
Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups

 , , , ,
, , , ,
Abstract
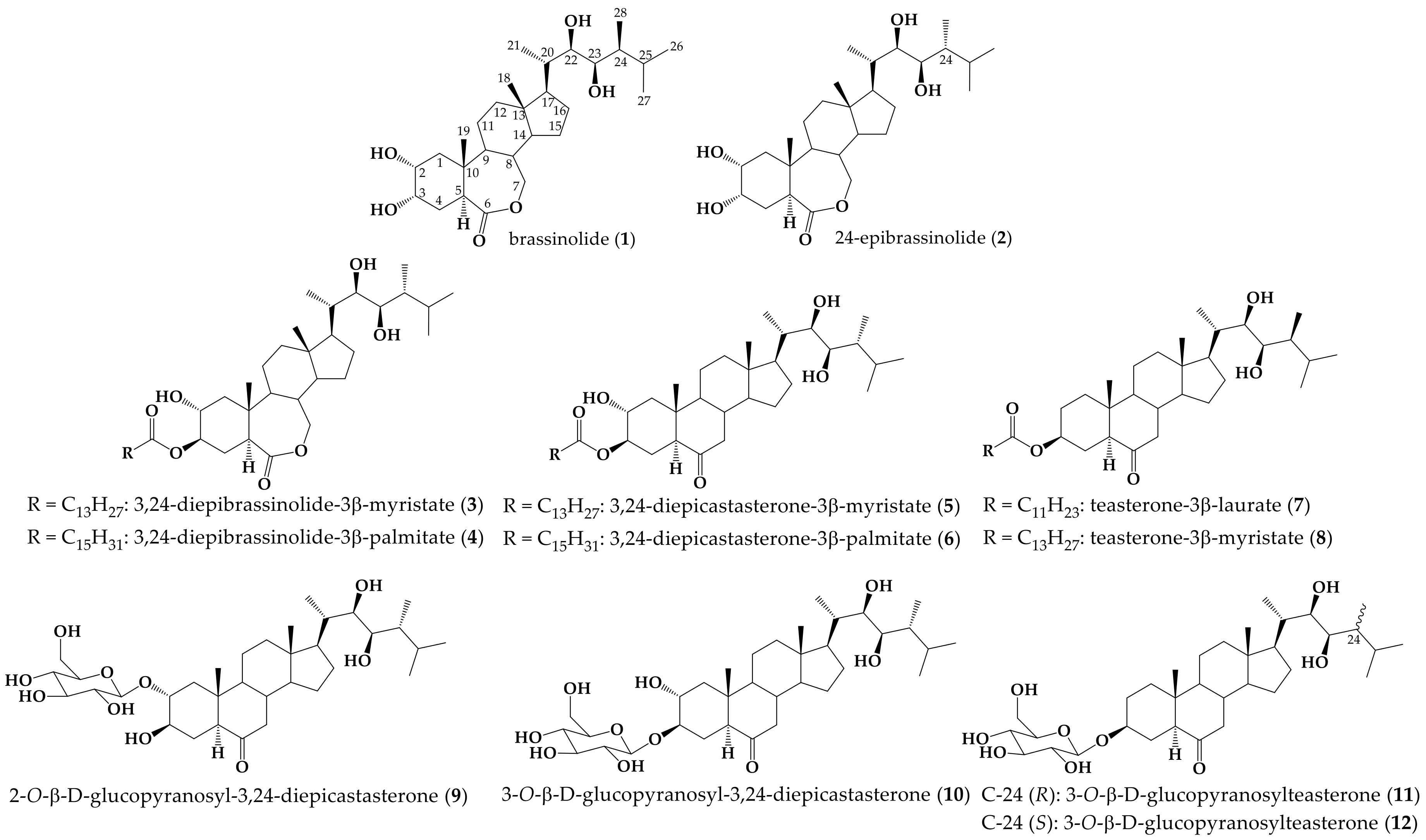
1. Introduction
2. Results and Discussion
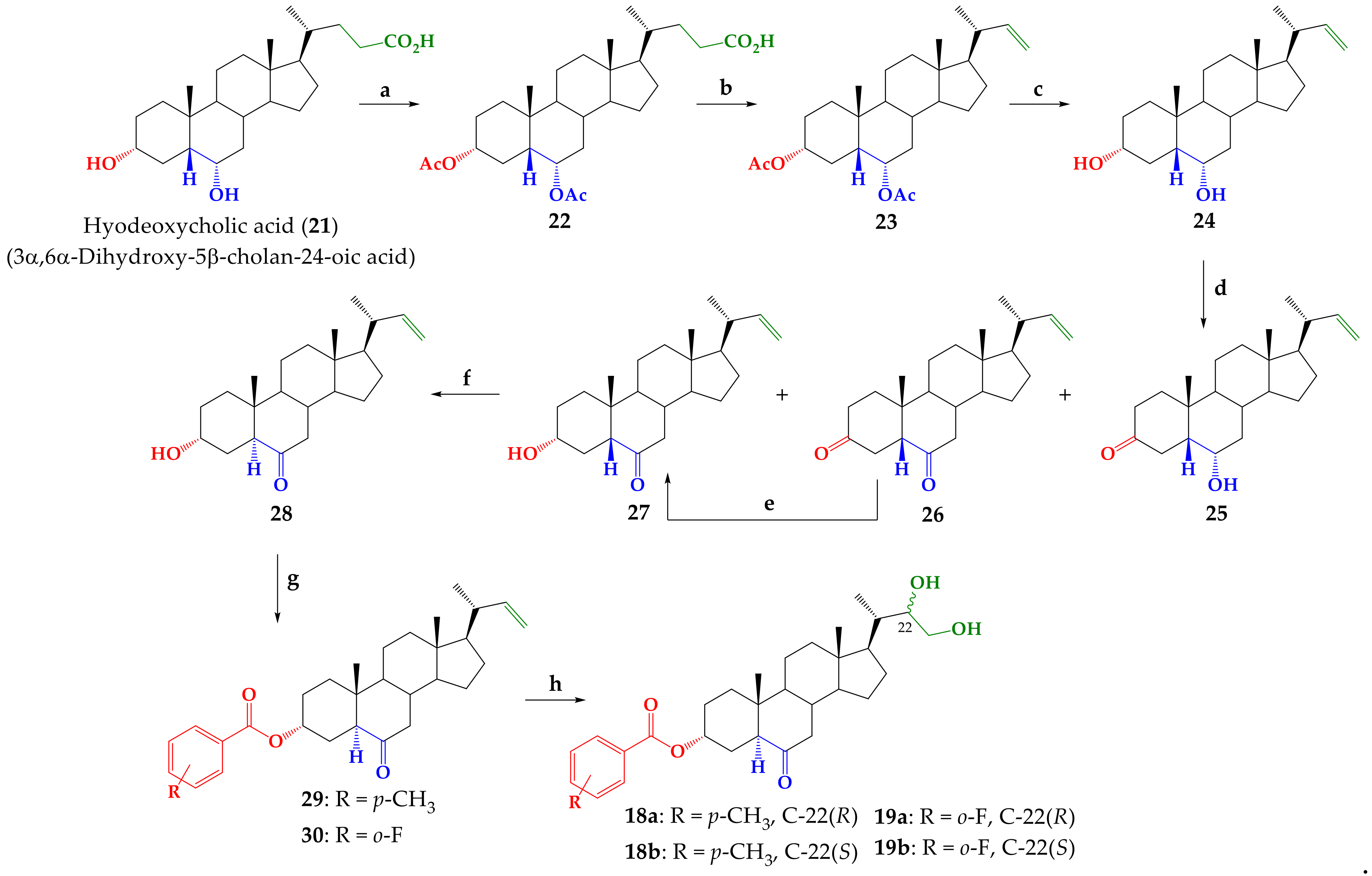
2.1. Chemistry
2.2. Biological
3. Materials and Methods
3.1. Chemistry
3.1.1. Synthesis
3α,6α-Diacetoxy-5β-cholan-24-oic acid (22)
24-Nor-5β-cholan-22-ene-3α,6α-diyl diacetate (23)
24-Nor-5β-chol-22-ene-3α,6α-diol (24)
6α-Hydroxy-24-nor-5β-chol-22-en-3-one (25), 24-nor-5β-chol-22-ene-3,6-dione (26) and 3α-hydroxy-24-nor-5β-chol-22-en-6-one (27)
3α-Hydroxy-24-nor-5β-chol-22-en-6-one (27) from 24-nor-5β-chol-22-ene-3,6-dione (26)
3α-Hydroxy-24-nor-5α-chol-22-en-6-one (28)
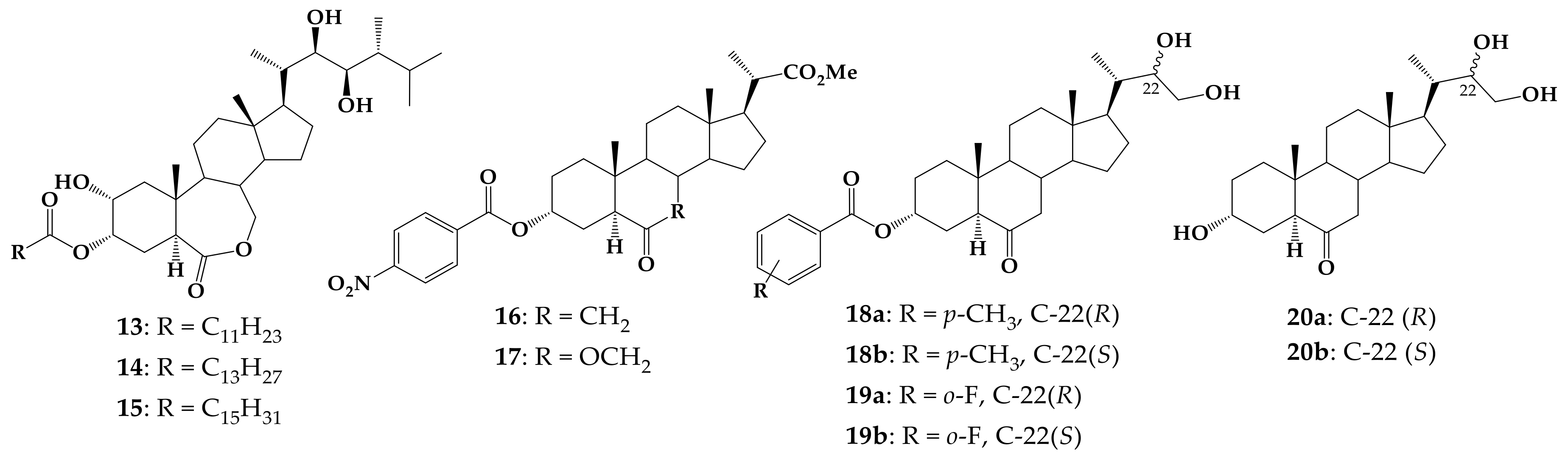
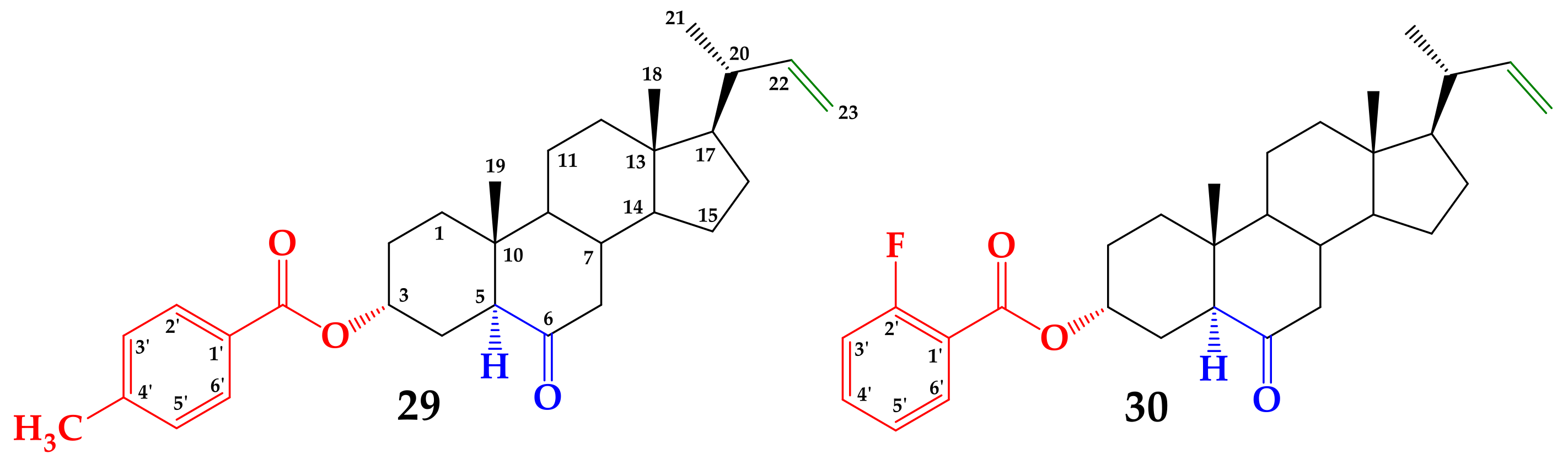
6-Oxo-24-nor-5α-chol-22-en-3α-yl 4-methylbenzoate (29)
6-Oxo-24-nor-5α-chol-22-en-3α-yl 2-fluorobenzoate (30)
(22R)-22,23-Dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl 4-methylbenzoate (18a) and (22S)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl 4-methylbenzoate (18b)
(22R)-22,23-Dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl 2-fluorobenzoate (19a) and (22S)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl 2-fluorobenzoate (19b)
3.2. Biological
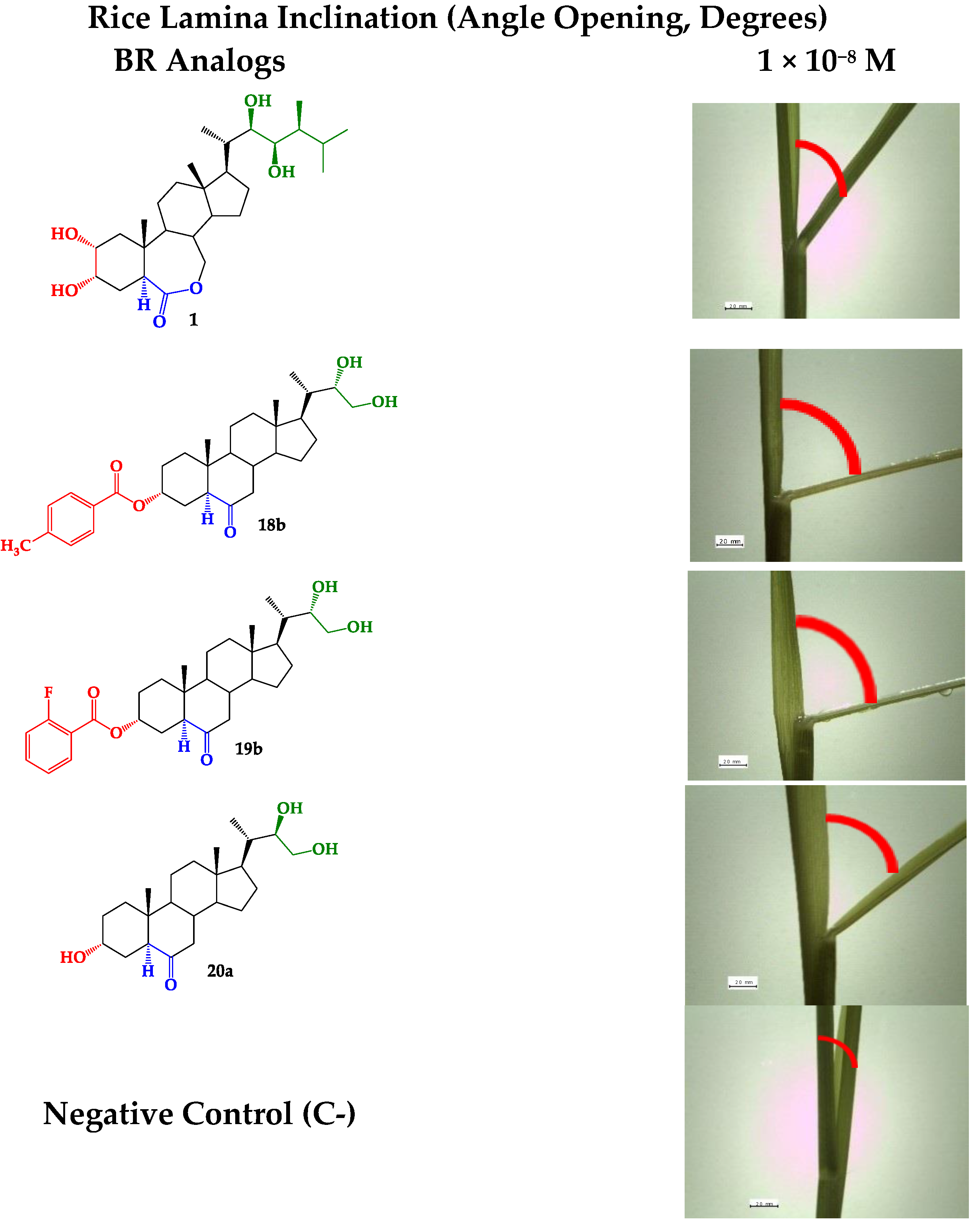
Rice Lamina Inclination Test (RLIT)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oh, M.-H.; Honey, S.H.; Tax, F.E. The Control of Cell Expansion, Cell Division, and Vascular Development by Brassinosteroids: A Historical Perspective. Int. J. Mol. Sci. 2020, 21, 1743. [Google Scholar] [CrossRef]
- Clouse, S.D.; Sasse, J.M. Brassinosteroids: Essential regulators of plant growth and development. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 427–451. [Google Scholar] [CrossRef]
- Yokota, T. Chapter 12-Brassinosteroids. In New Comprehensive Biochemistry; Hooykaas, P.J.J., Hall, M.A., Libbenga, K.R., Eds.; Elsevier: New York, NY, USA, 1999; Volume 33, p. 277293. [Google Scholar]
- Sasse, J.M. Physiological actions of brassinosteroids: An update. J. Plant Growth Regul. 2003, 22, 276–288. [Google Scholar] [CrossRef]
- Müssig, C. Brassinosteroid-promoted growth. Plant Biol. 2005, 7, 110–117. [Google Scholar] [CrossRef]
- Grove, M.D.; Spencer, G.F.; Rohwedder, W.K.; Mandava, N.; Worley, J.F.; Warthen, J.D.; Steffens, G.L.; Flippenanderson, J.L.; Cook, J.C. Brassinolide, a Plant Growth-Promoting Steroid Isolated from Brassica-Napus Pollen. Nature 1979, 281, 216–217. [Google Scholar] [CrossRef]
- Khripach, V.; Zhabinskii, V.; de Groot, A. Twenty years of brassinosteroids: Steroidal plant hormones warrant better crops for the XXI century. Ann. Bot. 2000, 86, 441–447. [Google Scholar] [CrossRef]
- Bajguz, A. Metabolism of brassinosteroids in plants. Plant Physiol. Biochem. 2007, 45, 95–107. [Google Scholar] [CrossRef]
- Abe, H.; Honjo, C.; Kyokawa, Y.; Asakawa, S.; Natsume, M.; Narushima, M. 3-Oxoteasterone and the Epimerization of Teasterone: Identification in Lily Anthers and Distylium racemosum Leaves and Its Biotransformation into Typhasterol. Biosci. Biotechnol. Biochem. 1994, 58, 986–989. [Google Scholar] [CrossRef]
- Kolbe, A.; Schneider, B.; Porzel, A.; Adam, G. Metabolism of 24-epi-castasterone and 24-epi-brassinolide in cell suspension cultures of Ornithopus sativus. Phytochemistry 1996, 41, 163–167. [Google Scholar] [CrossRef]
- Kolbe, A.; Schneider, B.; Porzel, A.; Schmidt, J.; Adam, G. Acyl-conjugated metabolites of brassinosteroids in cell suspension cultures of Ornithopus sativus. Phytochemistry 1995, 38, 633–636. [Google Scholar] [CrossRef]
- Kolbe, A.; Schneider, B.; Porzel, A.; Voigt, B.; Krauss, G.; Adam, G. Pregnane-type metabolites of brassinosteroids in cell suspension cultures of Ornithopus sativus. Phytochemistry 1994, 36, 671–673. [Google Scholar] [CrossRef]
- Soeno, K.; Asakawa, S.; Natsume, M.; Abe, H. Reversible Conversion between Teasterone and Its Ester Conjugates in Lily Cell Cultures. J. Pestic. Sci. 2000, 25, 117–122. [Google Scholar] [CrossRef][Green Version]
- Soeno, K.; Kyokawa, Y.; Natsume, M.; Abe, H. Teasterone-3-O-β-d-glucopyranoside, A New Conjugated Brassinosteroid Metabolite from Lily Cell Suspension Cultures and Its Identification in Lily Anthers. Biosci. Biotechnol. Biochem. 2000, 64, 702–709. [Google Scholar] [CrossRef]
- Hai, T.; Schneider, B.; Adam, G. Metabolic conversion of 24-epi-brassinolide into pentahydroxylated brassinosteroid glucosides in tomato cell cultures. Phytochemistry 1995, 40, 443–448. [Google Scholar] [CrossRef]
- Hai, T.; Schneider, B.; Porzel, A.; Adam, G. Metabolism of 24-epi-castasterone in cell suspension cultures of Lycopersicon esculentum. Phytochemistry 1996, 41, 197–201. [Google Scholar] [CrossRef]
- Kolbe, A.; Porzel, A.; Schneider, B.; Adam, G. Diglycosidic metabolites of 24-epi-teasterone in cell suspension cultures of Lycopersicon esculentum L. Phytochemistry 1997, 46, 1019–1022. [Google Scholar] [CrossRef]
- Kolbe, A.; Schneider, B.; Porzel, A.; Adam, G. Metabolic inversion of the 3-hydroxy function of brassinosteroids. Phytochemistry 1998, 48, 467–470. [Google Scholar] [CrossRef]
- Schneider, B.; Kolbe, A.; Porzel, A.; Adam, G. A metabolite of 24-epi-brassinolide in cell suspension cultures of Lycopersicon esculentum. Phytochemistry 1994, 36, 319–321. [Google Scholar] [CrossRef]
- Suzuki, H.; Kim, S.-K.; Takahashi, N.; Yokota, T. Metabolism of castasterone and brassinolide in mung bean explant. Phytochemistry 1993, 33, 1361–1367. [Google Scholar] [CrossRef]
- Khripach, V.A.; Zhabinskii, V.N.; Tsavlovskii, D.V. Synthesis of fatty acyl derivatives of 24-epibrassinolide. J. Steroid Biochem. Mol. Biol. 2013, 137, 345–354. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rarova, L.; Berka, K.; Strnad, M. Biological activities of new monohydroxylated brassinosteroid analogues with a carboxylic group in the side chain. Steroids 2014, 85, 58–64. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, D.; Sun, X.; Ding, T.; Lei, B.; Zhang, C. Structure-activity relationship of brassinosteroids and their agricultural practical usages. Steroids 2017, 124, 1–17. [Google Scholar] [CrossRef]
- Thompson, M.J.; Meudt, W.J.; Mandava, N.B.; Dutky, S.R.; Lusby, W.R.; Spaulding, D.W. Synthesis of Brassinosteroids and Relationship of Structure to Plant Growth-Promoting Effects. Steroids 1982, 39, 89–105. [Google Scholar] [CrossRef]
- Takatsuto, S.; Ikekawa, N.; Morishita, T.; Abe, H. Structure Activity Relationship of Brassinosteroids with Respect to the A/B-Ring Functional-Groups. Chem. Pharm. Bull. 1987, 35, 211–216. [Google Scholar] [CrossRef]
- Takatsuto, S.; Yazawa, N.; Ikekawa, N.; Takematsu, T.; Takeuchi, Y.; Koguchi, M. Structure Activity Relationship of Brassinosteroids. Phytochemistry 1983, 22, 2437–2441. [Google Scholar] [CrossRef]
- Zullo, M.A.T.; Adam, G. Brassinosteroid phytohormones: Structure, bioactivity and applications. Braz. J. Plant Physiol. 2002, 14, 143–181. [Google Scholar] [CrossRef]
- Peres, A.L.G.L.; Soares, J.S.; Tavares, R.G.; Righetto, G.; Zullo, M.A.T.; Mandava, N.B.; Menossi, M. Brassinosteroids, the Sixth Class of Phytohormones: A Molecular View from the Discovery to Hormonal Interactions in Plant Development and Stress Adaptation. Int. J. Mol. Sci. 2019, 20, 331. [Google Scholar] [CrossRef]
- Ferrer-Pertuz, K.; Espinoza, L.; Mella, J. Insights into the Structural Requirements of Potent Brassinosteroids as Vegetable Growth Promoters Using Second-Internode Elongation as Biological Activity: CoMFA and CoMSIA Studies. Int. J. Mol. Sci. 2017, 18, 2734. [Google Scholar] [CrossRef]
- Wang, Q.; Xu, J.; Liu, X.; Gong, W.; Zhang, C. Synthesis of brassinosteroids analogues from laxogenin and their plant growth promotion. Nat. Prod. Res. 2015, 29, 149–157. [Google Scholar] [CrossRef]
- Duran, M.I.; Gonzalez, C.; Acosta, A.; Olea, A.F.; Diaz, K.; Espinoza, L. Synthesis of Five Known Brassinosteroid Analogs from Hyodeoxycholic Acid and Their Activities as Plant-Growth Regulators. Int. J. Mol. Sci. 2017, 18, 516. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rárová, L.; Korinkova, P.; Mikulík, J.; Budesinsky, M.; Béres, T.; Berka, K.; Lu, Q.; et al. Design, synthesis and biological activities of new brassinosteroid analogues with a phenyl group in the side chain. Org. Biomol. Chem. 2016, 14, 8691–8701. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; Gil, R.P.; Martinez, C.S.P.; Manchado, F.C. Spirostanic analogues of teasterone. Synthesis, characterisation and biological activity of laxogenin, (23S)-hydroxylaxogenin and 23-ketolaxogenin (23-oxolaxogenin). J. Chem. Soc. Perkin Trans. 1 2001, 261–266. [Google Scholar] [CrossRef]
- Romero-Avila, M.; de Dios-Bravo, G.; Mendez-Stivalet, J.M.; Rodriguez-Sotres, R.; Iglesias-Arteaga, M.A. Synthesis and biological activity of furostanic analogues of brassinosteroids bearing the 5 alpha-hydroxy-6-oxo moiety. Steroids 2007, 72, 955–959. [Google Scholar] [CrossRef]
- Gomes, M.d.M.d.A.; Torres Netto, A.; Campostrini, E.; Bressan-Smith, R.; Zullo, M.A.T.; Ferraz, T.M.; Siqueira, L.d.N.; Leal, N.R.; Núñez-Vázquez, M. Brassinosteroid analogue affects the senescence in two papaya genotypes submitted to drought stress. Exp. Plant Physiol. 2013, 25, 186–195. [Google Scholar] [CrossRef]
- Brosa, C.; Soca, L.; Terricabras, E.; Ferrer, J.C.; Alsina, A. New synthetic brassinosteroids: A 5 alpha-hydroxy-6-ketone analog with strong plant growth promoting activity. Tetrahedron 1998, 54, 12337–12348. [Google Scholar] [CrossRef]
- Brosa, C.; Capdevila, J.M.; Zamora, I. Brassinosteroids: A new way to define the structural requirements. Tetrahedron 1996, 52, 2435–2448. [Google Scholar] [CrossRef]
- Diaz, K.; Espinoza, L.; Carvajal, R.; Conde-Gonzalez, M.; Niebla, V.; Olea, A.F.; Coll, Y. Biological Activities and Molecular Docking of Brassinosteroids 24-Norcholane Type Analogs. Int. J. Mol. Sci. 2020, 21, 1832. [Google Scholar] [CrossRef]
- Back, T.; Pharis, R. Structure-Activity Studies of Brassinosteroids and the Search for Novel Analogues and Mimetics with Improved Bioactivity. J. Plant Growth Regul. 2004, 22, 350–361. [Google Scholar] [CrossRef]
- Back, T.G.; Janzen, L.; Pharis, R.P.; Yan, Z. Synthesis and bioactivity of C-2 and C-3 methyl ether derivatives of brassinolide. Phytochemistry 2002, 59, 627–634. [Google Scholar] [CrossRef]
- Carvajal, R.; Gonzalez, C.; Olea, A.F.; Fuentealba, M.; Espinoza, L. Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules 2018, 23, 1306. [Google Scholar] [CrossRef]
- Oyarce, J.; Aitken, V.; Gonzalez, C.; Ferrer, K.; Olea, A.F.; Parella, T.; Espinoza, L. Synthesis and structural determination of new brassinosteroid 24-nor-5α-cholane type analogs. Molecules 2019, 24, 4612. [Google Scholar] [CrossRef]
- Huang, L.F.; Zhou, W.S. Studies on Steroidal Plant-Growth Regulators. Part 33. Novel Method for Construction of the Side-Chain of 23-Arylbrassinosteroids Via Heck Arylation and Asymmetric Dihydroxylation As Key Steps. J. Chem. Soc. Perkin Trans. 1 1994, 3579–3585. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. Studies on Steroidal Plant-Growth Hormones. 2. Stereoselective Synthesis of (22S, 23S)-Typhasterol from Hyodeoxycholic Acid. Acta Chim. Sin. 1985, 43, 1060–1067. [Google Scholar]
- Tian, W.S.; Zhou, W.S.; Jiang, B.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator. 9. The Preparation of 22R-Penta-Nor-Brassinolides and 22S-24,25,26,27,28-Penta-Nor-Brassinolides. Acta Chim. Sin. 1989, 47, 1017–1021. [Google Scholar]
- Yang, Y.X.; Zheng, L.T.; Shi, J.J.; Gao, B.; Chen, Y.K.; Yang, H.C.; Chen, H.L.; Li, Y.C.; Zhen, X.C. Synthesis of 5 alpha-cholestan-6-one derivatives and their inhibitory activities of NO production in activated microglia: Discovery of a novel neuroinflammation inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. The Synthesis of Steroids Containing Structural Unit of A, B Ring of Brassinolide and Ecdysone from Hyodeoxycholic Acid. Acta Chim. Sin. 1984, 42, 1173–1177. [Google Scholar]
- Zhou, W.S. The Synthesis of Brassinosteroid. Pure Appl. Chem. 1989, 61, 431–434. [Google Scholar] [CrossRef]
- Herrera, H.; Carvajal, R.; Olea, A.F.; Espinoza, L. Structural modifications of deoxycholic acid to obtain three known brassinosteroid analogues and full NMR spectroscopic characterization. Molecules 2016, 21, 1139. [Google Scholar] [CrossRef]
- Huang, B.; Du, D.; Zhang, R.; Wu, X.; Xing, Z.; He, Y.; Huang, W. Synthesis, characterization and biological studies of diosgenyl analogues. Bioorg Med. Chem Lett 2012, 22, 7330–7334. [Google Scholar] [CrossRef]
- Jones, S.R.; Selinsky, B.S.; Rao, M.N.; Zhang, X.; Kinney, W.A.; Tham, F.S. Efficient Route to 7α-(Benzoyloxy)-3-dioxolane Cholestan-24(R)-ol, a Key Intermediate in the Synthesis of Squalamine. J. Org. Chem. 1998, 63, 3786–3789. [Google Scholar] [CrossRef]
- Li, H.; Wang, H.; Jang, S. Rice Lamina Joint Inclination Assay. Bio-Protocol. 2017, 7, e2409. [Google Scholar] [CrossRef]
- Jang, S.; An, G.; Li, H.-Y. Rice Leaf Angle and Grain Size Are Affected by the OsBUL1 Transcriptional Activator Complex. Plant Physiol. 2017, 173, 688–702. [Google Scholar] [CrossRef]
- Bajguz, A.; Tretyn, A. The chemical characteristic and distribution of brassinosteroids in plants. Phytochemistry 2003, 62, 1027–1046. [Google Scholar] [CrossRef]
- Wada, K.; Marumo, S. Synthesis and Plant Growth-Promoting Activity of Brassinolide Analogs. Agric. Biol. Chem. 1981, 45, 2579–2585. [Google Scholar]
- Han, K.S.; Ko, K.W.; Nam, S.J.; Park, S.H.; Kim, S.K. Optimization of a rice lamina inclination assay for detection of brassinosteroids: I. effect of phytohormones on the inclination activity. J. Plant Biol. 1997, 40, 240–244. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | H-3 (δH ppm) | H-6 (δH ppm) | C-3 (δC ppm) | C-6 (δC ppm) |
|---|---|---|---|---|
| 24 | 3.48–3.42 | 4.02–3.96 | 71.72 | 67.63 |
| 25 | - | 4.15–4.10 | 212.63 | 67.73 |
| 26 | - | - | 208.65 | 210.82 |
| 27 | 3.70–3.62 | - | 70.18 | 213.89 |
| H/C Signal | Compound 18a | Compound 18b |
|---|---|---|
| H-21 | 0.921 ppm (3H, d, J = 6.7 Hz) | 0.953 ppm (3H, d, J = 7.0 Hz) |
| H-22 | 3.66–3.61 ppm (1H, m) | 3.51 ppm (1H, t, J = 10.2 Hz) |
| H-23a | 3.80 ppm (1H, ddd, J = 8.9, 3.3 and 1.2 Hz) | 3.83–3.76 ppm (1H, m) |
| H-23b | 3.52 ppm (1H, dd, J = 10.8 and 3.3 Hz) | 3.69–3.57 ppm (1H, m) |
| C21 | 12.73 ppm | 13.15 ppm |
| C22 | 74.14 ppm | 73.92 ppm |
| C23 | 66.16 ppm | 62.54 ppm |
| H/C Signal | Compound 19a | Compound 19b |
|---|---|---|
| H-21 | 0.917 ppm (3H, d, J = 6.4 Hz) | 0.954 ppm (3H, d, J = 6.7 Hz) |
| H-22 | 3.75–3.39 ppm (1H, m) | 3.51 ppm (1H, t, J = 10.2 Hz) |
| H-23a | 3.87–3.76 ppm (1H, m) | 3.84–3.76 ppm (1H, m) |
| H-23b | 3.75–3.39 ppm (1H, m) | 3.71–3.59 ppm (1H, m) |
| C21 | 12.73 ppm | 13.13 ppm |
| C22 | 75.56 ppm | 73.90 ppm |
| C23 | 61.15 ppm | 62.52 ppm |
| Bending Angle between Laminae and Sheaths (Degrees ± SD) | |||
|---|---|---|---|
| Compounds | 1 × 10−8 M | 1 × 10−7 M | 1 × 10−6 M |
| 1 (C+) | 31 ± 11 | 41 ± 4.5 | 70 ± 7.6 |
| 18a | 61 ± 6.3 * | 68 ± 9.6 * | 46 ± 7.5 * |
| 18b | 64 ± 3.3 * | 48 ± 2.9 | 14 ± 4.8 * |
| 19a | 43 ± 5.0 * | 58 ± 2.4 * | 68 ± 9.6 |
| 19b | 68 ± 5.0 * | 61 ± 2.5 * | 30 ± 0.0 * |
| 20a† | 45 ± 9.5 * | 31 ± 5.0 | 24 ± 5.8 * |
| 20b† | 35 ± 3.0 | 60 ± 3.0 * | 62 ± 12 |
| Control (C−) | 7 ± 5.0 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrer, K.; Díaz, K.; Kvasnica, M.; Olea, A.F.; Cuellar, M.; Espinoza, L. Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups. Molecules 2021, 26, 1173. https://doi.org/10.3390/molecules26041173
Ferrer K, Díaz K, Kvasnica M, Olea AF, Cuellar M, Espinoza L. Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups. Molecules. 2021; 26(4):1173. https://doi.org/10.3390/molecules26041173
Chicago/Turabian StyleFerrer, Karoll, Katy Díaz, Miroslav Kvasnica, Andrés F. Olea, Mauricio Cuellar, and Luis Espinoza. 2021. "Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups" Molecules 26, no. 4: 1173. https://doi.org/10.3390/molecules26041173
APA StyleFerrer, K., Díaz, K., Kvasnica, M., Olea, A. F., Cuellar, M., & Espinoza, L. (2021). Synthesis of New Brassinosteroid 24-Norcholane Type Analogs Conjugated in C-3 with Benzoate Groups. Molecules, 26(4), 1173. https://doi.org/10.3390/molecules26041173