
Optical Gain in Semiconducting Polymer Nano and Mesoparticles

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
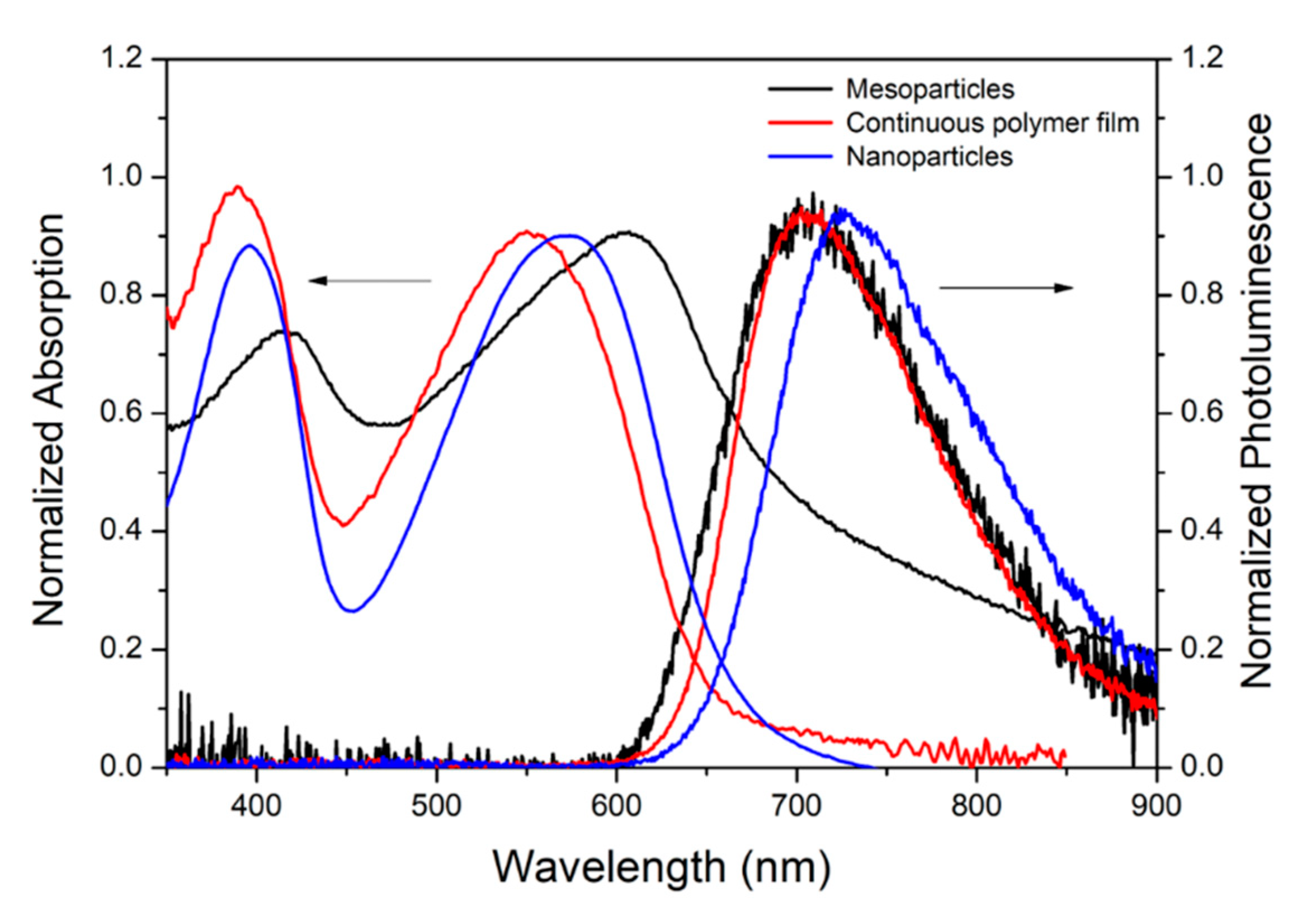
2. Results
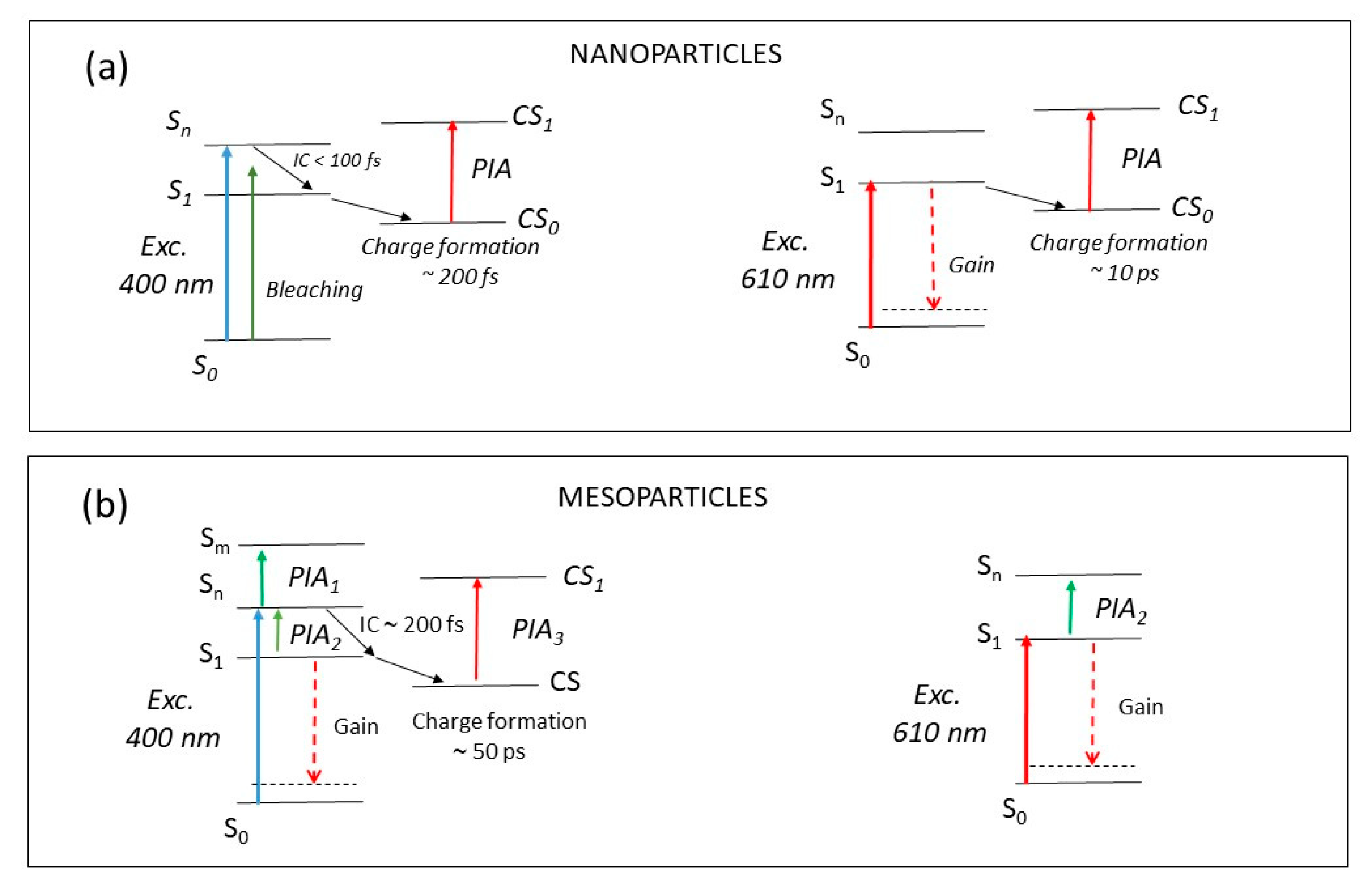
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pecher, J.; Mecking, S. Nanoparticles of Conjugated Polymers. Chem. Rev. 2010, 110, 6260–6279. [Google Scholar] [CrossRef]
- Geoghegan, M.; Hadziioannou, G. Polymer Electronics; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Murad, A.R.; Iraqi, A.; Aziz, S.B.; Abdullah, S.N.; Brza, M.A. Conducting Polymers for Optoelectronic Devices and Organic Solar Cells: A Review. Polymers 2020, 12, 2627. [Google Scholar] [CrossRef]
- Gao, H.; Poulsen, D.A.; Ma, B.; Unruh, D.A.; Zhao, X.; Millstone, J.E.; Fréchet, J.M.J. Site Isolation of Emitters within Cross-Linked Polymer Nanoparticles for White Electroluminescence. Nano Lett. 2010, 10, 1440–1444. [Google Scholar] [CrossRef]
- Piok, T.; Gamerith, S.; Gadermaier, C.; Plank, H.; Wenzl, F.; Patil, S.F.; Montenegro, R.; Kietzke, T.; Neher, D.; Scherf, U.; et al. Organic Light-Emitting Devices Fabricated from Semiconducting Nanospheres. Adv. Mater. 2003, 15, 800–804. [Google Scholar] [CrossRef]
- Kietzke, T.; Neher, D.; Landfester, K.; Montenegro, R.; Güntner, R.; Scherf, U. Novel approaches to polymer blends based on polymer nanoparticles. Nat. Mater. 2003, 2, 408–412. [Google Scholar] [CrossRef] [PubMed]
- Ganzer, L.; Zappia, S.; Russo, M.; Ferretti, A.M.; Vohra, V.; Diterlizzi, M.; Antognazza, M.R.; Destri, S.; Virgili, T. Ultrafast spectroscopy on water-processable PCBM: Rod–coil block copolymer nanoparticles. Phys. Chem. Chem. Phys. 2020, 22, 26583–26591. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Choi, J.; Bang, D.; Kim, E.; Lim, E.-K.; Park, H.; Suh, J.-S.; Lee, K.; Yoo, K.-H.; Huh, Y.-M.; et al. Convertible Organic Nanoparticles for Near-Infrared Photothermal Ablation of Cancer Cells. Angew. Chem. Int. Ed. 2010, 50, 441–444. [Google Scholar] [CrossRef]
- Zangoli, M.; Di Maria, F. Synthesis, characterization, and biological applications of semiconducting polythiophene-based nanoparticles. View 2021, 2, 20200086. [Google Scholar] [CrossRef]
- Jana, B.; Ghosh, A.; Patra, A. Photon Harvesting in Conjugated Polymer-Based Functional Nanoparticles. J. Phys. Chem. Lett. 2017, 8, 4608–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Liu, R.; Li, L. Nanoparticles made of π-conjugated compounds targeted for chemical and biological applications. Chem. Commun. 2015, 51, 16733–16749. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.R.; Shaikh, H.; Garcia-Hernandez, J.D.; Vespa, M.; Fukui, T.; Manners, I. Functional nanoparticles through π-conjugated polymer self-assembly. Nat. Rev. Mater. 2021, 6, 7–26. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lu, L.; Sreearunothai, P.; Seeley, A.; Yim, K.-H.; Petrozza, A.; Murphy, C.E.; Beljonne, D.; Cornil, J.; Friend, R.H. Optoelectronic and Charge Transport Properties at Organic−Organic Semiconductor Interfaces: Comparison between Polyfluorene-Based Polymer Blend and Copolymer. J. Am. Chem. Soc. 2008, 130, 13120–13131. [Google Scholar] [CrossRef]
- Cadby, A.J.; Lane, P.A.; Mellor, H.; Martin, S.J.; Grell, M.; Giebeler, C.; Bradley, D.D.C.; Wohlgenannt, M.; An, C.; Vardeny, Z.V. Film morphology and photophysics of polyfluorene. Phys. Rev. B 2000, 62, 15604–15609. [Google Scholar] [CrossRef]
- Khan, A.L.T.; Sreearunothai, P.; Herz, L.M.; Banach, M.J.; Köhler, A. Morphology-dependent energy transfer within polyfluorene thin films. Phys. Rev. B 2004, 69, 085201. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Liu, Y.-Y.; Liu, X.; Lin, H.; Gao, K.; Lai, W.-Y.; Huang, W. Organic solid-state lasers: A materials view and future development. Chem. Soc. Rev. 2020, 49, 5885–5944. [Google Scholar] [CrossRef] [PubMed]
- Moses, D. High quantum efficiency luminescence from a conducting polymer in solution: A novel polymer laser dye. Appl. Phys. Lett. 1992, 60, 3215–3216. [Google Scholar] [CrossRef]
- Virgili, T.; Marinotto, D.; Lanzani, G.; Bradley, D.D. Ultrafast resonant optical switching in isolated polyfluorenes chains. Appl. Phys. Lett. 2005, 86, 091113. [Google Scholar] [CrossRef]
- Martini, I.B.; Craig, I.M.; Molenkamp, W.C.; Miyata, H.; Tolbert, S.H.; Schwartz, B.J. Controlling optical gain in semiconducting polymers with nanoscale chain positioning and alignment. Nat. Nanotechnol. 2007, 2, 647–652. [Google Scholar] [CrossRef] [PubMed]
- Portone, A.; Ganzer, L.; Branchi, F.; Ramos, R.; Caldas, M.J.; Pisignano, D.; Molinari, E.; Cerullo, G.; Persano, L.; Prezzi, D.; et al. Tailoring optical properties and stimulated emission in nanostructured polythiophene. Sci. Rep. 2019, 9, 7370. [Google Scholar] [CrossRef] [PubMed]
- Yap, B.K.; Xia, R.; Campoy-Quiles, M.; Stavrinou, P.N.; Bradley, D.D.C. Simultaneous optimization of charge-carrier mobility and optical gain in semiconducting polymer films. Nat. Mater. 2008, 7, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wu, Y.; Lian, S.; Gao, J.; Zhang, S.; Hai, G.; Sun, C.; Li, X.; Xia, R.; Cabanillas-Gonzalez, J.; et al. Simultaneously Enhancing Photoluminescence Quantum Efficiency and Optical Gain of Polyfluorene via Backbone Intercalation of 2,5-Dimethyl-1,4-Phenylene. Adv. Opt. Mater. 2020, 8, 2000187. [Google Scholar] [CrossRef]
- Mroz, M.M.; Sforazzini, G.; Zhong, Y.; Wong, K.S.; Anderson, H.L.; Lanzani, G.; Cabanillas-Gonzalez, J. Amplified Spontaneous Emission in Conjugated Polyrotaxanes Under Quasi-cw Pumping. Adv. Mater. 2013, 25, 4347–4351. [Google Scholar] [CrossRef] [PubMed]
- Virgili, T.; Botta, C.; Mróz, M.M.; Parrenin, L.; Brochon, C.; Cloutet, E.; Pavlopoulou, E.; Hadziioannou, G.; Geoghegan, M. Size-Dependent Photophysical Behavior of Low Bandgap Semiconducting Polymer Particles. Front. Chem. 2019, 7, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.; Seo, J.H.; Park, S.H.; Beaupré, S.; Leclerc, M.; Heeger, A.J. A Thermally Stable Semiconducting Polymer. Adv. Mater. 2010, 22, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Aïch, R.B.; Blouin, N.; Bouchard, A.; Leclerc, M. Electrical and Thermoelectric Properties of Poly(2,7-Carbazole) Derivatives. Chem. Mater. 2009, 21, 751–757. [Google Scholar] [CrossRef]
- Banerji, N.R.; Cowan, S.; Leclerc, M.; Vauthey, E.; Heeger, A.J. Exciton Formation, Relaxation, and Decay in PCDTBT. J. Am. Chem. Soc. 2010, 132, 17459–17470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etzold, F.; Howard, I.A.; Mauer, R.; Meister, M.; Kim, T.-D.; Lee, K.-S.; Baek, N.S.; Laquai, F. Ultrafast Exciton Dissociation Followed by Nongeminate Charge Recombination in PCDTBT:PCBM Photovoltaic Blends. J. Am. Chem. Soc. 2011, 133, 9469–9479. [Google Scholar] [CrossRef]
- Brown, P.J.; Thomas, D.S.; Köhler, A.; Wilson, J.S.; Kim, J.-S.; Ramsdale, C.M.; Sirringhaus, H.; Friend, R.H. Effect of interchain interactions on the absorption and emission of poly(3-hexylthiophene). Phys. Rev. B 2003, 67, 064203. [Google Scholar] [CrossRef] [Green Version]
- Eggimann, H.J.; Le Roux, F.; Herz, L.M. How β-phase content moderates chain cenergy transfer in polyfluorene films. J. Phys. Chem. Lett. 2019, 10, 1729–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.; Bout, D.A.V. Probing the molecular weight dependent intramolecular interactions in single molecules of PCDTBT. J. Mater. Chem. C 2017, 5, 9786–9791. [Google Scholar] [CrossRef]
- McQuarrie, D.A. Statistical Mechanics; University Science Books: Sausalito, CA, USA, 2000. [Google Scholar]
- Baghgar, M.; Labastide, J.A.; Bokel, F.; Hayward, R.C.; Barnes, M.D. Effect of Polymer Chain Folding on the Transition from H- to J-Aggregate Behavior in P3HT Nanofibers. J. Phys. Chem. C 2014, 118, 2229–2235. [Google Scholar] [CrossRef]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Accounts Chem. Res. 2010, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Parrenin, L.; Laurans, G.; Pavlopoulou, E.; Fleury, G.; Pecastaings, G.; Brochon, C.; Vignau, L.; Hadziioannou, G.; Cloutet, E. Photoactive Donor–Acceptor Composite Nanoparticles Dispersed in Water. Langmuir 2017, 33, 1507–1515. [Google Scholar] [CrossRef]
- Wakim, S.; Beaupré, S.; Blouin, N.; Aich, B.-R.; Rodman, S.; Gaudiana, R.; Tao, Y.; Leclerc, M. Highly efficient organic solar cells based on a poly(2,7-carbazole) derivative. J. Mater. Chem. 2009, 19, 5351–5358. [Google Scholar] [CrossRef]
- Parrenin, L.; Brochon, C.; Hadziioannou, G.; Cloutet, E.; Hadziiannou, G. Low Bandgap Semiconducting Copolymer Nanoparticles by Suzuki Cross-Coupling Polymerization in Alcoholic Dispersed Media. Macromol. Rapid Commun. 2015, 36, 1816–1821. [Google Scholar] [CrossRef] [PubMed]
- Virgili, T.; Forni, A.; Cariati, E.; Pasini, D.; Botta, C. Direct Evidence of Torsional Motion in an Aggregation-Induced Emissive Chromophore. J. Phys. Chem. C 2013, 117, 27161–27166. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geoghegan, M.; Mróz, M.M.; Botta, C.; Parrenin, L.; Brochon, C.; Cloutet, E.; Pavlopoulou, E.; Hadziioannou, G.; Virgili, T. Optical Gain in Semiconducting Polymer Nano and Mesoparticles. Molecules 2021, 26, 1138. https://doi.org/10.3390/molecules26041138
Geoghegan M, Mróz MM, Botta C, Parrenin L, Brochon C, Cloutet E, Pavlopoulou E, Hadziioannou G, Virgili T. Optical Gain in Semiconducting Polymer Nano and Mesoparticles. Molecules. 2021; 26(4):1138. https://doi.org/10.3390/molecules26041138
Chicago/Turabian StyleGeoghegan, Mark, Marta M. Mróz, Chiara Botta, Laurie Parrenin, Cyril Brochon, Eric Cloutet, Eleni Pavlopoulou, Georges Hadziioannou, and Tersilla Virgili. 2021. "Optical Gain in Semiconducting Polymer Nano and Mesoparticles" Molecules 26, no. 4: 1138. https://doi.org/10.3390/molecules26041138
APA StyleGeoghegan, M., Mróz, M. M., Botta, C., Parrenin, L., Brochon, C., Cloutet, E., Pavlopoulou, E., Hadziioannou, G., & Virgili, T. (2021). Optical Gain in Semiconducting Polymer Nano and Mesoparticles. Molecules, 26(4), 1138. https://doi.org/10.3390/molecules26041138