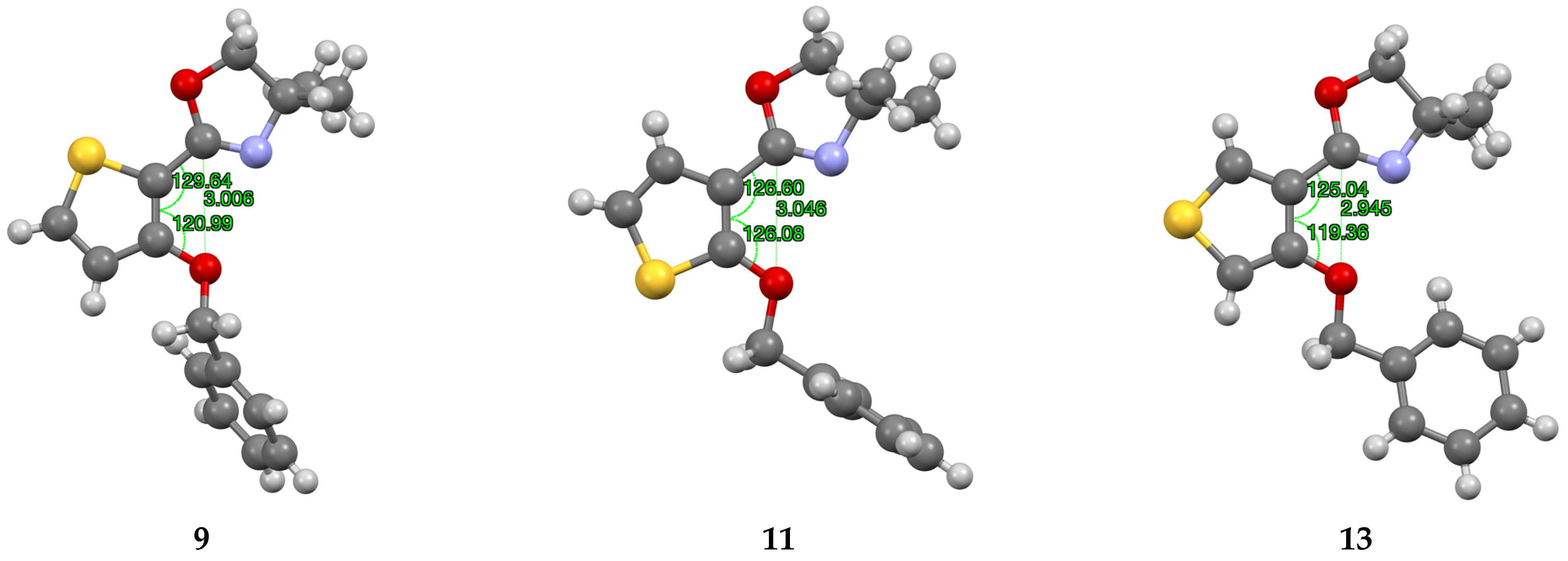
Compound 9
Slow evaporation of an MeCN solution gave crystals suitable for X-ray structure determination. Crystal data for 9: C16H17NO2S, M = 287.38, colourless plate, crystal dimensions 0.10 × 0.10 × 0.01 mm, monoclinic, space group P21/c, a = 15.9970(19), b = 7.5839(6), c = 12.4523(15) Å, β = 111.010(14)°, V = 1410.3(3) Å3, Z = 4, Dc = 1.353 Mg m–3, T = 93 K, R = 0.0584, RW = 0.1406 for 2462 reflections with I > 2σ(I) and 181 variables.
Compound 11
Slow evaporation of a CH2Cl2 solution gave crystals suitable for X-ray structure determination. Crystal data for 11: C16H17NO2S, M = 287.38, colourless prism, crystal dimensions 0.12 × 0.10 × 0.06 mm, monoclinic, space group P21/c, a = 14.8176(4), b = 8.72450(17), c = 11.9720(3) Å, β = 113.314(3)°, V = 1421.32(7) Å3, Z = 4, Dc = 1.343 Mg m–3, T = 93 K, R = 0.0274, RW = 0.0730 for 2906 reflections with I > 2σ(I) and 181 variables.
Compound 13
Slow evaporation of a toluene solution gave crystals suitable for X-ray structure determination. Crystal data for 13: C16H17NO2S, M = 287.38, colourless plate, crystal dimensions 0.20 × 0.20 × 0.01 mm, monoclinic, space group P21/c, a = 15.6319(4), b = 8.7152(3), c = 10.7572(3) Å, β = 93.909(3)°, V = 1462.10(7) Å3, Z = 4, Dc = 1.305 Mg m–3, T = 296 K, R = 0.0333, RW = 0.0888 for 2781 reflections with I > 2σ(I) and 181 variables.



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}