Abstract
A series of 30-diethylphosphate derivatives of betulin were synthesized and evaluated for their in vitro cytotoxic activity against human cancer cell lines, such as amelanotic melanoma (C-32), glioblastoma (SNB-19), and two lines of breast cancer (T47D, MDA-MB-231). The molecular structure and activities of the new compounds were also compared with their 29-phosphonate analogs. Compounds 7a and 7b showed the highest activity against C-32 and SNB-19 cell lines. The IC50 values for 7a were 2.15 and 0.91 μM, and, for 7b, they were 0.76 and 0.8 μM for the C-32 and SNB-19 lines, respectively. The most potent compounds, 7a and 7b, were tested for their effects on markers of apoptosis, such as H3, TP53, BAX, and BCL-2. For the whole series of phosphate derivatives, a lipophilicity study was performed, and the ADME parameters were calculated. The most active products were docked to the active site of the EGFR protein. The relative binding affinity of selected phosphate betulin derivatives toward EGFR was compared with standard erlotinib on the basis of ChemScore and KDEEP score. Positively, all derivatives docked inside the cavity and showed significant interactions. Moreover, a molecular dynamics study also reveals that ligands 7a,b form stable complexes and the plateau phase started after 7 ns.
1. Introduction
Cancer is a large group of diseases in which the mechanisms of cell growth control have been lost. Today, it is one of the leading causes of death in people all over the world (approximately 30% of all premature deaths among adults aged 30–69 years not caused by infectious diseases) [1]. Chemotherapy with synthetic drugs is burdened with numerous side effects. Therefore, substances of natural origin are being considered a potential source of new anticancer drugs.
Triterpenes are an important class of compounds synthesized by various plants, which show a broad spectrum of biological activity [2,3]. Betulin and betulinic acid are among the triterpenes of the lupane type. The structure of these compounds contains substituents such as isopropenyl moiety and hydroxyl and/or carboxyl groups that can be easily functionalized. This allows for the continuous expansion of the library of derivatives (terpenoids), and among these compounds, new substances with potential biological activity are sought. In the group of semi-synthetic derivatives of betulin, numerous compounds resulting from modification in the isopropenyl group have also been described. The products obtained in this way were tested for various activities, including, for example, anti-inflammatory, antiviral, and anticancer properties [4,5,6].
As a result of introducing a function containing phosphorus atoms into isopropenyl moiety of betulin diesters, compounds with antitumor (diethylphosphonate derivatives) and antischistosomal (triphenylphosphonium derivatives) activity were obtained [7,8]. Anti-HIV-1 studies performed with the series of phosphorus analogs of bevirimat showed that the 30-diethylphosphate derivative has comparable activity to bevirimat, but with higher selectivity [9].
The carbon–carbon triple bond occurs in drugs used in the pharmacotherapy of various diseases and is still the subject of many research studies related to the development of new pharmaceuticals [10]. Examples include the antiretroviral efavirenz, the contraceptive norethynodrel, oxybutynin for the treatment of overactive bladder, pargyline with the antidepressant effect, the antitumor erlotinib, and the antifungal terbinafine [11].
The literature review shows that the presence of an alkynyl moiety in a compound molecule may favorably influence its antitumor activity [12,13,14]. This is also confirmed by the studies conducted earlier by our team [15,16,17,18,19]. It, therefore, seemed interesting to obtain new betulin derivatives containing diethylphosphate group in position C30, and an alkynyl substituent in position C3 and/or C28 (Figure 1).
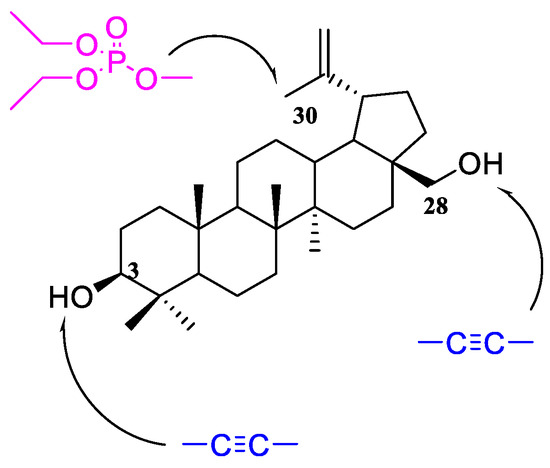
Figure 1.
Schematic structure of target compounds.
The aim of the present study was the synthesis of new betulin derivatives, analysis of the structure of these compounds, and evaluation of their in vitro cytotoxic activity. Human glioma (SNB-19), melanoma (C-32), and two breast cancer (T47D and MDA-MB-231) cell lines were used in the research. In addition, in silico techniques such as ADMET parameter prediction and molecular docking were used to more fully characterize novel substances with potential anticancer effects. The comparison of the structure and activity of phosphonate and phosphate derivatives carried out in the paper was to answer the question of how the replacement of the phosphonate with the phosphate group in the isopropenyl substituent affects the cytotoxic activity of betulin alkynyl derivative.
2. Results and Discussion
2.1. Synthesis
In order to modify the betulin molecule or its derivatives at position C30, first the functionalization of the allylic hydrogen is carried out, introducing a substituent of greater reactivity into it. The most common method for this purpose is bromination at the allyl position with N-bromosuccinimide (NBS) [20]. The 30-bromo- derivatives thus obtained are a convenient substrate for further transformations. They have been used in reactions with thiolates, aliphatic and alicyclic alcohols, potassium isocyanate, sodium azide, acid anhydrides, dimethylaminopyridine (DMAP), triphenylphosphine, and trialkyl phosphite. As a result of these reactions, new compounds were obtained that were evaluated for biological activity [6,7,8,20].
Another route for the preparation of the 30-substituted derivatives is the introduction of a hydroxyl group by oxidation of the isopropenyl moiety with m-chloroperoxybenzoic acid (m-CPBA) [21].
3,28-Diacetyl-30-hydroxybetulin 3 (Scheme 1) was used as the starting compound for this work. This compound has a free hydroxyl group at position 30, which can be converted into a phosphate substituent (derivative 4). The phosphorylation reaction was carried out with diethyl chlorophosphate in a pyridine solution. The next step was the hydrolysis of acetyl groups at C3 and C28 leading to deprotection of the hydroxyl groups, which enabled their further functionalization. Hydrolysis of the acetyl group is most often performed in an alkaline medium [22]. First, the substituent is unblocked at the C28 position (primary hydroxyl group), then at C3 (secondary hydroxyl group). Unfortunately, under these conditions, the C-O-P(O)(OEt)2 ester bond can also be hydrolyzed. Hydrolysis was carried out with sodium hydroxide in a water/tetrahydrofuran/methanol mixture in a volume ratio of 1:2:1. The course of the reaction was monitored by thin layer chromatography, and thus the optimal duration of 5 h was determined. Analysis of the spectra of the hydrolysis product 5 showed that no removal of the phosphate group had occurred under the reaction conditions used. This fact is confirmed by the presence in the 31P spectrum of the signal of the phosphorus atom at −0.91 ppm.
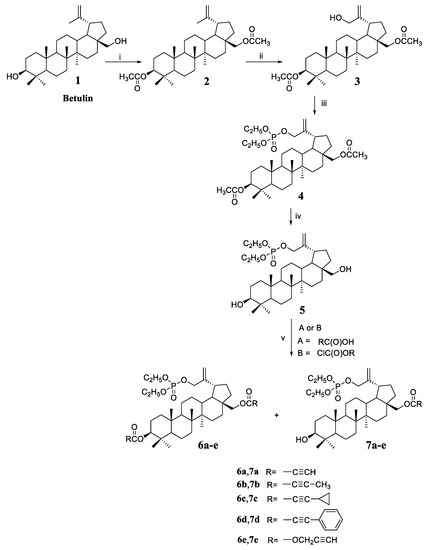
Scheme 1.
Synthesis of 30-diethoxyphosphoryloxybetulin 5 and its alkynyl derivatives 6a–6e and 7a–7e. Reaction conditions: (i) (CH3CO)2O, pyridine, room temp.; (ii) m-CPBA, CHCl3, reaction at boiling temp.; (iii) (C2H5O)2P(O)Cl, DMAP, pyridine, room temp.; (iv) NaOH, THF/MeOH/H2O, room temp.; (v) for A—DCC, DMAP, CH2Cl2, from −10 to room temperature, for B—pyridine, benzene, from −5 to room temp.
As already mentioned, the introduction of a substituent containing a triple bond to the betulin molecule may have a beneficial effect on the cytotoxic activity of the compound. Reactions of 30-diethoxyphosphoryloxybetulin 5 with four alkynyl acids were performed using free hydroxyl groups. Acids with different structures were selected for the synthesis, i.e., propiolic acid with a terminal triple bond and acids with a C≡C bond in the middle of the carbon chain, i.e., 2-butynoic, cyclopropylpropiolic, and phenylpropiolic acid.
Reactions with acids were performed using the Steglich method, which is suitable for substrates requiring mild conditions and a non-acidic environment [23]. The use of a 17% excess of alkynyl acid guarantees complete conversion of the hydroxyl group at the C28 position of the starting substrate 5, leading to the formation of monosubstituted derivatives 7a–7d that were isolated in 71–82% yields. Additionally, small amounts of diesters 6a–6d are formed in the reaction that were obtained in 5–12% yields.
The reaction of 5 with propargyl chloroformate was carried out in benzene in the presence of pyridine to give the alkynyloxycarbonyl derivatives 7e and 6e in yields 60% and 15%, respectively.
All products were purified by column chromatography. The structures of compounds 3–5, 6a–6e, and 7a–e were confirmed based on the analysis of 1H, 13C, and 31P NMR spectra and the mass spectrometry method (HR-MS).
In general, there is a wealth of information about the antitumor activity of betulinic acid. Taking this into account, phosphate derivatives of betulonic 8 and betulinic acid 9 were also included in this study (Scheme 2). In our earlier work, we described the synthesis and testing of anti-HIV-1 activity of these compounds, but so far they have not been tested as anticancer agents [9].
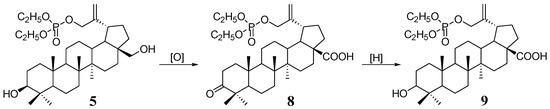
Scheme 2.
Synthesis of 30-phosphate derivatives of betulonic (8) and betulinic acid (9) from 30-diethoxyphosphoryloxybetulin (5).
2.2. Structural Studies
One of the steps in the synthesis of derivatives containing a phosphate group at the C30 position is the hydrolysis of acetyl groups at the C3 and C28 positions. This reaction is carried out in an alkaline medium. As previously described, for 3,28-diacetyl-30-diethylphosphonobetulin, this reaction proceeds with allyl–vinyl isomerization in the isopropenyl group [7]. The compound 4 contains a phosphate group in the allyl position (C30), so the substituent is connected via an oxygen atom. Based on 1H and 31P NMR spectra of the hydrolysis product (compound 5), isomerization was not observed. The structure of product 5 was confirmed based on the analysis of 31P NMR spectra (singlet at δ −0.91 ppm), and also 1H NMR (signals δ = 4.15 ppm and δ = 1.37 ppm), which prove the presence of the ethyl groups of the phosphate substituent (Figure S2a,c). The position of the H3 proton signals (δ = 3.21 ppm) and H28 (δ = 3.80 ppm and δ = 3.32 ppm) corresponds to the chemical shift of these protons in the spectra of unsubstituted betulin containing free hydroxyl groups. In the 13C NMR spectrum of compound 5 (Figure S2b), the signals of the C3 and C28 carbon atoms are shifted by about 3 ppm (δ = 77.9 ppm and δ = 59.2 ppm, respectively) compared to the position in the substrate 4 spectrum (δ = 80.9 ppm and δ = 62.4 ppm) including acetyl groups (Figure S1b). This shift is due to the absence of the adjacent electron-accepting carbonyl group, which had a revealing effect.
Using the X-ray structural study of a single-crystal and Raman spectra, the structure of new 30-phosphate derivatives was analyzed. Additionally, a comparison was made with selected 29-phosphonate analogs in terms of the position of the double bond in the isopropenyl group.
2.2.1. Raman Spectra
The experimental Raman spectra for phosphate and phosphonate derivatives were measured in the frequency range of 200–2000 cm−1 using a confocal alpha 300R (WITec, Germany) spectrometer.
Compounds for which experimental Raman spectra were made were grouped into three sets depending on the type of substituents, i.e., (5, 10), (7a, 11), and (7c, 13) (Figure 2).
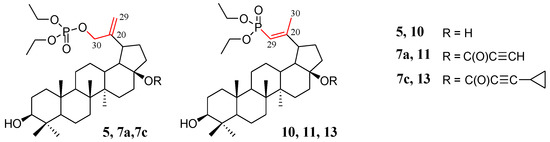
Figure 2.
Structure of allyl substituted compounds 5, 7a, and 7c (phosphate derivatives) and vinyl 10, 11, and 13 (phosphonate derivatives).
The two compounds in each set differ in the position of the C=C double bond and formation of the O=P-O-C (allyl substituted compounds 5, 7a, and 7c) or O=P-C=C (vinyl substituted compounds 10, 11, and 13) groups, respectively (Figure 2). In phosphate derivatives, this group is attached through an oxygen atom, and the distances of the phosphoryl group (P=O) from the C=C double bond are different.
Experimental frequencies and band assignments are collected in Table 1. The theoretical Raman spectra for selected compounds, i.e., 5 and 10, were calculated using the DFT method (see Materials and Methods). In Table S1, the calculated band frequencies and assignments for 5 and 10 are collected and included in the Supplementary Materials.

Table 1.
Experimental band frequencies and assignments of Raman spectra for compounds 5, 7a, 7c, 10, 11, and 13.
The band assignment of experimental spectra was performed by comparison with those calculated as well as using the literature databases of the IR and Raman spectra [24,25].
Comparing the intensities and positions of the individual theoretical and experimental peaks (Table 1 and Table S1), we can notice quite significant differences, which, however, do not affect the quality of band assignment to individual vibrations. This difference is mainly due to the approximations of the calculation model (the size of the base wave functions and harmonic vibrations approximation) and the fact that the calculations were performed for a single molecule and the interactions of molecules were not taken into account (see Materials and Methods). Despite the limitations of the model, the spectral calculations provided helpful additional information for the interpretation of the experimental data.
Raman spectra of all investigated compounds show intense bands in the range of 1664–1625 cm−1 assigned to the stretching vibration of the C=C double bound (Figure 3). According to literature data, the C=C stretching band in alkyl-substituted ethylenes is usually located in the neighborhood of 1650 cm−1. The position of the C=C stretching vibration band is influenced by the adjacent functional groups, so in the molecule 5, one of the carbon atoms is bound to two hydrogen atoms and the other one to hydrogen and the -CH2- group connected to the phosphate residue. In compound 10, however, one carbon atom is connected to a hydrogen atom and a methyl group, and the other to a hydrogen atom and a phosphorus atom of the phosphonate group (Figure 2).
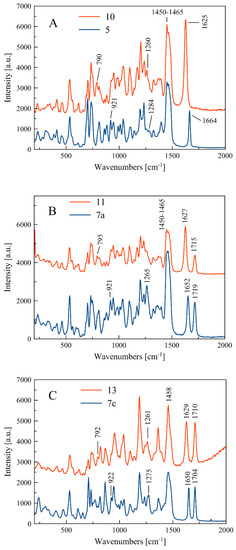
Figure 3.
Experimental Raman spectra in the range 200–2000 cm−1 for three sets of compounds: (5, 10) (A), (7a, 11) (B), and (7c, 13) (C).
The frequencies of the individual C=C stretching bands νc=c for the studied compounds are summarized in Table 2.
Table 2.
Bond length and selected group Raman frequencies for the double bond C20–C29 experimental and calculated. Band shift (ΔνC=C) was calculated as a difference between band frequencies for following sets of compounds: 5–10, 7a–11, and 7c–13.
Within each set of the compounds there is a band shift (Δνc=c)exp due to different positions of the C=C bond in the side chain. For compounds 5 and 10, the change in location of the C=C bond is accompanied by shortening of the bond length from 1.338(3) Å to 1.326(5) Å and causes the band shift towards larger wavelength by the value of (Δνc=c)exp = 39 cm−1. The calculated band shift (Δνc=c)cal for these compounds is much higher and equal to 68 cm−1 (see Table 2). For sets of compounds 7a–11 and 7c–13, the bond shifts (Δνc=c)exp are equal to 25 and 21 cm−1, respectively.
As shown in Table 1, the bands at 1284 and 1275 cm−1 relate to stretching vibration of the P-O bond in the O=P-O-C side chain, which appear in compounds 5 and 7c. In turn, the P-C stretching vibration in the O=P-C=C side chain at 1260 and 1261 cm−1 can be observed for compounds 10 and 13, respectively. It can be noticed that there are some deviations from this scheme, i.e., the P-O and P-C stretching bands do not appear clearly in the spectra of compounds 7a and 11, as shown in Figure 2.
Similarly, the bending vibrations of the P-O and P-C bonds are observed in ranges 921–926 and 790–795 cm−1, for 5, 7a, and 7c and 10, 11, and 13, respectively (see Table 1).
For compounds 7a, 7c, 11, and 13, the single peak of C=O stretching vibration in the range 1704–1719 cm−1 can be observed. The C-H bending vibration in the side chain and in the cycloalkane ring as well as the C-C cycloalkane ring vibrations appear mainly at low frequencies, e.g., about 700–500 cm−1, and they are confirmed by theoretical spectra (see Table S1).
2.2.2. Crystal Structure
The single crystal suitable for the X-ray diffraction analysis was grown by slow evaporation at room temperature technique from acetonitrile solution. The molecular structure of the compound 5 with atom numbering is shown in Figure 4. Additional information about the crystal can be found in Supplementary Materials, Tables S2 and S3.
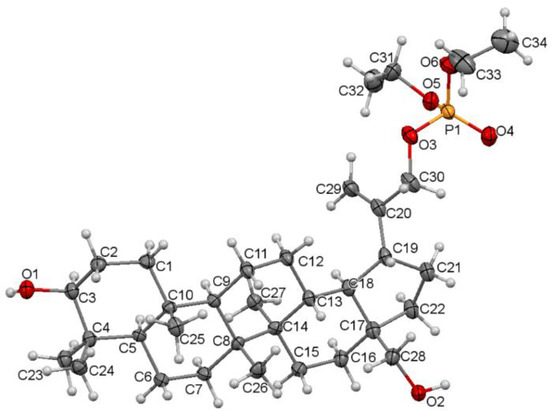
Figure 4.
Molecular structure of the compound 5 showing the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Compound 5 crystallizes in a monoclinic P21 space group. There are two molecules per asymmetric unit. The molecular structure of 5 is stabilized by hydrogen bonds as well as weak intermolecular C-H⋯O bonds (Table S4). In the pentacyclic scaffold of betulin, the six-membered rings adopt chair conformations and are joined similarly, as described previously for other betulin derivatives [7]. The five-membered ring assumes the conformation of the twisted envelope. Structure analysis confirms that in the derivative 5 the diethyl phosphate substituent is connected to the betulin molecule via an ester bond attached to a sp3 hybridized carbon (C30) atom. The length of the C20–C30 bond for compound 5 is 1.490 Å, and it is longer than the C20–C29 bond equal 1.326 Å (Table 3).
Table 3.
Selected bond lengths (Å) for the crystal structure of 30-diethoxyphosphoryloxybetulin 5 and 28-cyclopropylpropynoyl-29-diethoxyphosphorylbetulin 13.
The comparison of the structure of an isopropenyl group containing a phosphonate (13) and a phosphate (5) substituent is shown in Figure 5.
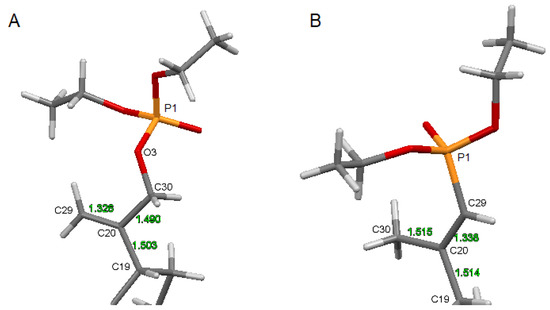
Figure 5.
The structure of the substituted isopropenyl group: (A)—for compound 5, (B)—for reference phosphonate derivative 13.
2.3. Anticancer Activity
The anticancer activity has been tested for various alkynyl derivatives of triterpenes, and the results obtained were promising [13,26,27]. On the other hand, the literature also contains information that the introduction of a dialkylphosphate group into a molecule of a known drug substance may improve its activity or even change it towards having an antitumor effect [28,29]. The combination of these two moieties in the betulin molecule could create a derivative with higher antitumor activity. Thus, the phosphate derivatives synthesized in this study 4, 5, 6a–6e, and 7a–7e were assessed for in vitro activity against four human tumor cell lines such as amelanotic melanoma (C-32), glioblastoma (SNB-19), and two lines of breast cancer (Luminal A class—T47D; Claudin—low class; a triple negative breast cancer—MDA-MB-231).
2.3.1. Cell Viability Studies
Evaluation of the effect of new betulin derivatives on the viability of the cultured cells was performed using the WST-1(water soluble tetrazolium salt) assay. Betulin 1 and cisplatin were used as the reference compounds. The results, expressed as half the maximum inhibitory concentration (IC50), are shown in Table 4.
Table 4.
Effect of 30-phosphate derivatives of betulin and reference compounds (betulin and cisplatin) on the viability of cells from four cancer cell lines studied.
For all cell lines tested, the 3,28-diester derivatives 6a-e were less active (IC50 range 4.65–31.5 μM) than the 28-monoester derivatives 7a–7e (IC50 range 0.76–15.40 μM). Among the monoester compounds, 7b and 7a were the most active. They contain, respectively, 2-butynoyl and propynoyl substituents. Monoester derivatives of acids with a more extensive structure, 7c (cyclopropylpropynoyl) and 7d (phenylpropynoyl), were significantly less active, but their activity was higher compared to betulin 1.
It can be seen that among the cell lines tested, cells of both breast tumors were characterized by the lowest sensitivity to alkynyl derivatives of betulin phosphate. The highest activity for the T47D (IC50 = 1.04 μM) and MDA-MB-231 (IC50 = 0.40 μM) lines was determined for the 30-phosphate of betulin 5. In the group of phosphate derivatives, only the 28-propynoyl derivative 7a showed a significant effect on T47D cells (IC50 = 2.61 μM). As previously described, compounds containing a phosphonate group at position C29, analogous betulin derivatives, were active against this cell line; however, the unsubstituted compound 10 (IC50 = 1.55 μM) was less active than the 28-propynoyl 11 (IC50 = 0.70 μM) analog [Table 4].
The C-32 and SNB-19 cells seem to be more sensitive to 30-phosphate betulin derivatives. Compound 7b showed the strongest activity for both lines (IC50 = 0.76 and 0.8 μM, respectively).
The derivative 7a with the terminal triple bond was slightly weaker in relation to SNB-19 cells (IC50 = 0.91 μM) and more than two times less active towards the C-32 line (IC50 = 2.15 μM). Among betulin 29-phosphonates, the activity of the corresponding propynoyl derivative 11 (IC50 = 0.6 μM) was significantly higher than that of the 2-butynoyl derivative 12 (IC50 = 7.70 μM). A meaningful difference in the cytotoxicity of 28-propargyloxycarbonyloxy derivatives was also observed. The structure–activity relationships observed in the 30-phosphate monoesters 7a–7e indicate that the order of the substituents in the cytotoxic effect on the SNB-19 cell line is as follows: 2-butynoyl > propynoyl > propargyloxycarbonyl > 3-cyclopropyl-2-propynoyl > 3-phenyl-2-propynoyl. Comparing the activities of compound 7a with those determined for 28-O-propynoylbetulin in relation to appropriate tumor cell lines (IC50 values 16.7, 4.2, and 110.8 μM for C-32, SNB-19, and T47D, respectively) [27], it seems that the introduction of a diethylphosphate substituent to the isopropenyl group may exert a beneficial effect on the cytotoxic activity of the obtained derivatives.
The phosphate derivatives of betulonic (8) and betulinic (9) acids showed no activity in relation to the examined breast cancer lines (T47D and MDA-MB-231). Activity against C-32 and SNB-19 cells determined for these compounds as well as the references (29 and 30 phosphonate derivatives and betulinic acid) are summarized in Table 5.
Table 5.
Effect of 30-phosphate derivatives of betulonic acid 8, betulinic acid 9, and reference compounds on the viability of cells from two cancer cell lines studied (C-32 and SNB-19).
Initial studies of the anticancer activity of betulinic acid were focused on human melanoma cells, and then its effects on numerous other cancer cell lines, including glioblastoma, have been reported [30,31]. The evaluation of the activity of phosphorus derivatives of betulonic acid and betulinic acid, carried out in our study, showed that these compounds are much less active against C-32 melanoma cells (IC50 range 30.74–47.46 μM) than betulinic acid (IC50 = 8.63 μM; R=H, R1=OH). The tested acid derivatives showed a much greater effect on SNB-19 cells. The strongest activity (IC50 = 1.29 μM) was presented in the 30-phosphonate derivative of 3-acetylbetulinic acid (R=30-P(O)(OC2H5)2, R1=CH3C(O)O). The IC50 value determined for 30-phosphate of betulinic acid 9 was more than 4.5-fold higher.
2.3.2. Analysis of the Expression of Genes Related to the Apoptosis Process
In the fight against cancer, it is especially important to understand the mechanisms involved in cell death. The effectiveness of anticancer therapy results not only from the inhibition of proliferation, but also from the ability to induce apoptosis in cancer cells. Apoptosis, also known as programmed cell death, is a fundamental physiological process that plays a key role in the development and maintenance of tissue and body homeostasis. A cell dying in the process of apoptosis undergoes both morphological changes (chromatin condensation, changes in the structure of the cell nucleus, and formation of apoptotic bodies) and biochemical changes (activation of many proteins classified as both transcription factors and enzymes and changes in the structure of the cell membrane) [32]. Changes in apoptotic cells are determined by the expression of relevant genes that are involved in this complex process and act both pro-and anti-apoptotically [33,34,35].
In our work, we assessed the influence of the tested compounds on the expression of selected genes related to apoptosis. The two most active derivatives 7a and 7b were selected for the study. We examined their influence on the transcriptional activity of genes encoding a proliferation marker (histone H3), a cell cycle regulator (p53), and proteins related to the mitochondrial apoptosis pathway (BCL-2 and BAX). The study was performed after 24-h exposure of the cultured cells to the test compounds (half of IC50).
The structural organization of DNA into chromatin is essential for maintaining cellular identity. In the S phase of the cell cycle, the chromatin structure is multiplied (related to, among others, the synthesis of histones), which is coupled with DNA replication. Histone H3, being one of the components of chromatin, undergoes several post-translational modifications, influencing gene expression. According to the literature data, the level of H3 mRNA quantified by QRT-PCR can serve as a marker in the assessment of proliferative activity and the development of brain glioma, for example [34,35].
In our study, compounds 7a and 7b reduced the number of mRNA copies of the histone H3 gene in the cells of both tumor lines compared to the level determined for the control (Figure 6). The antiproliferative effect of these compounds may therefore be due to the ability to reduce the histone H3 genes at the mRNA level.
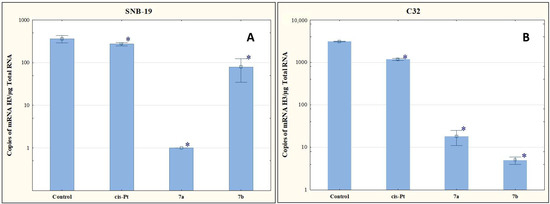
Figure 6.
The effect of compounds 7a and 7b on transcriptional activity of H3 for SNB-19 (A) and C-32 (B) cells. Cells were exposed to compound 7a and 7b for 24 h at a concentration equal to half of IC50 determined for SNB-19 (0.3 and 0.25 µg/mL, respectively) and for C-32 (0.7 and 0.25 µg/mL, respectively). For the reference compound, cisplatin, the appropriate concentrations were 0.8 µg/mL (SNB-19 line) and 0.6 µg/mL (C32 line). * p < 0.05 compared with the untreated control.
Most cancers have a mutation in the TP53 suppressor gene, which encodes the P53 protein. This leads to an inhibition of cell apoptosis and tumor growth. In apoptosis induced by DNA damage, the p53 protein is involved in various activation steps in both the internal and external pathways. It is related to its function as a transcription factor of pro-apoptotic genes and its direct interaction with anti-apoptotic proteins, which results in their blockade [36].
The results of our research presented in Figure 7 indicate that the tested compounds increase the number of mRNA copies of TP53. Compound 7b upregulated TP53 expression levels, but not as significantly as was observed for 7a.
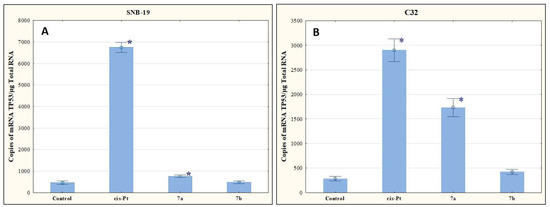
Figure 7.
The effect of compounds 7a and 7b on transcriptional activity of TP53 for SNB-19 (A) and C-32 (B) cells. Cells were exposed to compound 7a and 7b for 24 h at a concentration equal to half of IC50 determined for SNB-19 (0.3 and 0.25 µg/mL, respectively) and for C-32 (0.7and 0.25 µg/mL, respectively). For the reference compound, cisplatin, the appropriate concentrations were 0.8 µg/mL (SNB-19 line) and 0.6 µg/mL (C32 line). * p < 0.05 compared with the untreated control.
There is a strong genetic and biochemical link between the tumor suppressor p53 and the oncogene BCL-2 that is fundamental to cancer biology. The BCL-2 family of proteins includes both pro-apoptotic (e.g., BAX) and anti-apoptotic (e.g., BCL-2) proteins. The task of anti-apoptotic proteins from the BCL-2 family is to bind to the active form of BAX, which prevents it from penetrating into the outer membrane of the mitochondria and blocks apoptosis [33,37,38]. The effectiveness of anti-cancer therapies depends on the mechanisms of BCL-2/BAX-dependent cancer cell killing [38].
As shown in Figure 8, the tested compounds increased the mRNA copy number to BAX and BCL-2 ratio. Given this result, it can be assumed that the derivatives 7a and 7b affect apoptosis via the mitochondrial route.
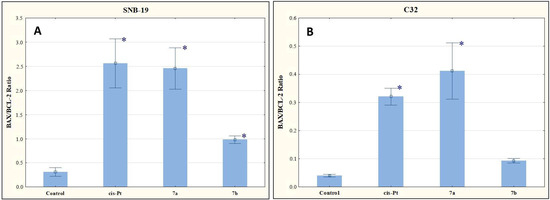
Figure 8.
The effect of compounds 7a and 7b on BAX/BCL-2 for SNB-19 (A) and C-32 (B) cells. Cells were exposed to compound 7a and 7b for 24 h at a concentration equal to half of IC50 determined for SNB-19 (0.3 and 0.25 µg/mL, respectively) and for C-32 (0.7 and 0.25 µg/mL, respectively). For the reference compound, cisplatin, the appropriate concentrations were 0.8 µg/mL (SNB-19 line) and 0.6 µg/mL (C32 line). * p < 0.05 compared with the untreated control.
2.4. Physicochemical Descriptors (Lipophilicity, ADME Properties)
For new derivatives, log PTLC parameters were determined, and correlations between the values of lipophilicity parameters determined experimentally and calculated with the use of computer programs were ascertained.
Lipophilicity is an important physicochemical parameter related to the biological activity of compounds. This descriptor, contained in Lipiński’s rule of five, concerns the transport of a compound through the biological membrane system and the formation of a compound–receptor complex [39]. To assess the lipophilic nature of triterpenoids 3–5, 6a–6e, and 7a–7e, the reverse phase chromatography (RP-TLC) method was used (chromatographic procedure by RP-TLC is included in the Supplementary Materials). The theoretical characteristics of the chromatographic parameters (RM0, log PTLC) of the tested derivatives were also performed. The results are presented in Table 6.
Table 6.
The experimental values of lipophilicity for tested compounds 3–5, 6a–6e, and 7a–7e.
The lowest value of experimental lipophilicity (log PTLC = 4.914) was shown for derivative 5 with two hydroxyl groups in positions C3 and C28. The conversion of these substituents into ester functions increased the lipophilicity of derivatives 6a–6e and 7a–7e. The highest increase in the log PTLC value in the group of tested triterpenoids was recorded for the 6d diester, which is related to the presence of two phenylpropynoyl groups.
The 7a–7e monoesters are characterized by lower log PTLC values than the compounds 6a–6e containing two ester groups. Depending on the structure of the substituent, the monoesters can be ranked in the following order of increasing lipophilicity propargyloxycarbonyl < propynoyl < 2-butynoyl < cyclopropylpropynoyl < phenylpropynoyl. The derivatives 7a and 7e with a terminal triple bond possess the lowest lipophilicity (log PTLC- 6.937 and 6.916, respectively) compared to the other alkynyl-substituted triterpenoids.
The experimentally determined lipophilicity of the tested compounds was compared with the theoretical values. Additionally, the partition coefficients (log P) were calculated by use of the seven theoretical programs such as ALOGPs, AC logP, miLogP, ALOGP, MLOGP, XLOGP2, and XLOGP3 [40]. The results (Table S5, Supplementary Materials) show that compound 5 has the lowest lipophilicity, which is consistent with its log PTLC value. In the case of the most lipophilic compound 6d, the same relationship was found.
The calculated theoretical values of log P of compounds 3–5, 6a–6e, and 7a–7e are in the range of 5.15–13.40. The highest log P values were obtained with the XLOGP3 and the lowest with the ALOGPs.
The correlation matrix for theoretical lipophilicity parameters of derivatives 3–5, 6a–6e, and 7a–7e are presented in Table S6 (Supplementary Materials). The calculated correlation coefficients are in the range 0.812–0.987, and the highest value of correlation is found in the cases of XLOGP2 and XLOGP3 (0.987). These programs calculate the value of the log P parameter using the same method based on the additive atom/group model.
The comparison of the experimental lipophilicity values (log PTLC) for the derivatives 3–5, 6a–6e, and 7a–7e, with the corresponding values obtained by different calculation methods, is depicted in Figure 9. The highest correlation between theoretically and experimentally received lipophilicity values were obtained in the case of the XLOGP2 program (0.861), while the lowest was in the case of MLOGP (0.763).
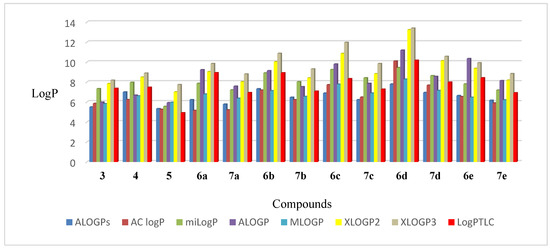
Figure 9.
The values of lipophilicity of triterpenoids 3–5, 6a–6e, and 7a–7e.
The Log PTLC values of the most active compounds 7a and 7b range from 6.937 to 7.982. This experimental lipophilicity does not comply with Lipiński’s rule (log P < 5). This means that lipophilicity is not the only possible factor influencing the biological activity of a compound.
For active monoesters 7a–7e, good correlation between antiproliferative activity against the C-32 and SNB-19 cell lines and lipophilicity could be observed. The obtained relationships described by the equations (A and B) are presented in Table 7.
Table 7.
The correlation between the cytotoxicity (IC50) and the log P values for monoesters 7a–7e.
In silico pharmacokinetic analysis is an important method for verifying and simulating the potential biological activity of a future drug substance. The main goal of early-phase pharmacokinetic studies is the preselection of newly synthesized chemical compounds and qualifying structures with optimal properties for further study. The bioavailability studies of compounds, in addition to the lipophilicity parameter, also include the determination of such descriptors as molecular weight (M), topological polar surface of the molecule (tPSA), log BB (Brain/blood partition coefficient), number of rotatable bonds (nROTB), and number of donor (nHBD) and acceptor (nHBA) sites for hydrogen bonds [41]. All ADME parameters for derivatives 3–5, 6a–6e, and 7a–7e were calculated using ACD/iLab software and tabulated in Table 8 [42].
Table 8.
Prediction of ADME properties of compounds 3–5, 6a–6e, and 7a–7e.
The values of nHBA (≤10), nHBD (≤5), and tPSA (<140 Å2) determined the oral bioavailability of the 30-diethylphosphate derivatives of betulin.
Essentially, the determination of the brain/blood barrier (BBB) permeability parameter has two aspects. The first is to identify neurotoxic substances that damage the brain. In addition, it may be helpful in designing new drugs that can achieve the goal in the CNS [43]. The presence of BBB makes the treatment of glioblastoma very difficult, as the chemotherapeutic agents used cannot be delivered to the target tumor cells. The triterpenes 3–5, 6a–6e, and 7a–7e have the log BB values that fit within an optimal range of the values for drug-likeness (log BB −3 to 1.2) [44]. On the other hand, the log BB values obtained for the tested phosphate derivatives (active in vitro against SNB-19 cells) are characteristic of drugs that are inactive in the CNS. However, only a small number of low molecular weight fat-soluble compounds have the ability to pass through the BBB. Other molecules need a special transport system for this. The use of such forms as liposomes, dendrimers, or polymeric micelles allows chemotherapeutic agents to overcome this barrier and reach the goal of action [45].
It is also known that in the cancer process, some primary tumor cancer cells may metastasize. Such cells have the ability to migrate and can colonize even distant tissues [46]. Metastasis is a significant problem in the treatment of neoplastic diseases and contributes to the worsening of the patient’s prognosis. Metastasis of glioblastoma beyond the nervous system is determined at the level of about 2% of cases, but the number of this type of metastasis is increasing. This is due to the greater effectiveness of the primary tumor treatment, which extends the survival time. Glioma metastases are most often diagnosed in the lungs and pleura (60%) [47].
2.5. In Silico Study
As already mentioned, abnormalities in the expression of genes regulating the cell cycle play an important role in the process of cancer development. Both the initiation and development of the neoplastic process are related not only to such factors as mutations of proto-oncogenes and suppressor genes, disturbances in the DNA repair process, and mutations of anti- and pro-apoptotic genes. Increased expression of growth factors and angiogenesis also plays an important role in these processes. The epidermal growth factor receptor (EGFR) has been observed to be elevated in stomach, brain, and breast tumors. This receptor belongs to the receptor tyrosine kinase (RTK), the pathway of which is the most disrupted signaling cascade in glioblastoma [48,49].
According to our previous article on 3-substituted derivatives of betulin as potential EGFR inhibitors [19], molecular modeling was carried out to predict the possible mode of action of the newly synthesized compounds 7a and 7b. EGFR structure was obtained from PDB (PDB ID: 1M17). The co-crystallized ligand for 1M17 was a known EGFR inhibitor (i.e., erlotinib) and was taken as the reference drug (Figure 10).
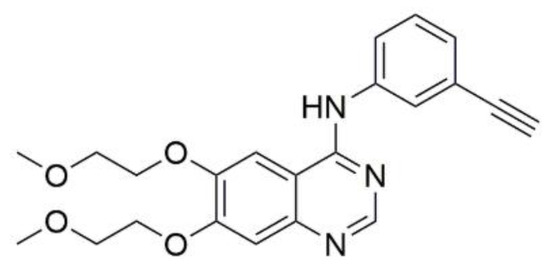
Figure 10.
Structure of reference drug erlotinib.
As illustrated in Figure 11, the crystal structure of 1M17 shows that N1 of the erlotinib quinazoline moiety receives a hydrogen bond from the amide nitrogen of Met769. Moreover, the water molecule (W10) essentially forms a hydrogen-bonding network, which involves nitrogen N3 of the erlotinib quinazoline moiety and the Thr766 side chain.
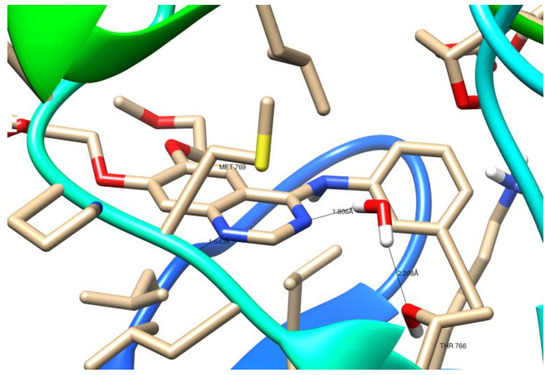
Figure 11.
Three-dimensional (3D) binding interaction of erlotinib with EGFR (PDB ID: 1M17).
Genetic Optimization for Ligand Docking (GOLD) ver. 2020.1 [50] was used for the hydrated docking analysis. GOLD is an automated ligand docking program that relies on the use of a genetic algorithm to study the full range of conformational flexibility of ligands with partial protein flexibility (flexibility of receptor hydrogens) [51]. The region of interest used for GOLD docking was defined as all the EGFR protein residues within the 10 Å of the reference ligand. For the remaining water molecules, the “Toggle” option was on (having GOLD decide whether the water should be present or absent, i.e., bound or displaced by the ligand during docking). The results of docking compounds 7a and 7b to the active site of EGFR tyrosine kinase obtained in the GOLD program are presented in Table 9. Ligand matches are presented in the arbitrary units [a.u.] of GOLD computer program. The higher the calculated value, the better the binding affinities of the ligand for its protein. The obtained results show that compound 7a has a slightly lower docking score compared to the reference erlotinib (30.87 a.u. and 31.06 a.u., respectively). In contrast, compound 7b shows a better binding affinity for EGFR compared to erlotinib (docking score 31.33 a.u.).
Table 9.
The top-ranked ChemScore fitness score function obtained with GOLD and binding energy obtained with KDEEP for betulin derivatives 7a and 7b.
Binding energy is an important parameter for assessing the stability of the ligand-protein complex. It is released when a ligand molecule binds to a protein, which lowers the total energy of the complex. Thus, the greater the energy released when binding a ligand to a protein, the greater the tendency of the ligand to associate with that protein. For this reason, a binding energy calculation has been made using the KDEEP predictor. KDEEP calculates binding energy ΔG [kcal/mol] using deep convolutional neural networks (DCNNs) model [52].
The KDEEP calculations for the crystal structure of the complex of erlotinib with EGFR (1M17) and the complex obtained by docking erlotinib to EGFR tyrosine kinase domain showed that both of these complexes achieved significantly higher binding energy values compared to compounds 7a and 7b (Table 9). According to the obtained in KDEEP results, compounds 7a,b show a better binding affinity to the active site of EGFR compared to erlotinib.
Docked poses of ligand 7a and their interactions with amino acid residues are presented in Figure 12 and Figure 13.
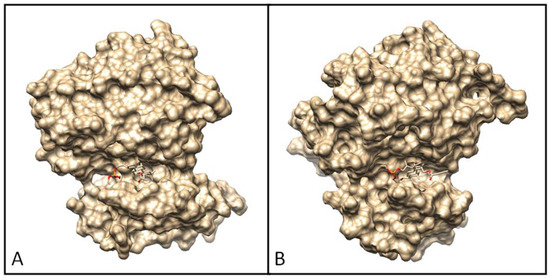
Figure 12.
Docking pose of EGFR tyrosine kinase complex with 7a (A) and 7b (B).
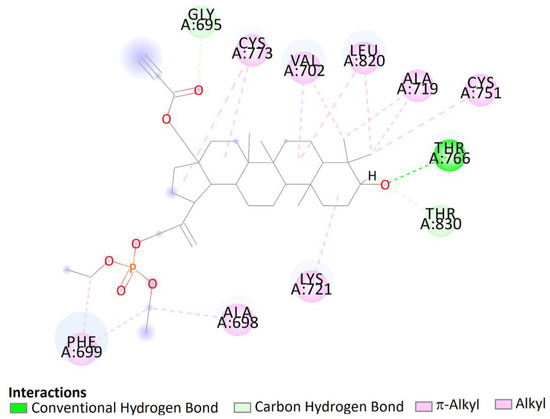
Figure 13.
Two-dimensional (2D) binding interaction of 7a with EGFR.
The top docked pose of compound 7a in the binding site showed hydrophobic interactions with Gly690, Thr830 (carbon hydrogen bond), Phe699 (π–alkyl), Ala698, Lys721, Cys751, Ala719, Leu820, Val702, and Cys773 (alkyl–alkyl). Moreover, hydroxy groups of triterpene in positions C3 were involved in hydrogen bonds to carboxyl oxygen atom of Thr766 (Figure 13).
The pose of compound 7b in the binding site showed hydrophobic interactions with 21 amino acids residues including interactions with Asp831, Phe832, and Gly833 (DFG motif). According to the obtained literature data, interactions with DFG motif play a significant role in tyrosine kinase activity [53]. Moreover, hydroxy groups of triterpene in positions C3 were involved in hydrogen bonds to carboxyl oxygen atom of Thr766, stabilizing the obtained complex (Figure 14).
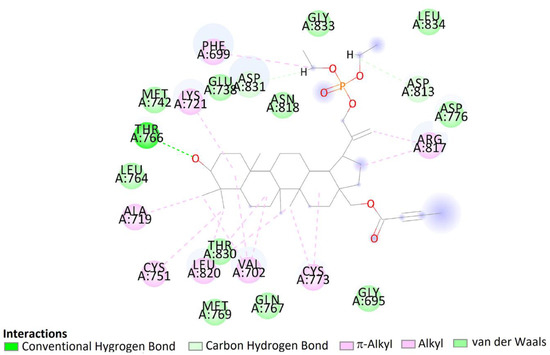
Figure 14.
Two-dimensional (2D) binding interaction of 7b with EGFR.
Molecular dynamics (MD) calculation provide a better understanding of structure–function relationships in motion and other conformational changes by the proteins. Therefore, an MD study was conducted in this paper to evaluate the dynamic behavior of the top-scoring complexes for a period of 10 ns. The molecular mechanics/generalized born surface area (MM/GBSA) method was selected for rescoring complexes, because it is the fastest force-field based method that computes the free energy of binding, as compared to the other computational free energy methods, such as free energy perturbation (FEP) or thermodynamic integration (TI) methods [54]. Comparison studies have also shown that MM/GBSA outperforms molecular mechanics/Poisson Boltzmann surface area MM/PBSA [55]. The MM/GBSA calculation was performed using MolAICal software [56]. MolAICal provides a way to calculate the MM/GBSA based on the output results of molecular dynamics simulations that are carried out by NAMD [57]. The calculated binding free energies are shown in Table 10.
Table 10.
Calculated binding free energies of tested compounds.
The binding free energies of 7a and 7b range from −46.53 to −43.51 kcal/mol, which are much smaller than those of erlotinib. According to the calculations performed, betulin derivatives 7a and 7b are better binders than the reference compound.
Analyzing the complexes ligand root-mean-square deviation (RMSD) and hydrogen bonding can provide insights into a structural conformation faced by the protein and ligand during simulation (Figure 15 and Figure 16).
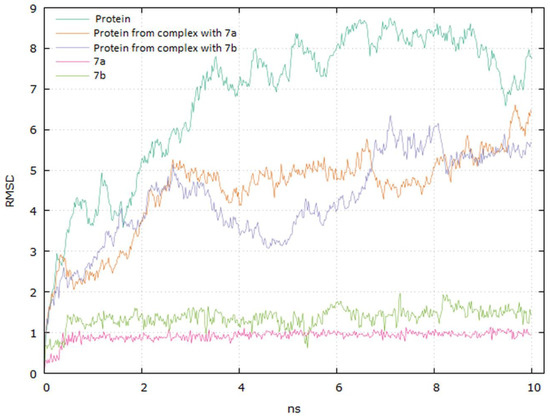
Figure 15.
Graphical representation of the plots showing proteins CαRMSD (Å) and ligands atoms versus time (10 ns).
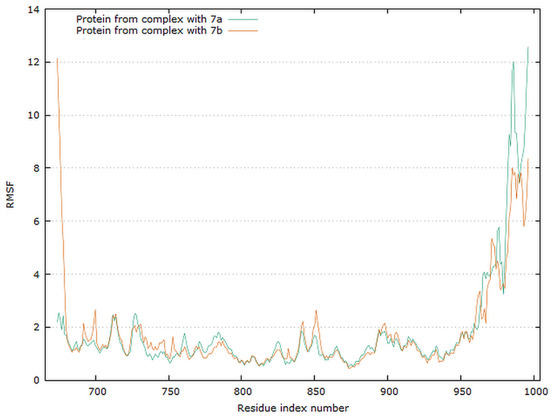
Figure 16.
Graphical representation of the plots showing proteins RMSF [Å] versus residue index number of protein for 7a and 7b complexes of EGFR tyrosine kinase.
The plots for EGFR proteins Cα versus time for the simulation with compounds 7a and 7b are shown in Figure 15. The visual analysis of the trajectory confirms the stability of the structure of protein at around 6 ns. It should be mentioned that the RMSD values obtained for the complexes were lower compared to the apo protein.
The plot for ligands RMSD versus time shows a plateau value of 1 Å and 1.3 Å for the ligands 7a and 7b, respectively, at around 0.3 ns. There was no a significant shift in RMSD values for the ligands backbone. The deviation value observed was within limits, which is quite stable for binding of the ligands with the EGFR tyrosine kinase. The higher value of RMSD for the 7b compound may be due to the butynyl group at carboxylate moiety.
The residue root mean square fluctuation (RMSF) is a measure of the flexibility of a residue. It is typically calculated for the Cα atom of each residue. The plot for protein RMSF versus residue number index shown in Figure 16 describes fluctuations in the range of 0.5–2.5 Å for complexes EGFR with 7a and 7b. The fluctuations of the C-terminals ends were higher. The catalytic residues are no longer higher than 0.5 Å for both complexes, and are suitable for stable binding.
The data presented in Figure 17 show the results of analysis of hydrogen bond contacts in the studied complexes. Compound 7a (Figure 17A) forms three hydrogen bonds, and the binding with Thr766 residue occurs in the complex at the 28% time of measurement. Ligand 7b forms two hydrogen bonds during simulation time, including a stable H-bond with Thr766 (Figure 17B).
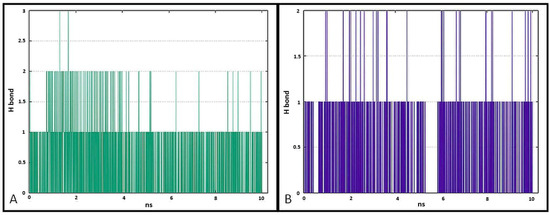
Figure 17.
Pictorial representation of the number of H-bond contacts formed by ligand 7a (A) and 7b (B) with EGFR tyrosine kinase.
From the discussion, it can be summarized that the results obtained in silico are consistent with the experimental data, and compounds 7a and 7b have an efficient binding inside the cavity of EGFR tyrosine kinase.
3. Materials and Methods
3.1. General Experimental Procedures
Materials and methods are included in the Supplementary Materials.
3,28-Diacetyl-30-hydroxybetulin 3 was obtained in 52% yield by oxidation of 3,28-diacetylbetulin 2 with m-chloroperbenzoic acid in chloroform. Melting point and 1H and 13C NMR spectra for compound 3 were consistent with the literature data [58].
Phosphate derivatives of betulinic 8 and betulonic 9 acid were synthesized according to the described method. Spectroscopic (1H, 13C, and 31P NMR) and melting point data were consistent with the literature values [9].
3.2. Procedures for Synthesizing Compounds
3.2.1. Synthesis of 3,28-Diacetoxy-30-diethoxyphosphoryloxybetulin 4
A flask with a solution of compound 3 (0.54 g, 1 mmol) and 12 mg (0.1 mmol) 4-dimethylaminopyridine (DMAP) in 6 mL pyridine was placed in an ice-water bath. Then, diethyl chlorophosphate (0.28 mL, 1.9 mmol) was added dropwise to the cooled mixture, and stirring was continued for 24 h under an inert gas (argon) atmosphere after the cooling bath was removed. The course of the reaction was monitored using thin layer chromatography (TLC). After completion of the reaction, the pyridine was evaporated under reduced pressure. Dichloromethane (30 mL) was added to the residue, and the resulting solution was washed with 10% HCl (2 × 10 mL), saturated NaHCO3 (12 mL), and then with water (2 × 10 mL). The organic phase was dried with anhydrous Na2SO4 and then concentrated. The product was isolated by column chromatography (SiO2, hexane:ethyl acetate, 3:2, v/v).
Yield: 72%, m. p. 61–63 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.48. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.06 (s, 1H, H-29), 5.00 (s, 1H, H-29), 4.51 (m, 2H, H-30), 4.48 (m, 1H, H-3), 4.26 (d, J = 10.8 Hz, 1H, H-28), 4.16 (m, 4H, 2× OCH2CH3), 3.84 (d, J = 10.8 Hz, 1H, H-28), 2.39 (m, 1H, H-19), 2.06 (s, 3H, C(O)CH3), 2.09 (s, 3H, C(O)CH3), 0.85–2.00 (m, CH, CH2), 1.38 (m, 6H, 2× OCH2CH3), 1.08 (s, 3H, CH3), 1.01 (s, 3H, CH3), 0.87 (s, 3H, CH3), 0.86 (s, 3H, CH3), 0.85 (s, 3H, CH3), 0.80 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 171.6; 171.0; 149.0; 110.5; 80.9; 63.8; 63.8; 62.4; 55.4; 50.2; 49.5; 46.3; 43.2; 42.7; 40.9; 38.4; 37.8; 37.5; 37.1; 34.3; 34.1; 31.3; 29.7; 28.0; 27.0; 26.6; 23.7; 21.3; 21.0; 20.9; 18.2; 16.2; 16.2; 16.1; 16.0; 14.9. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C38H62O8P [(M–H)−], Calc. 677.4182; Found 677.4180.
3.2.2. Synthesis of 30-Diethoxyphosphoryloxybetulin 5
A solution of 730 mg sodium hydroxide in 10 mL of methanol, 20 mL of tetrahydrofuran, and 10 mL of water was prepared in the flask. Then, 680 mg of compound 4 (1 mmol) was added. The reaction was carried out at room temperature while stirring with a magnetic stirrer. The progress of the reaction was monitored by TLC (SiO2, dichloromethane:ethanol, 15:1, v/v). After 5 h, the reaction mixture was evaporated to dryness. The residue was dissolved in dichloromethane and washed with 10 mL of hydrochloric acid and then three times with 30 mL of water. The organic phase was dried with anhydrous Na2SO4, and the product was purified using column chromatography (SiO2, dichloromethane:ethanol, 15:1, v/v).
Yield: 85%, m. p. 194–195 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.36. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.49 (d, J = 1.2 Hz, 1H, H-29); 4.99 (s, 1H, H-29), 4.51 (m, 2H, H-30), 4.15 (m, 4H, 2× OCH2CH3), 3.80 (d, J = 10.8 Hz, 1H, H-28), 3.32 (d, J = 10.8 Hz, 1H, H-28), 3.21 (m, 1H, H-3), 2.35 (m, 1H, H-19), 0.90–2.00 (m, CH, CH2), 1.37 (m, 6H, 2× OCH2CH3), 1.07 (s, 3H, CH3), 1.04 (s, 3H, CH3), 1.00 (s, 3H, CH3), 0.84 (s, 3H, CH3), 0.78 (s, 3H, CH3), 0.70 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 148.3; 109.2; 77.9; 68.0; 2 × 62.8; 59.2; 54.2; 49.3; 48.5; 46.7; 42.1; 41.6; 39.9; 37.8; 37.7; 36.2; 36.1; 33.2; 32.7; 30.5; 28.2; 27.0; 26.3; 26.0; 25.7; 19.9; 17.3; 2 × 15.2; 15.0; 14.3, 13.7. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C34H58O6P [(M–H)−], Calc. 593.3971; Found 593.3959.
3.2.3. General Procedure for Synthesis of Alkynyl Derivatives 6a–6d and 7a–7d
A solution of 595 mg of 30-diethoxyphosphoryloxybetulin 5 (1 mmol) in 5 mL of dichloromethane was cooled in an ice–salt bath to −10 °C, and 1.17 mmol of the corresponding alkynyl acid was added. Then, a solution of 240 mg of DCC and 10 mg of DMAP in 1 mL of dichloromethane was slowly added dropwise. The reaction was carried out under an argon atmosphere while stirring with a magnetic stirrer. The flask with the reaction mixture was kept in an ice–salt bath for the first 5 h, after which the reaction was carried out at room temperature. After 24 h, the precipitate was filtered off and the filtrate was concentrated to dryness in a vacuum evaporator. Subsequently, the reaction products were purified using column chromatography (SiO2, dichloromethane:ethanol, 15:1 or 40:1, v/v).
30-Diethoxyphosphoryloxy-3,28-dipropynoylbetulin6a
Yield: 5%, m. p. 127–130 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.75. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.07 (s, 1H, H-29), 5.00 (s, 1H, H-29), 4.62 (m, 1H, H-3), 4.51 (m, 2H, H-30), 4.40 (d, J = 10.8 Hz, 1H, H-28), 4.15 (m, 4H, 2× OCH2CH3), 3.98 (d, J = 10.8 Hz, 1H, H-28), 2.92 (s, 1H, C≡CH), 2.88 (s, 1H, C≡CH), 2.38 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.37 (m, 6H, 2× OCH2CH3), 1.05 (s, 3H, CH3), 1.00 (s, 3H, CH3), 0.91 (s, 3H, CH3), 0.90 (s, 3H, CH3), 0.88 (s, 3H, CH3), 0.74 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 153.2; 152.8; 148.8; 110.7; 83.6; 75.0; 74.6; 2 × 68.9; 64.4; 63.9; 63.9; 55.3; 50.2; 49.5; 46.4; 43.1; 42.7; 40.9; 38.3; 37.9; 37.7; 37.5; 37.0; 34.2; 34.1; 31.2; 29.6; 27.9; 26.9; 26.6; 23.5; 20.9; 18.1; 16.5; 2 × 16.2; 16.1; 14.7. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.93. HR-MS (APCI) m/z: C40H58O8P [(M–H)−], Calc. 697.3869; Found 697.3856.
3,28-Di(2-butynoyl)-30-diethoxyphosphoryloxybetulin6b
Yield: 10%, m. p. 66–70 °C.
TLC (dichloromethane:ethanol, 40:1, v/v): Rf = 0.73. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.06 (s, 1H, H-29), 4.99 (s, 1H, H-29), 4.59 (m, 1H, H-3), 4.50 (m, 2H, H-30), 4.34 (d, J = 10.8 Hz, 1H, H-28), 4.16 (m, 4H, 2× OCH2CH3), 3.93 (d, J = 10.8 Hz, 1H, H-28), 2.37 (m, 1H, H-19), 2.02 (s, 3H, CH3C≡C), 2.01 (s, 3H, C≡CCH3), 0.93–2.10 (m, CH, CH2), 1.36 (m, 6H, 2× OCH2CH3), 1.04 (s, 3H, CH3), 0.99 (s, 3H, CH3), 0.90 (s, 3H, CH3), 0.89 (s, 3H, CH3), 0.86 (s, 3H, CH3), 0.80 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 154.3; 153.9; 149.1; 111.4; 85.7; 85.0; 82.8; 72.5; 72.5; 69.0; 63.9; 63.8; 55.4; 50.2; 46.4; 45.5; 42.7; 40.9; 38.4; 37.9; 37.5; 37.0; 34.2; 34.1; 30.6; 29.6; 27.9; 27.3; 27.0; 23.6; 20.9; 19.7; 18.1; 16.5; 2 × 16.2; 16.1; 16.0; 14.7; 3.9; 3.9. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.90. HR-MS (APCI) m/z: C42H62O8P [(M-H)−], Calc. 725.4182; Found 725.4162.
3,28-Di(2-cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin6c
Yield: 8%, m. p. 67–70 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.70. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.05 (s, 1H, H-29), 4.98 (s, 1H, H-29), 4.58 (m, 1H, H-3), 4.50 (m, 2H, H-30), 4.31 (d, J = 10.8 Hz, 1H, H-28), 4.14 (m, 4H, 2× OCH2CH3), 3.91 (d, J = 10.8 Hz, 1H, H-28), 2.37 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.36 (m, 6H, 2× OCH2CH3), 1.02 (s, 3H, CH3), 0.97 (s, 3H, CH3), 0.95 (m, 2 × 2H, CH2C≡CH), 0.93 (m, 2 × 2H, CH2C≡CH), 0.88 (s, 3H, CH3), 0.87 (s, 3H, CH3), 0.85 (s, 3H, CH3), 0.77 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 154.9; 154.5; 149.4; 111.1; 94.0; 93.3; 83.1; 69.4; 2 × 69.0; 64.4; 2 × 64.2; 55.9; 50.7; 50.0; 47.0; 43.2; 41.4; 38.5; 38.3; 38.0; 37.5; 34.8; 34.6; 30.1; 28.4; 27.5; 27.1; 24.1; 21.4; 18.7; 17.1; 16.7; 16.7; 16.5; 15.2; 2 × 9.7; 0.0. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C37H58O6P [(M–H)−], Calc. 629.3971; Found 629.3965.
30-Diethoxyphosphoryloxy-3,28-di(2-phenylpropynoyl)betulin6d
Yield: 12%, m. p. 89–91 °C.
TLC (dichloromethane:ethanol, 40:1, v/v): Rf = 0.77. 1H-NMR (600 MHz, CDCl3) δ (ppm): 7.62 (m, 4H, Harom), 7.47 (m, 2H, Harom), 7.40 (m, 4H, Harom), 5.07 (s, 1H, H-29), 5.01 (s, 1H, H-29), 4.68 (m, 1H, H-3), 4.52 (m, 2H, H-30), 4.43 (d, J = 10.8 Hz, 1H, H-28), 4.17 (m, 4H, 2× OCH2CH3), 4.02 (d, J = 10.8Hz, 1H, H-28), 2.41 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.38 (m, 6H, 2× OCH2CH3), 1.08 (s, 3H, CH3), 1.02 (s, 3H, CH3), 0.95 (s, 3H, CH3), 0.94 (s, 3H, CH3), 0.90 (s, 3H, CH3), 0.84 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 154.7; 154.2; 149.0; 2 × 133.0; 2 × 130.5; 2 × 129.7; 2 × 128.5; 2 × 119.9; 110.7; 86.5; 85.8; 83.2; 81.1; 80.7; 68.9; 64.1; 2 × 63.8; 55.4; 50.2; 46.5; 42.7; 40.9; 38.4; 38.0; 37.6; 37.1; 34.3; 34.1; 31.2; 29.7; 28.0; 17.3; 27.0; 26.6; 23.6; 20.9; 18.2; 16.6; 2 × 16.2; 16.1; 14.8. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.88. HR-MS (APCI) m/z: C37H58O6P [(M–H)−], Calc. 849.4495; Found 849.4483.
30-Diethoxyphosphoryloxy-28-propynoylbetulin7a
Yield: 72%, m. p. 177–180 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.51. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.07 (s, 1H, H-29), 5.00 (s, 1H, H-29), 4.51 (m, 2H, H-30), 4.40 (d, J = 10.8Hz, 1H, H-28), 4.15 (m, 4H, 2× OCH2CH3), 3.99 (d, J = 10.8 Hz, 1H, H-28), 3.20 (m, 1H, H-3), 2.92 (s, 1H, C≡CH), 2.38 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.37 (m, 6H, 2× OCH2CH3), 1.04 (s, 3H, CH3), 1.01 (s, 3H, CH3), 0.99 (s, 3H, CH3), 0.84 (s, 3H, CH3), 0.79 (s, 3H, CH3), 0.69 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 153.2; 148.8; 110.7; 78.9; 74.8; 74.7; 68.9; 64.5; 2 × 63.9; 55.3; 50.3; 49.5; 46.4; 43.1; 42.7; 40.9; 38.9; 38.7; 37.6; 37.1; 34.2; 33.6; 31.2; 29.6; 28.0; 27.4; 27.0; 26.7; 20.8; 18.4; 18.3; 2 × 16.2; 16.0; 15.4; 14.8. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.94. HR-MS (APCI) m/z: C37H58O7P [(M–H)−], Calc. 645.3920; Found 645.3918.
28-(2-Butynoyl)-30-diethoxyphosphoryloxybetulin7b
Yield: 81%, m. p. 133–136 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.52. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.06 (s, 1H, H-29), 4.99 (s, 1H, H-29), 4.50 (m, 2H, H-30), 4.35 (d, J = 10.8Hz, 1H, H-28), 4.15 (m, 4H, 2× OCH2CH3), 3.94 (d, J = 10.8 Hz, 1H, H-28), 3.20 (m, 1H, H-3), 2.38 (m, 1H, H-19), 2.01 (s, 3H, C≡CCH3), 0.93–2.10 (m, CH, CH2), 1.37 (m, 6H, 2× OCH2CH3), 1.04 (s, 3H, CH3), 0.99 (s, 3H, CH3), 0.98 (s, 3H, CH3), 0.83 (s, 3H, CH3), 0.78 (s, 3H, CH3), 0.70 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 154.3; 148.9; 110.5; 85.5; 78.9; 72.5; 68.8; 63.8; 63.8; 55.3; 50.3; 49.5; 46.4; 43.1; 42.7; 40.9; 38.9; 38.7; 37.5; 37.1; 34.2; 31.2; 29.6; 28.0; 27.4; 27.0; 26.6; 20.8; 18.3; 2 × 16.2; 16.1; 15.4; 14.8, 3.9. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C38H60O7P [(M–H)-], Calc. 659.4077; Found 659.4079.
28-(2-Cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin7c
Yield: 82%, m. p. 133–134 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.54. 1H-NMR (600 MHz, CDCl3) δ (ppm): 5.05 (s, 1H, H-29), 4.95 (s, 1H, H-29), 4.50 (m, 2H, H-30), 4.32 (d, J = 10.8Hz, 1H, H-28), 4.15 (m, 4H, 2× OCH2CH3), 3.92 (d, J = 10.8 Hz, 1H, H-28), 3.20 (m, 1H, H-3), 2.38 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.36 (m, 6H, 2× OCH2CH3), 1.03 (s, 3H, CH3), 0.99 (s, 3H, CH3), 0.98 (s, 3H, CH3), 0.95 (s, 2H, C≡CCHCH2), 0.93 (s, 2H, C≡CCHCH2), 0.83 (s, 3H, CH3), 0.77 (s, 3H, CH3), 0.69 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 155.0; 149.4; 111.0; 94.0; 79.5; 69.3; 69.0; 2 × 64.4; 64.2; 55.8; 50.8; 50.0; 43.5; 43.2; 41.4; 39.4; 39.2; 39.0; 38.1; 37.7; 34.8; 34.7; 34.4; 31.7; 30.2; 28.5; 27.9; 27.5; 26.0; 21.4; 18.8; 2 × 16.7; 16.6; 15.9; 15.3; 9.75. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.93. HR-MS (APCI) m/z: C40H62O7P [(M–H)−], Calc. 685.4233; Found 685.4229.
30-Diethoxyphosphoryloxy-28-(2-phenylpropynoyl)betulin7d
Yield: 71%, m. p. 131–134 °C.
TLC (dichloromethane:ethanol, 40:1, v/v): Rf = 0.52. 1H-NMR (600 MHz, CDCl3) δ (ppm): 7.62 (m, 2H, Harom), 7.47 (m, 1H, Harom), 7.40 (m, 2H, Harom), 5.07 (s, 1H, H-29), 5.01 (s, 1H, H-29), 4.51 (m, 2H, H-30), 4.42 (d, J = 10.8 Hz, 1H, H-28), 4.16 (m, 4H, 2× OCH2CH3), 4.03 (d, J = 10.8Hz, 1H, H-28), 3.20 (m, 1H, H-3), 2.41 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.37 (m, 6H, 2× OCH2CH3), 1.06 (s, 3H, CH3), 1.01 (s, 3H, CH3), 0.99 (s, 3H, CH3), 0.84 (s, 3H, CH3), 0.78 (s, 3H, CH3), 0.69 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 154.6; 148.9; 133, 130.6; 129.7; 128.6; 119.7; 110.6; 86.4; 80.7; 78.9; 68.8; 64.1; 2 × 63.8; 55.3; 50.3; 49.5; 46.5; 43.2; 42.7; 40.9; 38.9; 38.2; 37.6; 37.1; 34.3; 34.2; 31.62; 29.7; 28.0; 27.4; 27.0; 26.7; 20.9; 19.7; 18.3; 2 × 16.2; 16.1; 15.4; 14.8. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C43H62O7P [(M–H)−], Calc. 721.4233; Found 721.4226.
3.2.4. Synthesis of Alkynyl Derivatives 6e and 7e
A solution of compound 5 (595 mg, 1mmol) in 4.5 mL of dry benzene and 3.75 mL of pyridine was cooled to about −5 °C. Then, 1.5 mmol of propargyl chloroformate in 4 mL of benzene was gradually added dropwise while stirring with a magnetic stirrer. Stirring was continued for 18 h at room temperature. The volatiles were evaporated, 6 mL of chloroform was added to the residue, and the resulting solution was washed with 10% sulfuric acid and then with water. After drying over anhydrous sodium sulfate, the chloroform was removed in a vacuum evaporator. The mono- and disubstituted derivatives, 6e and 7e, were isolated using column chromatography (SiO2, dichloromethane:ethanol 15: 1, v/v).
30-Diethoxyphosphoryloxy-3,28-dipropargyloxycarbonyloxybetulin6e
Yield: 15%, m. p. 129–131 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.82. 1H-NMR (600 MHz, CDCl3) δ (ppm): 4.98 (s, 1H, H-29), 4.91 (s, 1H, H-29), 4.67 (m, 2H, OCH2-C≡C), 4.65 (m, 2H, OCH2-C≡C), 4.29 (d, J = 6 Hz, 2H, H-30), 4.30 (d, J = 10.8 Hz, 1H, H-28), 4.27 (m, 1H, H-3), 4.07 (m, 4H, 2× OCH2CH3), 3.85 (d, J = 10.8 Hz, 1H, H-28), 2.47 (t, J = 2.4 Hz, 1H, C≡CH), 2.45 (t, J = 2.4 Hz, 1H, C≡CH), 2.28 (m, 1H, H-19), 1.0–2.10 (m, CH, CH2), 1.29 (m, 6H, 2× OCH2CH3), 0.96 (s, 3H, CH3), 0.91 (s, 3H, CH3), 0.85 (s, 3H, CH3), 0.78 (s, 3H, CH3), 0.77 (s, 3H, CH3), 0.70 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 155.1; 154.6; 148.8; 110.7; 86.2; 75.7; 75.5; 68.9; 66.9; 63.9; 63.8; 2 × 55.3; 55.0; 53.5; 50.2; 49.5; 46.6; 42.7; 40.9; 38.3; 38.1; 37.5; 37.0; 34.1; 31.2; 29.5; 27.6; 26.9; 26.6; 23.6; 20.9; 18.1; 16.4; 2 × 16.2; 16.1; 16.0; 14.8. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.9. HR-MS (APCI) m/z: C42H62O10P [(M–H)−], Calc. 757.4081; Found 757.4075.
30-Diethoxyphosphoryloxy-28-propargyloxycarbonyloxybetulin7e
Yield: 60%, m. p. 172–174 °C.
TLC (dichloromethane:ethanol, 15:1, v/v): Rf = 0.64. 1H-NMR (600 MHz, CDCl3) δ (ppm): 4.97 (s, 1H, H-29), 4.91 (s, 1H, H-29), 4.67 (dd, J = 1.2 Hz, J = 2.4 Hz, 2H, OCH2-C≡C), 4.42 (d, J = 6 Hz, 2H, H-30), 4.30 (d, J = 10.8 Hz, 1H, H-28), 4.07 (m, 4H, 2× OCH2CH3), 3.85 (d, J = 10.8 Hz, 1H, H-28), 3.11 (m, 1H, H-3), 2.47 (t, J = 2.4 Hz, 1H, C≡CH), 2.47 (m, 1H, H-19), 0.93–2.10 (m, CH, CH2), 1.28 (m, 6H, 2× OCH2CH3), 0.96 (s, 3H, CH3), 0.91 (s, 3H, CH3), 0.90 (s, 3H, CH3), 0.75 (s, 3H, CH3), 0.69 (s, 3H, CH3), 0.61 (m, 1H, H-5). 13C-NMR (150 MHz, CDCl3) δ (ppm): 155.1; 148.9; 110.6; 78.9; 75.7; 68.9; 66.9; 63.9; 63.8; 55.3; 55.2; 50.3; 49.5; 46.6; 42.7; 40.9; 38.9; 38.7; 37.5; 37.1; 34.2; 34.1; 31.2; 29.5; 28.0; 27.4; 27.0; 26.7; 20.8; 18.3; 2 × 16.2; 16.1; 16.0; 15.4; 14.8. 31P-NMR (243 MHz, CDCl3) δ (ppm): −0.91. HR-MS (APCI) m/z: C38H60O8P [(M–H)−], Calc. 675.4026; Found 675.4019.
3.3. Raman Spectroscopy
Raman spectra were measured using the Raman confocal microscope Alfa 300R (WITec Instruments Corp., Germany) equipped with an Olympus 50×/0.50 long working distance objective and 532 nm wavelength diode laser. Averaged spectra were accumulated 100 times with a 2 s integration time, at the range 200–3800 cm−1 and with a spectral resolution about 1.5 cm−1 (at 2000 cm−1). The power of the laser beam on the sample did not exceed 10 mW, which prevented heating of the samples. OriginPro 9.1 software was used to analyze the spectra. The spectra were smoothed (Savitzky–Golay method, seven points) and background subtracted. The band positions were determined by the curve fitting (Gaussian function shape) with accuracy of about 1 cm−1.
The theoretical vibrational spectra of compounds 5 and 10 were calculated using Gaussin09 software. Structure optimization and spectra calculations were performed with DFT B3LYP hybrid functional and 6–31G(d) basis set. No imaginary frequencies were found for the optimized structures, which means that at least local energy minima were obtained. All calculated frequencies were scaled by a scaling factor 0.960 in order to correct the calculated value to experimentally observable.
3.4. Crystal Structure
The methodology for X-ray analysis [59,60,61] and additional information about the crystal are included in the Supplementary Materials. Crystal structure was deposited at the Cambridge Crystallographic Data Center, with deposit number CCDC 2,039,827, and is available free of charge via www.ccdc.cam.ac.uk/data_request/cif.
3.5. Biological Activity
3.5.1. Cell Culture and Medium
The synthesized compounds and references were tested for cytotoxic activity in vitro against human cancer cell lines such as amelanotic melanoma (C-32, ATCC, Rockville, MD, USA), glioblastoma (SNB-19, DSMZ, Braunschweig, Germany), and breast cancer (T47D, and MDA-MB-231, ATCC, Rockville, MD, USA). Cell cultures were maintained using DMEM (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (FBS) (Biological Industries Cromwell, CT, USA) and penicillin (10,000 U/mL) –streptomycin (10 mg/mL) mix (Lonza, Basel, Switzerland).
3.5.2. Cell Viability Studies
Cultures were performed in 96-well plates (Nunc Thermo Fisher Scientific, Waltham, MA, USA). Cells were seeded at 5 × 104/well and incubated for 24 h (at 37 °C, 5% CO2, constant humidity). Then, the medium was replaced with a fresh one with the addition of test compounds in the concentration: 25; 12.5; 6.25; 3.12; 1.56; 0.78; 0.39; 0 µg/mL and incubated again for a further 72 h. After this time, the WST-1 test (Roche Molecular Biochemicals, Mannheim, Germany) was performed to assess the metabolic activity of the cells. The principle of the WST-1 colorimetric test is based on the ability of live cells to decompose bright red tetrazole salts of WST-1 to dark red formazan. The reaction is mediated by mitochondrial dehydrogenases and correlates with the number of cells and their metabolic activity. The absorbance was measured at λ = 450 nm with a UVM340 microplate reader (Biogenet, Józefów, Poland). Results were expressed as a mean value of at least three independent experiments performed in triplicate.
3.5.3. Analysis of the Expression of H3, TP53, BCL-2, and BAX by qRT-PCR
The influence of tested compounds on the transcriptional activity of cells of the genes H3, TP53, BCL-2, and BAX was investigated. Assays were performed by real-time qRT-PCR on an Opticon TM DNA Engine (MJ Research, USA) using the QuantTect® SYBR® Green qRT-PCR Kit (Qiagen, Valencia, Spain). Cells were exposed to compound 7a and 7b for 24 h at a concentration equal to half of IC50 for appropriate cell line. For the reference compound, cisplatin, concentrations were 0.8 µg/mL (SNB-19 line) and 0.6 µg/mL (C-32 line). RNA extraction was performed on spin columns using the Quick RNATM MiniPrep kit (Zymo Research, Irvine, CA, USA). RNA extracts were assessed qualitatively and quantitatively. Total RNA integrity was checked electrophoretically on a 1.2% agarose gel with ethidium bromide. The amount and purity of total RNA in the extracts were determined spectrophotometrically with the HP8452A spectrophotometer (Hewlett Packard, Waldbronn, Germany).
3.5.4. Statistical Analysis
All experiments were repeated in triplicate. Control treatment refers to untreated cells. As a positive control, used cells were treated with cisplatin. The data were expressed as mean ± standard deviation (SD). One-way ANOVA with post-hoc Tukey’s test was used to compare the data from different groups, p < 0.05 was considered statistically significant.
3.6. Physicochemical Descriptors (Lipophilicity, ADME Properties)
Chromatographic procedure by RP-TLC [62] is included in the Supplementary Materials. The ADME parameters for all derivatives were calculated using ACD/iLab software (Advanced Chemistry Development, Inc., Toronto, ON, Canada) [42].
3.7. Molecular Docking
The three-dimensional (3D) structures of studied compounds were generated in their low-energy conformation using Gaussian 16 (revision A.03) computer code [63] at the density functional theory (DFT, B3LYP) and 6–311 + G(d,p) basis sets. Target macromolecule for molecular docking studies was obtained from the Protein Data Bank (https://www.rcsb.org/). We used 3D crystal structures of EGFR (PDB ID: 1M17). Protein Preparation Wizard of UCSF Chimera (DockPrep) software [64] was used to process and prepare the protein. Missing chains were added, the implicit hydrogen atoms also were added to the atoms to satisfy their appropriate valences, and the ligand of no significance present in the protein structure was deleted. Then the structure was optimized by assigning the bond orders, bond angles, and topology. The formal atomic charges were fixed for the amino acid residues, and energy minimization was carried out.
Genetic Optimization for Ligand Docking (GOLD) 2020.1 [50] was used for the docking analysis. The region of interest used for GOLD docking was defined as the EGFR protein residues within the 10 Å of the reference erlotinib (X = 22.01, Y = 0.25, Z = 52.79 Å). For remaining water molecules, the “Toggle” option was on. Default values of all other parameters were used, and the complexes were submitted to 100 genetic algorithm runs using the ChemScore fitness function. After calculations, only the ten highest scored pose was returned as a docking result for ligand–cavity configuration. All obtained results were ranked according to their score value and presented in GOLD arbitrary units (a.u.). Calculation of protein–ligand binding free energy was performed using the KDEEP predictor based on DCNNs (https://playmolecule.org/Kdeep) [52]. Molecular docking details were visualized using the BIOVIA Discovery Studio virtual environment [65] and Chimera [63]. Molecular dynamics simulation was performed with Nanoscale Molecular Dynamics software ver. 2.13 (NAMD, https://www.ks.uiuc.edu/Research/namd/) [57]. All input files were prepared using QwikMD [66] computer program based on GOLD output complexes. Protein–ligand systems have been solvated with 0.15 mol/L NaCl water box. Then system was minimized, annealed, and equilibrated. After that, 10 ns production simulation was performed. The MolAICal [56] is used to calculate the MM/GBSA between ligands and EGFR protein based on molecular dynamical (MD) simulated results by NAMD.
4. Conclusions
In the presented study, new 30-phosphate derivatives of betulin were synthesized, and evaluated as anticancer agents. Taking into account all the tested neoplastic cell lines, it can be seen that in the examined group of alkynyl derivatives, monoesters 7a–7e (IC50 in range 0.76–15.40 µM) showed higher activity than diesters 6a–6e (IC50 in range 5.65–31.46 µM). Compounds 7a and 7b were characterized by potent in vitro activity against the C-32 and SNB-16 cell lines. In addition, derivative 7a characterized antitumor activity higher than that of the reference drug cisplatin against C-32, SND-19, and T47D cells. Comparing the activity of 28-propynoylbetulin with its 30-phosphate analogue 7a in relation to appropriate tumor cell lines shows that the introduction of diethylphosphate substituent to the isopropenyl moiety resulted in an improvement in activity. With respect to T47D cells, alkynyl esters with 30-phosphate substituent (7a–7c) showed weaker activity than their phosphonate analogs (11–13). In relation to the SNB-16 and C-32 lines, no relationship between the activity and the type of the phosphorus substituent was observed.
The most active in vitro compounds, 7a and 7b, were tested on expression of H3, TP53, BAX, and BCL-2 genes. The results have shown that 7a and 7b can probably activate the apoptosis process in SNB-19 and C-32 cells. Further research is necessary to determine the exact path of cell death of the studied lines. The antitumor activity of triterpenes against the tested cell lines was more dependent on their chemical structure and less on lipophilicity. It was observed that the correlation between lipophilicity and antitumor activity was significant for the monoesters 7a–7e containing a free hydroxyl group at the C3 position. The obtained results suggest that the relationship between ADME properties and antitumor activity is a complex issue.
Taking into account the potential mechanisms of antitumor activity, molecular docking of the most active in vitro derivatives, 7a and 7b, to the selected receptor, EGFR, was also carried out. In the library of designed phosphate derivatives of betulin, compounds 7a and 7b are reported to have good binding interactions as compared to standard erlotinib. Compound 7b was found to have the highest binding affinity and form a perfect binding pose with EGFR, justified by KDEEP score. Furthermore, the molecular dynamic study of the most effective ligands 7a,b has proved the stability of ligand–protein complexes with little deviation and fluctuation from 7 ns up to 10 ns. Compounds 7a and 7b demonstrated comparable degree of fit to the reference ligand erlotinib. Such a result may indicate a potential molecular target with which the antitumor activity of the test compounds is related.
Supplementary Materials
Synthesis—Materials and methods, Figure S1a. 1H NMR, 3,28-diacetoxy-30-diethoxyphosphoryloxybetulin 4, Figure S1b. 13C NMR, 3,28-diacetoxy-30-diethoxyphosphoryloxybetulin 4, Figure S1c. 31P NMR, 3,28-diacetoxy-30-diethoxyphosphoryloxybetulin 4, Figure S1d. HR-MS, 3,28-diacetoxy-30-diethoxyphosphoryloxybetulin 4, Figure S2a. 1H NMR, 30-diethoxyphosphoryloxybetulin 5, Figure S2b. 13C NMR, 30-diethoxyphosphoryloxybetulin 5, Figure S2c. 31P NMR, 30-diethoxyphosphoryloxybetulin 5, Figure S2d. HR-MS, 30-diethoxyphosphoryloxybetulin 5, Figure S3a. 1H NMR, 30-diethoxyphosphoryloxy-28-propynoylbetulin 7a, Figure S3b. 13C NMR, 30-diethoxyphosphoryloxy-28-propynoylbetulin 7a, Figure S3c. 31P NMR, 30-diethoxyphosphoryloxy-28-propynoylbetulin 7a, Figure S3d. HR-MS, 30-diethoxyphosphoryloxy-28-propynoylbetulin 7a, Figure S4a. 1H NMR, 28-(2-butynoyl)-30-diethoxyphosphoryloxybetulin 7b, Figure S4b. 13C NMR, 28-(2-butynoyl)-30-diethoxyphosphoryloxybetulin 7b, Figure S4c. 31P NMR, 28-(2-butynoyl)-30-diethoxyphosphoryloxybetulin 7b, Figure S4d. HR-MS, 28-(2-butynoyl)-30-diethoxyphosphoryloxybetulin 7b, Figure S5a. 1H NMR, 28-(2-cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin 7c, Figure S5b. 13C NMR, 28-(2-cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin 7c, Figure S5c. 31P NMR, 28-(2-cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin 7c, Figure S5d. HR-MS, 28-(2-cyclopropylpropynoyl)-30-diethoxyphosphoryloxybetulin 7c, Table S1. Calculated Raman band frequencies and assignments for compounds 5 and 10, X-ray diffraction experiment, Table S2 Crystal parameters, data collection and refinement details for compound 5, Table S3 Monocrystal characteristic, Table S4 Selected hydrogen bonds in the crystal structure of compound 5, Chromatographic procedure by RP-TLC, Table S5. The theoretical values of lipophilicity for tested compounds 3–5, 6a–6e and 7a–7e, Table S6. The correlation matrix for theoretically obtained lipophilicity parameters of compounds 3–5, 6a–6e and 7a–7e.
Author Contributions
E.C. and S.B. developed the concept of the work. E.C. performed experiments, analyzed spectra, and wrote the paper. M.J. and R.W. made measurements and calculations of Raman spectra. K.M. performed and described the molecular docking study and the molecular dynamics simulations. M.L. conducted a study of the biological activity. E.B. and M.K.-T. contributed to the synthesis and purification of all compounds. J.K.: X-ray diffraction measurement. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This work was supported by the Medical University of Silesia Grants nos. PCN-1-004/K/0/F and KNW-1-106/K/9/B. Energy-minimization calculations have been carried out using resources provided by Wroclaw Centre for 818 Networking and Supercomputing (https://wcss.pl), Grant no. 382. The authors are grateful to Czestochowa University of Technology for granting access to HPC platform provided by the MICLAB project no. POIG.02.03.00.24-093/13.
Conflicts of Interest
The authors declare no conflict of interest.
References
- World Health Organization. WHO Report on Cancer, Setting Priorities, Investing Wisely and Providing Care for All. 2020. Available online: https://apps.who.int/iris/handle/10665/330745 (accessed on 30 January 2021).
- Rastogi, S.; Pandey, M.M.; Kumar, A.; Rawat, S. Medicinal plants of the genus Betula—traditional uses and a phytochemical–pharmacological review. J. Ethnopharmacol. 2015, 159, 62–83. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ji, M.-Y.; Qiu, B.; Li, Q.-Y.; Zhang, K.-Y.; Liu, J.-C.; Dang, L.-S.; Li, M.-H. Phytochemicals and biological activities of species from the genus Maytenus. Med. Chem. Res. 2020, 29, 575–606. [Google Scholar] [CrossRef]
- Tolmacheva, I.A.; Shelepen’Kina, L.N.; Vikharev, Y.B.; Anikina, L.V.; Grishko, V.V.; Tolstikov, A.G. Synthesis and biological activity of S-containing betulin derivatives. Chem. Nat. Compd. 2005, 41, 701–705. [Google Scholar] [CrossRef]
- Kazakova, O.; Medvedeva, N.; Tolstikov, G.A.; Kukovinets, O.S.; Yamansarov, E.Y.; Spirikhin, L.V.; Gubaidullin, A.T. Synthesis of terminal acetylenes using POCl3 in pyridine as applied to natural triterpenoids. Mendeleev Commun. 2010, 20, 234–236. [Google Scholar] [CrossRef]
- Sidova, V.; Zoufaly, P.; Pokorny, J.; Dzubak, P.; Hajduch, M.; Popa, I.; Urban, M. Cytotoxic conjugates of betulinic acid and substituted triazoles prepared by Huisgen Cycloaddition from 30-azidoderivatives. PLoS ONE 2017, 12, e0171621. [Google Scholar] [CrossRef]
- Chrobak, E.; Bębenek, E.; Kadela-Tomanek, M.; Latocha, M.; Jelsch, C.; Wenger, E.; Boryczka, S. Betulin phosphonates; synthesis, structure, and cytotoxic activity. Molecules 2016, 21, 1123. [Google Scholar] [CrossRef]
- Spivak, A.Y.; Keiser, J.; Vargas, M.; Gubaidullin, R.R.; Nedopekina, D.A.; Shakurova, E.R.; Khalitova, R.R.; Odinokov, V.N. Synthesis and activity of new triphenylphosphonium derivatives of betulin and betulinic acid against Schistosoma mansoni in vitro and in vivo. Bioorganic Med. Chem. 2014, 22, 6297–6304. [Google Scholar] [CrossRef]
- Chrobak, E.; Marciniec, K.; Dąbrowska, A.; Pęcak, P.; Bębenek, E.; Kadela-Tomanek, M.; Bak, A.; Jastrzębska, M.; Boryczka, S. New phosphorus analogs of bevirimat: Synthesis, evaluation of anti-HIV-1 activity and molecular docking study. Int. J. Mol. Sci. 2019, 20, 5209. [Google Scholar] [CrossRef]
- Talele, T.T. Acetylene group, friend or foe in medicinal chemistry. J. Med. Chem. 2020, 63, 5625–5663. [Google Scholar] [CrossRef]
- DrugBank. Available online: https://go.drugbank.com/drugs/ (accessed on 15 September 2020).
- Csuk, R.; Barthel, A.; Sczepek, R.; Siewert, B.; Schwarz, S. Synthesis, encapsulation and antitumor activity of new betulin derivatives. Arch. Pharm. Chem. Life Sci. 2011, 1, 37–49. [Google Scholar] [CrossRef]
- Bi, Y.; Xu, J.; Wu, X.; Ye, W.; Yuan, S.; Zhang, L. Synthesis and cytotoxic activity of 17-carboxylic acid modified 23-hydroxy betulinic acid ester derivatives. Bioorganic Med. Chem. Lett. 2007, 17, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Yamansarov, E.Y.; Saltykova, I.V.; Kovalev, S.V.; Petrov, R.A.; Shkil’, D.O.; Seleznev, E.I.; Beloglazkina, E.K.; Majouga, A.G. Synthesis and cytotoxicity of new alkyne derivatives of pentacyclic triterpenoids. Russ. Chem. Bull. 2019, 68, 855–861. [Google Scholar] [CrossRef]
- Marciniec, K.; Pawełczak, B.; Latocha, M.; Skrzypek, L.; Maciążek-Jurczyk, M.; Boryczka, S. Synthesis, anti-breast cancer activity, and molecular docking study of a new group of acetylenic quinolinesulfonamide derivatives. Molecules 2017, 22, 300. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Latocha, M.; Boryczka, S.; Kurczab, R. Synthesis, molecular docking study, and evaluation of the antiproliferative action of a new group of propargylthio- and propargylselenoquinolines. Med. Chem. Res. 2014, 23, 3468–3477. [Google Scholar] [CrossRef]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kempińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, structure and cytotoxic activity of new acetylenic derivatives of betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Kadela-Tomanek, M.; Jastrzębska, M.; Pawełczak, B.; Bębenek, E.; Chrobak, E.; Latocha, M.; Książek, M.; Kusz, J.; Boryczka, S. Alkynyloxy derivatives of 5,8-quinolinedione: Synthesis, in vitro cytotoxicity studies and computational molecular modeling with NAD(P)H: Quinone oxidoreductase 1. Eur. J. Med. Chem. 2017, 126, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Chrobak, E.; Kadela-Tomanek, M.; Bębenek, E.; Marciniec, K.; Wietrzyk, J.; Trynda, J.; Pawełczak, B.; Kusz, J.; Kasperczyk, J.; Chodurek, E.; et al. New phosphate derivatives of betulin as anticancer agents: Synthesis, crystal structure, and molecular docking study. Bioorganic Chem. 2019, 87, 613–628. [Google Scholar] [CrossRef]
- Evers, M.; Poujade, C.; Soler, F.; Ribeill, Y.; James, C.; Lelièvre, Y.; Gueguen, J.-C.; Reisdorf, D.; Morize, I.; Pauwels, R.; et al. Betulinic acid derivatives: A new class of human immunodeficiency virus type 1 specific inhibitors with a new mode of action. J. Med. Chem. 1996, 39, 1056–1068. [Google Scholar] [CrossRef]
- Lan, P.; Wang, J.; Zhang, D.-M.; Shu, C.; Cao, H.-H.; Sunab, P.; Wu, X.-M.; Ye, W.-C.; Chen, W.-M. Synthesis and antiproliferative evaluation of 23-hydroxybetulinic acid derivatives. Eur. J. Med. Chem. 2011, 46, 2490–2502. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis; John Wiley & Sons: New Jersey, NJ, USA, 2007; pp. 230–232. [Google Scholar]
- Neises, B.; Steglich, W. Esterification of carboxylic acids with dicyclohexylcarbodiimide/4-Dimethylaminopyridine:tert-butyl ethyl fumarate. Org. Synth. 1985, 63, 183. [Google Scholar] [CrossRef]
- Lin-Vien, D.; Fateley, W.G.; Colthup, N.B.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: Cambridge, MA, USA, 1991. [Google Scholar]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies, 3rd ed.; John Wiley & Sons, LTD.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Csuk, R.; Barthel, A.; Schwarz, S.; Kommera, H.; Paschke, R. Synthesis and biological evaluation of antitumor-active γ-butyrolactone substituted betulin derivatives. Bioorganic Med. Chem. 2010, 18, 2549–2558. [Google Scholar] [CrossRef] [PubMed]
- Bębenek, E.; Jastrzębska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolińska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel triazole hybrids of betulin: Synthesis and biological activity profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Greene, L.M.; Keely, N.O.; Wang, S.; Cotter, T.S.; Zisterer, D.M.; Meegan, M.J. Synthesis and biochemical activities of antiproliferative amino acid and phosphate derivatives of microtubule-disrupting β-lactam combretastatins. Eur. J. Med. Chem. 2013, 62, 705–721. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Zhang, H.; Qingbin Kong, Q.; Wang, J.; Jiang, Y. Complex roles of the old drug aspirin in cancer chemoprevention and therapy. Med. Res. Rev. 2019, 39, 114–145. [Google Scholar] [CrossRef] [PubMed]
- Pisha, E.; Chai, H.; Lee, I.-S.; Chagwedera, T.E.; Farnsworth, N.R.; Cordell, G.A.; Beecher, C.W.; Fong, H.H.; Kinghorn, A.D.; Brown, D.M.; et al. Discovery of betulinic acid as a selective inhibitor of human melanoma that functions by induction of apoptosis. Nat. Med. 1995, 1, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S.; Jeremias, I.; Steiner, H.H.; Pietsch, T.; Debatin, K.-M. Betulinic acid: A new cytotoxic agent against malignant brain-tumor cells. Int. J. Cancer 1999, 82, 435–441. [Google Scholar] [CrossRef]
- Prokhorova, E.A.; Zamaraev, A.V.; Kopeina, G.S.; Zhivotovsky, B.; Lavrik, I.N. Role of the nucleus in apoptosis: Signaling and execution. Cell. Mol. Life Sci. 2015, 72, 4593–4612. [Google Scholar] [CrossRef]
- Hemann, M.T.; Lowe, S.W. The p53–Bcl-2 connection. Cell Death Differ. 2006, 13, 1256–1259. [Google Scholar] [CrossRef]
- Polo, S.E.; Almouzni, G. Histone metabolic pathways and chromatin assembly factors as proliferation markers. Cancer Lett. 2005, 220, 1–9. [Google Scholar] [CrossRef]
- Orchel, J.; Slowinski, J.; Mazurek, U.; Wilczok, T. H3 mRNA level as a new proliferative marker in astrocytomas. Biochim Biophys Acta 2004, 1689, 42–46. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2012, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yip, K.W.; Reed, J.C. Bcl-2 family proteins and cancer. Oncogene 2008, 27, 6398–6406. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Inglot, T.; Gumieniczek, A.; Komsta, Ł.; Kasińska, A. LC and principal component analysis in the lipophilicity study of seven angiotensin II-AT1 receptor antagonists (sartans). Chromatographia 2008, 68, 977–983. [Google Scholar] [CrossRef]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—design and description. J. Comput. Aided Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar] [CrossRef] [PubMed]
- ACD Labs. Available online: https://www.acdlabs.com (accessed on 21 August 2019).
- Schyman, P.; Liu, R.; Desai, V.; Wallqvist, A. vNN web server for ADMET predictions. Front. Pharmacol. 2017, 8, 889. [Google Scholar] [CrossRef]
- Dincela, E.D.; Gürsoya, E.; Yilmaz-Ozdenb, T.; Ulusoy-Güzeldemirci, N. Antioxidant activity of novel imidazo[2,1-b]thiazole derivatives: Design, synthesis, biological evaluation, molecular docking study and in silico ADME prediction. Bioorganic Chem. 2020, 103, 104220. [Google Scholar] [CrossRef]
- Desai, V.; Bhushan, A. Natural bioactive compounds: Alternative approach to the treatment of glioblastoma multiforme Hindawi. BioMed Res. Int. 2017, 2017, 1–10. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Chowdhury, F.A.; Hossain, K.; Mostofa, A.; Akbor, M.M.; Bin Sayeed, M.S. Therapeutic potential of thymoquinone in glioblastoma treatment: Targeting major gliomagenesis signaling pathways. BioMed Res. Int. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.C.; Nissink, J.W.M.; Taylor, R. Protein-ligand docking and virtual screening with GOLD in virtual screening in drug discovery. In Virtual Screening in Drug Discovery; Shoichet, B., Alvarez, J., Eds.; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Jiménez, J.; Škalič, M.; Martínez-Rosell, G.; De Fabritiis, G. KDEEP: Protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model. 2018, 58, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Abdelatef, S.A.; El-Saadi, M.T.; Amin, N.H.; Abdelazeem, A.H.; Omar, H.A.; Abdellatif, K.R. Design, synthesis and anticancer evaluation of novel spirobenzo[h]chromene and spirochromane derivatives with dual EGFR and B-RAF inhibitory activities. Eur. J. Med. Chem. 2018, 150, 567–578. [Google Scholar] [CrossRef]
- Beveridge, D.L.; DiCapua, F.M. Free Energy Via Molecular Simulation: Applications to Chemical and Biomolecular Systems. Annu. Rev. Biophys. Biophys. Chem. 1989, 18, 431–492. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef]
- Bai, Q.; Tan, S.; Xu, T.; Liu, H.; Huang, J.; Yao, X. MolAICal: A soft tool for 3D drug design of protein targets by artificial intelligence and classical algorithm. Briefings Bioinform. 2020. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef]
- Chrobak, E.; Bębenek, E.; Marciniec, K.; Kadela-Tomanek, M.; Siudak, S.; Latocha, M.; Boryczka, S. New 30-substituted derivatives of pentacyclic triterpenes: Preparation, biological activity, and molecular docking study. J. Mol. Struct. 2021, 1226, 129394. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis PRO; Agilent Technologies Ltd: Yarnton, UK, 2014.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Bębenek, E.; Bober-Majnusz, K.; Siudak, S.; Chrobak, E.; Kadela-Tomanek, M.; Wietrzyk, J.; Boryczka, S. Application of TLC to Evaluate the Lipophilicity of Newly Synthesized Betulin Derivatives. J. Chromatogr. Sci. 2020, 58, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision A. 03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Dassault Systèmes. BIOVIA Discovery Studio 2020; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Ribeiro, J.V.; Bernardi, R.C.; Rudack, T.; Stone, J.E.; Phillips, J.C.; Freddolino, P.L.; Schulten, K. QwikMD—integrative molecular dynamics toolkit for novices and experts. Sci. Rep. 2016, 6, 26536. [Google Scholar] [CrossRef] [PubMed]




















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).