
Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
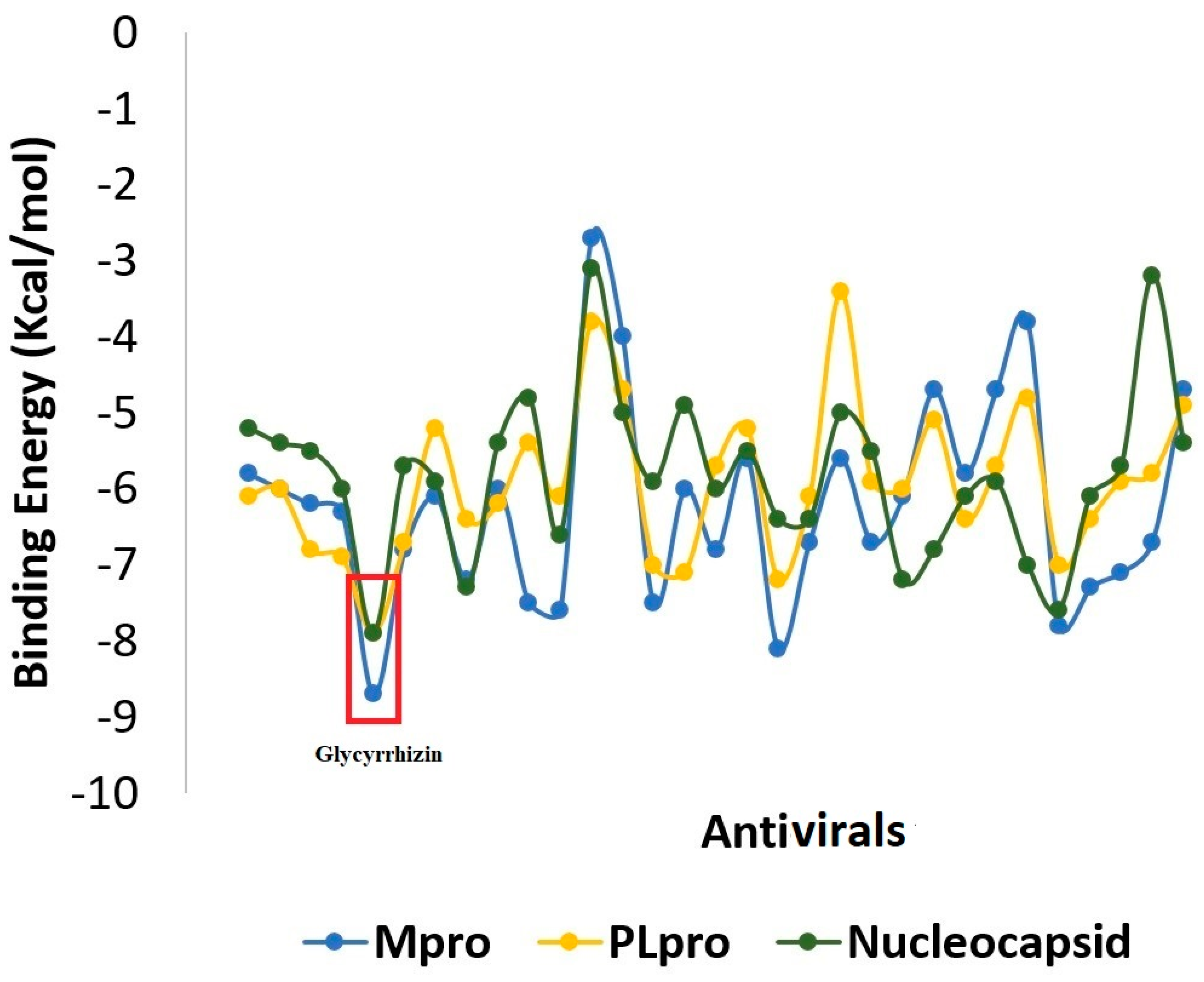
2.1. Molecular Docking
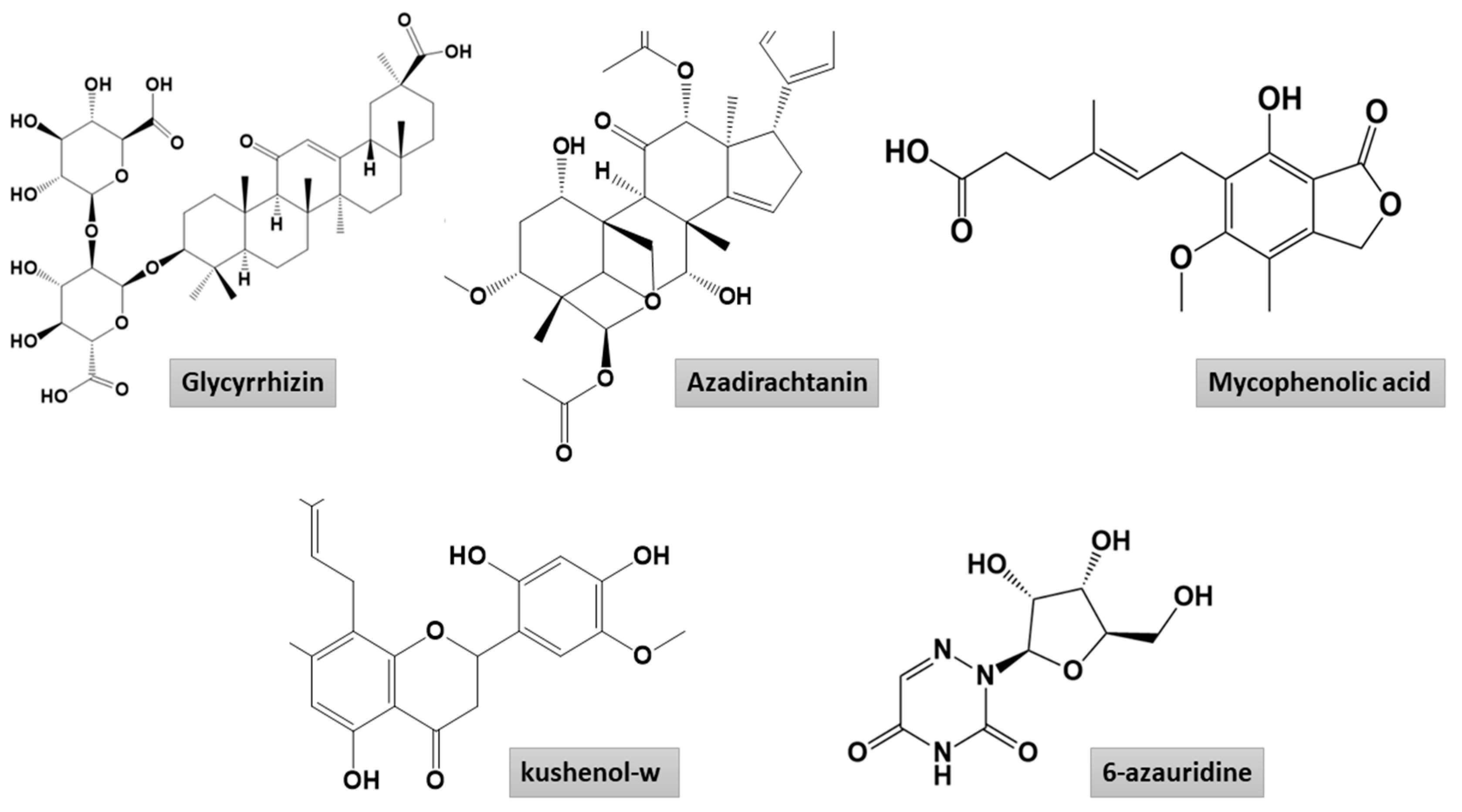
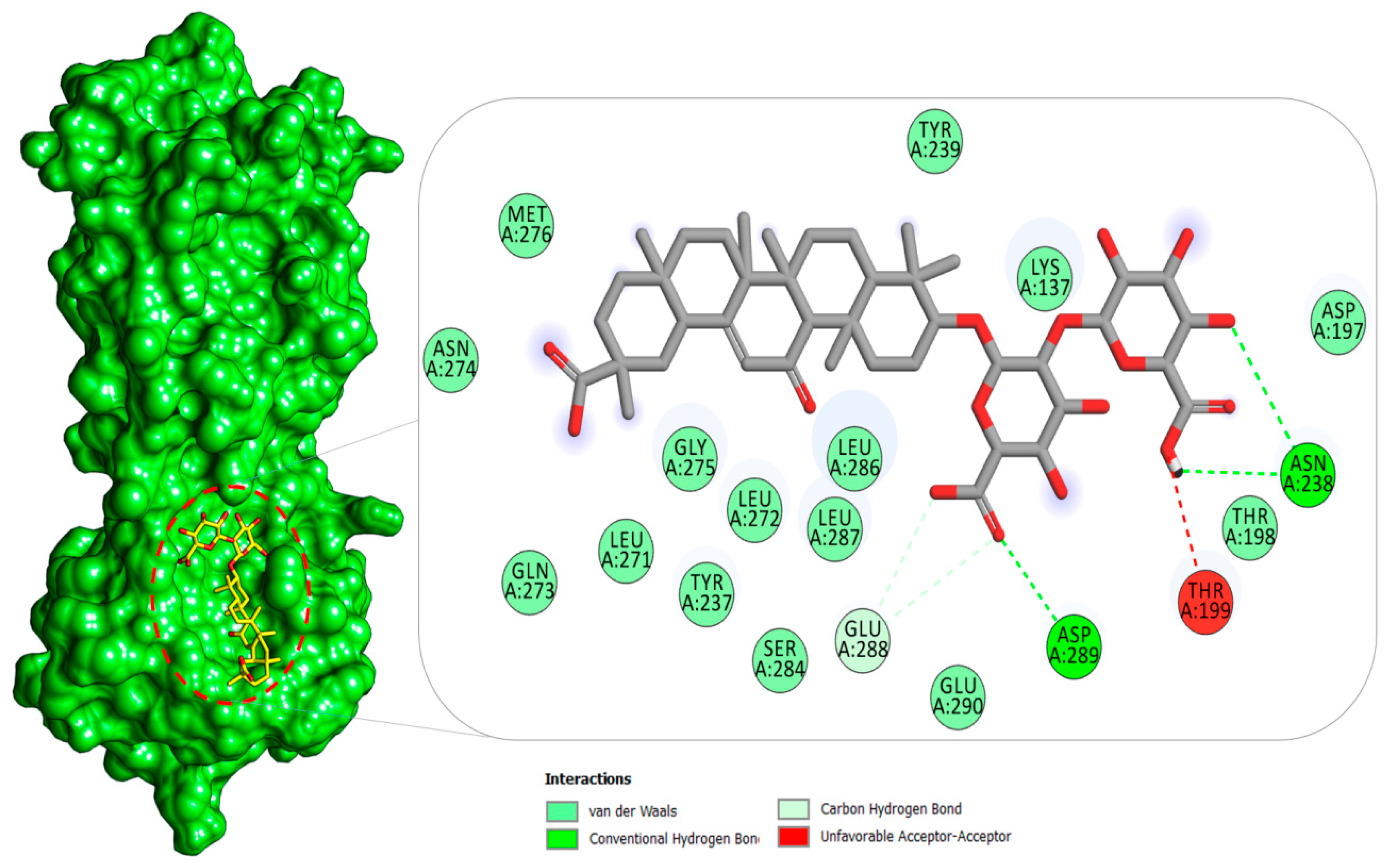
2.1.1. Mpro–Glycyrrhizin Complex
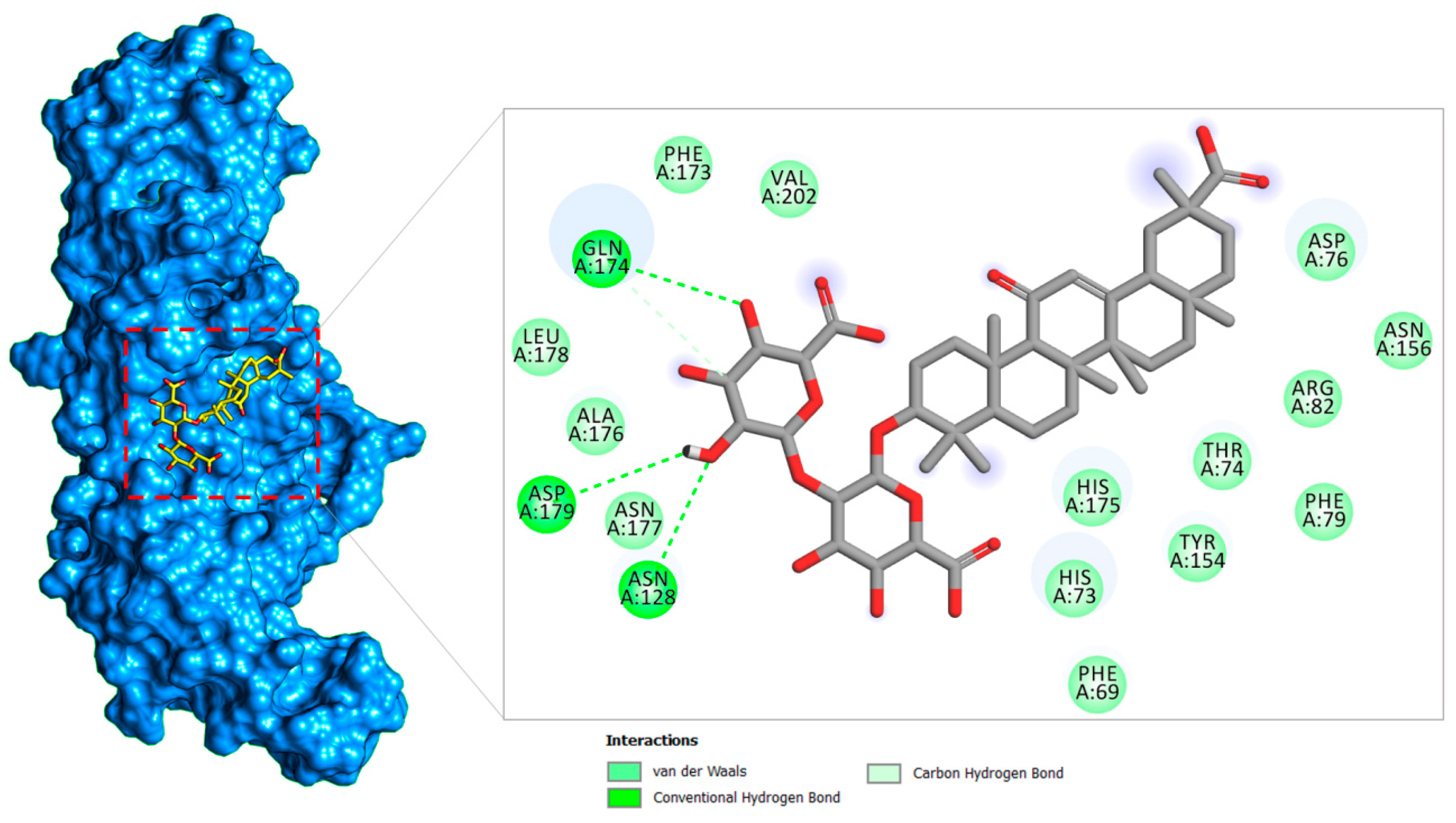
2.1.2. PLpro–Glycyrrhizin Complex
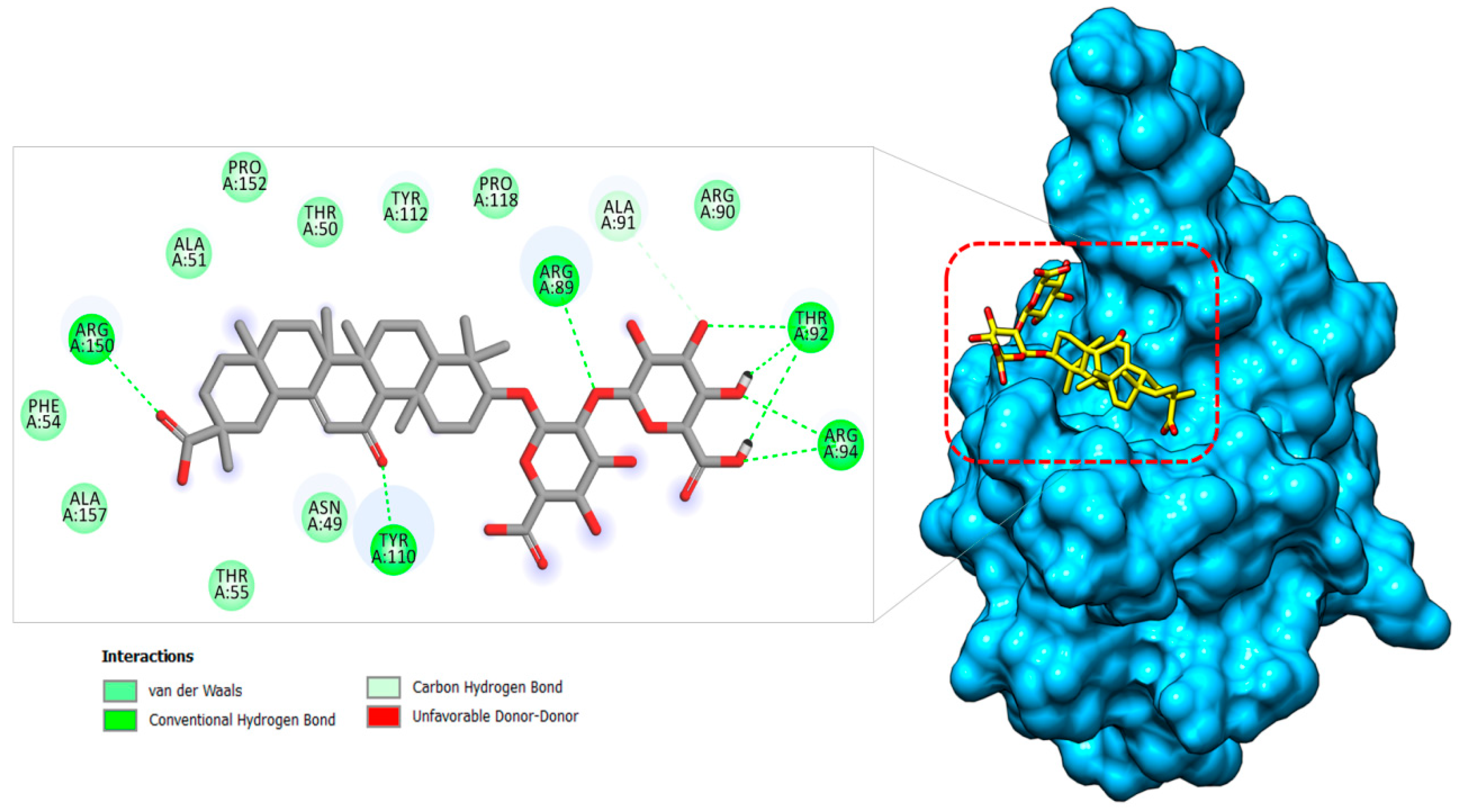
2.1.3. Nucleocapsid–Glycyrrhizin Complex
2.2. MD Simulation Analysis
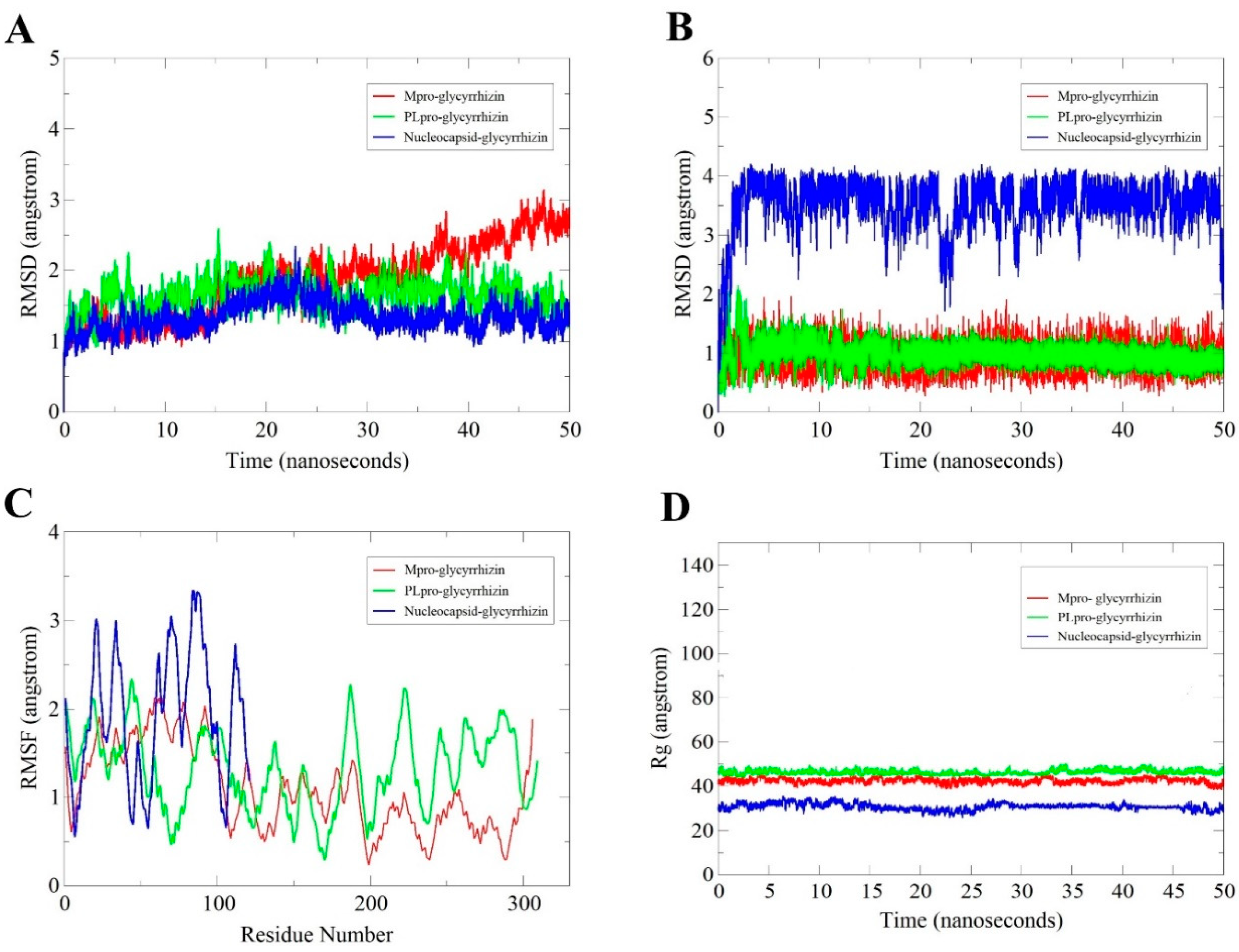
2.2.1. Root-Mean-Square Deviation (RMSD) Analysis
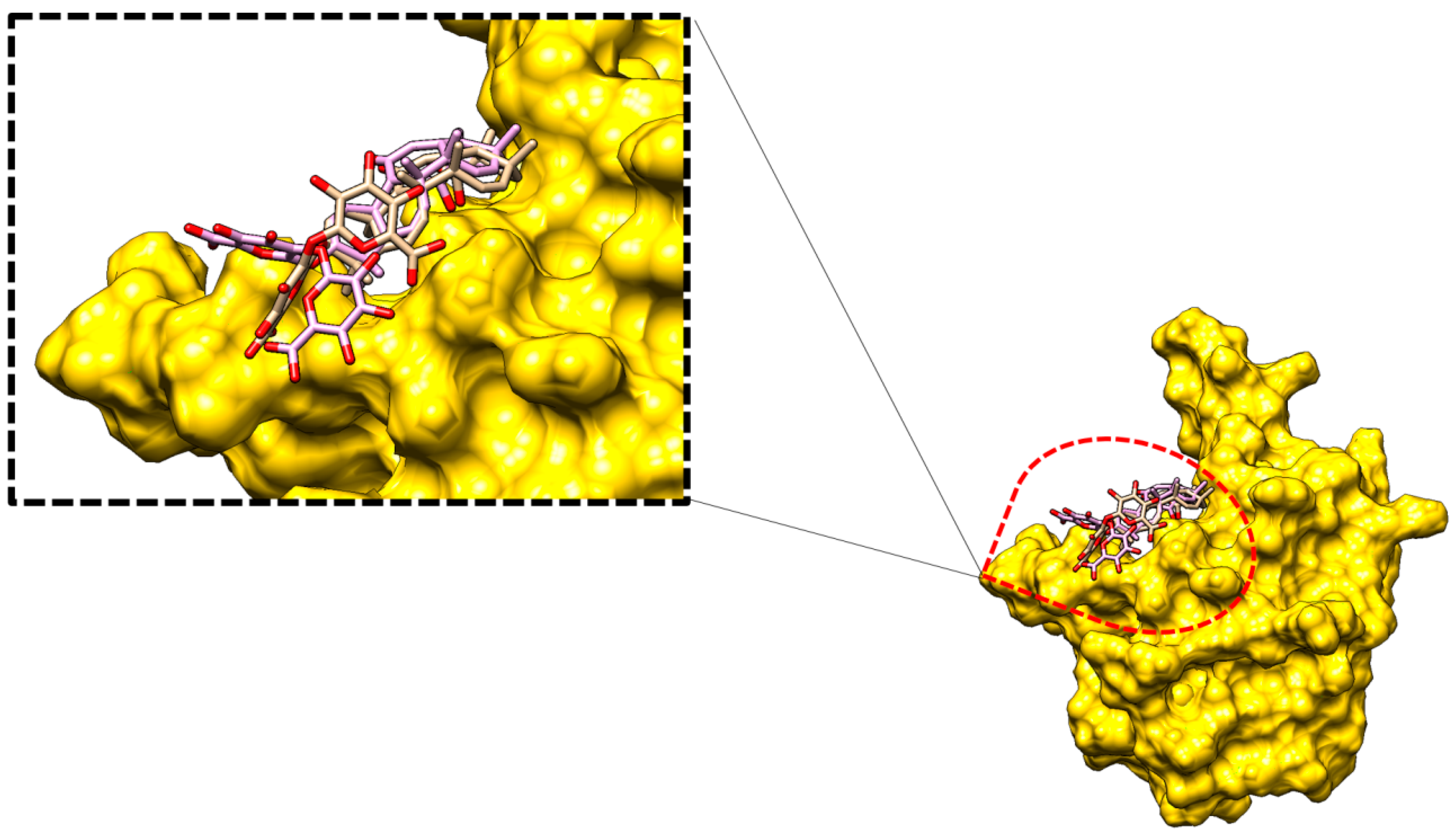
2.2.2. Glycyrrhizin Conformation Stability
2.2.3. Root-Mean-Square Fluctuation (RMSF) Analysis
2.2.4. Radius of Gyration (Rg) Analysis
2.3. MMGB/PBSA Analysis
2.4. Per-Residue Decomposition
2.5. WaterSwap Binding Energy
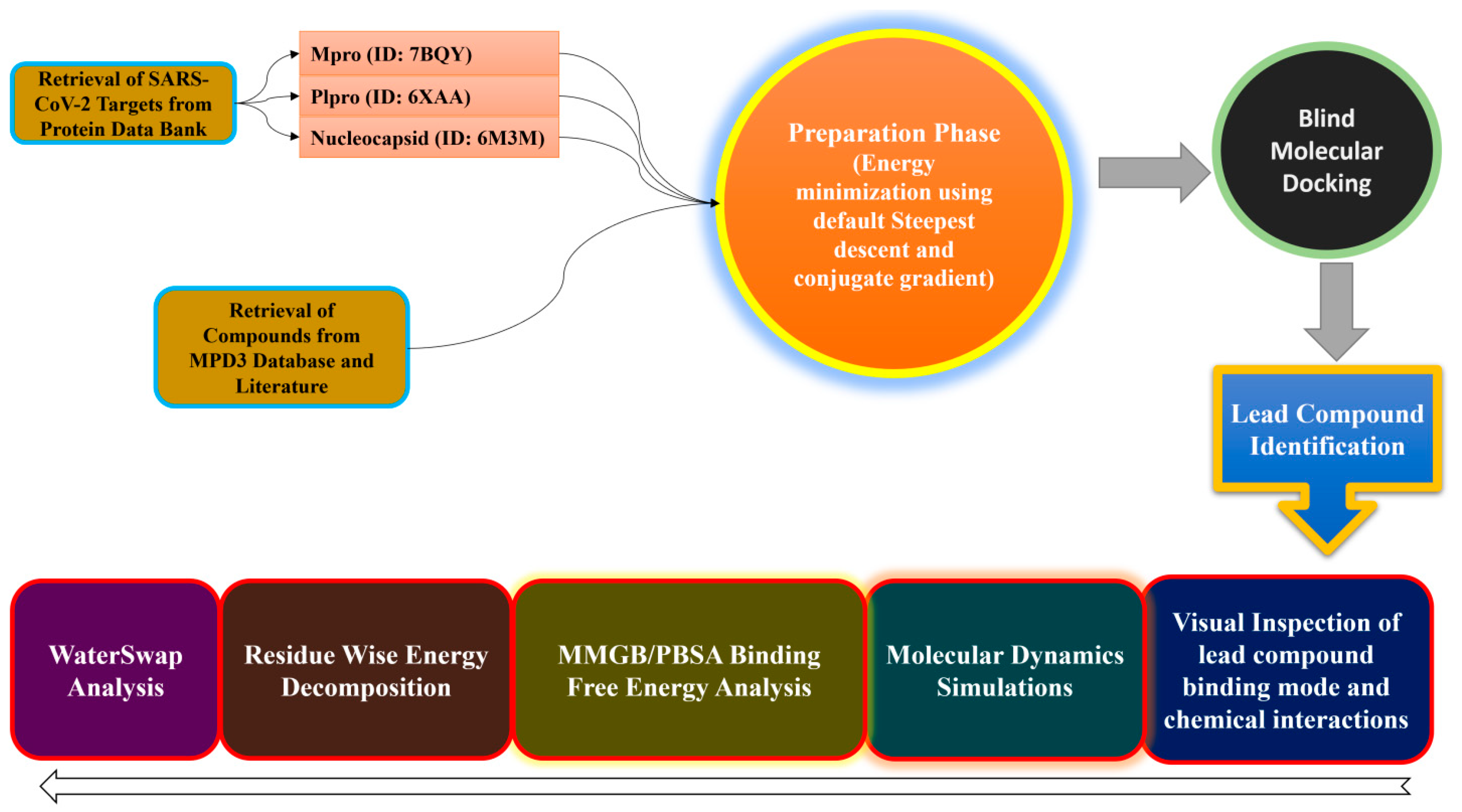
3. Materials and Methods
3.1. Target Proteins Preparation
3.2. Compound Preparation
3.3. Structure-Based Virtual Screening
3.4. MD Simulations
3.5. MMGB/PBSA Analysis
3.6. WaterSwap Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The Deadly Coronaviruses: The 2003 SARS Pandemic and the 2020 Novel Coronavirus Epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, D.S.; Azhar, E.I.; Madani, T.A.; Ntoumi, F.; Kock, R.; Dar, O.; Ippolito, G.; Mchugh, T.D.; Memish, Z.A.; Drosten, C.; et al. The Continuing 2019-nCoV Epidemic Threat of Novel Coronaviruses to Global Health—The latest 2019 Novel Coronavirus Outbreak in Wuhan, China. Int. J. Infect. Dis. 2020, 91, 264–266. [Google Scholar] [CrossRef] [Green Version]
- Ye, Z.-W.; Yuan, S.; Yuen, K.-S.; Fung, S.-Y.; Chan, C.-P.; Jin, D.-Y. Zoonotic Origins of Human Coronaviruses. Int J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [Green Version]
- Tahir ul Qamar, M.; Saleem, S.; Ashfaq, U.A.; Bari, A.; Anwar, F.; Alqahtani, S. Epitope-Based Peptide Vaccine Design and Target Site Depiction against Middle East Respiratory Syndrome Coronavirus: An Immune-Informatics Study. J. Transl. Med. 2019, 17, 362. [Google Scholar] [CrossRef]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Alqahtani, S.M.; Alamri, M.A.; Chen, L.-L. Structural Basis of SARS-CoV-2 3CLpro and Anti-COVID-19 Drug Discovery from Medicinal Plants. J. Pharm. Anal. 2020, 10, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-Z.; Holmes, E.C. A Genomic Perspective on the Origin and Emergence of SARS-CoV-2. Cell 2020, 181, 223–227. [Google Scholar] [CrossRef]
- Fahmi, M.; Kubota, Y.; Ito, M. Nonstructural Proteins NS7b and NS8 are Likely to be Phylogenetically Associated with Evolution of 2019-nCoV. Infect. Genet. Evol. 2020, 81, 104272. [Google Scholar] [CrossRef]
- Tahir ul Qamar, M.; Rehman, A.; Tusleem, K.; Ashfaq, U.A.; Qasim, M.; Zhu, X.; Fatima, I.; Shahid, F.; Chen, L.-L. Designing of a Next Generation Multiepitope Based Vaccine (MEV) against SARS-COV-2: Immunoinformatics and in Silico Approaches. PLoS ONE 2020, 15, e0244176. [Google Scholar] [CrossRef]
- Lenzen, M.; Li, M.; Malik, A.; Pomponi, F.; Sun, Y.-Y.; Wiedmann, T.; Faturay, F.; Fry, J.; Gallego, B.; Geschke, A.; et al. Global Socio-Economic Losses and Environmental Gains from the Coronavirus Pandemic. PLoS ONE 2020, 15, e0235654. [Google Scholar] [CrossRef]
- Boopathi, S.; Poma, A.B.; Kolandaivel, P. Novel 2019 Coronavirus Structure, Mechanism of Action, Antiviral drug Promises and Rule Out against Its Treatment. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Tahir ul Qamar, M.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Aslam, S.; Fatima, I.; Fareed, M.M.; Zohaib, A.; Chen, L.-L. Reverse Vaccinology Assisted Designing of Multiepitope-Based Subunit Vaccine Against SARS-CoV-2. Infect. Dis. Poverty 2020, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef]
- Chen, R.; Fu, J.; Hu, J.; Li, C.; Zhao, Y.; Qu, H.; Wen, X.; Cao, S.; Wen, Y.; Wu, R.; et al. Identification of the Immunodominant Neutralizing Regions in the Spike Glycoprotein of Porcine Deltacorona-Virus. Virus Res. 2020, 276, 197834. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Song, W.; Zhou, H.; Xu, J.; Chen, S.; Xiang, Y.; Wang, X. Cryo-Electron Microscopy Structures of the SARS-CoV Spike Glycoprotein Reveal a Prerequisite Conformational State for Receptor Binding. Cell Res. 2017, 27, 119–129. [Google Scholar] [CrossRef]
- Jin, Z.; Zhao, Y.; Sun, Y.; Zhang, B.; Wang, H.; Wu, Y.; Zhu, Y.; Zhu, C.; Hu, T.; Du, X.; et al. Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 2020, 27, 529–532. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal Structure of SARS-CoV-2 Nucleocapsid Protein RNA Binding Domain Reveals Potential Unique Drug Tar-Geting Sites. Acta Pharm. Sin. B 2020, 19, 1228–1238. [Google Scholar] [CrossRef]
- Elfiky, A.A. SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) Targeting: An in Silico Perspective. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-Like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Gyebi, G.A.; Ogunro, O.B.; Adegunloye, A.P.; Ogunyemi, O.M.; Afolabi, S.O. Potential Inhibitors of Coronavirus 3-Chymotrypsin-Like Protease (3CLpro): An in Silico Screening of Alkaloids and Terpenoids from African Medicinal Plants. J. Biomol. Struct. Dyn. 2020, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Tomar, P.P.S.; Arkin, I.T. SARS-CoV-2 E Protein Is a Potential Ion Channel That Can Be Inhibited by Gliclazide and Memantine. Biochem. Biophys. Res. Commun. 2020, 530, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by full-Length Human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Du, L.; Zhao, G.; Lin, Y.; Chan, C.; He, Y.; Jiang, S.; Wu, C.; Jin, D.-Y.; Yuen, K.-Y.; Zhou, Y.; et al. Priming with rAAV Encoding RBD of SARS-CoV S Protein and Boosting with RBD-Specific Peptides for T Cell Epitopes Elevated Humoral and Cellular Immune Responses Against SARS-CoV Infection. Vaccine 2008, 26, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved $α$-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Kumar, Y.; Singh, H.; Patel, C.N. In Silico Prediction of Potential Inhibitors for the Main Protease of SARS-CoV-2 Using Molecular Docking and Dynamics Simulation Based Drug-Repurposing. J. Infect. Public Health 2020, 13, 1210–1223. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and Molecular Dynamics Simulation Studies Reveal Potential Covalent and FDA-Approved Inhibitors of SARS-CoV-2 Main Protease 3CLpro. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.-M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-Based Design of Antiviral Drug Candidates Targeting the SARS-CoV-2 Main Protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, R.S.; Jagdale, S.S.; Bansode, S.B.; Shankar, S.S.; Tellis, M.B.; Pandya, V.K.; Chugh, A.; Giri, A.P.; Kulkarni, M.J. Discovery of Potential Multi-Target-Directed Ligands by Targeting Host-Specific SARS-CoV-2 Structurally Conserved Main Proteases. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havranek, B.; Islam, S.M. An in Silico Approach for Identification of Novel Inhibitors as Potential Therapeutics Targeting COVID-19 Main Protease. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Tahir ul Qamar, M.; Mirza, M.U.; Alqahtani, S.M.; Froeyen, M.; Chen, L.-L. Discovery of Human Coronaviruses Pan-Papain-like Protease Inhibitors Using Computational Approaches. J. Pharm. Anal. 2020, 10, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Ashfaq, U.A.; Qasim, M.; Yasmeen, E.; Tahir ul Qamar, M.; Anwar, F. Screening of Medicinal Plant Phytochemicals as Natural Antagonists of p53-MDM2 Interaction to Reactivate p53 Functioning. Anticancer Drugs 2017, 28, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Rehan Khalid, R.; Tahir ul Qamar, M.; Maryam, A.; Ashique, A.; Anwar, F.H.; Geesi, M.; Siddiqi, A.R. Comparative Studies of the Dynamics Effects of BAY60-2770 and BAY58-2667 Binding with Human and Bacterial H-NOX Domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durdagi, S.; Tahir ul Qamar, M.; Salmas, R.E.; Tariq, Q.; Anwar, F.; Ashfaq, U.A. Investigating the Molecular Mechanism of Staphylococcal DNA Gyrase Inhibitors: A Combined Ligand-Based and Structure-Based Resources Pipeline. J. Mol. Graph. Model. 2018, 85, 122–129. [Google Scholar] [CrossRef]
- Muneer, I.; Tusleem, K.; Abdul Rauf, S.; Hussain, H.M.J.; Siddiqi, A.R. Others Discovery of Selective Inhibitors for Cyclic AMP Response Element-Binding Protein: A Combined Ligand and Structure-Based Resources Pipeline. Anti-Cancer Drugs 2019, 30, 363–373. [Google Scholar]
- Yu, W.; MacKerell, A.D. Computer-Aided Drug Design Methods. In Antibiotics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 85–106. [Google Scholar]
- Tahir ul Qamar, M.; Ashfaq, U.A.; Tusleem, K.; Mumtaz, A.; Tariq, Q.; Goheer, A.; Ahmed, B. In-Silico Identification and Evaluation of Plant Flavonoids as Dengue NS2B/NS3 Protease Inhibitors Using Molecular Docking and Simulation Approach. Pak. J. Pharm. Sci. 2017, 30, 2119–2137. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA Methods to Estimate Ligand-Binding Affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Mumtaz, A.; Ashfaq, U.A.; Qamar, U.M.T.; Anwar, F.; Gulzar, F.; Ali, M.A.; Saari, N.; Pervez, M.T. MPD3: A Useful Medicinal Plants Database for Drug Designing. Nat. Prod. Res. 2017, 31, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Santos, I.d.A.; Grosche, V.R.; Bergamini, F.R.G.; Sabino-Silva, R.; Jardim, A.C.G. Antivirals against Coronaviruses: Candidate Drugs for SARS-CoV-2 Treatment? Front. Microbiol. 2020, 11, 1818. [Google Scholar] [CrossRef] [PubMed]
- LuoLiu, P.; Li, J. Pharmacologic Perspective: Glycyrrhizin May Be an Efficacious Therapeutic Agent for COVID-19. Int. J. Antimicrob. Agents 2020, 105995. [Google Scholar]
- Elshabrawy, H.A. SARS-CoV-2: An Update on Potential Antivirals in Light of SARS-CoV Antiviral Drug Discoveries. Vaccines 2020, 8, 335. [Google Scholar] [CrossRef]
- Kato, F.; Matsuyama, S.; Kawase, M.; Hishiki, T.; Katoh, H.; Takeda, M. Antiviral Activities of Mycophenolic Acid and IMD-0354 Against SARS-CoV-2. Microbiol. Immunol. 2020, 64, 635–639. [Google Scholar] [CrossRef]
- Case, D.; Ben-Shalom, I.; Brozell, S.; Cerutti, D.; Cheatham III, T.; Cruzeiro, V.; Darden, T.; Duke, R.; Ghoreishi, D.; Gilson, M.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters From ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Trott, O.; Olson, A.J. Auto Dock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multi-Threading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar]
- Maiorov, V.N.; Crippen, G.M. Significance of Root-Mean-Square Deviation in Comparing Three-Dimensional Structures of Globular Proteins. J. Mol. Biol. 1994, 235, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Andleeb, S.; Imtiaz-Ud-Din; Rauf, M.K.; Azam, S.S.; Badshah, A.; Sadaf, H.; Raheel, A.; Tahir, M.N.; Raza, S. A One-Pot Multicomponent Facile Synthesis of Dihydropyrimidin-2(1: H)-Thione Derivatives Using Triphenyl-Germane as a Catalyst and Its Binding Pattern Validation. RSC Adv. 2016, 6, 79651–79661. [Google Scholar] [CrossRef]
- Abro, A.; Azam, S.S. Binding Free Energy Based Analysis of Arsenic (+3 Oxidation State) Methyltransferase with S-Adenosylmethionine. J. Mol. Liq. 2016, 220, 375–382. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE Algorithm to Solve Distance Constraint Equations for Small Molecules in Molecular Dynamics Simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Woods, C.J.; Malaisree, M.; Michel, J.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Rapid Decomposition and Visualisation of Protein-Ligand Binding Free Energies by Residue and by Water. Faraday Discuss. 2014, 169, 477–499. [Google Scholar] [CrossRef] [Green Version]
- Woods, C.J.; Malaisree, M.; Hannongbua, S.; Mulholland, A.J. A Water-Swap Reaction Coordinate for the Calculation of Absolute Protein-Ligand Binding Free Energies. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [Green Version]
- Kiani, Y.S.; Ranaghan, K.E.; Jabeen, I.; Mulholland, A.J. Molecular Dynamics Simulation Framework to Probe the Binding Hypothesis of CYP3A4 Inhibitors. Int. J. Mol. Sci. 2019, 20, 4468. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, B.; Ashfaq, U.A.; Tahir ul Qamar, M.; Ahmad, M. Anticancer Potential of Phytochemicals against Breast Cancer: Molecular Docking and Simulation Approach. Bangladesh J. Pharmacol. 2014, 9, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Tahir ul Qamar, M.; Mumtaz, A.; Ashfaq, U.A.; Adeel, M.M.; Fatima, T. Potential of Plant Alkaloids as Dengue ns3 Protease Inhibitors: Molecular Docking and Simulation Approach. Bangladesh J. Pharmacol. 2014, 9, 262–267. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, U.A.; Jalil, A.; Tahir ul Qamar, M. Antiviral Phytochemicals Identification from Azadirachta Indica Leaves against HCV NS3 Protease: An in Sili-Co Approach. Nat. Prod. Res. 2016, 30, 1866–1869. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Kiran, S.; Ashfaq, U.A.; Javed, M.R.; Anwar, F.; Ali, M.A.; Gilani, A. ul H. Discovery of Novel Dengue NS2B/NS3 Protease Inhibitors Using Pharmacophore Modeling and Molecular Docking Based Virtual Screening of the Zinc Database. Int. J. Pharmacol. 2016, 12, 621–632. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. [Google Scholar]
- Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.H.; Tahir ul Qamar, M.; Li, G. Promising Terpenes as SARS-CoV-2 Spike Receptor-Binding Domain (RBD) Attachment Inhibitors to the Human ACE2 Receptor: Integrated Computational Approach. J. Mol. Liq. 2020, 320, 114493. [Google Scholar] [CrossRef] [PubMed]
- Ton, A.-T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inform. 2020, 39, 2000028. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M Pro from SARS-CoV-2 and Discovery of its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Yu, H.; Yang, H.; Xue, F.; Wu, Z.; Shen, W.; Li, J.; Zhou, Z.; Ding, Y.; Zhao, Q.; et al. Structures of Two Coronavirus Main Proteases: Implications for Substrate Binding and Antiviral Drug Design. J. Virol. 2008, 82, 2515–2527. [Google Scholar] [CrossRef] [Green Version]
- Kneller, D.W.; Phillips, G.; O’Neill, H.M.; Jedrzejczak, R.; Stols, L.; Langan, P.; Joachimiak, A.; Coates, L.; Kovalevsky, A. Structural Plasticity of the SARS-CoV-2 3CL Mpro Active Site Cavity Revealed by Room Temperature X-ray Crystallography. Nat. Commun. 2020, 11, 3202. [Google Scholar] [CrossRef]
- Sacco, M.D.; Ma, C.; Lagarias, P.; Gao, A.; Townsend, J.A.; Meng, X.; Dube, P.; Zhang, X.; Hu, Y.; Kitamura, N.; et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv. 2020, 6, eabe0751. [Google Scholar] [CrossRef]
- Alberto, J.-A.; Ribas-Aparicio, R.M.; Ozores, A.G.; Vega, C.J.A. Virtual Screening of Approved Drugs as Potential SARS-CoV-2 Main Protease Inhibitors. Comput. Biol. Chem. 2020, 88, 107325. [Google Scholar] [CrossRef]
- Ngo, S.T.; Pham, Q.A.N.; Le, T.L.; Pham, D.-H.; Vu, V.V. Computational Determination of Potential Inhibitors of SARS-CoV-2 Main Protease. J. Chem. Inf. Model. 2020, 60, 5771–5780. [Google Scholar] [CrossRef]
- Wang, J. Fast Identification of Possible Drug Treatment of Coronavirus Disease-19 (COVID-19) through Computational Drug Repurposing Study. J. Chem. Inf. Model. 2020, 60, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2020, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Cao, D.; Li, J.; et al. Structural Basis for the Inhibition of the Papain-Like Protease of SARS-CoV-2 by Small Molecules. Biorxiv 2020. [Google Scholar] [CrossRef]
- Petushkova, A.I.; Zamyatnin, A. A Papain-Like Proteases as Coronaviral Drug Targets: Current Inhibitors, Opportunities, and Limitations. Pharmaceuticals 2020, 13, 277. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Kim, J.H.; Kim, Y.M.; Jeong, H.J.; Kim, D.W.; Park, K.H.; Kwon, H.-J.; Park, S.-J.; Lee, W.S.; Ryu, Y.B. Tanshinones as Selective and Slow-Binding Inhibitors for SARS-CoV Cysteine Proteases. Bioorg. Med. Chem. 2012, 20, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.-F.; Chen, C.-C.; Moses, D.C.; Chen, Y.-H.; Lin, C.-H.; Tsai, Y.-C.; Chou, C.-Y. Porcine Epidemic Diarrhea Virus Papain-Like Protease 2 Can Be Noncompetitively Inhibited by 6-Thioguanine. Antiviral Res. 2018, 158, 199–205. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Zhang, A.; Tatineni, M.; Miller, M.A.; Tsigelny, I.F. Potential COVID-19 Papain-like Protease PLpro Inhibitors: Repurposing FDA-Approved Drugs. PeerJ 2020, 8, e9965. [Google Scholar] [CrossRef]
- Amin, S.A.; Ghosh, K.; Gayen, S.; Jha, T. Chemical-Informatics Approach to COVID-19 Drug Discovery: Monte Carlo based QSAR, vIrtual Screening and Molecular Docking Study of Some in-House Molecules as Papain-Like Protease (PLpro) Inhibitors. J. Biomol. Struct. Dyn. 2020, 1–10. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ahmad, S.; Abdullah, I.; Froeyen, M. Identification of Novel Human USP2 Inhibitor and its Putative Role in Treatment of COVID-19 by Inhibiting SARS-CoV-2 Papain-Like (PLpro) Protease. Comput. Biol. Chem. 2020, 89, 107376. [Google Scholar] [CrossRef]
- Bhati, S. Structure-Based Drug Designing of Naphthalene Based SARS-CoV PLpro Inhibitors for the Treatment of COVID-19. Heliyon 2020, 6, e05558. [Google Scholar] [CrossRef]
- Bhowmik, D.; Nandi, R.; Jagadeesan, R.; Kumar, N.; Prakash, A.; Kumar, D. Identification of Potential Inhibitors against SARS-CoV-2 by Targeting Proteins Responsible for Envelope for-Mation and Virion Assembly Using Docking Based Virtual Screening, and Pharmacokinetics Approaches. Infect. Genet. Evol. 2020, 84, 104451. [Google Scholar] [CrossRef] [PubMed]
- Tahir ul Qamar, M.; Maryam, A.; Muneer, I.; Xing, F.; Ashfaq, U.A.; Khan, F.A.; Anwar, F.; Geesi, M.H.; Khalid, R.R.; Rauf, S.A.; et al. Computational Screening of Medicinal Plant Phytochemicals to Discover Potent Pan-Serotype Inhibitors against Dengue Virus. Sci. Rep. 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; McCammon, J.A. Molecular Dynamics Simulations of Biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Glycyrrhizin, an Active Component of Liquorice Roots, and Replication of SARS-Associated Coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef] [Green Version]
- Ashfaq, U.A.; Masoud, M.S.; Nawaz, Z.; Riazuddin, S. Glycyrrhizin as Antiviral Agent against Hepatitis C Virus. J. Transl. Med. 2011, 9, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Du, Q. Potential Natural Compounds for Preventing SARS-CoV-2 (2019-nCoV) Infection. Preprints 2020. [Google Scholar] [CrossRef]
- Prasad, A.; Muthamilarasan, M.; Prasad, M. Synergistic Antiviral Effects against SARS-CoV-2 by Plant-Based Molecules. Plant Cell Rep. 2020, 39, 1109–1114. [Google Scholar] [CrossRef]
- Chrzanowski, J.; Chrzanowska, A.; Graboń, W. Glycyrrhizin: An Old Weapon against a Novel Coronavirus. Phyther. Res. 2020. [Google Scholar] [CrossRef]
- Pham, M.Q.; Vu, K.B.; Pham, T.N.H.; Tran, L.H.; Tung, N.T.; Vu, V.V.; Nguyen, T.H.; Ngo, S.T. Rapid Prediction of Possible Inhibitors for SARS-CoV-2 Main Protease Using Docking and FPL Simulations. RSC Adv. 2020, 10, 31991–31996. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Energy Component | Mpro–Glycyrrhizin | PLpro–Glycyrrhizin | Nucleocapsid–Glycyrrhizin |
|---|---|---|---|---|
| MMGBSA | Van der Waals Energy | −36.50 | −61.10 | −37.97 |
| Electrostatic Energy | −13.92 | −8.53 | 8.75 | |
| Polar Solvation Energy | 30.19 | 26.79 | 3.15 | |
| Nonpolar Solvation Energy | −4.19 | −5.85 | −3.97 | |
| Gas Phase Energy | −50.42 | −69.63 | −29.22 | |
| Solvation Energy | 25.99 | 20.93 | −0.82 | |
| Total Binding Energy | −24.42 | −48.69 | −30.05 | |
| MMPBSA | Van der Waals Energy | −36.50 | −61.10 | −37.97 |
| Electrostatic Energy | −13.92 | −8.53 | 8.75 | |
| Polar Solvation Energy | 42.56 | 35.77 | 6.65 | |
| Nonpolar Solvation Energy | −2.94 | −4.31 | −3.38 | |
| Gas Phase Energy | −50.42 | −69.63 | −29.22 | |
| Solvation Energy | 39.62 | 31.46 | 3.27 | |
| Total Binding Energy | −10.80 | −38.17 | −25.95 |
| Complex | Residues | MMGBSA | MMPBSA |
|---|---|---|---|
| Mpro–Glycyrrhizin | Lys137 | −1.74 | −1.51 |
| Asp197 | −1.76 | −0.45 | |
| Thr198 | −1.50 | −1.76 | |
| Thr199 | −1.18 | −2.84 | |
| Tyr237 | −1.46 | −1.89 | |
| Asn238 | −2.98 | −3.45 | |
| Tyr239 | −1.54 | −1.48 | |
| Leu271 | −1.24 | −1.69 | |
| Leu272 | −1.65 | −3.48 | |
| Gln273 | −1.14 | −1.24 | |
| Asn274 | −1.56 | −1.42 | |
| Met276 | –1.73 | −1.98 | |
| Ser284 | −1.89 | −1.51 | |
| Leu286 | −1.98 | −1.47 | |
| Leu287 | −1.48 | −2.84 | |
| Glu288 | −1.44 | −1.84 | |
| Asp289 | −3.74 | −3.54 | |
| Glu290 | −1.88 | −5.45 | |
| PLpro–Glycyrrhizin | Phe69 | −2.54 | −3.54 |
| His73 | −2.11 | −2.45 | |
| Thr74 | −1.82 | −1.45 | |
| Asp76 | −1.99 | −1.68 | |
| Phe79 | −1.47 | −1.46 | |
| Arg82 | −1.82 | −1.12 | |
| Asn128 | –4.41 | −1.39 | |
| Tyr154 | −1.61 | −1.48 | |
| Asn156 | −1.61 | −5.24 | |
| Phe173 | −1.11 | −1.58 | |
| Gln174 | −5.48 | −3.61 | |
| His175 | −1.94 | −1.48 | |
| Ala176 | −1.69 | −1.12 | |
| Asn177 | −1.81 | −1.62 | |
| Leu178 | −1.64 | −1.11 | |
| Asp179 | −2.47 | −1.83 | |
| Val202 | −1.43 | −1.19 | |
| Nucleocapsid–Glycyrrhizin | Asn49 | −1.99 | −2.54 |
| Thr50 | −1.81 | −1.42 | |
| Ala51 | −1.25 | −2.45 | |
| Phe54 | −1.66 | −3.15 | |
| Thr55 | −1.65 | −1.12 | |
| Arg89 | −2.74 | −1.84 | |
| Thr92 | −2.45 | −3.65 | |
| Arg94 | −4.66 | −2.48 | |
| Tyr112 | −1.45 | −1.24 | |
| Tyr110 | −3.74 | −3.51 | |
| Pro118 | −1.89 | −1.48 | |
| Arg150 | −2.78 | −3.58 |
| Algorithm | Mpro–Glycyrrhizin | PLpro–Glycyrrhizin | Nucleocapsid–Glycyrrhizin |
|---|---|---|---|
| Bennett’s | −22.39 | −25.84 | −22.34 |
| Free energy perturbation | −22.48 | −25.94 | −23.83 |
| Thermodynamic integration | −22.47 | −24.61 | −23.45 |
| Mean | −22.44 | −25.46 | −23.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhseen, Z.T.; Hameed, A.R.; Al-Hasani, H.M.H.; Ahmad, S.; Li, G. Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules 2021, 26, 674. https://doi.org/10.3390/molecules26030674
Muhseen ZT, Hameed AR, Al-Hasani HMH, Ahmad S, Li G. Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules. 2021; 26(3):674. https://doi.org/10.3390/molecules26030674
Chicago/Turabian StyleMuhseen, Ziyad Tariq, Alaa R. Hameed, Halah M. H. Al-Hasani, Sajjad Ahmad, and Guanglin Li. 2021. "Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2" Molecules 26, no. 3: 674. https://doi.org/10.3390/molecules26030674
APA StyleMuhseen, Z. T., Hameed, A. R., Al-Hasani, H. M. H., Ahmad, S., & Li, G. (2021). Computational Determination of Potential Multiprotein Targeting Natural Compounds for Rational Drug Design Against SARS-COV-2. Molecules, 26(3), 674. https://doi.org/10.3390/molecules26030674