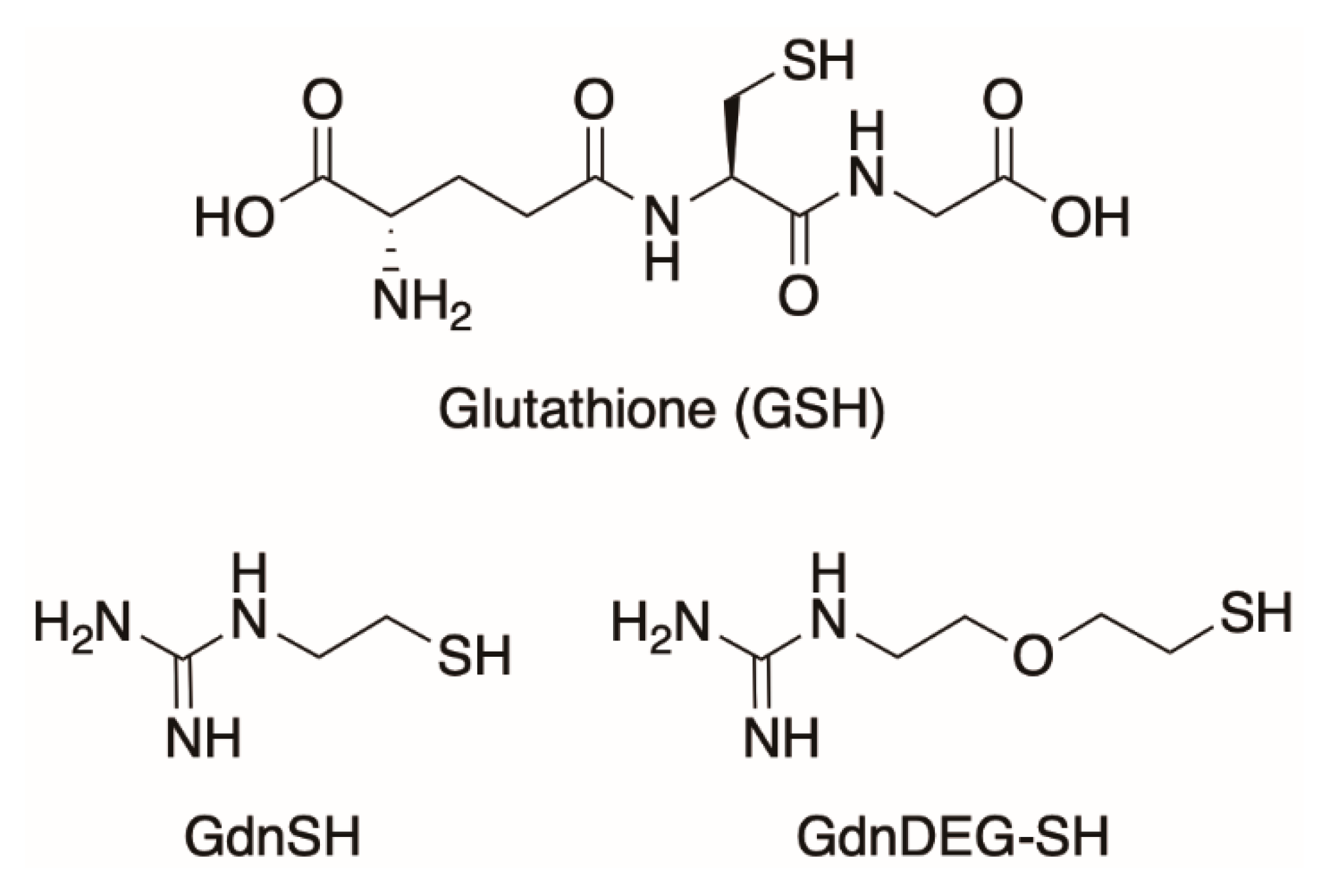
4. Materials and Methods
4.1. General
Nuclear magnetic resonance (NMR) spectra were recorded on a JNM-ECX 400 spectrometer (1H/400 MHz, 13C/100 MHz) of JEOL (Tokyo, Japan), where the chemical shifts were determined with respect to a 1H signal corresponding to the nondeuterated solvent and a 13C signal corresponding to the solvent as an internal standard (1H-NMR: 7.24 ppm for CDCl3, 4.67 ppm for D2O, 3.29 ppm for CD3OD; 13C-NMR: 77.16 ppm for CDCl3, 47.68 ppm for CD3OD). Matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF MS) was performed on autoflex speed spectrometer of Bruker (Bremen, Germany). Electrospray ionization (HR ESI) TOF MS spectra were recorded on micrOTOF-Q II-S1 of Bruker with MeOH as a solvent. Analytical thin layer chromatography (TLC) was performed on precoated, glass-backed silica gel Merck 60 F254. Visualization of the developed chromatogram was performed by UV absorbance, Hanessian’s stain or iodine. UV absorption spectra and temperature-dependent transmittance changes were recorded on V-650 UV–VIS spectrophotometer of JASCO (Tokyo, Japan) or U-3310 spectrophotometer of Hitachi High-Technologies (Tokyo, Japan). Reversed-phase high-performance liquid chromatography (RP-HPLC) for BPTI assay was conducted with GL7400 HPLC system of GL Sciences (Tokyo, Japan) using TSKgel Protein C4-300 column of Tosoh Bioscience (φ4.6 × 150 mm, Tokyo, Japan). RP-HPLC for redox potential measurement was conducted with HPLC system of JASCO (Tokyo, Japan) using YMC Triart C18 column (φ4.6 × 250 mm, Tokyo, Japan).
4.2. Materials
Deuterated solvents and di-tert-butyl decarbonate were purchased from Kanto Chemicals (Tokyo, Japan). maleimide-PEG (Mw 2000) was purchased from NOF America Corporation (White Plains, NY, USA). Acetonitrile, 35% hydrochloric acid, sodium hydroxide, tosyl chloride, sodium sulfate, potassium carbonate, ammonium chloride, sodium iodide, 30% hydrogen peroxide, sodium hydrogen carbonate, trifluoroacetic acid, and 2-propanol were purchased from Kishida Chemical (Tokyo, Japan). Coomassie brilliant blue, 5,5′-dithiobis(2-nitrobenzoic Acid) (DTNB), 1,4-dithiothreitol (DTT), l-glutathione oxidized (GSSG), l-glutathione reduced (GSH), and guanidine hydrochloride (GdnHCl) were purchased from Nacalai Tesque (Kyoto, Japan). α-Cyano-4-hydroxycinnamic acid, cytidine 2′:3′-cyclic monophosphate (cCMP) monosodium salt, triethylamine (Et3N), and ribonuclease A (RNase A) from bovine pancreas were purchased from Sigma-Aldrich (St. Louis, MO, USA). N,N′-Bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine, 1,1′-carbonyldiimidazole, 2-(2-aminoethoxy)ethanol, potassium thioacetate, and gentisic acid were purchased from Tokyo Chemical Industry (Tokyo, Japan). Bovine pancreatic trypsin inhibitor (BPTI) was purchased from Takara Bio (Shiga, Japan). Hen egg white lysozyme was purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). Dry N,N-dimethylformamide (DMF), dry CH2Cl2, and dry tetrahydrofuran (THF) were purchased from Kanto Chemicals. Column chromatography was carried out with Silica Gel 60 (spherical, neutral, particle size: 63–210 μm) purchased from Kanto Chemicals. Deionized water (filtered through a 0.22 μm membrane filter, >18.2 MΩ cm) was purified in Purelab DV35 of ELGA (Buckinghamshire, UK) and a Milli-Q system of Merck Millipore (Burlington, MA, USA).
4.3. Determination of Thiol Concentration for Oxidative Protein Folding Assay
To a solution of thiol (40 mM) in 10 mM HCl aqueous solution, Ellman’s reagent [5,5-dithio-bis-(2-nitrobenzoic acid), 0.50 mM] [
29] was added. Concentration of the thiol group was determined by measuring the absorbance at 412 nm at 30 °C with U-3310 spectrophotometer prior to its use for the oxidative protein folding assay. Amounts of free thiol groups were determined by Ellman’s reagent. Each amount includes both the free thiol of 1.0 mM reducing agent (GdnDEG-SH) and 8.0 μM reduced substrate, RNase A.
4.4. Preparation of Reduced and Denatured RNase A
RNase A was dissolved in a buffer (200 mM Tris-HCl, pH 8.7) containing 6.0 M GdnHCl and 100 mM DTT and incubated for 2 h at 25 °C. The resulting sample was dialyzed by 10 mM HCl aq. to remove the denaturing and reducing reagents.
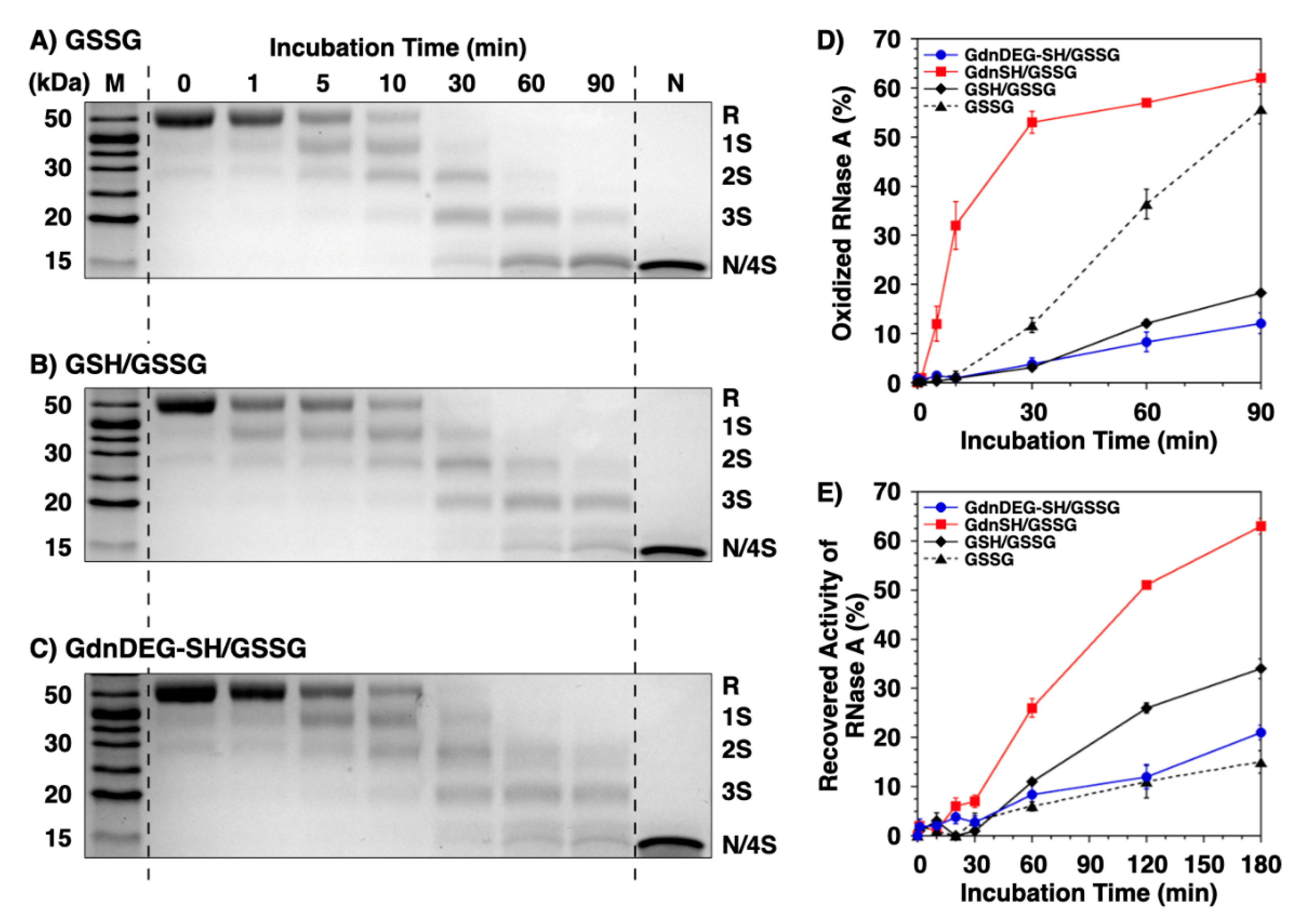
4.5. RNase A Refolding Assay
Fully reduced and denatured RNase A (8.0 μM) was incubated for 3 h at 30 °C in the presence of 200 μM GSSG and 1.0 mM reducing agent in a buffer (50 mM Tris-HCl, 300 mM NaCl, pH 7.5) [
30]. At selected time points during this incubation, aliquots (50 μL each) were taken from the reaction solution, which were immediately added to a buffer (150 μL, 50 mM Tris-HCl, 300 mM NaCl, pH 7.5) containing cCMP (final concentration of cCMP = 0.60 mM), followed by the measurement of the linear increase in absorbance at 284 nm at 30 °C with U-3310 spectrophotometer [
18,
31]. Values represent means ± SEM from three independent experiments.
4.6. Gel Shift Assay of RNase A Disulfide Bond Formation
Oxidative folding of RNase A (8.0 μM) was carried out in a buffer (50 mM Tris-HCl, 300 mM NaCl, pH 7.5) containing 200 μM GSSG with/without 1.0 mM reducing agent. At selected time points, free thiols were blocked by the addition of Laemmli’s 4 × SDS-loading buffer [
32] containing 10 mM maleimide-PEG (Mw 2000). Redox states of RNase A were separated by nonreducing 14% SDS-PAGE using WIDE RANGE gel (Nacalai Tesque). Proteins were detected by Coomassie brilliant blue G250 staining. The band intensities were analyzed by a ChemiDoc Touch imaging system and Image Lab (Bio-Rad).
4.7. BPTI Folding Assay
Reduction and denaturation of BPTI was carried out as described previously [
25]. Fully reduced and denatured BPTI (30 μM) was incubated at 30 °C in the presence of 200 μM GSSG and 1.0 mM reducing agent in a buffer (50 mM Tris-HCl, 300 mM NaCl, pH 7.5), where the buffer was degassed by flushing N
2 prior to use. At selected time points, the reaction was quenched by adding an equal volume of 1 M HCl aq., which was then analyzed by RP-HPLC at a flow rate of 1.0 mL min
−1 monitoring at 229 nm with a linear gradient elution (solvent A: 0.05% trifluoroacetic acid in water and solvent B: 0.05% trifluoroacetic acid in acetonitrile; percentages of solvent A: 95% at 0 min, 80% at 15 min, and 30% at 115 min). The molecular mass values of the folding intermediates were determined by MALDI-TOF MS in a linear positive-ion mode using α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) as the matrix. The molecular mass was calculated using Protein-Prospector web server [
33].
4.8. Determination of Thiol pKa Values
Stock solutions of citric buffer (sodium citrate and HCl for pH 2.0–4.0), phosphate buffer (Na
2HPO
4 and KH
2PO
4 for pH 5.0–8.0), and borate buffers (Na
2B
4O
7 and HCl for pH 8.5–9.0 and Na
2B
4O
7 and NaOH for pH 10.0–12.0) were prepared, and these buffers were degassed with N
2 for 1 h immediately prior to use. A stock solution of a thiol compound in degassed water (5.0 mM) were then prepared. Immediately after the aqueous solution of the thiol compound (20 μL) and a buffer (1.98 mL) were combined in a 1-cm thick quartz cuvette, the UV absorption spectrum of the sample was measured. The pH value of the sample was measured by a HORIBA pH meter (9618S-10D), which had been calibrated prior to use with pH 4.01, 6.86, and 9.18 standard solutions (HORIBA 101-S). Absorbance at 240 nm was plotted in the function of the pH values, and the p
Ka value of the thiol compound was calculated with KaleidaGraph software (version 4.5.0) by a curve fitting analysis using the following equation:
where a is the absorbance below the pH 3 and b is the difference of absorbances above the pH 11 and below the pH 3, and curve fitting calculation provides m2 = p
Ka. For all analyses,
r2 values were higher than 0.99.
4.9. Redox Potential E0′ Measurements
E0′ value of a thiol compound was determined by following the protocol described in a previous paper [
6]. Buffer (100 mM Tris-HCl, 1.0 mM EDTA, pH 7.0) was treated by bubbling high purity N
2 for longer than 1 h prior to use. DTT
red (60 μM, 4.5 mL) in the buffer was added to a disulfide (GdnSS or UreaSS, 60 μM, 4.5 mL) in the buffer under N
2, which was stirred at 25 ± 0.1 °C for 24 h. To quench the reaction, an aliquot of the reaction mixture (1 mL) was added to 1 M HCl aq. (200 μL), and the obtained sample solution was immediately analyzed by RP-HPLC (YMC Triart C18 column,
φ4.6 × 250 mm). The column was equilibrated with water containing 0.1% TFA at a flow rate of 1.0 mL min
−1. The RP-HPLC analysis was conducted with water containing 0.1% TFA (eluent A) and CH
3CN containing 0.1% TFA (eluent B) with a linear gradient (percentage of eluent B: 0% in 0–8 min, 0–6% in 8–15 min, 6–10% in 15–30 min). The concentrations of the species at equilibrium were calculated from the observed peak areas and corresponding calibration curves.
The equilibrium constant
Keq for the reaction (Equation (2)), described as Equation (3), was determined by averaging three times of individual experiments following the above procedure.
The redox potential
E0′ was calculated by the Nernst’s equation (Equation (4))
where
n is the number of transferred electrons (
n = 2),
F is Faraday’s constant (96,500 C mol
−1),
R is the universal gas constant (8.314 J K
−1 mol
−1),
T is the temperature (298 K), and
E0′
DTT is the redox potential of DTT (−327 mV).
4.10. Aggregation Inhibition Assay
Lysozyme (20 μM) was dissolved in Tris-HCl buffer (50 mM, pH 7.5) containing NaCl (300 mM) and an additive, and the mixture was incubated at 96 °C for 20 min followed by air cooling to room temperature.
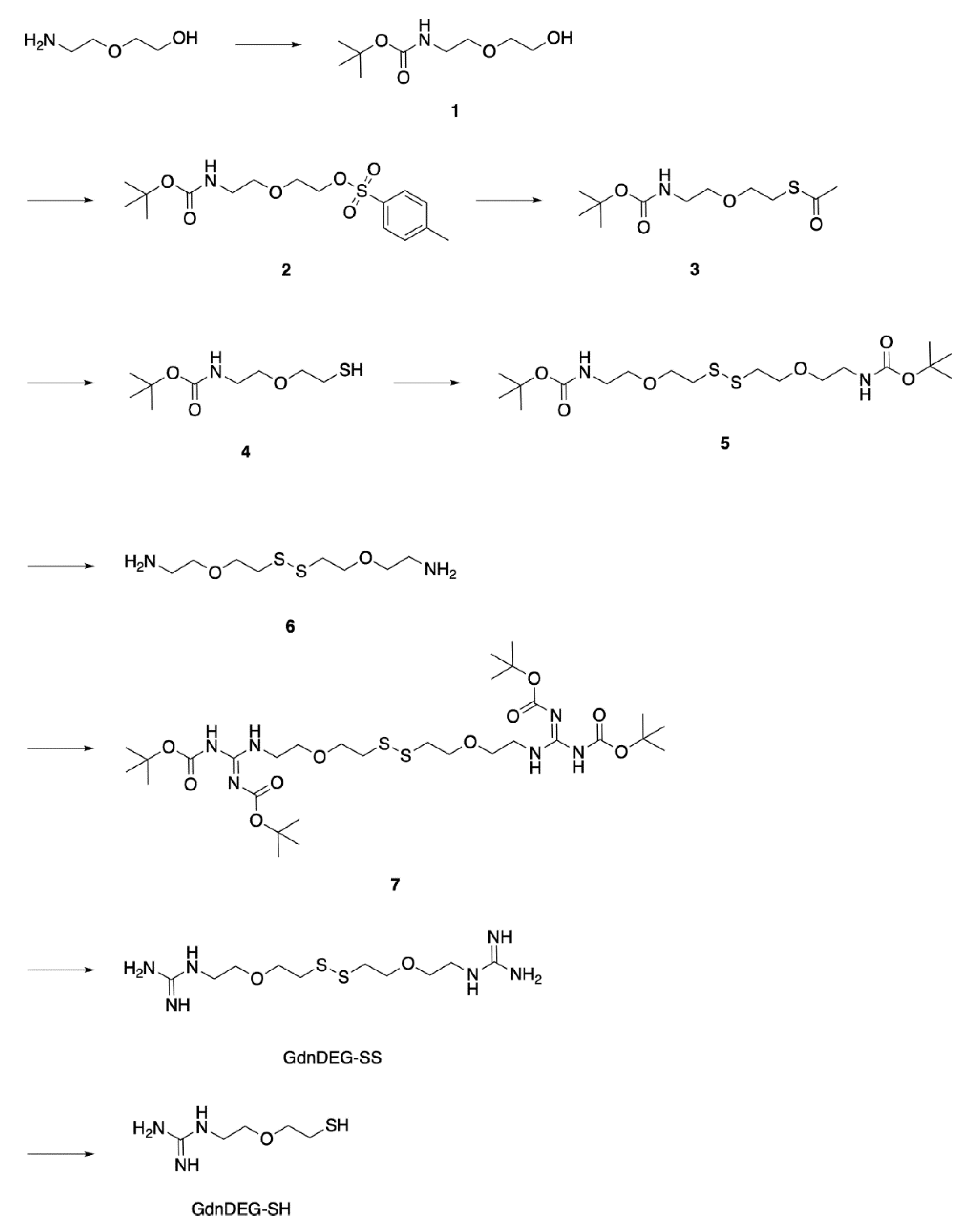
4.11. Synthesis of 1
To a dry EtOH solution (31 mL) of 2-(2-aminoethoxy)ethanol (1.52 g, 14.5 mmol), di-tert-butyl decarbonate (3.28 g, 15.0 mmol) was added at 0 °C under N2. After being stirred for 4 h at 25 °C, CH2Cl2 (100 mL) was added to the reaction mixture, and the mixture was washed with water (100 mL, twice) and brine (50 mL, once). The organic extract was dried over anhydrous Na2SO4 and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 30 °C, and the residue was chromatographed on silica gel (Silica Gel 60) with CH2Cl2/MeOH (100/0 to 90/10 v/v) to allow isolation of 1 (2.81 g, 13.7 mmol) in 94% yield.
TLC Rf (Merck 60 F254, CH2Cl2/MeOH = 90/10 v/v): 0.56; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 5.18 (brs, 1H), 3.73 (brs, 2H), 3.59–3.54 (m, 4H), 3.33 (q, J = 5.0 Hz, 2H), 2.84 (brs, 1H), 1.45 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 156.24, 79.41, 72.33, 70.37, 61.70, 40.44, 28.46 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 228.132 (calculated m/z on the basis of the monoisotopic mass of C9H19NNaO4 [M + Na]+ = 228.121).
4.12. Synthesis of 2
To a dry THF solution (31 mL) of 1 (2.68 g, 13.1 mmol), 15% NaOH aq. (10 mL) and a THF solution (10 mL) of TsCl (2.94 g, 15.4 mmol) were added dropwise at 0 °C under N2. After being stirred for 12 h at 25 °C, water (100 mL) was added to the reaction mixture. The resulting mixture was extracted with CH2Cl2 (50 mL, three times). The collected organic extract was washed with brine (50 mL, once), dried over anhydrous Na2SO4, and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 30 °C to allow isolation of 2 (4.06 g, 11.3 mmol) in 86% yield.
TLC Rf (Merck 60 F254, CH2Cl2): 0.25; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 7.80 (d, J = 8.7 Hz, 2H), 7.36 (d, J = 8.7 Hz, 2H), 4.86 (brs, 1H), 4.18–4.15 (m, 2H), 3.63 (t, J = 4.6 Hz, 2H), 3.45 (t, J = 5.0 Hz, 2H), 3.24 (q, J = 5.0 Hz, 2H), 2.45 (s, 3H), 1.45 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 156.24, 144.98, 133.09, 129.91, 128.01, 79.36, 70.40, 69.18, 68.41, 40.29, 28.46, 21.68 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 382.114 (calculated m/z on the basis of the monoisotopic mass of C16H25NNaO6S [M + Na]+ = 382.130).
4.13. Synthesis of 3
To a dry DMF solution (35 mL) of 2 (3.98 g, 11.1 mmol), AcSK (4.03 g, 35.3 mmol) was added at 25 °C under N2. After being stirred for 13 h at 90 °C, the reaction mixture was cooled to 25 °C. To the resulting mixture, water (300 mL) was added, and the mixture was extracted with CH2Cl2 (100 mL, four times). The collected organic extract was washed with brine (100 mL, four times), dried over anhydrous Na2SO4, and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 30 °C, and the residue was chromatographed on silica gel (Silica Gel 60) with AcOEt/hexane (20/80 v/v) to allow isolation of 3 (2.69 g, 10.2 mmol) in 92% yield.
TLC Rf (Merck 60 F254, EtOAc/hexane = 50/50 v/v): 0.54; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 4.87 (brs, 1H), 3.57 (t, J = 6.4 Hz, 2H), 3.51 (t, J = 5.0 Hz, 2H), 3.30 (q, J = 5.0 Hz, 2H), 3.08 (t, J = 6.4 Hz, 2H), 2.35 (s, 3H), 1.45 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 195.46, 155.99, 79.36, 69.99, 69.53, 40.42, 30.62, 28.95, 28.48 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 286.113 (calculated m/z on the basis of the monoisotopic mass of C11H21NNaO4S [M + Na]+ = 286.109).
4.14. Synthesis of 4
To a dry EtOH solution (19 mL) of 3 (2.69 g, 10.2 mmol), K2CO3 (3.54 g, 25.6 mmol) was added at 25 °C under N2. After being stirred for 22 h at 25 °C, the reaction mixture was neutralized with saturated NH4Cl aq. To the mixture, water (100 mL) was added, and the mixture was extracted with AcOEt (50 mL, three times). The collected organic extract was dried over anhydrous Na2SO4, and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 30 °C, and the residue was chromatographed on silica gel (Silica Gel 60) with AcOEt/MeOH (20/80 v/v) to allow isolation of 4 (948 mg, 4.28 mmol) in 42% yield.
TLC Rf (Merck 60 F254, EtOAc/hexane = 20/80 v/v): 0.28; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 4.90 (brs, 1H), 3.59 (t, J = 6.4 Hz, 2H), 3.52 (t, J = 5.0 Hz, 2H), 3.32 (q, J = 5.0 Hz, 2H), 2.69 (dt, J = 7.8, 8.2 Hz, 2H), 1.55 (t, J = 8.2 Hz, 2H), 1.45 (s, 9H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 155.91, 78.98, 72.42, 69.73, 40.26, 28.34, 24.11 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 244.098 (calculated m/z on the basis of the monoisotopic mass of C9H19NNaO3S [M + Na]+ = 244.099).
4.15. Synthesis of 5
To an EtOAc solution (11 mL) of 4 (942 mg, 4.26 mmol), NaI (6.0 mg, 40 μmol) and 30% H2O2 aq. (480 μL) were added at 25 °C under air. After being stirred for 1 h at 25 °C, to the reaction mixture, saturated NaHCO3 aq. (100 mL) was added, and the mixture was extracted with EtOAc (50 mL, three times). The collected organic extract was washed with brine (50 mL, once), dried over anhydrous Na2SO4, and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 25 °C to allow isolation of 5 (753 mg, 1.71 mmol) in 80% yield.
TLC Rf (Merck 60 F254, EtOAc/hexane = 20/80 v/v): 0.32; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 5.13 (brs, 1H), 3.71 (t, J = 6.4 Hz, 2H), 3.53 (t, J = 5.0 Hz, 2H), 3.30 (q, J = 5.0 Hz, 2H), 2.88 (t, J = 6.4 Hz, 2H), 1.44 (s, 18H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 155.94, 79.14, 69.98, 69.14, 40.36, 38.57, 28.42 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 463.192 (calculated m/z on the basis of the monoisotopic mass of C18H36N2NaO2S2 [M + Na]+ = 463.191).
4.16. Synthesis of 6
To a CH2Cl2 solution (11 mL) of 5 (752 mg, 1.71 mmol), TFA (2.5 mL) was added at 0 °C under N2. After being stirred for 2 h at 0 °C, the reaction mixture was evaporated to dryness under reduced pressure at 25 °C. To the residue, water (50 mL) was added, and the mixture was washed with CH2Cl2 (50 mL, three times). The aqueous extract was evaporated to dryness under reduced pressure to allow isolation of 6 (738 mg, 1.69 mmol) in 99% yield.
TLC Rf (Merck 60 F254, EtOAc): 0.05; 1H-NMR (400 MHz, CD3OD, 25 °C): δ = 3.79 (t, J = 6.4 Hz, 4H), 3.71 (t, J = 5.0 Hz, 4H), 3.13 (t, J = 5.0 Hz, 4H), 2.94 (t, J = 6.4 Hz, 4H) ppm; 13C-NMR (100 MHz, CD3OD, 25 °C): δ = 69.10, 66.24, 39.25, 37.55 ppm; MALDI-TOF MS (gentisic acid, positive mode): m/z = 241.111 (calculated m/z on the basis of the monoisotopic mass of C8H21N2O2S2 [M + Na]+ = 241.104).
4.17. Synthesis of 7
To a dry DMF solution (15 mL) of 6 (770 mg, 1.77 mmol), N,N′-bis(tert-butoxycarbonyl)-1H-pyrazole-1-carboxamidine (1.209 g, 3.898 mmol) and triethylamine (990 μL, 7.09 mmol) were added at 25 °C under N2. After being stirred for 16 h at 25 °C, to the reaction mixture, CH2Cl2 (30 mL) was added, and the mixture was washed with water (100 mL, three times) and brine (50 mL, once). The organic extract was dried over anhydrous Na2SO4 and filtered off from insoluble substances. The filtrate was evaporated to dryness under reduced pressure at 30 °C, and the residue was chromatographed on silica gel (Silica Gel 60) with AcOEt/hexane (20/80 to 50/50 v/v) to allow isolation of 7 (846 mg, 1.16 mmol) in 66% yield.
TLC Rf (Merck 60 F254, EtOAc/hexane = 20/80 v/v): 0.15; 1H-NMR (400 MHz, CDCl3, 25 °C): δ = 11.47 (brs, 2H), 8.62 (brs, 2H), 3.74 (t, J = 6.4 Hz, 4H), 3.61 (m, 8H), 2.90 (t, J = 6.4 Hz, 4H), 1.50 (s, 36H) ppm; 13C-NMR (100 MHz, CDCl3, 25 °C): δ = 163.61, 156.37, 153.08, 83.10, 79.35, 69.40, 69.16, 40.66, 38.45, 28.37, 28.16 ppm; ESI-TOF MS (MeOH, positive mode): m/z = 725.3609 (calculated m/z on the basis of the monoisotopic mass of C30H57N6O10S2 [M + Na]+ = 725.3578).
4.18. Synthesis of GdnDEGSS
To a MeOH solution (3 mL) of 7 (198 mg, 0.272 mmol), 1 M HCl aq. (10 mL) was added at 25 °C under air. After being stirred for 2 h at 25 °C, the reaction mixture was evaporated to dryness under reduced pressure at 25 °C. To the residue, water (10 mL) was added, and the mixture was washed with CH2Cl2 (30 mL, three times). The aqueous extract was evaporated to dryness under reduced pressure to allow isolation of GdnDEGSS (86 mg, 0.22 mmol) in 76% yield.
TLC Rf (Merck 60 F254, MeOH): 0.01; 1H-NMR (400 MHz, D2O, 25 °C): δ = 3.73 (t, J = 6.0 Hz, 4H), 3.60 (t, J = 5.0 Hz, 4H), 3.31 (t, J = 5.0 Hz, 4H), 2.85 (t, J = 6.0 Hz, 4H) ppm; 13C-NMR (100 MHz, D2O, 25 °C): δ = 157.37, 68.77, 68.34, 41.30, 37.43 ppm; ESI-TOF MS (MeOH, positive mode): m/z = 325.1487 (calculated m/z on the basis of the monoisotopic mass of C10H25N6O2S2 [M + H]+ = 325.1480).
4.19. Synthesis of GdnDEG-SH
To a degassed aqueous solution (10 mL) of GdnDEGSS (86 mg, 0.22 mmol), dithiothreitol (249 mg, 1.61 mmol) was added at 25 °C under N2. After being stirred for 6 h at 25 °C, to the reaction mixture, water (10 mL) was added, and the mixture was washed with a mixture of 2-propanol and CHCl3 (10/50 v/v, 20 mL, four times). The aqueous extract was evaporated to dryness under reduced pressure to allow isolation of GdnDEGSH (71 mg, 0.35 mmol) in 82% yield.
1H-NMR (400 MHz, D2O, 25 °C): δ = 3.58–3.55 (m, 4H), 3.29 (t, J = 5.0 Hz, 2H), 2.61 (t, J = 6.0 Hz, 2H) ppm; 13C-NMR (100 MHz, D2O, 25 °C): δ = 72.22, 68.54, 68.34, 41.21, 23.24 ppm; ESI-TOF MS (MeOH, positive mode): m/z = 164.0859 (calculated m/z on the basis of the monoisotopic mass of C5H14N3OS [M + H]+ =164.0858.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}