Abstract
Epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) are two extensively studied membrane-bound receptor tyrosine kinase proteins that are frequently overexpressed in many cancers. As a result, these receptor families constitute attractive targets for imaging and therapeutic applications in the detection and treatment of cancer. This review explores the dynamic structure and structure-function relationships of these two growth factor receptors and their significance as it relates to theranostics of cancer, followed by some of the common inhibition modalities frequently employed to target EGFR and VEGFR, such as tyrosine kinase inhibitors (TKIs), antibodies, nanobodies, and peptides. A summary of the recent advances in molecular imaging techniques, including positron emission tomography (PET), single-photon emission computerized tomography (SPECT), computed tomography (CT), magnetic resonance imaging (MRI), and optical imaging (OI), and in particular, near-IR fluorescence imaging using tetrapyrrolic-based fluorophores, concludes this review.
1. Introduction
EGFR and VEGFR overexpression is frequently found in several types of tumors, including breast, lung, colon, and ovarian, and therefore it is a very attractive therapeutic and imaging target for cancer treatment research [1,2]. The complete detection of tumors, as well as tumor infiltrative areas and tumor metastasis, has an enormous impact on treatment planning, tumor response to treatment, and on overall treatment outcomes, and significantly increases the success of cancer treatment and the patient’s quality of life. Since molecular imaging allows noninvasive and repetitive imaging of dynamic processes, it has advantages over other conventional detection techniques that use biopsies and surgical procedures for cancer diagnosis, surgical guidance, and treatment monitoring. Therefore, molecular imaging plays a pivotal role in medicine, especially in the field of cancer diagnosis and treatment, as it provides accurate information regarding the stage and location of cancer by visualizing tumor properties at an early stage, evaluating therapeutic targets, and monitoring treatment outcomes. The most common molecular imaging modalities currently employed include positron emission tomography (PET), single-photon emission computerized tomography (SPECT), computed tomography (CT), magnetic resonance imaging (MRI), ultrasound imaging (US), and optical imaging (OI). In this review, we will summarize the contrast agents used in common molecular imaging modalities that specifically target the EGFR and VEGFR receptor families. We will focus on optical imaging methodologies, particularly the use of fluorescence imaging, as this technique provides superior sensitivity and resolution even at the subcellular level, giving real-time information on tumor cell properties and location. In addition, optical imaging techniques have become essential tools in the fundamental study of small animal models and in drug development.
The use of fluorescent dyes conjugated to biomolecules that are selective for over-expressed cell receptors, such as EGFR and VEGFR, has many advantages, as it enables visualization and detection of primary and metastatic lesions as well as fluorescence-guided surgery. Photostable dyes and nanoparticles with emissions within the near-IR region (600–900 nm) are particularly attractive since near-IR fluorescence displays low Raman scattering cross sections associated with the use of low energy excitation photons, larger Raman-free observation windows, and reduced absorption and fluorescence from other molecules. Furthermore, light penetration through human tissue typically increases with the wavelength. Therefore, conjugation of fluorescent dyes to antibodies or to peptide sequences directed at EGFR and VEGFR receptors, as well as to tyrosine kinase inhibitors (TKIs), is an effective strategy currently used in medical imaging and cancer treatment.
It is of great importance to enhance our understanding of the structure and structure-function relationship of the targeted receptors in order to design and create effective drug targets. To this end, we will first discuss the structure of the EGFR and VEGFR receptor tyrosine kinase (RTK) domains and their relevant function in kinase activity. We also review currently used imaging modalities for EGFR and VEGFR family proteins and currently used drugs for EGFR and VEGFR targeting and inhibition, with emphasis on low molecular weight molecules including peptides, peptoids, and TKIs.
Receptor Tyrosine Kinases (RTKs)
The human protein kinase family is one of the largest gene families owing to their regulatory role in virtually every facet of cell biology [3]. Protein kinase enzymes catalyze the transphosphorylation of hydroxyl-containing substrates (typically proteins) via the conversion of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) facilitated by a divalent cation (typically Mg2+) as depicted in the following substitution reaction centered on phosphorus:
MgATP− + protein-O:H → protein-O:PO32− + MgADP + H+, where the γ-phosphoryl group (PO32−), as opposed to a phosphate group (OPO32−), is transferred from ATP to the substrate protein [4]. The transfer of the phosphoryl group depicts the initiation of intracellular signaling cascades to transduce the signal from the cell surface, through intracellular vesicles, to the nucleus in order to activate gene expression. Of the 518 identified protein kinase family members (2% of all human genes), 385 are serine/threonine protein kinases, 90 are protein-tyrosine kinases, and 43 are tyrosine-kinase like proteins. Of the 90 protein-tyrosine kinases, 58 are transmembrane receptors (containing extracellular, transmembrane, and intracellular domains), and 32 are intracellular non-receptors [4]. The human epidermal growth factor receptor (HER)/erythroblastic leukemia viral oncogene homolog (ErbB) and the vascular endothelial growth factor (VEGF) RTKs are among the most extensively studied cell signaling families in biology.
2. EGFR Family of Receptor Tyrosine Kinases (RTKs)
2.1. EGFR/ErbB/HER
The EGFR/ErbB/HER family of RTKs consists of four homologous transmembrane proteins that are ubiquitously expressed in epithelial, cardiac, neuronal and mesenchymal cells: (1) ErbB1, also referred to as epidermal growth factor receptor (EGFR) or human epidermal growth factor receptor 1 (HER1), (2) ErbB2/HER2/Neu, (3) ErbB3/HER3, and (4) ErbB4/HER4 [4]. The ErbB nomenclature is derived from the name of avian viral erythroblastosis oncogene to which EGFR is related. The epidermal growth factor (EGF) was first discovered in 1962 [5], followed by its receptor (EGFR) in 1978 [6], by the late Stanley Cohen was the first receptor to be characterized as a protein-tyrosine kinase. The ErbB/HER family exerts critical functions in cell physiology through canonical signaling pathways to regulate transcription, differentiation, proliferation, cell cycle progression, and apoptosis [2]. Noncanonical signaling pathways, generally induced by external factors (e.g., xenobiotics) or cellular and environmental stresses, are consequently activated in cancer cells conferring upon them a survival advantage and therapeutic resistance via induction of anti-apoptotic effects, aberrant angiogenesis, and initiation of metastatic growth, and promotion of tumor cell proliferation and growth [2].
2.2. Structure of EGFR
Each ErbB family member protein consists of a large glycosylated N-terminal extracellular domain (ECD) consisting of ~620 amino acids with a ligand-binding region [7]. ECD is subdivided into four domains (I–IV), where domains I and II have a beta-helix/solenoid structure, and domains II and IV are cysteine-rich domains that contain disulfide modules [4,8,9,10,11,12]. A single anchoring hydrophobic transmembrane domain (TMD) links the extracellular region to the cytoplasmic region via a juxtamembrane domain (JMD). Detailed structural analysis suggests two juxtamembrane regions, (a) an extracellular juxtamembrane, and (b) an intracellular cytosolic juxtamembrane. The extracellular juxtamembrane (eJM) has a short stretch of seven amino acids that links the TMD to the C-terminal domain of IV of ECD, while the intracellular juxtamembrane (iJM) separates the TMD from the kinase domain (Figure 1). The intracellular regions consist of nearly 540 amino acids that consist of a tyrosine kinase domain (TKD) and a carboxyterminal tail (CTT) of ~230 amino acids [4,13]. The C-terminal carboxy tail contains tyrosine amino acids that are important in phosphorylation and its structure is not yet well defined due to the flexibility of the structure.
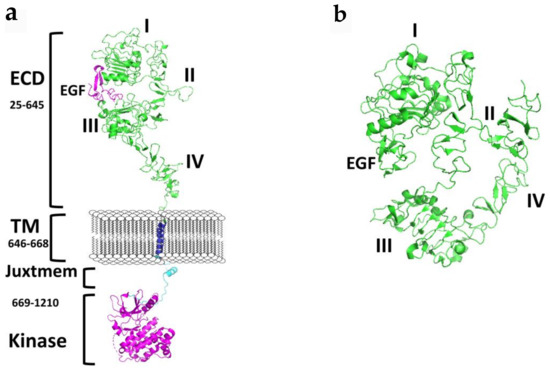
Figure 1.
A schematic diagram of the structure EGFR. (a) Full-length EGFR showing extracellular domain (ECD) in an open conformation, transmembrane (TM), and the cytoplasmic kinase domain. Published crystal structures of EGFR were used to prepare the schematic. ECD (PDB ID: 3NJP); transmembrane (PDB ID:2KS1), Kinase domain with juxtamembrane domain (PDB ID: 3GOP). Notice the distance between domains II and IV in an open conformation. Dimerization arm II is open for dimerization interaction. (b) Structure of ECD of EGFR in the closed conformation (PDB ID: 1NQL). Domains IV is folded and interacts with II. In the closed conformation EGFR dimerization arm II is not available for dimerization. PyMol (Schrodinger LLC. OR) was used to generate the structures of EGFR. ECD, extracellular domain; eJM, extracellualr juxtamembrane; TMD, transmembrane domain; iJM, intracellular juxtamembrane; TKD, tyrosine kinase domain [11,14,15,16].
2.3. Epidermal Growth Factor (EGF) Ligand Family
Eleven homologous growth factor ligands that activate ErbB receptors have been identified and can be categorized into three classes: (1) high-affinity ligands, (2) low-affinity ligands, and (3) neuregulins [17]. Structurally, all of the EGF-family members contain a central B-sheet hairpin surrounded by three highly conserved intramolecular disulfide bridges that form tightly coiled structural loops essential for receptor binding [17]. EGFR ligands are shown in Table 1.

Table 1.
EGFR Ligands and their Classification.
It has been shown that seven of the EGF ligands can stimulate different downstream signaling effects: EGF, HB-EGF, and BTC promote receptor downregulation and a shorter signaling pulse, in contrast to TGF-a, AREG, EREG, and EPGN, which promote receptor recycling and a sustained signal [17].
2.4. Importance of EGFR Dimerization in Cell Signaling and Cancer
Receptor tyrosine kinases of the EGFR family control the cell growth and proliferation in normal cells, and any dysregulation of this process leads to cancer [18]. The binding of ligands to the extracellular domain initiates the signaling process that changes the conformation of ECD of EGFR, leading to homo- and heterodimers (with other members of EGFR), resulting in passing the signal from outside the cells to inside the cell by phosphorylation of the kinase domain. This process is facilitated by a change in the structure of EGFR that helps dimerization. Details of the extra- and intracellular domains structure have provided insights into the signal transduction process. However, most of the models proposed are based on crystal or NMR structures of individual domains as an elucidation of the entire intact protein receptor is not feasible. In the inactive state, EGFR is known to be in a “closed” conformation where there is a large gap between domains I and III, and domain IV is folded in such a way to “tether” to domain II [17] (Figure 1b). In this conformation, EGFR cannot form dimers as dimerization arm II and IV are not able to interact with other receptors. Upon binding of the ligand to ECD between domains I and III, a conformation change occurs, resulting in the opening of the tethered conformation where domains II and IV move far away from each other (Figure 1a). Domains II and IV of two EGFR molecules come into contact with one another, forming a dimer. It is known that among the EGFR family of receptors, EGFR, HER3, and HER4 have known ligands and are known to exist in an open and closed conformation. Although the overall structure of HER2 is similar to other EGFRs, HER2 does not have a known ligand and is always in the open conformation [19]. Thus, HER2 is the preferred dimerization partner for other EGFRs. Dimerization of ECD activates the intracellular kinase domains of the receptor resulting in autophosphorylation of a ~230 residue C-terminal tail and initiation of downstream signaling. Transmembrane domain and juxtamembrane domains are known to participate in the dimerization process that helps kinase activation [8,9].
On the cytoplasmic side, the kinase domains form an asymmetric dimer in which one kinase acts as an activator of the other dimer partner by transphosphorylation. For details of structural aspects of this process, readers can refer to reviews [13,16,20,21]. The C-lobe of one kinase (the ‘activator’) interacts with the N-lobe of another kinase (the “receiver”). The carboxyl-terminal tail of EGFR has autophosphorylation sites and acts as a binding region to SH2 domains. The tyrosine residues phosphorylated by EGF’s addition to cells include Y703, Y920, Y992, Y1045, Y1068, Y1086, Y1148, and Y1173. In addition to these autophosphorylated sites, there are also residues that are phosphorylated by other kinases, which interestingly appear downstream in the EGFR-activation cascade. For example, Y845 is phosphorylated by c-SRC [17], and T654 is phosphorylated by PKC [15]. EGFRs work together in a concerted manner to generate the signaling for cell growth. HER2 does not have a known ligand and hence is always ready to partner with EGFR/HER2/HER3. On the other hand, HER3 is known to have weak enzyme activity and hence does not participate in phosphorylation activity significantly. However, with EGFR, HER2, and HER4, HER3 dimerizes and participates in phosphorylation activity and signaling.
2.5. The Tyrosine Kinase Domain (TKD)
The homologous kinase domains of eukarytoic protein kinases (ePK) are responsible for catalytic activity with three distinct functions: (1) binding and proper orientation of the ATP phosphoryl donor-divalent cation complex, (2) binding and proper orientation of the phosphoryl acceptor protein substrate, and (3) transfer of the γ-phosphate from ATP to the hydroxyl or phenol containing residue (Ser, Thr or Tyr) of the protein substrate [22]. The ErbB/HER tyrosine kinase domain (TKD) is comprised of about 270 amino acids and, similar to all other protein kinases, has a small N-terminal lobe and a large C-terminal lobe [4]. Activation of the TKD is essential for cellular responses to ligand binding and is achieved via the juxtaposition of two monomer catalytic kinase domains to form an asymmetric dimer, where the donor TKD C-lobe abuts the acceptor TKD N-lobe. This leads to the trans-autophosphorylation of critical tyrosine residues in the C-terminal tails and triggers the signaling cascade [16].
Taking a closer look at the ATP binding pocket, the space between the N- and C-terminal lobes (Figure 2) can be subdivided into the front cleft, gate area, and back cleft [4]. The gate area and back cleft comprise hydrophobic pocket II (HPII). The Sh2 residue is termed the gatekeeper due to its ability to control access to the back cleft. The front cleft contains the hinge residues and adenine-binding pocket, the flexible glycine-rich P-loop (named for its proximity to the phosphate groups of the ATP substrate), the catalytic loop motif (HRD(X)4N), and the portion connecting the hinge residues to the αD-helix [4]. The β1- and β2-strands of the N-terminal lobe dock with the ATP adenine moiety. The gate area consists of the β3-strand of the N-terminal lobe and the proximal section of the activation segment, including the DFG (Asp-Phe-Gly) motif. The back-cleft projects to the αC-helix to a portion of the αE-helix within the C-terminal lobe and to the N-terminal lobe segments β4- and β5-strands of the small lobe [4]. One of the hurdles in the development of protein kinase inhibitors is to increase selectivity to reduce unwanted side effects, a process that is facilitated by characterizing drug-kinase interactions [4,23,24].
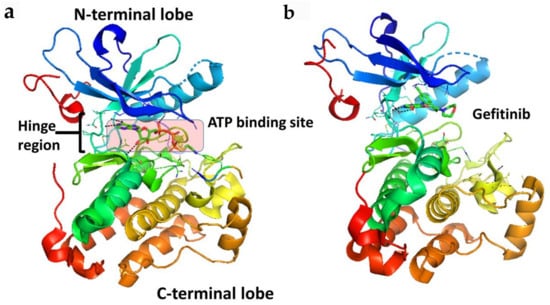
Figure 2.
Crystal structure of the kinase domain of EGFR (a) with ATP binding site highlighted (PDB ID: 2GS6). TKIs of EGFR bind to EGFR in the ATP binding pocket, forming 1 to 3 hydrogen bonds to the hinge region. (b) EGFR kinase domain with gefitinib bound in the ATP binding pocket (PDB ID: 3UG2). PyMol (Schrodinger LLC. OR, New York, NY, USA) was used to generate the figure [33,34].
Although ligand binding is needed for EGFR dimerization and activity in normal as well as cancer cells, ligand binding is not necessary in some cancer types. Mutation or overexpression of receptor proteins can lead to dimerization of receptors. Most of the proposed models of the mechanism of EGFR activation involve monomer to dimer transition. However, the existence of higher-order multimers was suggested in cells [25,26]. Using molecular modeling and single-molecule tracking studies it was shown that higher-order multimers are proposed [27,28,29]. Single-molecule tracking studies of EGFR in live cells shown that EGFR forms large clusters after activation [10,30,31,32].
3. VEGFR Family of Receptor Tyrosine Kinases (RTKs)
3.1. VEGFR
The vascular endothelial growth factor (VEGF) family of RTKs is comprised of three structurally similar transmembrane proteins: (1) VEGF receptor-1 (VEGFR-1), first called feline McDonough sarcoma (fms)-related tyrosine kinase-1 (Flt-1), (2) VEGFR-2, also known as kinase insert domain receptor (KDR) or the murine homolog fetal liver kinase-1 (Flk-1), and (3) VEGFR-3, or fms-related tyrosine kinase-4 (Flt-4) [1]. VEGFRs, initially reported to be expressed solely on endothelial cells, are found to be expressed on both endothelial and non-endothelial cells, including tumor cells1. These receptors, along with their ligands and co-receptors neuropilin-1 (NRP-1), neuropilin-2 (NRP-2), and the heparan sulfate proteoglycans (HSPGs) (glycoproteins containing glycosaminoglycan (GAG) chains(s)), mediate critical interactions in vasculogenesis, lymphangiogenesis, and angiogenesis [1]. Vasculogenesis is the de novo formation of blood vessels that typically occurs during embryonic development versus angiogenesis, which is the sprouting and splitting of pre-existing vasculator to form new blood vessels. The flux of vascular networks is mediated by local and systemic tissue demands, thus, angiogenesis aids in the repair of tissue (would healing) and in the general growth and maintenance of the organism [35]. Lymphangiogenesis, as the name suggests, involves the formation of new lymph vessels from pre-existing lymphatics.
As the diffusion limit of oxygen in mammalian tissue is around 100–200 μM, surrounding metabolically active tissue outside that range can become hypoxic without an adequate supply of blood and thus, oxygen [36]. Reflecting the extent to which oxygen can diffuse through tissue, in the absence of neo-vascularization, solid tumor growth is limited to about 0.2–2.0 mm in diameter lest it becomes hypoxic [36]. The complex and tightly regulated process of angiogenesis is dependent upon 30 pro- and 30 anti-angiogenic factors held in balance. A shift in the balance of these factors favoring the increase of pro-angiogenic factors, called an angiogenic switch, is critical in the malignant growth and progression of solid tumors [1].
3.2. Structure of VEGFR
Members of the VEGFR family typically consist of an extracellular ligand-binding domain (ECD) with a seven immunoglobulin (Ig)-like motif (with connecting linkers), a single transmembrane domain, and an intracellular region containing a juxtamembrane domain (JMD) and a tyrosine kinase domain (TKD) split by a kinase insert and a carboxyl terminus (Figure 3). VEGFRs may induce intracellular signal transduction as homo- and/or heterodimers with different phosphorylation patterns occuring in heterodimers compared to homodimers.
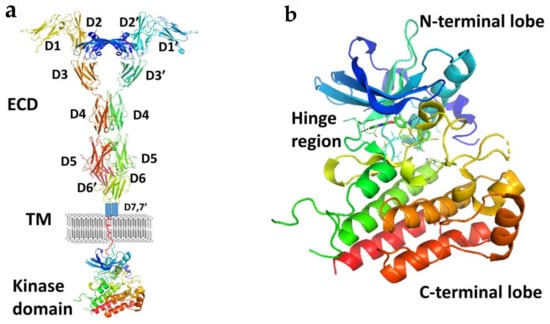
Figure 3.
(a) A schematic representation of full length VEGFR-1 with VEGF-A. Extracellular domains D1 to D6 are from the crystal structure of VEGFR-1 (PDB ID: 5T89). D7 and TM are shown in schematic. Kinase domain was from the crystal structure of VEGF (PDB ID: 3VHE). (b) Kinase domain of VEGF (PDB ID: 3VHE) showing hinge region and N and C-terminal lobes. PyMol (Schrodinger LLC. OR) was used to generate the figure [37,38].
VEGFRs are activated upon ligand-mediated dimerization. It is suggested that ligand-induced receptor dimers are stabilized by both ligand-receptor as well as homotypic receptor-receptor interactions. Ligand binding in the extracelullar domain induces transmembrane signaling that results in cross-phosphorylation in the intracellular kinase domains. It is interesting to note that while VEGFR1 and VEGFR2 share 44% homology, they differ vastly with respect to their structure and function. For example VEGF-A binds to VEGFR1 and VEGFR2 but does so via different domains (Ig domain 2 and 3, respectively) of the VEGF-A ligand. Thus, mutations that disrupt the binding of VEGF-A with VEGFR1 may not necessarily affect the binding with VEGFR2 and vice versa.
The neuropilins (NRPs) are 120–140 kDa transmembrane non-protein-tyrosine kinase glycoproteins that act as co-receptors for both the semaphorin family and the VEGF family. Neuropilins contain large (~250 kDa) transmembrane plexins that transduce semaphorin signaling as co-receptors with VEGFR1, VEGFR2, and VEGFR3 that transduce VEGF family signaling. Neuropilins also function as receptors for VEGF isoforms independently of VEGFR1, VEGFR2, or VEGFR3. NRPs are pleiotropic receptors, and therefore, other molecules may interfere with the signaling of the NRP-VEGF receptor complexes. They consist of a large extracellular domain, a transmembrane segment, and an intracellular domain composed of approximately ~40 amino acid residues, which are relatively short to perform a catalytic function. Therefore, it is possible that the intracellular domain serves as a docking site for downstream molecules, either alone or in conjunction with co-receptors.
3.3. Vascular Endothelial Growth Factor (VEGF) Ligand Family
There are seven VEGF family members that have been identified thus far that specifically interact with VEGFRs 1–3, neuropilin-1 and -2, and heparin sulfate proteoglycan co-receptors (see Table 2): vascular endothelial growth factor A (VEGF-A), VEGF-B, VEGF-C, VEGF-D, viral homolog VEGF-E, snake venom homolog VEGF-F, and placenta growth factor (PlGF). Generally, these ligands are structurally similar disulfide-linked homodimeric glycoproteins that can undergo alternative splicing to express biologically relevant isoforms, namely VEGF-A and VEGF-B (i.e., VEGF-A121, VEGF-A145, VEGF-A165, VEGF-A189, VEGF-A206, and VEGF-B167 and VEGF-B186).
Table 2.
VEGF ligands and their receptor binding partner(s).
Produced predominantly by endothelial, stromal, and hematopoietic cells, VEGF glycoproteins are secreted upon stimulation by growth factor [i.e., transforming growth factor β (TGF-β), interleukins, and platelet-derived growth factors (PDGFs)]. Originally identified as vascular permeability factor (VPF), the ~35 kDa homodimeric glycoprotein VEGF-A is the most potent/prevalent angiogenic growth factor/protein influencing angiogenesis.
4. Targeting EGFR and VEGFR with Different Inhibition Modalities
4.1. Tyrosine Kinase Inhibitors (TKIs)
Many biologically and pharmacologically significant drugs are heterocyclic compounds that are also highly relevant in medicinal chemistry. In particular, quinazoline-based compounds are an important class of heterocyclic pharmacophores that have been shown to possess a wide range of biological activities, including analgesic, anti-inflammatory, anti-hypertensive, anti-cancer, antimicrobial, antibacterial, antifungal, anti-HIV, anti-malarial, and anti-viral properties [40]. The quinazoline scaffold is considered a privileged structure and serves as the framework core for several small-molecule tyrosine kinase inhibitors (TKIs), as shown in Figure 4. The quinazoline structure itself is a mancude heterobicyclic planar molecule containing a benzene ring fused to pyrimidine via two adjacent carbon atoms (a.k.a., 1,3-diazanaphthalene). The total synthesis and antitumor activity of 4-aminoquinazolines have been recently reviewed [41].
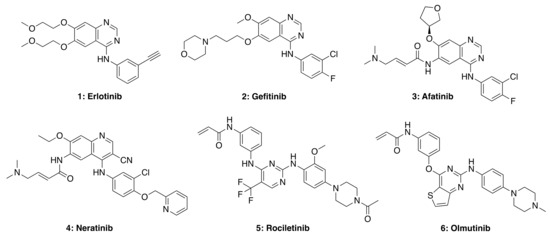
Figure 4.
Representative structures of first (1 and 2), second (3 and 4), third (5), and fourth (6) generation EGFR TKIs. Structures were generated using Chemdraw based on structures of TKIs available in selleckchem.com.
Small molecule TKIs function by preventing phosphorylation through the obstruction of protein kinase activity via (1) competitive binding with ATP, (2) competitive binding with the substrate, (3) competitive binding with both ATP and substrate, or (4) allosteric inhibition (e.g., affecting protein conformational change) [42,43]. TKIs are typically utilized in targeted therapy, selectively identifying and attacking cells that express mutations or overexpress tyrosine kinase receptors. First-generation TKIs, such as Erlotinib (1) and Gefitinib (2), are reversible inhibitors that compete for binding to the kinase domain with endogenous ATP, therefore preventing kinase phosphorylation, blocking downstream signaling and cell regulation. Both these drugs are FDA-approved for the treatment of non-small cell lung carcinoma (NSCLC). However, due to acquired patient resistance to these drugs, second-, third- and fourth-generation TKIs were developed. Afatinib (3) and Neratinib (4) are second-generation TKIs that bind irreversibly to cysteine residues in the kinase domain; such stronger interaction with the ATP binding site can also cause side-effects in patients treated with these second-generation TKIs, including skin rashes and diarrhea. Third- and fourth-generation TKIs, such as rociletinib (5) and olmutinib (6), are selective against certain mutants (e.g., EGFR T790M, and C797S, respectively) but show limited wild-type EGFR inhibition. Therefore these drugs do not show as many side-effects observed in first- and second-generation TKIs. Nevertheless, most third-generation TKIs also develop resistance in patients due to receptor alterations and mutations, while fourth-generation TKIs are currently being investigated for C797S mutation-acquired resistance.
Improved tumor delivery of TKIs by using various formulations and nanoparticle delivery systems have been reported. For example, cyclodextrin-modified PLGA nanoparticles loaded with erlotinib were shown to improve the therapeutic efficacy against non-small cell lung cancer (NSCLC) [44]. Chitosan-based nanoparticles increased the biocompatibility and biodegradability of erlotinib and resulted in enhanced cytotoxicity against A549 lung cancer cells [45]. Superparamagnetic iron oxide nanoparticles conjugated with erlotinib [46] and gold-based nanoparticles bearing erlotinib and doxorubicin [47] have also been reported. Lipid-based nanoparticles have been used to deliver erlotinib [48] and afatinib [49] and are shown to have enhanced therapeutic efficacy against NSCLC. Similarly, hyaluronic acid-based nanoparticles have been explored for the delivery of erlotinib [50] and afatinib [51] to target tumors.
4.2. EGFR Antibodies, Nanobodies, and Small Peptide Ligands
As mentioned in the introduction, EGFR alteration such as mutation, overexpression, and gene amplification is found in many cancer types such as lung, breast, colon, and rectum as well as head and neck cancer [52,53,54]. Antibodies have been targeted to EGFR to reduce the signaling for cancer cell growth and are used as therapeutic agents [54]. Antibodies trastuzumab and pertuzumab are used in HER2-related breast cancer. Nearly 80% of colorectal tumors overexpress EGFR, and hence EGFR is one of the major targets in colorectal cancer (CRC). Antibodies cetuximab and panitumumab are used as therapeutic agents for metastatic colorectal cancer (mCRC). Approximately 40% of patients develop metastatic disease [55]. However, only 10% of advanced metastatic colorectal cancers respond to EGFR antibody therapy [56,57]. Cetuximab and panitumumab both bind to ECD domain III of EGFR and prevent the binding of EGFR ligands resulting in locking the EGFR in the “closed” or autoinhibitory conformation. This leads to the prevention of dimerization of EGFR and hence downstream signaling. It is postulated that the antibody-receptor complex is then internalized and either degraded or recycled, and this turnover is controlled by the ubiquitin-proteasome system [58,59,60,61].
Detailed binding analysis of cetuximab and panitumumab revealed that both cetuximab and panitumumab compete with EGF for its binding site to bind to EGFR (Figure 5). However, both antibodies have slightly different binding sites, and the binding epitope on EGFR is in proximity. In terms of binding affinity, both have different affinities to bind to EGFR, with panitumumab binding having an 8-fold higher affinity than cetuximab. Panitumumab binds near residues P349, P362 D355, F412, and I438 on EGFR, whereas cetuximab binds near residues Q384, Q408, H409, K443, K465, I467, and S468, as well as F352, D355, and P387. Panitumumab’s binding epitope overlaps with the EGF binding site in two locations (D355 and K443), whereas cetuximab overlaps with EGF’s binding site in five locations (D355, Q408, H409, K443, and S468) [62,63]. In addition to inhibiting the formation of open conformation, these antibodies induce antibody-dependent cellular cytotoxicity in vivo by recruiting immune cells to tumor cells (Figure 5) [64,65].
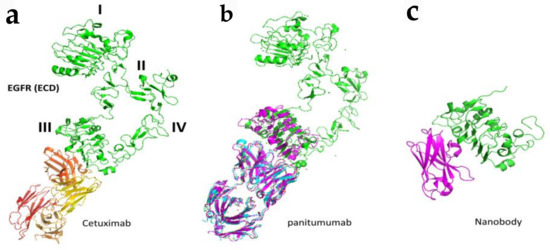
Figure 5.
Binding modes of antibodies and nanobodies to EGFR extracellular domain. (a) Fab region of antibody cetuximab (red and yellow) bound to EGFR (green) domain III of ECD (PDB ID: 1YY9). (b) Panitumumab bound to EGFR ECD at domain III (PDB ID: 5SX4). (c) Crystal structure of nanobody bound to domain III of EGFR ECD. Only domain III is shown (PDB ID: 4KRl). PyMol (Schrodinger LLC. OR) was used to generate the figure [72,73].
Resistance is a common mechanism after treatment with cancer therapeutic agents in most cancers. However, in colorectal cancer, resistance to antibody treatment is not common but found in few cases. The development of resistance was due to acquired mutations in the antibody binding region of EGFR [66,67]. It was found that resistance develops due to the substitution of amino acid arginine at S468 (S468/492R) in vitro as well as in clinical samples [68].
In cetuximab-resistant colorectal tumors, the following somatic mutations are observed that map to the cetuximab epitope on EGFR: G441/465R, G441/465E, and K443/467T. Other mutations, including S440/464L and I467/491M, are also seen in colorectal cancer cell lines. Substitutions at G441/465 and S440/464 have also been observed in panitumumab-resistant colorectal tumors [59,61,66,67,68,69,70]. However, another antibody, necitumumab, is known to bind to EGFR with cetuximab/panitumumab resistance EGFR [24]. Crystal structure of necitumumab Fab (Fab11F8) in complex with isolated domain III from EGFR (sEGFRd3) with S468R substitution (sEGFRd3-S468R) indicated that R substitution does not affect the binding of antibody necitumumab. Antibody matuzumab binds to domain III of ECD of EGFR, but the action of the mechanism is different from cetuximab and panitumumab. It binds to a different epitope on domain III compared to cetuximab and inhibits EGFR by preventing the activating conformational transition [71]. Structural analysis studies of matuzumab: EGFR indicate that the binding of matuzumab to domain III sterically blocks the conformational changes of domain III of EGFR, decreasing the ligand affinity for binding to EGFR. Therefore, it does not competitively bind with other antibodies, such as cetuximab.
Antibodies are effective in binding to specific target receptor molecules, however, they have limitations in terms of delivery, tumor penetration, and production because of their size [73]. The antigen-recognition region in antibodies consists of the variable regions of both heavy (VH) and light chains (VL). An antibody fragment consisting of a single monomeric variable region fragment is capable of binding selectively to an antigen and is called nanobodies. This single Ig domain is stable and can be generated rapidly and cheaply with simple expression systems [74] and are called VHH domains. These VHH domains are being developed for a range of research applications [75]. For therapeutic use, VHH domains (monomeric or multivalent) can be modified to extend serum half-life and/or functionality [76]. Such nanobodies are investigated for EGFR binding [77,78,79,80] (Figure 5).
Several small peptides have been proposed that bind to the EGFR extracellular domain, inspired by the natural ligands and identified from the combinatorial screening of various peptide libraries. Such peptides benefit from their easy and low-cost synthesis, high specificity, and high flexibility regarding their sequence, derivatization, and conjugation possibilities. In particular, two EGFR small peptide ligands, LARLLT (7) designated EGFR-L1 and YHWYGYTPQNVI (8) designated EGFR-L2, have been intensely investigated due to their low immunogenicity, ease of conjugation to various molecules, and superior EGFR-targeting ability [81,82] (Figure 6). The EGFR-L1 peptide was selected from the computational screening of a large peptide library and shown to target EGFR in vitro (using H1299 cells overexpressing EGFR) and in vivo (using H1299 tumor-bearing mice). This peptide was shown to bind specifically to the domain I on the ECD of the EGFR protein, as shown in Figure 7. On the other hand, EGFR-L2 was selected from screening a phage display peptide library and also shown to specifically bind to the EGF binding pocked of EGFR, in vitro (using SMMC-7721 cells) and in vivo (using SMMC-7721 tumor-bearing mice).
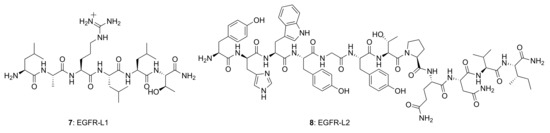
Figure 6.
Structures of EGFR-targeting peptides, EGFR-L1 and EGFR-L2.
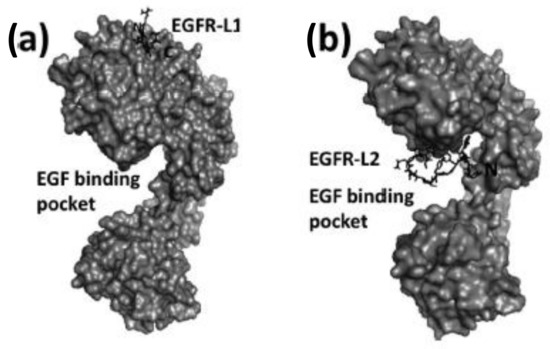
Figure 7.
Structures of peptide ligands on their binding sites: (a) EGFR-L1 in domain I, and (b) EGFR-L2 in the EGF binding pocket (PDB ID: 1NQL) proposed based on docking studies. Peptides are shown as sticks, and EGFR is shown in surface representation. This figure was reprinted with permission from Reference [83].
EGFR-L1 peptide was known to bind domain I of EGFR whereas, EGFR-L2 was known to bind to EGF binding pocket [81,82]. We have used docking methods to investigate the binding of EGFR-L1 and L2 peptides. The binding of these peptides was calculated based on low energy docked structure. The proposed models of binding of these peptides to EGFR ECD are shown in Figure 7 [83]. Docking studies were further confirmed by competitive binding studies and surface plasmon resonance (SPR) analysis. The peptide EGFR-L1 exhibited a β-turn structure upon binding to EGFR. Based on this β-turn structure, we further modified the peptide for its conformational as well as chemical stability and designed a cyclic version of the peptide [84]. The cyclic version of the peptide exhibited a higher affinity to bind to EGFR compared to the linear version of the same peptide.
Recently a derivative of EGFR-L2, bearing a glutamic acid residue in place of glutamine, showed enhanced cellular uptake in EGFR-overexpressing cells as a result of stronger binding to the EGF binding site [85].
Other peptides targeting different domains of EGFR were studied either for inhibition of dimerization or downregulation of phosphorylation. As explained above, EGFR extracellular domain II is important in stabilizing the dimerization. Peptides and small molecules have been targeted to inhibit the domain II dimerization arm. Mizuguchi et al. have described a cyclic peptide with conformational constraints such as a β-turn that inhibits EGFR dimers. The structure–activity relationship of differently designed peptides indicated that retro-inverso sequences of the dimerization arm exhibited antiproliferative activity in A431 cells and inhibited the dimerization and phosphorylation of EGFR [86]. Based on the peptides designed from the dimerization arm, bivalent ligands with optimized linkers (connected by poly(L-proline) or poly[(glycine)4(L-serine)]) were used to target EGFR. These bivalent ligands were proposed to bind to two EGFRs simultaneously and inhibit phosphorylation of the EGFR kinase domain. These bivalent ligands exhibited increased inhibition of phosphorylation compared to the monomeric peptide [87]. Peptides developed from the dimerization arm were also used to target adeno-associated virus (AAV) to EGFR expressing cells [88].
Apart from the extracellular domain, the juxtamembrane region of EGFR (Figure 1) is targeted by peptides to reduce the EGFR signaling for cell growth in cancer cells. A linear peptide was designed first and was shown to inhibit EGFRs activation [89]. Later, a stapled peptide was designed to stabilize the conformation and in vivo stability of the helical peptide. The stapled peptide was shown to be more effective in vivo in cancer models to reduce tumor growth [90]. Furthermore, the substrate binding site of the kinase was also targeted by peptides to EGFR resistant cancer cell lines. Tavakoli et al. [91] designed a library of peptides based on computation work to target mutant EGFR kinase. Such a library of peptides will be helpful to design conjugates of fluorophores to target EGFR. Peptides were also designed to inhibit the interaction of EGFR with EGF. Based on the binding surface of EGFR with EGF, Foy et al. have designed peptides for therapeutic purposes as well as for vaccine development for cancer [92].
Peptidomimetics have been designed and investigated that effectively target the EGFR ECD inhibiting protein-protein interactions and phosphorylation of the kinase domain, therefore modulating the signal for cell growth [93]. Based on the dimerization site of domain IV of extracellular domains of EGFR peptidomimetics that target EGFR dimerization inhibition were designed by our group. The designed peptidomimetics target HER2 protein rather than EGFR. However, the peptidomimetics designed to inhibit both EGFR:HER2 and HER2:HER3 dimerization [94,95,96].
5. Targeting EGFR and VEGFR for Molecular Imaging
Molecular imaging plays a pivotal role in medicine, especially in the field of cancer diagnosis and treatment, as it accurately provides information regarding the stage and location of cancer by visualizing the tumor properties, evaluating therapeutic targets, and monitoring treatment and outcomes [97]. Modern molecular imaging modalities currently in use for the detection of EGFR- and VEGFR-overexpressed cancers include PET, SPECT, CT, MRI, US, and OI (see Table 3). Molecular imaging provides information on receptor status not only at the tumor site but also at the sites where the tumor reaches the vital organs and biopsy is not possible. It is an important tool for early screening and diagnosis of disease, for focused and personalized patient therapy, and for measuring the effectiveness of therapy so that adjustments can be made and treatment based on the patient’s response can be designed. Moreover, it can also prevent false positive or false negative results due to the heterogeneity of receptor expression that occurs in individual biopsy specimens [98].
Table 3.
Imaging modalities and contrast agents are commonly used for EGFR and VEGFR families of receptors.
In order to develop an effective therapeutic agent, the targeting moiety or ligand must be attached with the appropriate labeling agent depending upon the imaging modality employed. For instance, PET and SPECT imaging modalities utilize radionuclides; optical imaging requires fluorescent dyes, quantum dots (QDs), or nanoparticles; MRI uses paramagnetic or superparamagnetic metal oxides; and ultrasound molecular imaging requires microbubbles, which are microspheres filled with perfluorobutane gas; CT utilizes emulsions, liposomes, lipoproteins and polymeric nanoparticles for imaging. The purpose of these imaging techniques is to provide real-time visuals of the receptors, over-expressed on tumor cells, clearly differentiating them from the normal cells to aid in designing novel therapeutics against cancer. Therefore, the high affinity of the targeting moiety must not be compromised even after binding with the labeling agent in order to obtain excellent resolution and quantitative diagnostic accuracy. Moreover, the process of contrast agent production should be cost-effective without compromising its quality.
5.1. Antibody-Based EGFR Imaging Agents
As discussed above, antibodies, nanobodies, and peptides have been targeted at EGFR for therapeutic purposes. Some of these have limitations in terms of the development of resistance and new antibodies that overcome the resistance have been designed using epitope mapping. Since these antibody molecules have high affinity and specificity to bind to EGFR (wild and mutated), antibodies can be labeled with imaging agents as indicated in Table 3. Apart from antibodies to EGFR, EGF-based ligand imaging probes are also designed. Some of these agents have been reviewed by Chen et al. [124]. Antibody-based imaging agents can be classified as whole antibody imaging agents, nanobodies, Fab fragments, and affibodies.
EGF, a natural ligand for EGFR, is labeled with gallium-68 using DOTA chelating agent for PET imaging. Li et al. have used fluorine-18 to label EGF. However, these agents have limitations in terms of imaging due to rapid clearance from the body. Since antibodies have slow clearance from the body, cetuximab (65 to 95 h half-life) was labeled with 111In or 99mTc for SPECT imaging (Table 3). Apart from radiolabeling, fluorescent-based imaging agents were also developed. Cetuximab was labeled with commercially-available IRDye800CW, a cyanine-based near-IR dye. These fluorescent imaging agents are suitable for analysis of tissue layers at the cellular level in vitro, in vivo animal models, as well as in patient tissue samples [125].
Molecular imaging agents developed with full-length monoclonal antibodies have several limitations, including relatively high molecular weight and size, which limits their penetration into tissues in the tumor area. Furthermore, they tend to produce high imaging background and poor imaging quality. To overcome these limitations, Fab, the antigen-binding fragment of an antibody, is used. These fragments have high specificity of the whole IgG but have superior pharmacokinetic and nonimmunogenic properties compared with antibodies. These Fab fragments can be linked to radionucleotides or to fluorescent imaging probes, making them useful imaging agents with the ability to target certain proteins in cells and tissues. Cetuximab Fab was labeled with 111In for imaging purposes using this technology [126,127].
Another class of molecules that are derived from antibodies are designated affibodies, which are mimics of antibodies. The general structure of these molecules consists of three-helical bundles that are derived from the natural receptor for the Fc-portion of IgG. The amino acids in the helical bundles of the two helices of the three can be randomized and large libraries of affibody structures can be generated. Among the several generated sequence of structures, potential binding affibody to a particular receptor can be chosen. Since these molecules are relatively small proteins (~7 kDa), the designed peptides can be made to obtain suitable physicochemical properties for solubility and route of administration. Moreover, these small proteins can be conjugated to different imaging probes [128,129]. Zhao et al. [130] conjugated EGFR-targeting affibody to Ac-Cys label and Alexa680 (Cys-ZEGFR:1907). Affibodies have also been labeled with a fluorine-18 probe and to an iron oxide nanoparticle for PET, optical, and MRI imaging [131,132,133,134,135].
5.2. Antibody-Based Imaging Agents for VEGFR
As mentioned above, VEGF and its receptors play a critical role in the progression of metastasis in many cancers. High expression of VEGFs and their receptors were found in tumor tissue samples [136]. VEGF is also a well-known therapeutic target in CRC. Monoclonal antibodies that are used in the treatment of angiogenesis can be grouped into two categories, those that (a) bind to VEGF and inhibit VEGF receptor interactions, and (b) bind to VEGF receptors and activate the immune response. There is another class of molecule, VEGF-Trap, a fusion protein that consists of VEGFR-1 and VEGFR-2 binding domains and Fc region of IgG1 antibody. This fusion protein binds to VEGF-A, VEGF-B, inhibiting the activation of VEGFR-1 and 2 and hence angiogenesis [137,138]. Antibody bevacizumab reduces angiogenesis by blocking VEGF-A and is used as a therapeutic agent for treating CRC [139,140,141,142]. Ranibizumab is another antibody that has a high affinity for VEGF-A isoforms [143]. 89Zr-Df-Ranibizumab was used as VEGF-PET imaging agent in different types of cancers to analyze angiogenesis changes following treatment of TKIs [144]. Luo et al. have used 64Cu-NOTA-RamAb, a ramucirumab-based PET imaging agent for mapping VEGF-2 expression in vivo [145]. VEGF-trap (Zif-aflibercept) is also used for the treatment of CRC. Zr-89-labeled bevacizumab was used as an imaging agent for visualizing VEGF expression. However, because of the limitations in PK properties of Zr-89 labeled antibody, the agent could not be efficiently used for CRC imaging [146]. Zhang et al. developed a 64Cu-based imaging agent using bevacizumab. Tetrazine was conjugated with bevacizumab and pre-targeted immune-PET near-IR fluorescence was used for imaging VEGF expression. They showed that using biorthoganol chemistry VEGF-overexpressing CRC tumors could be imaged using pre-targeted immune-PET and near-IR fluorescence imaging. The conjugate was tumor-specific, and tumor to background contrast of the image could be achieved using this technique [147]. VEGF was also used for targeting a liposome with imaging agents to tumors. Zanganeh et al. developed an agent for near-infrared fluorescence diffuse optical tomography (FDOT) using indocyanine green (ICG) and a single-chain version of VEGF (scVEGF), the conjugate scVEGF-Lip/ICG [148]. Higher tumor accumulation of the tracer and longer clearance (t1/2 ~ 90 min) was found compared to the ICG-based tracer alone.
5.3. Fluorophore-Peptide Conjugates for Targeting EGFR and VEGFR
Among the different types of small-molecule ligands with the ability to bind to tyrosine kinases, it is peptides, peptomimetics, and TKIs that have been the most used for conjugation to fluorophores. Our group has selected two peptide sequences, EGFR-L1, and EGFR-L2 (Figure 6), which have been shown to bind to the ECD of EGFR with high affinity for conjugation with phthalocyanine [83], porphyrin [149], and BODIPY fluorophores [150,151,152,153]. Such fluorophore-peptide conjugates linked via a short or a longer tripegylated linker, as shown in Figure 8, showed enhanced cell-targeting ability compared with the unconjugated fluorophore. Surface plasmon resonance (SPR) and computational (Autodock) studies showed that the conjugates were able to bind to the known binding sites for the peptides (in domain I for LARLLT and within the EGF binding pocket for YHWYGYTPQNVI, as shown in Figure 7), and that the fluorophore further stabilized the structure of the conjugates in the peptide-binding sites due to additional interactions with hydrophobic residues on the EGFR protein. In the case of phthalocyanine (Pc) conjugates 9, the most promising were found to be 9a, in part due to their enhanced solubility and EGFR-targeting ability compared with 9b, bearing the more hydrophobic peptide. The Pc conjugates were evaluated in vitro using several cell lines with different EGFR expressions (A431, HT-29, HEp2, and Vero cells) and shown to accumulate within the high EGFR expressing cells up to 17-fold compared with unconjugated Pc. [83] Studies in nude mice bearing A431 and HT-29 human tumor xenografts clearly showed tumor-localized fluorescence 24 h after i.v. administration.
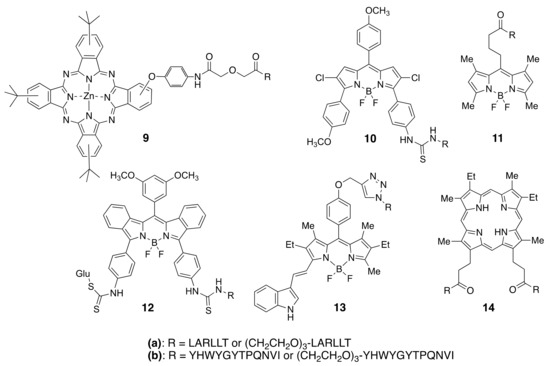
Figure 8.
Structures of fluorophores conjugated to EGFR-L1 and EGFR-L2 peptides.
BODIPYs 10a,b and 11a,b were tested in human carcinoma HEp2 cells, overexpressing EGFR, and shown to accumulate within cells up to 90-fold compared with unconjugated BODIPY [150]. When a smaller fluorophore was used, enhanced uptake was observed for the conjugate bearing the EGFR-L2 peptide (e.g., 11b) relative to EGFR-L1 (11a) [151]. This indicates that conjugation of a fluorophore to a peptide increases the hydrophobicity of the conjugate, decreasing its solubility. To increase solubility, BODIPY 12a bearing a D-glucose moiety, or en-modified biotin, were investigated [152]. In vitro studies using SW480, HT-29, DLD-1, and LoVo cells with different EGFR overexpression showed that 12a effectively accumulated in the high EGFR expressing cells. Furthermore, in vivo studies in HT-29 tumor-bearing mice showed tumor-localized near-IR fluorescence signal at 24 h after i.v. administration, which persisted up to 96 h, suggesting continuous uptake and slow clearance of the conjugate. More recently, to increase the in vivo stability of the fluorophore-peptide conjugate, 13a and its derivative containing a cyclic version of EGFR-L1 were investigated in HEp2, HT-29, DLD-1, and LOVO cells [153]. These studies showed that the conjugate bearing the cyclo(K(N3)larllt) [84] peptide had enhanced binding affinity for the ECD of EGFR compared with 13a, accumulating 5-fold in cells overexpressing EGFR.
We have also reported the conjugation of EGFR-L1 and EGFR-L2 to mesoporphyrin IX, via the propionic acid chains. These studies showed that conjugate 14a, bearing two EGFR-L1 peptides, had a much higher ECD EGFR-binding affinity compared with the single peptide conjugates [149]. This might be a result of the ability of 14a to simultaneously bind to two EGFR proteins, both in the open and closed conformations, therefore increasing its binding affinity relative to the single peptide conjugates.
The heptapeptide ATWLPPR has been the most used for targeting VEGFR. This peptide was conjugated with a tetraphenylchlorin fluorophore via a 6-aminohexanoic spacer and shown to bind to neuropilin-1 (NRP-1) recombinant chimeric protein, although not to the VEGF-2 receptor. This conjugate showed a 25-fold enhanced uptake in human umbilical vein endothelial cells (HUVEC) compared with an unconjugated fluorophore, and in vivo studies demonstrated preferential accumulation in nude mice xenografted bearing U87 human malignant gliomas [154,155,156]. The ATWLPPR peptide was also conjugated to verteporfin and investigated in a rat laser-injury model of choroidal neovascularisation (CNV) [157]. PDT treatment 1 h after the administration of the conjugate caused the complete closure of the rat lesions, while no damage was observed to the normal cells. In addition, we reported the conjugation of the ATWLPPR peptide to protoporphyrin IX via one of the propionic side chains [158]. The conjugate was observed to have 5-fold higher accumulation in human myeloid leulemia HL-60 cells versus the human carcinoma HEp2 cells, localizing preferencially in the mitochondria and lysosomes.
Several hexa- and penta-peptides devived from ATWLPPR were shown to have an affinity for binding the NRP-1 receptor, including KDKPPR, DKPRR and TKPRR. The two later pentapetides were conjugated with a tetraphenylchlorin fluorophore using three different spacers, and investigated both in vitro and in vivo. The conjugates were observed to have favorable biodistribution in an animal model, particularly the DKPPR conjugate [159].
Peptidomimetics have been investigated that effectively target the EGFR ECD inhibiting protein-protein interactions. One of such peptomimetics, designated 5-1, shown to bind to HER2 ECD and to inhibit protein-protein interactions, was conjugated with a BODIPY [160]. This conjugate accumulated in HER2-overexpressing cell lines and was observed to inhibit protein-protein interactions in vitro via a PLA assay. These peptidomimetics were also conjugated with fluorophores and doxorubicin to target the EGFR related cancer [161,162].
5.4. TKI Conjugates for EGFR and VEGFR Imaging
Many EGFR TKIs have been labeled with a radioactive isotope and used as EGFR or VEGFR imaging agents. The most used positron-emitting nuclides for PET imaging are 11C (t1/2 = 20.4 min), 18F (t1/2 = 110 min), and 124I (t1/2 = 20.4 mi4.2 days). For example, 11C-labelled erlotinib [163] and 11C-gefitinib [164] have been reported, as well as 18F-gefitinib [165] and 18F-afatinib [166] for in vivo imaging of tumors. Neto et al. [167] reported the synthesis and investigation of three radio-fluorinated and iodinated 4-anilinoquinazolines as potent anti-tumor imaging agents.
2-[Fluorine-18]-fluoro-2-deoxy-D-glucose (18F-FDG) has also been widely used for monitoring tumor treatment. Li et al. [168] demonstrated a significant correlation between EGFR-TKI treatment sensitivity in non-small cell lung cancer (NSCLC) with CD147-mediated glucose metabolic regulation using (18 F-FDG)-PET/CT imaging. Another potent PET tracer, N-(3-chloro-4-fluorophenyl)-7-(2-(2-(2-(2-18F-fluoroethoxy) ethoxy) ethoxy) ethoxy)-6-methoxyquinazolin-4-amine (18F-MPG) was used by Chen et al. [169] as a powerful imaging tool for quantitative detection of EGFR activating mutations.
4-Anilinoquinazoline moieties, derivatives of the TKI gefitinib, were complexed with Ru(II) via an amine side chain, and shown to have enhanced ability for causing cellular apoptosis [170]. This is not surprising, as several RuII and RuIII based complexes have shown promising anticancer and antimetastasis activities [171]. The anilinoquinazoline moieties were responsible for inducing apoptosis, while the RuII center preserved the reactivity towards the DNA model compound 9-ethylguanine [170].
Bourkoula [172] and coworkers synthesized 99mTc and Re complexes with derivatized 6-amino-4-[(3-bromophenyl)amino]quinazoline to generate potent small-molecule TKIs for reversibly binding to EGFR. In vitro biological studies of both the complexes indicated their ability to inhibit EGFR phosphorylation, a key step for controlling tumor growth.
Several fluorescent Ru(II) organometallic complexes have been conjugated to 4-aminoquinazoline derivatives and used in tumor imaging and treatment [173]. Among a series of derivatives, those conjugated with 4-(3′-chloro-4′-fluoroanilino)-6-(2-(2-aminoethyl)aminoethoxy)-7-methoxyquinazoline exhibited the highest inhibitory activity against MCF-7 cancer cells, and induced enhanced cell apoptosis [173]. More recently, a fluorescent Ru(II)-bipyridine complex was conjugated with a small library of aminoquinazolines in 1:1 and 1:2 ratios [174]. The resulting conjugates accumulated in EGFR-overexpressing cells, and the most promising (4-bromophenyl)-aminoquinazoline-containing conjugate was used in fluorescence imaging of U87MG glioma cells.
A near-IR phthalocyanine (Pc) was conjugated with erlotinib to give conjugates 15a bearing either a tri-ethylene glycol or penta-ethyleneglycol linker [175] (Figure 9). Both conjugates were highly phototoxic to HEpG2 cells with EGFR-overexpression, and there was no observed influence on the size of the oligoethylene glycol linker. Fluorescence imaging of nude mice bearing A431 tumors showed 5-fold higher tumor-localized fluorescence using 15a compared with fluorophore alone.
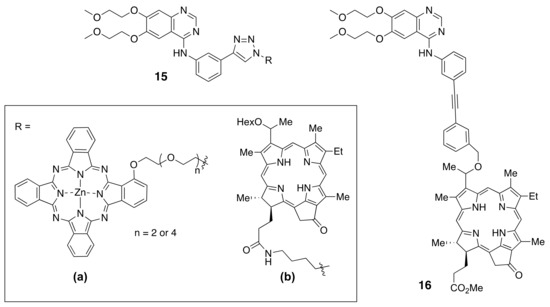
Figure 9.
Structures of fluorophores conjugated to TKIs.
Afatinib has been conjugated with cyanine-based fluorophores (Cy3 and Cy5) and the resulting conjugates shown to be efficient theranostics agents for HER1 and HER2-overexpressing tumor cells and for imaging A549 xenograft animal tumors [176]. A quinazoline derivative has also been conjugated with a 5(6)-carboxytetramethylrhodamine fluorophore and shown to have an efficiency of 34% for labeling EGFR [177].
Recently, Pandey and coworkers [178,179] reported the conjugation of pheophorbide a derivatives to erlotinib at different positions around the ring, including the synthesis of 15b and 16 (Figure 9). A series of near-IR conjugates were obtained and investigated, including derivatives of pyropheophorbide, λmax ~ 600 nm, purpurinimide, λmax ~ 700 nm, bacteriopupurinimide, λmax ~ 782 nm, and chlorin e6, λmax ~ 660 nm were conjugated with erlotinib using various linkers. The conjugates were evaluated in cellular uptake, cytotoxicity, and in SCID-tumor bearing mice studies. A radioactive analog of conjugate 15b bearing 124I was shown to be a promising dual agent for imaging (PET, fluorescence) and phototherapy (PDT) of bladder tumors. However, the most promising conjugate based on its higher tumor cell accumulation and in vivo tumor control was found to be 16. This conjugate had the highest uptake into SCID mice bearing either UMUC3 bladder or FaDu head and neck tumors and faster clearance from the liver and normal tissues compared with the photosensitizer alone and other conjugates with erlotinib at different points of attachment. Conjugate 16 also showed improved long-term tumor control compared with the photosensitizer alone at a similar administered dose and light PDT treatment parameters.
6. Conclusions
EGFR and VEGFR are important targets for cancer diagnosis and treatment. A large number of ligands have been developed that target these families of receptors, from high molecular weight antibodies, affibodies and nanobodies, to low molecular weight TKIs and small peptides. The targeting of these receptors has allowed for efficient imaging and targeted therapy options for various tumors that overexpress these proteins, advancing the field of focused and personalized medicine of cancer. Research in this field requires the interdisciplinary work of chemists, radiologists, and biologists to achieve the conjugation of an impressive variety of imaging agents, ranging from fluorophores to radioisotopes and QDs, to various antiEGFR, antiVEGFR, TKIs, and peptides. While antibody-based molecules have high affinity and specificity to bind to EGFR, small molecule TKI and peptides have lower cost synthesis, lower immunogenicity, faster clearance, better intratumoral diffusion, and easier functionalization with imaging probes compared with antibodies, while showing high specificity for EGFR-targeting. The structures of the ligands, the nature of the imaging agent, the modes of attachment, and linkers used all determine the tumor specificity and therapeutic efficacy of the resulting conjugates. As more structural details of EGFR and VEGFR with their target peptides and natural ligands are elucidated, new generations of stable peptide molecules can be used for conjugation with imaging probes, particularly with pyrrole-based fluorophores, and used for tumor detection, diagnostic imaging, and cancer treatment.
Author Contributions
Conceptualization, N.E.M.K. and S.D.; methodology, N.E.M.K. and S.D.; software, M.d.G.H.V. and S.D.J.; validation, N.E.M.K. and S D.; formal analysis, N.E.M.K. and S.D.; investigation, N.E.M.K. and S.D.; resources, M.d.G.H.V.; data curation, M.d.G.H.V. and S.D.J.; writing—original draft preparation, N.E.M.K. and S.D.; writing—review and editing, N.E.M.K., S.D., S.D.J. and M.d.G.H.V.; supervision, M.d.G.H.V.; project administration, M.d.G.H.V. and S.D.J.; funding acquisition, M.d.G.H.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Institutes of Health, grant number R01 CA179902, and the National Science Foundation, grant number 1800126.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moser, C.; Lang, S.A.; Stoeltzing, O. The direct effects of anti-vascular endothelial growth factor therapy in tumor cells. Clin. Colorectal Cancer 2007, 6, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Phamacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Phamacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef]
- Cohen, S. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J. Biol. Chem. 1962, 237, 1555–1562. [Google Scholar] [CrossRef]
- Carpenter, G.; King, L., Jr.; Cohen, S. Epidermal growth factor stimulates phosphorylation in membrane preparations in vitro. Nature 1978, 276, 409–410. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.E. EGFR Inhibition in the treatment of non-small cell lung cancer. Drug Dev. Res. 2008, 69, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 2016, 5, e14107. [Google Scholar] [CrossRef]
- Lu, C.; Mi, L.-Z.; Grey, M.J.; Zhu, J.; Graef, E.; Yokoyama, S.; Springer, T.A. Structural evidence for loose linkage between binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 2010, 30, 5432–5443. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87, 42–59. [Google Scholar] [CrossRef]
- Mineev, K.S.; Bocharov, E.V.; Pustovalova, Y.E.; Bocharova, O.V.; Chupin, V.V.; Arseniev, A.S. Spatial structure of the transmembrane domain heterodimer of erbb1 and erbb2 receptor tyrosine kinases. J. Mol. Biol. 2010, 400, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The juxtamembrane region of the EGF receptor functions as an activator domain. Mol. Cell 2009, 34, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Mitchell, R.; Luwor, R.B.; Burgess, A.W. Epidermal growth factor receptor: Structure-function informing the design of anticancer therapeutics. Exp. Cell Res. 2018, 371, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Chia, C.M.; Winston, R.M.; Handyside, A.H. EGF, TGF-alpha and EGFR expression in human preimplantation embryos. Development 1995, 121, 299–307. [Google Scholar]
- Lee-Hoeflich, S.T.; Crocker, L.; Yao, E.; Pham, T.; Munroe, X.; Hoeflich, K.P.; Sliwkowski, M.X.; Stern, H.M. A central role for HER3 in HER2-amplified breast cancer: Implications for targeted therapy. Cancer Res. 2008, 68, 5878–5887. [Google Scholar] [CrossRef]
- Mattoon, D.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc. Natl. Acad. Sci. USA 2004, 101, 923–928. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynana, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C.; et al. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimer and oligomers. Nat. Comm. 2018, 9, 4325. [Google Scholar] [CrossRef]
- Wang, Z.; Cole, P.A. Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 2014, 548, 1–21. [Google Scholar]
- Liao, J.J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. [Google Scholar] [CrossRef] [PubMed]
- van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate kinase-ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.H.; Walker, F.; Orchard, S.G.; Henderson, C.; Fuchs, D.; Rothacker, J.; Nice, E.C.; Burgess, A.W. Ligand-induced dimer-tetramer transition during the activation of the cell surface epidermal growth factor receptor–A multidimensional microscopy analysis. J. Biol. Chem. 2005, 280, 30392–30399. [Google Scholar] [CrossRef]
- Clayton, A.H.; Orchard, S.G.; Nice, E.C.; Posner, R.G.; Burgess, A.W. Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors 2008, 26, 316–324. [Google Scholar] [CrossRef]
- Kozer, N.; Barua, D.; Orchard, S.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Exploring higher-order EGFR oligomerisation and phosphorylation–a combined experimental and theoretical approach. Mol. Biosyst. 2013, 9, 1849–1863. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Xu, X.; Choe, G.; Chung, D.H.; Seo, J.W.; Lee, J.H.; Lee, C.T.; Jheon, S.; Sung, S.W.; Chung, J.H. Protein overexpression and gene amplification of epidermal growth factor receptor in non-small cell lung carcinomas: Comparison of four comercially available antibodies by immunohistochemistry and fluorescence in situ hybridization study. Lung Cancer 2010, 68, 375–382. [Google Scholar] [CrossRef]
- Huang, F.; Kirkpatrick, D.; Jiang, X.; Gygi, S.; Sorkin, A. Differential regulation of EGF receptor internalization and degradation by multiubiquitination within the kinase domain. Mol. Cell 2006, 21, 737–748. [Google Scholar] [CrossRef]
- Arkhipov, A.; Shan, Y.; Das, R.; Endres, N.F.; Eastwood, M.P.; Wemmer, D.E.; Kuriyan, J.; Shaw, D.E. Architecture and membrane interactions of the EGF receptor. Cell 2013, 152, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Kukimoto-Niino, M.; Parker, L.; Handa, N.; Terada, T.; Fujimoto, T.; Terazawa, S.; Wakiyama, M.; Sato, M.; Sano, S.; et al. Structural basis for the altered drug sensitivities of non-small cell lung cancer-associated mutants of human epidermal growth factor receptor. Oncogene 2013, 32, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Kircher, S.M.; Nimeiri, H.S.; Benson, A.B., 3rd. Target angiogenesis in colorectal cancer: Tyrosine kinase inhibitors. Cancer J. 2016, 22, 182–189. [Google Scholar] [CrossRef]
- Adair, T.H.; Montani, J.P. Angiogenesis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Markovic-Mueller, S.; Stuttfeld, E.; Asthana, M.; Weinert, T.; Bliven, S.; Goldie, K.N.; Kisko, K.; Capitani, G.; Ballmer-Hofer, K. Structure of the full-length VEGFR-1 extracellular domain in complex with VEGF-a. Structure 2017, 25, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Oguro, Y.; Miyamoto, N.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; Mik, H.; Hori, A.; Kamiyama, K.; Imamura, S. Design, synthesis, and evaluation of 5-methyl-4-phenoxy-5h-pyrrolo [3,2-d]pyrimidine derivatives: Novel VEGFR2 kinase inhibitors binding to inactive kinase conformation. Bioorg. Med. Chem. 2010, 18, 7260–7273. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, Y.; Matsunaga, Y.; Tokunaga, Y.; Obayashi, S.; Saito, M.; Morita, T. Snake venom vascular endothelial growth factors (VEGF-Fs) exclusively vary their structure and functions among species. J. Biol. Chem. 2009, 284, 9885–9891. [Google Scholar] [CrossRef] [PubMed]
- Jafari, E.; Khajouei, M.R.; Hassanzadeh, F.; Hakimelahi, G.H.; Khodarahmi, G.A. Quinazoline and quinazoline derivatives: Recent structures with potent antimicrobial and cytotoxic activities. Res. Pharm. Sci. 2016, 11, 1–14. [Google Scholar]
- Lipunova, G.N.; Nosova, E.V.; Charushin, V.N.; Chupakhin, O.N. Synthesis and antitumour activity of 4-aminoquinazoline derivatives. Russ. Chem. Rev. 2016, 85, 759–793. [Google Scholar] [CrossRef]
- Dietel, E.; Brobeil, A.; Tag, C.; Gattenloehner, S.; Wimmer, M. Effectiveness of EGFR/HER2-targeted drugs is influenced by the downstream interaction shifts of PTPIP51 in HER2-amplified breast cancer cells. Oncogenesis 2018, 7, 64. [Google Scholar] [CrossRef]
- Singh, S.S.; Dahal, A.; Shrestha, L.; Jois, S.D. Genotype driven therapy for non-small cell lung cancer: Resistance, pan inhibitors and immunotherapy. Curr. Med. Chem. 2020, 27, 5274–5316. [Google Scholar] [CrossRef]
- Vaidya, B.; Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Damon, J.K.; Sarode, A.; Kanabar, D.; Garcia, J.V.; Mitragotri, S.; Muth, A.; et al. Cyclodextrin modified erlotinib loaded PLGA nanoparticles for improved therapeutic efficacy against non-small cell lung cancer. Int. J. Biol. Macromol. 2019, 122, 338–347. [Google Scholar] [CrossRef]
- Pandey, P.; Dua, K.; Dureja, H. Erlotinib loaded chitosan nanoparticles: Formulation, physicochemical characterization and cytotoxic potential. Int. J. Biol. Macromol. 2019, 139, 1304–1316. [Google Scholar] [CrossRef]
- Ali, A.A.; Hsu, F.T.; Hsieh, C.L.; Shiau, C.Y.; Chiang, C.H.; Wei, Z.H.; Chen, C.Y.; Huang, H.S. Erlotinib-conjugated iron oxide nanoparticles as a smart cancer-targeted theranostic probe for MRI. Sci. Rep. 2016, 6, 36650. [Google Scholar] [CrossRef]
- Feng, Y.; Cheng, Y.; Chang, Y.; Jian, H.; Zheng, R.; Wu, X.; Xu, K.; Wang, L.; Ma, X.; Li, X.; et al. Time-staggered delivery of erlotinib and doxorubicin by gold nanocages with two smart polymers for reprogrammable release and synergistic with photothermal therapy. Biomaterials 2019, 217, 119327. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiary, Z.; Barar, J.; Aghanejad, A.; Saei, A.A.; Nemati, E.; Ezzati, N.; Dolatabadi, J.; Omidi, Y. Microparticles containing erlotinib-loaded solid lipid nanoparticles for treatment of non-small cell lung cancer. Drug Dev. Ind. Pharm. 2017, 43, 1244–1253. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Huang, Z.; Li, J.; Mo, Z.; Huang, Y.; Ma, C.; Wang, W.; Pan, X.; Wu, C. PLGA porous microspheres dry powders for codelivery of afatinib-loaded solid lipid nanoparticles and paclitaxel: Novel therapy for EGFR tyrosine kinase inhibitors resistant nonsmall cell lung cancer. Adv. Health. Mater. 2019, 8, e1900965. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, W. HA/HSA co-modified erlotinib-albumin nanoparticles for lung cancer treatment. Drug Des. Devel. Ther. 2018, 12, 2285–2292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, X.; Cai, S.; Mei, H.; He, Y.; Huang, D.; Shi, W.; Li, S.; Cao, J.; He, B. Photo-induced specific intracellular release EGFR inhibitor from enzyme/ROS-dual sensitive nano-platforms for molecular targeted-photodynamic combinational therapy of non-small cell lung cancer. J. Mater. Chem. B 2020, 8, 7931–7940. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Santarius, T.; Shipley, J.; Brewer, D.; Stratton, M.R.; Cooper, C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 2010, 10, 59–64. [Google Scholar] [CrossRef]
- Thomas, R.; Weihua, Z. Rethink of EGFR in cancer with its kinase independent function on board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Kim, G.P.; Grothey, A. Targeting colorectal cancer with human anti-EGFR monoclonal antibodies: Focus on panitumumab. Biologics 2008, 2, 223–228. [Google Scholar]
- Van Cutsem, E.; Peeters, M.; Siena, S.; Humblet, Y.; Hendlisz, A.; Neyns, B.; Canon, J.L.; Van Laethem, J.L.; Maurel, J.; Richardson, G.; et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J. Clin. Oncol. 2007, 25, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Cohen, R.B.; Jones, C.U.; Sur, R.K.; Raben, D.; Baselga, J.; Spencer, S.A.; Zhu, J.; et al. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010, 11, 21–28. [Google Scholar] [CrossRef]
- Garcia-Foncillas, J.; Sunakawa, Y.; Aderka, D.; Wainberg, Z.; Ronga, P.; Witzler, P.; Stintzing, S. Distinguishing features of cetuximab and panitumumab in colorectal cancer and other solid tumors. Front. Oncol. 2019, 9, 849. [Google Scholar] [CrossRef]
- Zhou, Y.; Goenaga, A.-L.; Harms, B.D.; Zou, H.; Lou, J.; Conrad, F.; Adams, G.P.; Schoeberl, B.; Nielsen, U.B.; Marks, J.D. Impact of intrinsic affinity on functional binding and biological activity of EGFR antibodies. Mol. Cancer Ther. 2012, 11, 1467–1476. [Google Scholar] [CrossRef]
- Lu, Y.; Li, X.; Liang, K.; Luwor, R.; Siddik, Z.H.; Mills, G.B.; Mendelsohn, J.; Fan, Z. Epidermal growth factor receptor (EGFR) ubiquitination as a mechanism of acquired resistance escaping treatment by the anti-EGFR monoclonal antibody cetuximab. Cancer Res. 2007, 67, 8240–8247. [Google Scholar] [CrossRef] [PubMed]
- Sickmier, E.A.; Kurzeja, R.J.M.; Michelsen, K.; Vazir, M.; Yang, E.; Tasker, A.S. The panitumumab EGFR complex reveals a binding mechanism that overcomes cetuximab induced resistance. PLoS ONE 2016, 11, e0163366. [Google Scholar] [CrossRef]
- Voigt, M.; Braig, F.; Göthel, M.; Schulte, A.; Lamszus, K.; Bokemeyer, C.; Binder, M. Functional dissection of the epidermal growth factor receptor epitopes targeted by anitumumab and cetuximab. Neoplasia 2012, 14, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, M.L.; Kikhney, J.; Hannewald, J.; Metzger, A.U.; Steffens, K.J.; Bomke, J.; Krah, A.; Wegener, A. In-depth biophysical analysis of interactions between therapeutic antibodies and the extracellular domain of the epidermal growth factor receptor. Anal. Biochem. 2011, 421, 138–151. [Google Scholar] [CrossRef]
- Brand, T.M.; Iida, M.; Wheeler, D.L. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol. Ther. 2011, 11, 777–792. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef]
- Braig, F.; März, M.; Schieferdecker, A.; Schulte, A.; Voigt, M.; Stein, A.; Grob, T.; Alawi, M.; Indenbirken, D.; Kriegs, M.; et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget. 2015, 6, 12035–12047. [Google Scholar] [CrossRef]
- Montagut, C.; Dalmases, A.; Bellosillo, B.; Crespo, M.; Pairet, S.; Iglesias, M.; Salido, M.; Gallen, M.; Marsters, S.; Tsai, S.P.; et al. Identification of a mutation in the extracellular domain of the epidermal growth factor receptor conferring cetuximab resistance in colorectal cancer. Nat. Med. 2012, 18, 221–223. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular basis for necitumumab inhibition of EGFR variants associated with acquired cetuximab resistance. Mol. Cancer Ther. 2018, 17, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockage in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef]
- Schmiedel, J.; Blaukat, A.; Li, S.; Knöchel, T.; Ferguson, K.M. Matuzumab binding to EGFR prevents the conformational rearrangement required for dimerization. Cancer Cell 2008, 13, 365–373. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef]
- Schmitz, K.R.; Bagchi, A.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Ferguson, K.M. Structural evaluation of EGFR inhibition mechanisms for nanobodies/vhh domains. Structure 2013, 21, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Harmsen, M.M.; De Haard, H.J. Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 2007, 77, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Vaneycken, I.; D’huyvetter, M.; Hernot, S.; De Vos, J.; Xavier, C.; Devoogdt, N.; Caveliers, V.; Lahoutte, T. Immuno-imaging using nanobodies. Curr. Opin. Biotechnol. 2011, 22, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Saerens, D.; Ghassabeh, G.H.; Muyldermans, S. Single-domain antibodies as building blocks for novel therapeutics. Curr. Opin. Pharmacol. 2008, 8, 600–608. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, L.; Liu, S.; Chen, Q.; Zeng, L.; Chen, X.; Zhang, Q. Targeted nanobody complex enhanced photodynamic therapy for lung cancer by overcoming tumor microenvironment. Cancer Cell Int. 2020, 20, 570. [Google Scholar] [CrossRef]
- Sharifi, J.; Khirehgesh, M.R.; Safari, F.; Akbari, B. EGFR and anti-EGFR nanobodies: Review and update. J. Drug Target. 2020, 1–16. [Google Scholar] [CrossRef]
- Mashayekhi, V.; Xenaki, K.T.; van Bergen en Henegouwen, P.M.P.; Oliveira, S. Dual targeting of endothelial and cancer cells potentiates in vitro nanobody-targeted photodynamic therapy. Cancers 2020, 12, 2732. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Qin, L.; Ye, H.; Wang, Y.; Long, B.; Jiao, Z. Anti-EGFR binding nanobodies delivery system to improve the diagnosis and treatment of solid tumours. Recent Pat Anticancer Drug Discov. 2020, 15, 200–211. [Google Scholar] [CrossRef]
- Song, S.; Liu, D.; Peng, J.; Deng, H.; Guo, Y.; Xu, L.X.; Miller, A.D.; Xu, Y. Novel peptide ligand directs liposomes toward EGF-R high-expressing cancer cells in vitro and in vivo. FASEB J. 2009, 23, 1396–1404. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, R.; Wu, X.; Sun, Y.; Yao, M.; Li, J.; Xu, Y.; Gu, J. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Ongarora, B.G.; Fontenot, K.R.; Hu, X.; Sehgal, I. Satyanarayana-Jois, S.D.; Vicente, M.G.H. Phthalocyanine-peptide conjugates for epidermal growth factor receptor targeting. J. Med. Chem. 2012, 55, 3725–3738. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Sable, R.; Singh, S.S.; Vicente, M.G.H.; Jois, S.D. Peptide ligands for targeting the extracellular domain of EGFR: Comparison between linear and cyclic peptides. Chem. Biol. Drug Des. 2018, 91, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Hossein-Nejad-Ariani, H.; Althagafi, E.; Kaur, K. Small peptide ligands for targeting EGFR in triple negative breast cancer cells. Sci. Rep. 2019, 9, 2723. [Google Scholar] [CrossRef]
- Mizuguchi, T.; Ohara, N.; Iida, M.; Ninomiya, R.; Wada, S.; Kiso, Y.; Saito, K.; Akaji, K. Evalutaion of dimerization-inhibitors activities of cyclic peptides containinga beta-hairpin loop sequence of the EGF receptor. Bioorg. Med. Chem. 2012, 20, 5730–5737. [Google Scholar] [CrossRef]
- Toyama, K.; Kobayakawa, T.; Nomura, W.; Tamamura, H. Inhibition of EGFR activation by bivalent ligands based on a cyclic peptide mimicking the dimerization arm structure of EGFR. Chem. Pharm. Bull. (Tokyo) 2018, 66, 1083–1089. [Google Scholar] [CrossRef]
- Feiner, R.C.; Kemker, I.; Krutzke, L.; Allmendinger, E.; Mandell, D.J.; Sewald, N.; Kochanek, S.; Muller, K.M. EGFR-binding peptides: From computational design towards tumor-targeting of adeno-associated virus capsids. Int. J. Mol. Sci. 2020, 21, 9535. [Google Scholar] [CrossRef]
- Hart, M.R.; Su, H.Y.; Broka, D.; Goverdhan, A.; Schroeder, J.A. Inactive ERBB receptors cooperate with reactive oxygen species to suppress cancer progression. Mol. Ther. 2013, 21, 1996–2007. [Google Scholar] [CrossRef]
- Maisel, S.A.; Broka, D.; Atwell, B.; Bunch, T.; Kupp, R.; Singh, S.K.; Mehta, S.; Schroeder, J. Stapled EGFR peptide reduces inflammatory breast cancer and inhibits additional her-driven models of cancer. J. Transl. Med. 2019, 17, 201. [Google Scholar] [CrossRef]
- Tavakoli, F.; Ganjalikhany, M.R. Structure-based inhibitory peptide design targeting peptide-substrate binding site in EGFR tyrosine kinase. PLoS ONE 2019, 14, e0217031. [Google Scholar] [CrossRef]
- Foy, K.C.; Wygle, R.M.; Miller, M.J.; Overholser, J.P.; Bekaii-Saab, T.; Kaumaya, P.T.P. Peptide vaccines and peptidomimetics of EGFR (HER-1) ligand binding domain inhibit cancer cell growth in vitro and in vivo. J. Immunol. 2013, 191, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Berezov, A.; Chen, J.; Liu, Q.; Zhang, H.T.; Greene, M.I.; Murali, R. Disabling receptor ensembles with rationally designed interface peptomimetics. J. Biol. Chem. 2002, 277, 28330–28339. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayanajois, S.; Villalva, S.; Jianchao, L.; Lin, G.M. Design, synthesis, and docking studies of peptidomimetics based on HER2-herceptin binding site with potential antiproliferative activity against breast cancer cell lines. Chem. Biol. Drug Des. 2009, 74, 246–257. [Google Scholar] [CrossRef]
- Kanthala, S.; Gauthier, T.; Satyanarayanajois, S. Structure-activity relationships of peptidomimetics that inhibit PPI of HER2-HER3. Biopolymers 2014, 101, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Kanthala, S.P.; Liu, Y.Y.; Singh, S.; Sable, R.; Pallerla, S.; Jois, S.D. A peptidomimetic with a chiral switch is an inhibitor of epidermal growth factor receptor heterodimerization. Oncotarget. 2017, 8, 74244–74262. [Google Scholar] [CrossRef]
- Mankoff, D.A. A definition of molecular imaging. J. Nucl. Med. 2007, 48, 18N–21N. [Google Scholar]
- Kramer-Marek, G.; Longmire, M.R.; Choyke, P.L.; Kobayashi, H. Recent advances in optical cancer imaging of EGF receptors. Curr. Med. Chem. 2012, 19, 4759–4766. [Google Scholar] [CrossRef]
- Saga, T.; Endo, K.; Akiyama, T.; Sakahara, H.; Koizumi, M.; Watanabe, Y.; Nakai, T.; Hosono, M.; Yamamoto, T.; Toyoshima, K.; et al. Scintigraphic detection of overexpressed c-ERBB-2 protooncogene products by a class-switched murine anti-c-ERBB-2 protein monoclonal antibody. Cancer Res. 1991, 51, 990–994. [Google Scholar] [PubMed]
- Allan, S.M.; Dean, C.J.; Eccles, S.; Sacks, N.P. Clinical radioimmunolocalization with a rat monoclonal antibody directed against c-ERBB-2. Cell Biophys. 1994, 24–25, 93–98. [Google Scholar] [CrossRef]
- Meenakshi, A.; Kumar, R.S.; Ganesh, V.; Kumar, N.S. Preliminary study on radioimmunodiagnosis of experimental tumor models using technetium-99m-labeled anti-C-ERBB-2 monoclonal antibody. Tumor. 2002, 88, 507–512. [Google Scholar] [CrossRef]
- Bakir, M.A.; Eccles, S.; Babich, J.W.; Aftab, N.; Styles, J.; Dean, C.J.; Lambrecht, R.M.; Ott, R.J. c-ERBB2 protein overexpression in breast cancer as a target for PET using iodine-124-labeled monoclonal antibodies. J. Nucl. Med. 1992, 33, 2154–2160. [Google Scholar] [PubMed]
- Wiercioch, R.; Balcerczak, E.; Byszewska, E.; Mirowski, M. Uptake of radiolabelled herceptin by experimental mammary adenocarcinoma. Nucl. Med. Rev. Cent. East Eur. 2003, 6, 99–103. [Google Scholar]
- Kotts, C.E.; Su, F.M.; Leddy, C.; Dodd, T.; Scates, S.; Shalaby, M.R.; Wirth, C.M.; Giltinan, D.; Schroff, R.W.; Fritzberg, A.R.; et al. 186Re-Labeled antibodies to p185HER2 as HER2-targeted radioimmunopharmaceutical agents: Comparison of physical and biological characteristics with 125I and 131I-labeled counterparts. Cancer Biother. Radiopharm. 1996, 11, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Tolmachev, V.; Andersson, K.; Gedda, L.; Sandström, M.; Carlsson, J. [(177)Lu]pertuzumab: Experimental studies on targeting of HER-2 positive tumour cells. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 1457–1462. [Google Scholar] [CrossRef]
- Palm, S.; Enmon, R.M., Jr.; Matei, C.; Kolbert, S.K..; Xu, S.; Zanzonico, P.B.; Finn, R.L.; Koutcher, J.A.; Larson, S.M.; Sgouros, G. Pharmacokinetics and biodistribution of (86)Y-trastuzumab for (90)Y dosimetry in an ovarian carcinoma model: Correlative microPET and MRI. J. Nucl. Med. 2003, 44, 1148–1155. [Google Scholar]
- Artemov, D.; Mori, N.; Okollie, B.; Bhujwalla, Z.M. MR molecular imaging of the HER-2/neu receptor in breast cancer cells using targeted iron oxide nanoparticles. Magn. Reson. Med. 2003, 49, 403–408. [Google Scholar] [CrossRef]
- Chen, T.-J.; Cheng, T.-H.; Chen, C.-Y.; Hsu, S.C.; Cheng, T.-L.; Liu, G.-C.; Wang, Y.-M. Targeted Herceptin-dextran iron oxide nanoparticles for noninvasive imaging of HER2/neu receptors using MRI. J. Biol. Inorg. Chem. 2009, 14, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Scollard, D.; Chen, P.; Wang, J.; Holloway, C.; Reilly, R.M. Imaging of HER2/neu expression in BT-474 human breast cancer xenografts in athymic mice using [(99m)Tc]-HYNIC-trastuzumab (Herceptin) Fab fragments. Nucl. Med. Commun. 2005, 26, 427–432. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, J.; Scollard, D.A.; Mondal, H.; Holloway, C.; Kahn, H.J.; Reilly, R.M. Imaging of HER2/neu-positive BT-474 human breast cancer xenografts in athymic mice using (111)In-trastuzumab (Herceptin) Fab fragments. Nucl. Med. Biol. 2005, 32, 51–58. [Google Scholar] [CrossRef]
- Pysz, M.A.; Foygel, K.; Rosenberg, J.; Gambhir, S.S.; Schneider, M.; Willmann, J.K. Antiangiogenic cancer therapy: Monitoring with molecular US and a clinically translatable contrast agent (BR55). Radiology 2010, 256, 519–527. [Google Scholar] [CrossRef]
- Kaluzova, M.; Bouras, A.; Machaidze, R.; Hadjipanayis, C.G. Targeted therapy of glioblastoma stem-like cells and tumor non-stem cells using cetuximab-conjugated iron-oxide nanoparticles. Oncotarget 2015, 6, 8788–8806. [Google Scholar] [CrossRef]
- Hadjipanayis, C.G.; Machaidze, R.; Kaluzova, M.; Wang, L.; Schuette, A.J.; Chen, H.; Wu, X.; Mao, H. EGFRvIII antibody-conjugated iron oxide nanoparticles for magnetic resonance imaging-guided convection-enhanced delivery and targeted therapy of glioblastoma. Cancer Res. 2010, 70, 6303–6312. [Google Scholar] [CrossRef]
- Liu, X.; Du, C.; Li, H.; Jiang, T.; Luo, Z.; Pang, Z.; Geng, D.; Zhang, J. Engineered superparamagmetic iron oxide nanoparticles (SPIONs) for dual-modality imaging of intracranial glioblastoma via EGFRvIII targeting. Beilstein J. Nanotechnol. 2019, 10, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Mu, K.; Zhang, S.; Ai, T.; Jiang, J.; Yao, Y.; Jiang, L.; Zhou, Q.; Xiang, H.; Zhu, Y.; Yang, X.; et al. Monoclonal antibody-conjugated superparamagnetic iron oxide nanoparticles for imaging of epidermal growth factor receptor-targeted cells and gliomas. Mol. Imaging 2015, 14, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Shevtsov, M.A.; Nikolaev, B.P.; Yakovleva, L.Y.; Marchenko, Y.Y.; Dobrodumov, A.V.; Mikhrina, A.L.; Martynova, M.G.; Bystrova, O.A.; Yakovenko, I.V.; Ischenko, A.M. Superparamagnetic iron oxide nanoparticles conjugated with epidermal growth factor (SPION-EGF) for targeting brain tumors. Int. J. Nanomedicine 2014, 9, 273–287. [Google Scholar] [CrossRef]
- Wu, X.; Liu, H.; Liu, J.; Haley, K.N.; Treadway, J.A.; Larson, J.P.; Ge, N.; Peale, F.; Bruchez, M.P. Immunofluorescent labeling of cancer marker HER2 and other cellular targets with semiconductor quantum dots. Nat. Biotechnol. 2003, 21, 41–46. [Google Scholar] [CrossRef]
- Loo, C.; Lowery, A.; Halas, N.; West, J.; Drezek, R. Immunotargeted nanoshells for integrated cancer imaging and therapy. Nano Lett. 2005, 5, 709–711. [Google Scholar] [CrossRef]
- Singh, A.; Xu, J.; Mattheolabakis, G.; Amiji, M. EGFR-targeted gelatin nanoparticles for systemic administration of gemcitabine in an orthotopic pancreatic cancer model. Nanomedicine 2016, 12, 589–600. [Google Scholar] [CrossRef]
- Qian, Y.; Qiu, M.; Wu, Q.; Tian, Y.; Zhang, Y.; Gu, N.; Li, S.; Xu, L.; Yin, R. Enhanced cytotoxic activity of cetuximab in EGFR-positive lung cancer by conjugating with gold nanoparticles. Sci. Rep. 2014, 4, 7490. [Google Scholar] [CrossRef] [PubMed]
- Eghtedari, M.; Liopo, A.; Coplang, J.A.; Oraevsky, A.A.; Motamedi, M. Engineering of hetero-functional gold nanorods for the in vivo molecular targeting of breast cancer cells. Nano Lett. 2009, 9, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Piktel, E.; Niemirowicz, K.; Watek, M.; Wollny, T.; Deptula, P.; Bucki, R. Recent insights in nanotechnology-based drugs and formulations designed for effective anti-cancer therapy. J. Nanobiotechnol. 2016, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Puvanakrishnan, P.; Diagaradjane, P.; Kazmi, S.M.; Dunn, A.K.; Krishnan, S.; Tunnell, J.W. Narrow band imaging of squamous cell carcinoma tumors using topically delivered anti-EGFR antibody conjugated gold nanorods. Lasers Surg. Med. 2012, 44, 310–317. [Google Scholar] [CrossRef]
- Chen, W.; Shen, B.; Sun, X. Analysis of progress and challenges of EGFR-targeted molecular imaging in cancer with a focus on affibody molecules. Mol. Imaging 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Day, K.E.; Sweeny, L.; Kulbersh, B.; Zinn, K.R.; Rosenthal, E.L. Preclinical comparison of near-infrared-labeled cetuximab and panitumumab for optical imaging of head and neck squamous cell carcinoma. Mol. Imaging Biol. 2013, 15, 722–729. [Google Scholar] [CrossRef]
- Xu, N.; Cai, G.; Ye, W.; Wang, X.; Li, Y.; Zhao, P.; Zhang, A.; Zhang, R.; Cao, B. Molecular imaging application of radioiodinated anti-EGFR human Fab to EGFR-overexpressing tumor xenografts. Anticancer Res. 2009, 29, 4005–4011. [Google Scholar]
- van Dijk, L.K.; Boerman, O.C.; Franssen, G.M.; Kaanders, J.H.A.M.; Bussink, J. 111In-Cetuximab-F(ab’)2 SPECT and 18F-FDG PET for prediction and response monitoring of combined-modality treatment of human head and neck carcinomas in a mouse model. J. Nucl. Med. 2015, 56, 287–292. [Google Scholar] [CrossRef]
- Frejd, F.Y.; Kim, K.T. Affibody molecules as engineered protein drugs. Exp. Mol. Med. 2017, 49, e306. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Ståhl, S.; Uhlén, M.; Nygren, P.A. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, X.; Qi, S.; Liu, H.; Jiang, H.; Hoppmann, S.; Cao, Q.; Chua, M.S.; So, S.K.; Cheng, Z. Molecular imaging of hepatocellular carcinoma xenografts with epidermal growth factor receptor targeted affibody probes. Biomed. Res. Int. 2013, 2013, 759057. [Google Scholar] [CrossRef]
- Ehlerding, E.B.; Sun, L.; Lan, X.; Zeng, D.; Cai, W. Dual-targeted molecular imaging of cancer. J. Nucl. Med. 2018, 59, 390–395. [Google Scholar] [CrossRef]
- Garousi, J.; Andersson, K.G.; Dam, J.H.; Olsen, B.B.; Mitran, B.; Orlova, A.; Buijs, J.; Ståhl, S.; Löfblom, J.; Thisgaard, H.; et al. The use of radiocobalt as a label improves imaging of EGFR using DOTA-conjugated Affibody molecule. Sci. Rep. 2017, 7, 5961. [Google Scholar] [CrossRef]
- Gong, H.; Sampath, L.; Kovar, J.L.; Olive, D.M. Targeting EGFR and HER2 for Molecular Imaging of Cancer; Molecular Imaging InTech: Berlin, Germany, 2012; pp. 351–374. [Google Scholar]
- Garousi, J.; Andersson, K.G.; Mitran, B.; Pichl, M.L.; Ståhl, S.; Orlova, A.; Löfblom, J.; Tolmachev, V. PET imaging of epidermal growth factor receptor expression in tumours using 89Zr-labelled ZEGFR:2377 affibody molecules. Int. J. Oncol. 2016, 48, 1325–1332. [Google Scholar] [CrossRef]
- Wei, W.; Ni, D.; Ehlerding, E.B.; Luo, Q.-Y.; Cai, W. PET imaging of receptor tyrosine kinases in cancer. Mol. Cancer Ther. 2018, 17, 1625–1636. [Google Scholar] [CrossRef]
- Taurone, S.; Galli, F.; Signore, A.; Agostinelli, E.; Dierckx, R.A.; Minni, A.; Pucci, M.; Artico, M. [Corrigendum] VEGF in nuclear medicine: Clinical application in cancer and future perspectives (Review). Int. J. Oncol. 2016, 49, 437–447. [Google Scholar] [CrossRef][Green Version]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-Trap: A VEGF blocker with potent antitumour effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef]
- Gardner, V.; Madu, C.O.; Lu, Y. Physiologic and Pathologic Angiogenesis. Signaling Mechanisms and Targeted Therapy; Anti-VEGF Therapy in Cancer: A Double-Edged Sword; TechOpen: London, UK, 2017; pp. 385–410. [Google Scholar]
- Ishigami, S.I.; Arii, S.; Furutani, M.; Niwano, M.; Harada, T.; Mizumoto, M.; Mori, A.; Onodera, H.; Imamura, M. Predictive value of vascular endothelial growth factor (VEGF) in metastasis and prognosis of human colorectal cancer. Br. J. Cancer 1998, 78, 1379–1384. [Google Scholar] [CrossRef]
- Ranieri, G.; Patruno, R.; Ruggieri, E.; Montemurro, S.; Valerio, P.; Ribatti, D. Vascular endothelial growth factor (VEGF) as a target of bevacizumab in cancer: From the biology to the clinic. Curr. Med. Chem. 2006, 13, 1845–1857. [Google Scholar] [CrossRef]
- Rosen, L.S.; Jacobs, I.A.; Burkes, R.L. Bevacizumab in colorectal cancer: Current role in treatment and the potential of biosimilars. Target Oncol. 2017, 12, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Modest, D.P.; Pant, S.; Sartore-Bianchi, A. Treatment sequencing in metastatic colorectal cancer. Eur. J. Cancer 2019, 109, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Dedania, V.S.; Bakri, S.J. Current perspectives on ranibizumab. Clin. Ophthalmol. 2015, 9, 533–542. [Google Scholar] [PubMed]
- Nagengast, W.B.; Lub-de Hooge, M.N.; Oosting, S.F.; den Dunnen, W.F.; Warnders, F.J.; Brouwers, A.H.; de Jong, J.R.; Price, P.M.; Hollema, H.; Hospers, G.A.; et al. VEGF-PET imaging is a noninvasive biomarker showing differential changes in the tumor during sunitinib treatment. Cancer Res. 2011, 71, 143–153. [Google Scholar] [CrossRef]
- Luo, H.; England, C.G.; Graves, S.A.; Sun, H.; Liu, G.; Nickles, R.J.; Cai, W. PET imaging of VEGFR-2 expression in lung cancer with 64Cu-labeled ramucirumab. J. Nucl. Med. 2016, 57, 285–290. [Google Scholar] [CrossRef]
- Jansen, M.H.; Lagerweij, T.; Sewing, A.C.; Vugts, D.J.; van Vuurden, D.G.; Molthoff, C.F.; Caretti, V.; Veringa, S.J.; Petersen, N.; Carcaboso, A.M.; et al. Bevacizumab targeting diffuse intrinsic pontine glioma: Results of 89Zr-bevacizumab PET imaging in brain tumor models. Mol. Cancer Ther. 2016, 15, 2166–2174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hong, H.; Engle, J.W.; Yang, Y.; Barnhart, T.E.; Cai, W. Positron emission tomography and near-infrared fluorescence imaging of vascular endothelial growth factor with dual-labeled bevacizumab. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 1–13. [Google Scholar]
- Zanganeh, S.; Xu, Y.; Hamby, C.V.; Backer, M.V.; Backer, J.M.; Zhu, Q. Enhanced fluorescence diffuse optical tomography with indocyanine green-encapsulating liposomes targeted to receptors for vascular endothelial growth factor in tumor vasculature. J. Biomed. Opt. 2013, 18, 126014. [Google Scholar] [CrossRef]
- Fontenot, K.R.; Ongarora, B.G.; LeBlanc, L.E.; Zhou, Z.; Jois, S.D.; Vicente, M.G.H. Targeting of the epidermal growth factor receptor with mesoporphyrin IX-peptide conjugates. J. Porphyr. Phthalocyanines 2016, 20, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Williams, T.M.; Zhou, Z.; Fronczek, F.R.; Sibrian-Vazquez, M.; Jois, S.D.; Vicente, M.G.H. Synthesis of BODIPY-peptide conjugates for fluorescence labeling of EGFR overexpressing cells. Bioconjugate Chem. 2017, 28, 1566–1579. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Zhou, Z.; Singh, S.S.; Sibrian-Vazquez, M.; Jois, S.D.; Vicente, M.G.H. Targeting EGFR overexpression at the surface of colorectal cancer cells by exploiting amidated BODIPY-peptide conjugates. Photochem. Photobiol. 2020, 96, 581–595. [Google Scholar] [CrossRef]
- Kaufman, N.E.M.; Meng, Q.; Griffin, K.E.; Singh, S.S.; Dahal, A.; Zhou, Z.; Fronczek, F.R.; Mathis, J.M.; Jois, S.D.; Vicente, M.G.H. Synthesis, characterization and evaluation of near-IR boron-dipyrromethene (BODIPY) bioconjugates for labeling of adenocarcinomas by selectively targeting the epidermal growth factor receptor (EGFR). J. Med. Chem. 2019, 62, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Kaufman, N.E.M.; Zhou, Z.; Singh, S.S.; Jois, S.D.; Vicente, M.G.H. Click conjugation of boron dipyrromethene (BODIPY) fluorophores to EGFR-targeting linear and cyclic peptides. Molecules 2021, 26, 593. [Google Scholar] [CrossRef]
- Thomas, N.; Tirand, L.; Chatelut, R.; Plenat, F.; Frochot, C.; Dodeller, M.; Guillemin, F.; Barberi-Heyob, M. Tissue distribution and pharmacokinetics of an ATWLPPR-conjugated chlorin-type photosensitizer targeting neuropilin-1 in glioma-bearing nude mice. Photochem. Photobiol. Sci. 2008, 7, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.; Bechet, D.; Becuwe, P.; Tirand, L.; Vanderesse, R.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Peptide-conjugated chlorin-type photosensitizer binds neuropilin-1 in vitro and in vivo. J. Photochem. Photobiol. B Biol. 2009, 96, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bechet, D.; Tirand, L.; Faivre, B.; Plenat, F.; Bonnet, C.; Bastogne, T.; Frochot, C.; Guillemin, F.; Barberi-Heyob, M. Neuropilin-1 targeting photosensitization-induced early stages of thrombosis via tissue factor release. Pharm. Res. 2010, 27, 468–479. [Google Scholar] [CrossRef]
- Renno, R.Z.; Terada, Y.; Haddadin, M.J.; Michaud, N.A.; Gragoudas, E.S.; Miller, J.W. Selective photodynamic therapy by targeted verteporfin delivery to experimental choroidal neovascularization mediated by a homing peptide to vascular endothelial growth factor receptor-2. Arch. Ophthalmol. 2004, 122, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Sibrian-Vazquez, M.; Hu, X.; Jensen, T.J.; Vicente, M.G.H. Synthesis and cellular studies of PPIX-homing peptide conjugates. J. Porphyr. Phthalocyanines 2012, 16, 603–615. [Google Scholar] [CrossRef]
- Kamarulzaman, E.E.; Gazzali, A.M.; Acherar, S.; Frochot, C.; Barberi-Heyob, M.; Boura, C.; Chiambault, P.; Sibile, E.; Wahab, H.A.; Vanderesse, R. New peptide-conjugated chlorin-type photosensitizer targeting neuropilin-1 for anti-vascular targeted photodynamic therapy. Int. J. Mol. Sci. 2015, 16, 24059–24080. [Google Scholar] [CrossRef]
- Banappagari, S.; McCall, A.; Fontenot, K.; Vicente, M.G.H.; Gujar, A.; Jois, S.D. Design, synthesis and characterization of peptidomimetic conjugate of BODIPY targeting HER2 protein extracellular domain. Eur. J. Med. Chem. 2013, 65, 60–69. [Google Scholar] [CrossRef]
- Pallerla, S.; Gauthier, T.; Sable, R.; Jois, S.D. Design of a doxorubicin-peptidomimetic conjugate that targets HER2-positive cancer cells. Eur. J. Med. Chem. 2017, 125, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Naik, H.; Gauthier, T.; Singh, S.; Jois, S. Design of novel lipidated peptidomimetic conjugates for targeting EGFR heterodimerization in HER2+ cancer. Bioorg. Med. Chem. Lett. 2018, 28, 3506–3513. [Google Scholar] [CrossRef]
- Petrulli, J.R.; Sullivan, J.M.; Zheng, M.-Q.; Bennett, D.C.; Charest, J.; Huang, Y.; Morris, E.D.; Contessa, J.N. Quantitative analysis of [11C]-Erlotinib PET demonstrates specific binding for activating mutations of the EGFR kinase domain. Neoplasia 2013, 15, 1347–1353. [Google Scholar] [CrossRef]
- Abourbeh, G.; Itamar, B.; Salnikov, O.; Beltsov, S.; Mishani, E. Identifying erlotinib-sensitive non-small cell lung carcinoma tumors in mice using [11C] erlotinib PET. Eur. J. Nucl. Med. Mol. Imaging Res. 2015, 5, 4. [Google Scholar] [CrossRef]
- Su, H.; Seimbille, Y.; Ferl, G.Z.; Bodenstein, C.; Fueger, B.; Kim, K.J.; Hsu, Y.-T.; Dubinett, S.M.; Phelps, M.E.; Czernin, J.; et al. Evaluation of [18F] gefitinib as a molecular imaging probe for the assessment of the epidermal growth factor receptor status in malignant tumors. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Slobbe, P.; Windhorst, A.D.; Stigter-van Walsum, M.; Schuit, R.C.; Smit, E.F.; Niessen, H.G.; Solca, F.; Stehle, G.; van Dongen, G.A.; Poot, A.J. Development of [18F] afatinib as new TKI-PET tracer for EGFR positive tumors. Nucl. Med. Biol. 2014, 41, 749–757. [Google Scholar] [CrossRef]
- Neto, C.; Fernandes, C.; Oliveira, M.C.; Gano, L.; Mendes, F.; Kniess, T.; Santos, I. Radiohalogenated 4-anilinoquinazoline-based EGFR-TK inhibitors as potential cancer imaging agents. Nucl. Med. Biol. 2012, 39, 247–260. [Google Scholar] [CrossRef]
- Li, X.; Fu, Q.; Zhu, Y.; Wang, J.; Liu, J.; Yu, X.; Xu, W. CD147-mediated glucose metabolic regulation contributes to the predictive role of 18F-FDG PET/CT imaging for EGFR-TKI treatment sensitivity in NSCLC. Mol. Carcinog. 2019, 58, 247–257. [Google Scholar] [CrossRef]
- Sun, X.; Xiao, Z.; Chen, G.; Han, Z.; Liu, Y.; Zhang, C.; Sun, Y.; Song, Y.; Wang, K.; Fang, F.; et al. A PET imaging approach for determining EGFR mutation status for improved lung cancer patient management. Sci. Transl. Med. 2018, 10, eaan8840. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Luo, Q.; Lin, Y.; Zhao, Y.; Wang, X.; Du, Z.; Hao, X.; Yu, Y.; Lu, S.; Ji, L.; et al. Complexation with organometallic ruthenium phamacophores enhances the ability of 4-anilinoquinazolines inducing apoptosis. Chem. Commun. 2013, 49, 10224–10226. [Google Scholar] [CrossRef]
- Aird, R.E.; Cummings, J.; Ritchie, A.A.; Muir, M.; Morris, R.E.; Chen, H.; Sadler, P.J.; Jodrell, D.I. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar] [CrossRef] [PubMed]
- Bourkoula, A.; Paravatou-Petsotas, M.; Papadopoulos, A.; Santos, I.; Pietzsch, H.J.; Livaniou, E.; Pelecanou, M.; Papadopoulos, M.; Pirmettis, I. Synthesis and characterization of rhenium and technetium-99 m tricarbonyl complexes bearing the 4-[3-bromophenyl]quinazoline moiety as a biomarker for EGFR-TK imaging. Eur. J. Med. Chem. 2009, 44, 4021–4027. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zheng, W.; Lin, Y.; Wang, X.; Lu, S.; Hao, X.; Luo, Q.; Li, X.; Yang, L.; Wang, F. Novel ruthenium complexes ligated with 4-anilinoquinazoline derivatives: Synthesis, characterisation and preliminary evaluation of biological activity. Eur. J. Med. Chem. 2014, 77, 110–120. [Google Scholar] [CrossRef]
- Ilmi, R.; Tseriotou, E.; Stylianou, P.; Christou, Y.A.; Ttofi, I.; Dietis, N.; Pitris, C.; Odysseos, A.D.; Georgiades, S.N. A novel conjugate of bis[((4-bromophenyl)amino)quinazoline], a EGFR-TK ligand, with a fluorescent Ru(II)-bipyridine complex exhibits specific subcellular localization in mitochondria. Mol. Pharm. 2019, 16, 4260–4273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-L.; Huang, Q.; Zheng, K.; Li, J.; Liu, J.-Y.; Xue, J.-P. A novel strategy for targeting photodynamic therapy. Molecular combo of photodynamic agent zinc(II) phthalocyanine and small molecule target-based anticancer drug erlotinib. Chem. Commun. 2013, 49, 9570–9572. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Song, W.; Gao, X.; Su, Y.; Gao, E.; Gao, Q. Discovery of nonpeptide, reversible HER1/HER2 dual-targeting small molecule inhibitors as near-infrared fluorescent probes for efficient tumor detection, diagnostic imaging, and drug screening. Anal. Chem. 2019, 91, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Shreder, K.R.; Wong, M.S.; Nomanbhoy, T.; Leventhal, P.S.; Fuller, S.R. Synthesis of AX7593, a quinazoline-derived photoaffinity probe for EGFR. Org. Lett. 2004, 6, 3715–3718. [Google Scholar] [CrossRef]
- Cheruku, R.R.; Cacaccio, J.; Durrani, F.A.; Tabaczynski, W.A.; Watson, R.; Marko, A.; Kumar, R.; El-Khouly, M.E.; Fukuzumi, S.; Missert, J.R.; et al. Epidermal growth factor receptor-targeted multifunctional photosensitizers for bladder cancer imaging and photodynamic therapy. J. Med. Chem. 2019, 62, 2598–2617. [Google Scholar] [CrossRef]
- Cheruku, R.R.; Cacaccio, J.; Durrani, F.A.; Tabaczynski, W.A.; Watson, R.; Siters, K.; Missert, J.R.; Tracy, E.C.; Dukh, M.; Guru, K.; et al. Synthesis, tumor specificity and photosensitizing efficacy of erlotinib-conjugated chlorins and bacteriochlorins: Identification of a highly effective candidate for photodynamic therapy of cancer. J. Med. Chem. 2021, 64, 741–767. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).