
Simultaneous Quantification of 3′- and 6′-Sialyllactose in Rat Plasma Using Liquid Chromatography-Tandem Mass Spectrometry and Its Application to a Pharmacokinetic Study


Abstract
1. Introduction
2. Results and Discussion
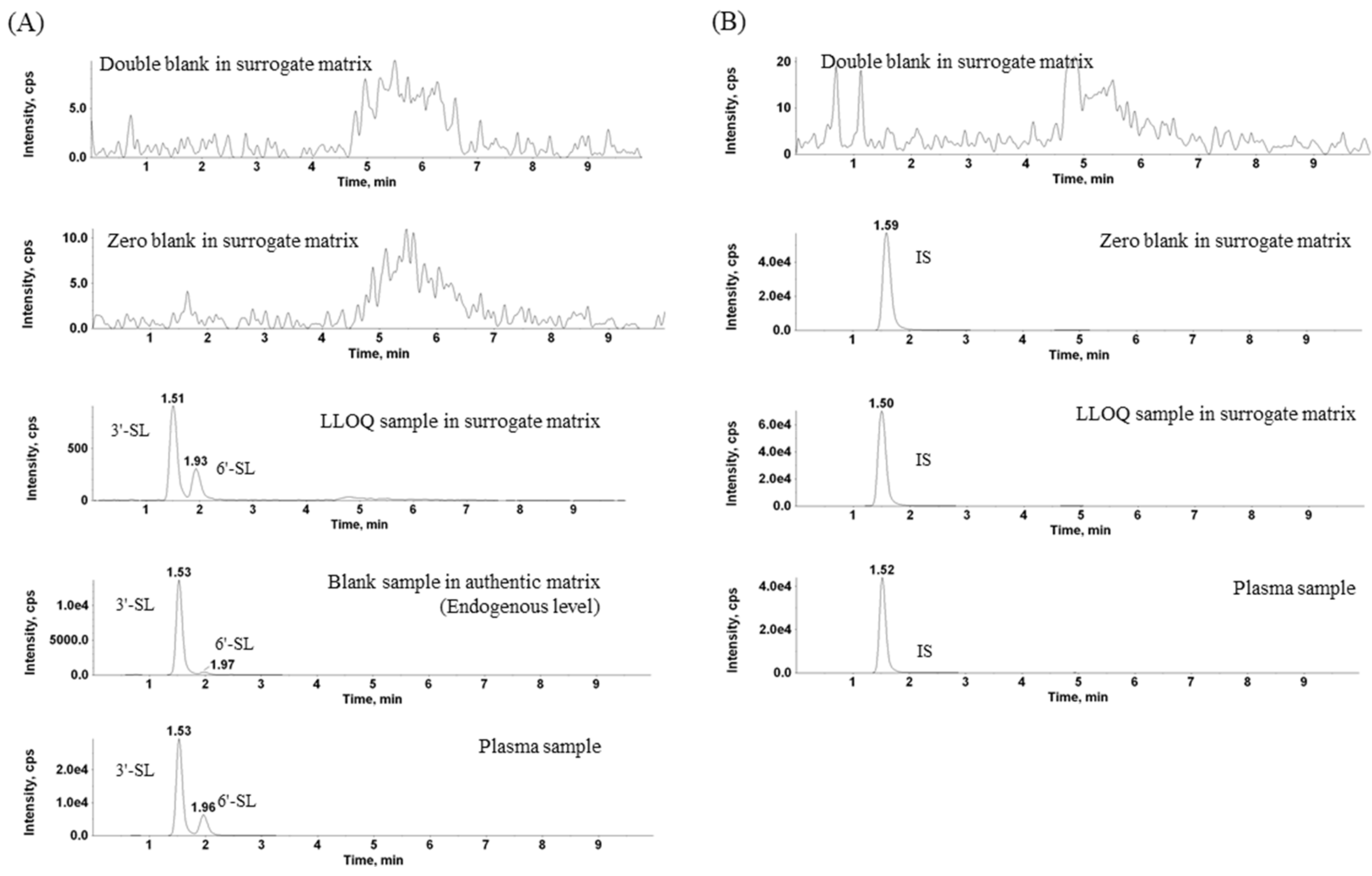
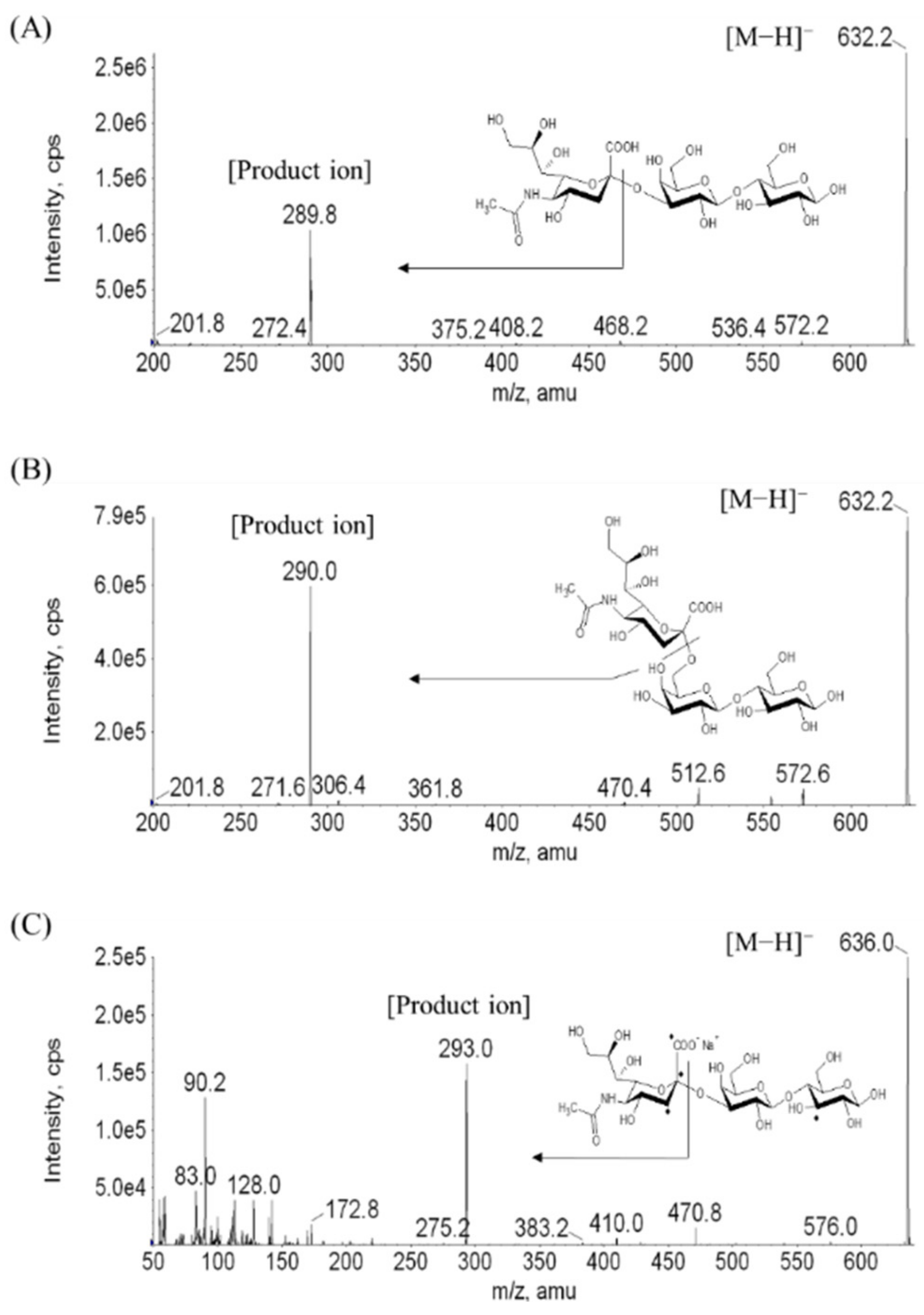
2.1. Separation of 3′-SL and 6′-SL and Extraction
2.2. Method Validation
2.2.1. Selectivity
2.2.2. Linearity, Accuracy and Precision
2.2.3. Matrix Effect and Recovery
2.2.4. Parallelism
2.2.5. Carryover and Dilution Integrity
2.2.6. Stability
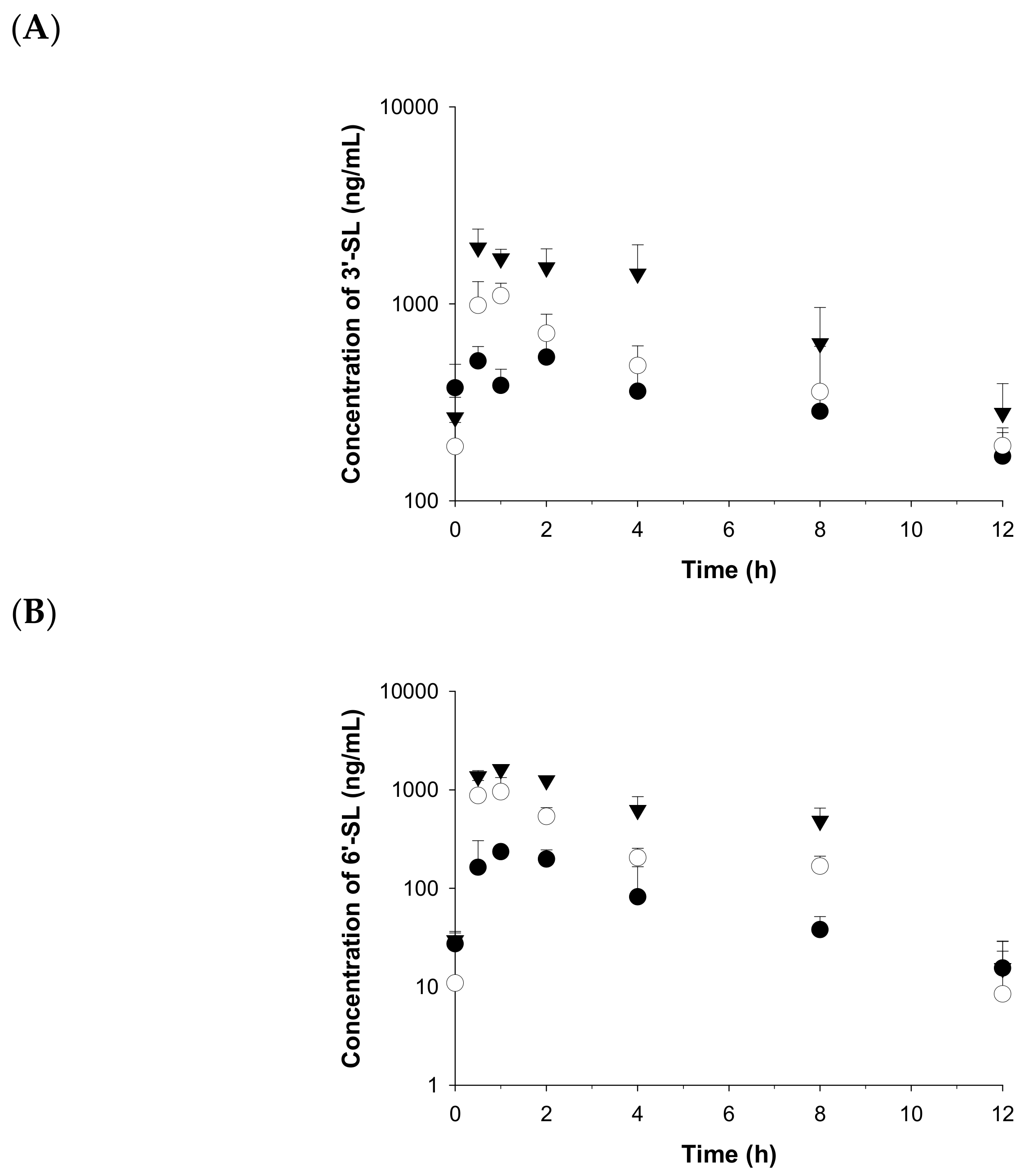
2.3. Application to a Pharmacokinetic Study in Rats
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. HPLC–MS
3.3. Preparation of Standard Stock Solutions and Quality Control Samples
3.4. Sample Preparation
3.5. Method Validation
3.5.1. Selectivity
3.5.2. Linearity, Accuracy, and Precision
3.5.3. Matrix Effect and Recovery
3.5.4. Parallelism
3.5.5. Carryover and Dilution Integrity
3.5.6. Stability
3.6. Application to a Pharmacokinetic Study in Rats
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Martín-Sosa, S.; Martín, M.-J.; García-Pardo, L.-A.; Hueso, P. Sialyloligosaccharides in Human and Bovine Milk and in Infant Formulas: Variations with the Progression of Lactation. J. Dairy Sci. 2003, 86, 52–59. [Google Scholar] [CrossRef]
- Kim, D.H.; Gurung, R.B.; Seo, W.; Lee, A.W.; Woo, J. Toxicological evaluation of 3′-sialyllactose sodium salt. Regul. Toxicol. Pharm. 2018, 94, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Gurung, R.B.; Kim, D.H.; Kim, L.; Lee, A.W.; Wang, Z.; Gao, Y. Toxicological evaluation of 6′-sialyllactose (6′-SL) sodium salt. Regul. Toxicol. Pharm. 2018, 95, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Thakare, R.; Chhonker, Y.S.; Gautam, N.; AlAmoudi, J.A.; Alnouti, Y. Quantitative analysis of endogenous compounds. J. Pharm. Biomed. Anal. 2016, 128, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Santos-Fandila, A.; Zafra-Gómez, A.; Vázquez, E.; Navalón, A.; Rueda, R.; Ramirez, M. Ultra high performance liquid chromatography–tandem mass spectrometry method for the determination of soluble milk glycans in rat serum. Talanta 2014, 118, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Chen, C.; Newburg, D.S. Quantification of neutral human milk oligosaccharides by graphitic carbon high-performance liquid chromatography with tandem mass spectrometry. Anal. Biochem. 2013, 433, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Tao, N.; DePeters, E.J.; German, J.B.; Grimm, R.; Lebrilla, C.B. Variations in bovine milk oligosaccharides during early and middle lactation stages analyzed by high-performance liquid chromatography-chip/mass spectrometry. J. Dairy Sci. 2009, 92, 2991–3001. [Google Scholar] [CrossRef] [PubMed]
- Niñonuevo, M.R.; Perkins, P.D.; Francis, J.; Lamotte, L.M.; Locascio, R.G.; Freeman, S.L.; Mills, D.A.; German, J.B.; Grimm, R.; Lebrilla, C.B. Daily variations in oligosaccharides of human milk determined by microfluidic chips and mass spectrometry. J. Agric. Food Chem. 2008, 56, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Dreisewerd, K.; Kölbl, S.; Peter-Katalinić, J.; Berkenkamp, S.; Pohlentz, G. Analysis of native milk oligosaccharides directly from thin-layer chromatography plates by matrix-assisted laser desorption/ionization orthogonal-time-of-flight mass spectrometry with a glycerol matrix. J. Am. Soc. Mass Spectrom. 2006, 17, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry Bioanalytical Method Validation. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM); 2018. Available online: http://www.fda.gov/downloads/Drugs/Guidances/ucm070107.pdf (accessed on 24 May 2018).
- Wakamatsu, A.; Ochiai, S.; Suzuki, E.; Yokota, Y.; Ochiai, M.; Kotani, Y.; Sasahara, S.; Nakanaga, K.; Hashimoto, Y.; Ueno, S.; et al. Proposed selection strategy of surrogate matrix to quantify endogenous substances by Japan Bioanalysis Forum DG2015-15. Bioanalysis 2018, 10, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tabatabaei, A.; Zook, D.; Wang, Y.; Danks, A.; Stauber, K. A surrogate analyte-based liquid chromatography-tandem mass spectrometry method for the determination of endogenous cyclic nucleotides in rat brain. J. Pharm. Biomed. Anal. 2017, 146, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; James, C.A.; Wong, P. Bioanalytical method development and validation for the determination of glycine in human cerebrospinal fluid by ion-pair reversed-phase liquid chromatography–tandem mass spectrometry. J. Pharm. Biomed. Anal. 2016, 128, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cui, Z.; Deng, Y.; Dean, B.; Hop, C.E.; Liang, X. Surrogate analyte approach for quantitation of endogenous NAD+ in human acidified blood samples using liquid chromatography coupled with electrospray ionization tandem mass spectrometry. J. Chromatogr. B 2016, 1011, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Virág, D.; Király, M.; Drahos, L.; Édes, A.E.; Gecse, K.; Bagdy, G.; Juhász, G.; Antal, I.; Klebovich, I.; Dalmadi-Kiss, B.; et al. Development, validation and application of LC–MS/MS method for quantification of amino acids, kynurenine and serotonin in human plasma. J. Pharm. Biomed. Anal. 2020, 180, 113018. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Perrier, D. Pharmacokinetics. In Revised and Expanded, 2nd ed.; Informa HealthCare: New York, NY, USA, 1982. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| 3′-SL | 6′-SL | |
|---|---|---|
| PA_Quant a/PA_Qual b | PA_Quant/PA_Qual | |
| MQC | 108.6 | 1.745 |
| Rat plasma 1 | 106.2 | 1.721 |
| Rat plasma 2 | 106.5 | 1.747 |
| Rat plasma 3 | 105.4 | 1.737 |
| Rat plasma 4 | 111.1 | 1.683 |
| Rat plasma 5 | 120.4 | 1.728 |
| Rat plasma 6 | 101.6 | 1.742 |
| Mean | 108.5 | 1.729 |
| CV(%) c | 5.5 | 1.3 |
| Nominal Concentration (ng/mL) | ||||||||
|---|---|---|---|---|---|---|---|---|
| 3′-SL | 6′-SL | |||||||
| LLOQ | LQC | MQC | HQC | LLOQ | LQC | MQC | HQC | |
| 20 | 60 | 600 | 7500 | 20 | 60 | 600 | 7500 | |
| (A) Intra-day (n = 6) | ||||||||
| Mean concentration | 19.3 | 57.9 | 552 | 6970 | 19.9 | 58.5 | 641 | 7960 |
| CV (%) a | 2.8 | 2.8 | 1.4 | 3.0 | 2.3 | 5.0 | 2.3 | 2.5 |
| RE (%) b | −3.8 | −3.4 | −8.1 | −7.1 | −0.3 | −2.5 | 6.8 | 6.1 |
| (B) Inter-day (n = 18) | ||||||||
| Mean concentration | 18.9 | 58.2 | 542 | 6998 | 19.5 | 58.5 | 615 | 7550 |
| CV (%) | 3.1 | 2.6 | 2.3 | 3.0 | 3.9 | 4.2 | 3.6 | 4.6 |
| RE (%) | −5.4 | −3.1 | −9.7 | −6.7 | −2.7 | −2.4 | 2.5 | 0.7 |
| Nominal Concentration (ng/mL) | ||||
|---|---|---|---|---|
| 3′-SL | 6′-SL | |||
| MQC | HQC | MQC | HQC | |
| 600 | 7500 | 600 | 7500 | |
| (A) Matrix effect (IS normalized, n = 6) | ||||
| Mean concentration (%) | 113.9 | 103.1 | 101.9 | 108.4 |
| CV (%) a | 6.4 | 4.5 | 8.8 | 4.2 |
| (B) Recovery (n = 6) | ||||
| Mean concentration (%) | 88.6 | 94.7 | 89.0 | 91.4 |
| CV (%) | 10.3 | 7.5 | 6.3 | 6.2 |
| Nominal Concentration (ng/mL) | ||||||
|---|---|---|---|---|---|---|
| 3′-SL | 6′-SL | |||||
| LQC | MQC | HQC | LQC | MQC | HQC | |
| 60 | 600 | 7500 | 60 | 600 | 7500 | |
| Endogenous concentration (ng/mL) a | 483 | 483 | 483 | 45.5 | 45.5 | 45.5 |
| Adjusted QC concentration (ng/mL) b | 543 | 1083 | 7983 | 106 | 646 | 7546 |
| Measured concentration (ng/mL) | 501 | 1024 | 7554 | 97.7 | 651 | 7756 |
| CV (%) c | 1.4 | 2.2 | 2.2 | 7.6 | 2.7 | 3.4 |
| RE (%) d | −7.8 | −5.5 | −5.4 | −7.4 | 0.9 | 2.8 |
| Nominal Concentration (ng/mL) | ||||
|---|---|---|---|---|
| 3′-SL | 6′-SL | |||
| LQC | HQC | LQC | HQC | |
| 481 | 7500 | 37.5 | 7500 | |
| (A) Benchtop stability at room temperature (24 °C, 4 h; n = 3) | ||||
| Mean concentration (ng/mL) | 463 | 6890 | 37.0 | 7490 |
| CV (%) a | 1.6 | 4.3 | 4.6 | 7.1 |
| RE (%) b | −3.7 | −8.1 | −1.3 | −0.1 |
| (B)Post-preparative stability (10 °C, 26 h; n = 3) | ||||
| Mean concentration (ng/mL) | 482 | 7090 | 34.7 | 7210 |
| CV (%) | 3.3 | 1.6 | 2.8 | 0.6 |
| RE (%) | 0.1 | −5.5 | −7.4 | −3.9 |
| (C)Freeze–thaw stability (4 cycles; n = 3) | ||||
| Mean concentration (ng/mL) | 465 | 6900 | 38.9 | 7180 |
| CV (%) | 3.2 | 0.5 | 11.5 | 2.2 |
| RE (%) | −3.5 | −8.0 | 3.9 | −4.3 |
| (D)Long-term stability (−80 °C, 37 days; n = 3) | ||||
| Mean concentration (ng/mL) | 465 | 6750 | 35.4 | 7020 |
| CV (%) | 5.1 | 2.3 | 1.3 | 3.7 |
| RE (%) | −3.4 | −10.0 | −5.4 | −6.4 |
| 3′-SL (mg/kg) | 6′-SL (mg/kg) | |||||
|---|---|---|---|---|---|---|
| 100 | 500 | 2000 | 100 | 500 | 2000 | |
| t1/2 (h) | 6.5 | 4.9 | 3.4 | 2.8 | 1.8 | 1.8 |
| Cmax (ng/mL) | 536 ± 105 a | 1100 ± 101 | 1930 ± 273 | 235 ± 2 | 958 ± 216 | 1620 ± 85 |
| Tmax (h) | 2.0 | 1.0 | 0.5 | 1.0 | 1.0 | 1.0 |
| AUClast (ng·h/mL) | 4000 ± 438 | 5700 ± 698 | 12000 ± 1390 | 991 ± 190 | 3270 ± 316 | 7620 ± 622 |
| AUCinf (ng·h/mL) | 5570 | 7030 | 13,300 | 1050 | 3290 | 7660 |
| CL (mL/h/kg) | 18,000 | 71,100 | 150,000 | 95,000 | 152,000 | 261,000 |
| Vd (mL/kg) | 168,000 | 499,000 | 737,000 | 383,000 | 400,000 | 687,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, S.-I.; Eom, H.Y.; Hwang, J.H.; Kim, L.; Lee, J.-H. Simultaneous Quantification of 3′- and 6′-Sialyllactose in Rat Plasma Using Liquid Chromatography-Tandem Mass Spectrometry and Its Application to a Pharmacokinetic Study. Molecules 2021, 26, 1177. https://doi.org/10.3390/molecules26041177
Jang S-I, Eom HY, Hwang JH, Kim L, Lee J-H. Simultaneous Quantification of 3′- and 6′-Sialyllactose in Rat Plasma Using Liquid Chromatography-Tandem Mass Spectrometry and Its Application to a Pharmacokinetic Study. Molecules. 2021; 26(4):1177. https://doi.org/10.3390/molecules26041177
Chicago/Turabian StyleJang, Seok-In, Han Young Eom, Jeong Ho Hwang, Lila Kim, and Jong-Hwa Lee. 2021. "Simultaneous Quantification of 3′- and 6′-Sialyllactose in Rat Plasma Using Liquid Chromatography-Tandem Mass Spectrometry and Its Application to a Pharmacokinetic Study" Molecules 26, no. 4: 1177. https://doi.org/10.3390/molecules26041177
APA StyleJang, S.-I., Eom, H. Y., Hwang, J. H., Kim, L., & Lee, J.-H. (2021). Simultaneous Quantification of 3′- and 6′-Sialyllactose in Rat Plasma Using Liquid Chromatography-Tandem Mass Spectrometry and Its Application to a Pharmacokinetic Study. Molecules, 26(4), 1177. https://doi.org/10.3390/molecules26041177