
Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer’s Disease and Parkinson’s Disease Treatment (2011–2020)


Abstract
:1. Introduction
2. The Synthesis of Pyrazole Scaffold Molecules
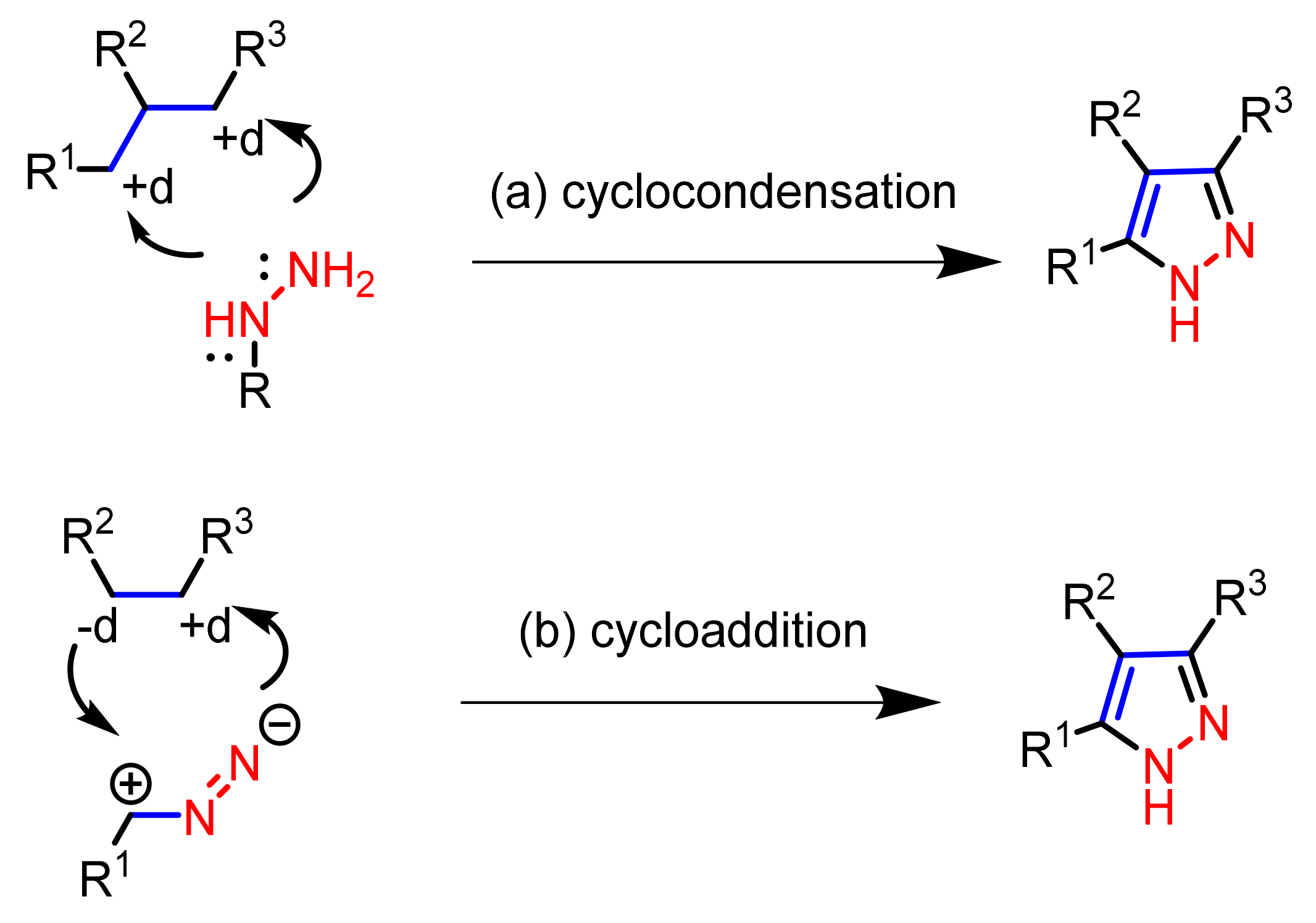
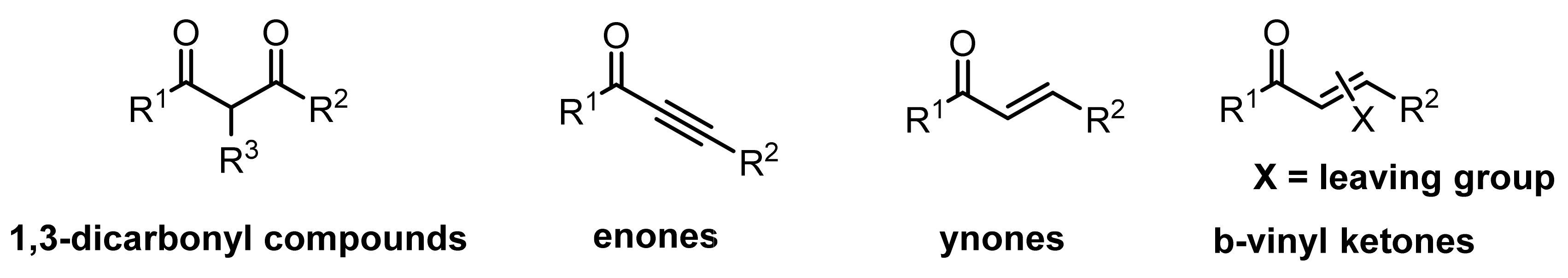
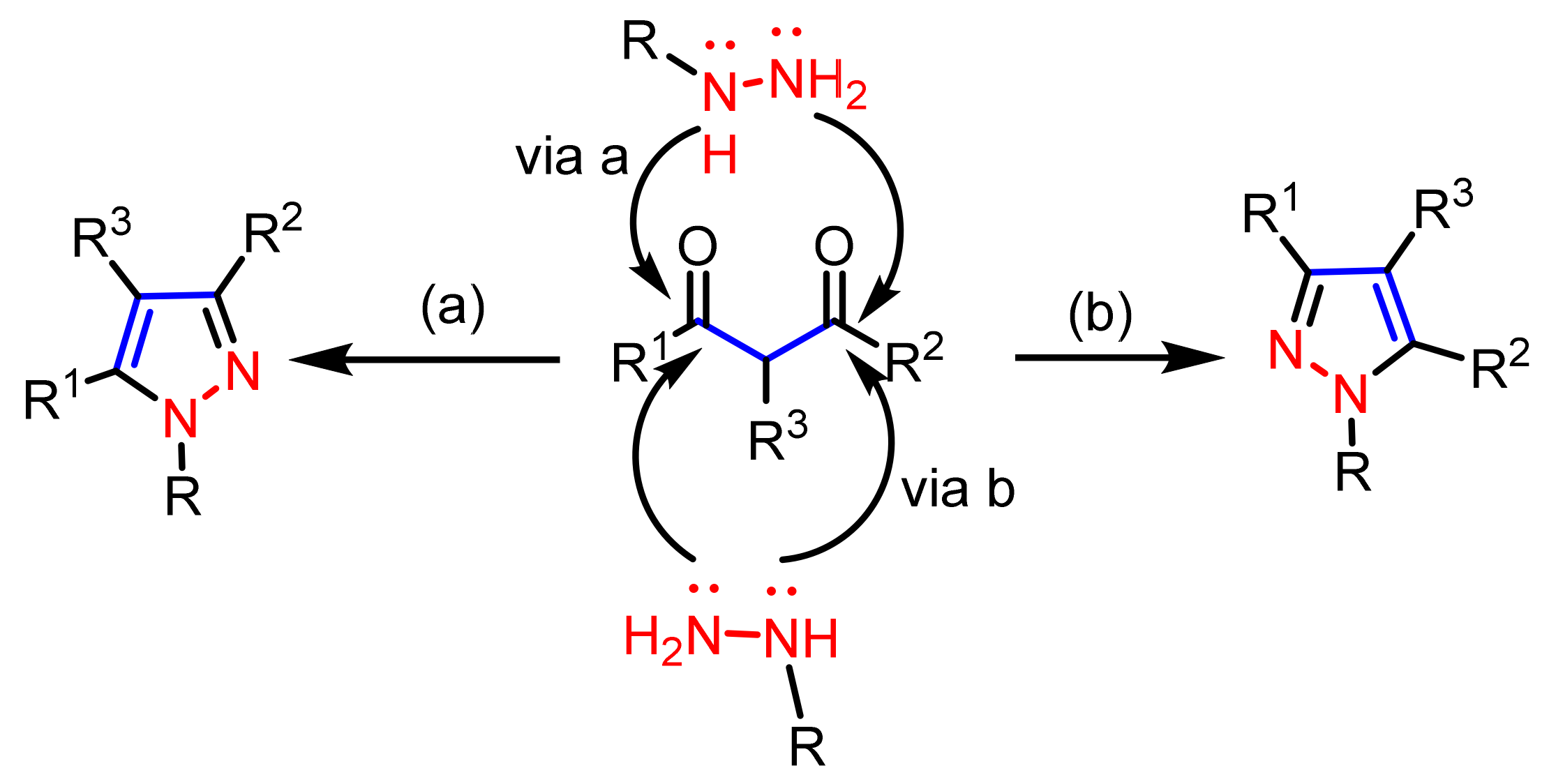
2.1. Cyclocondensation of Hydrazines with 1,3-Dielectrophilic Derivatives
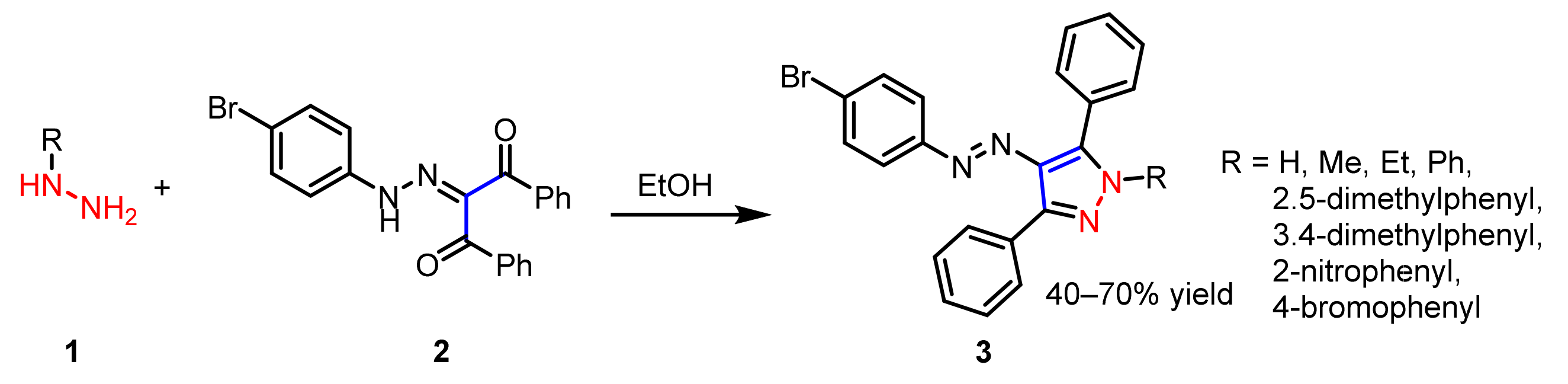
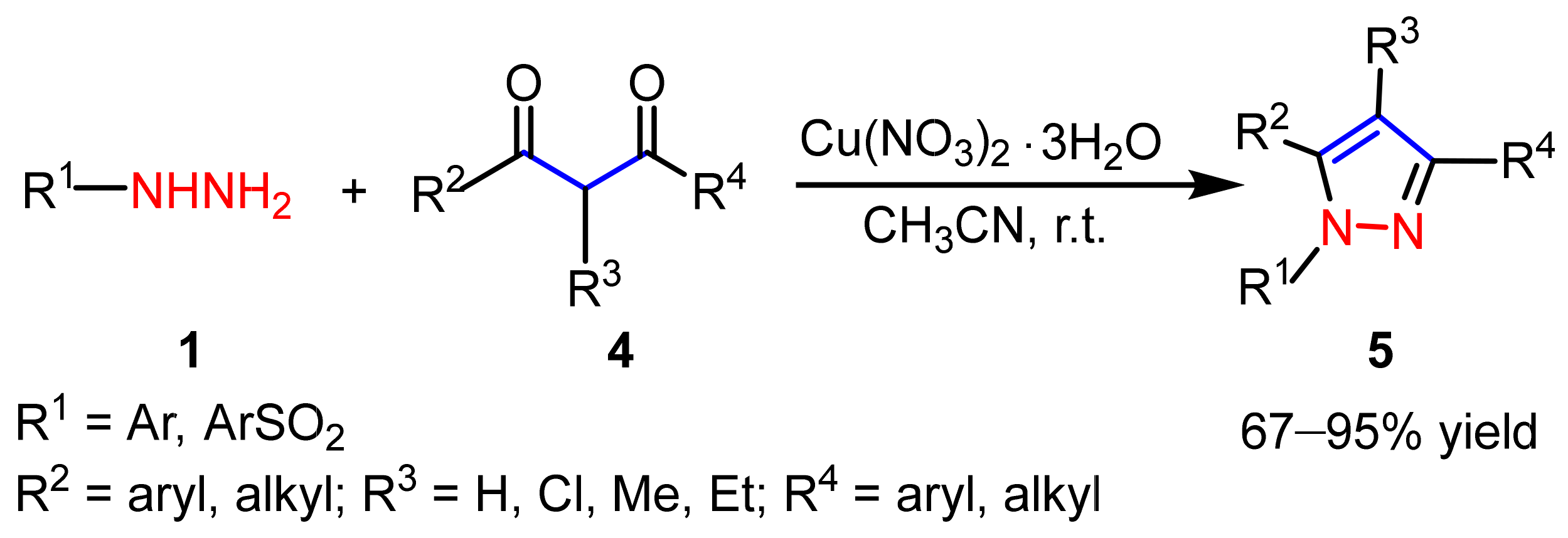
2.1.1. Cyclocondensation of Hydrazines with 1,3-Dicarbonyl and Related Compounds
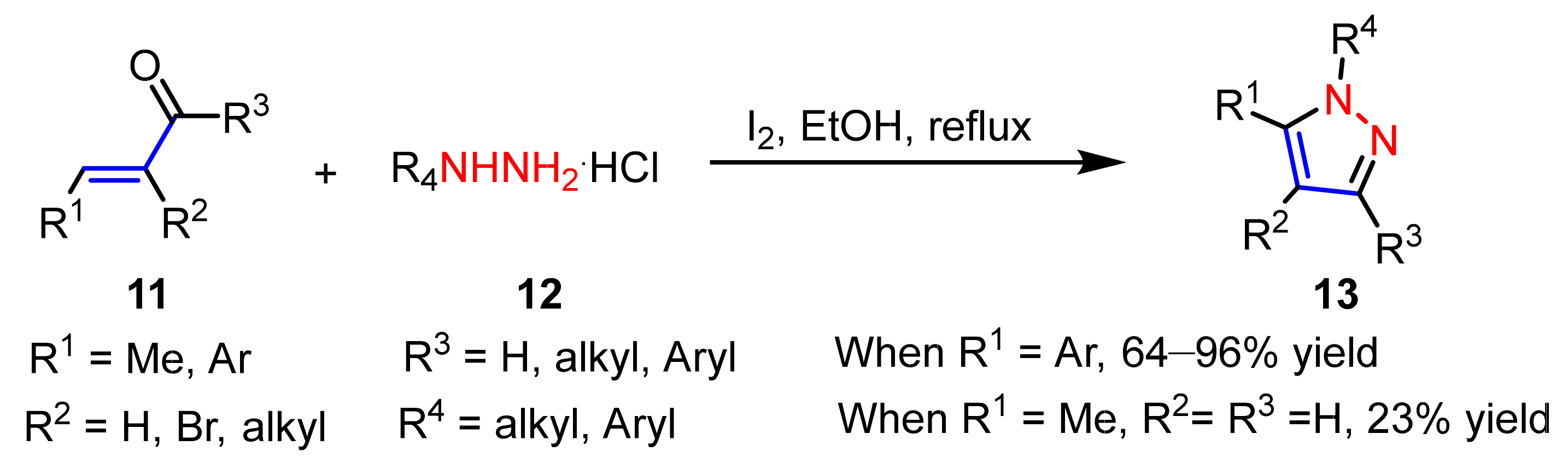
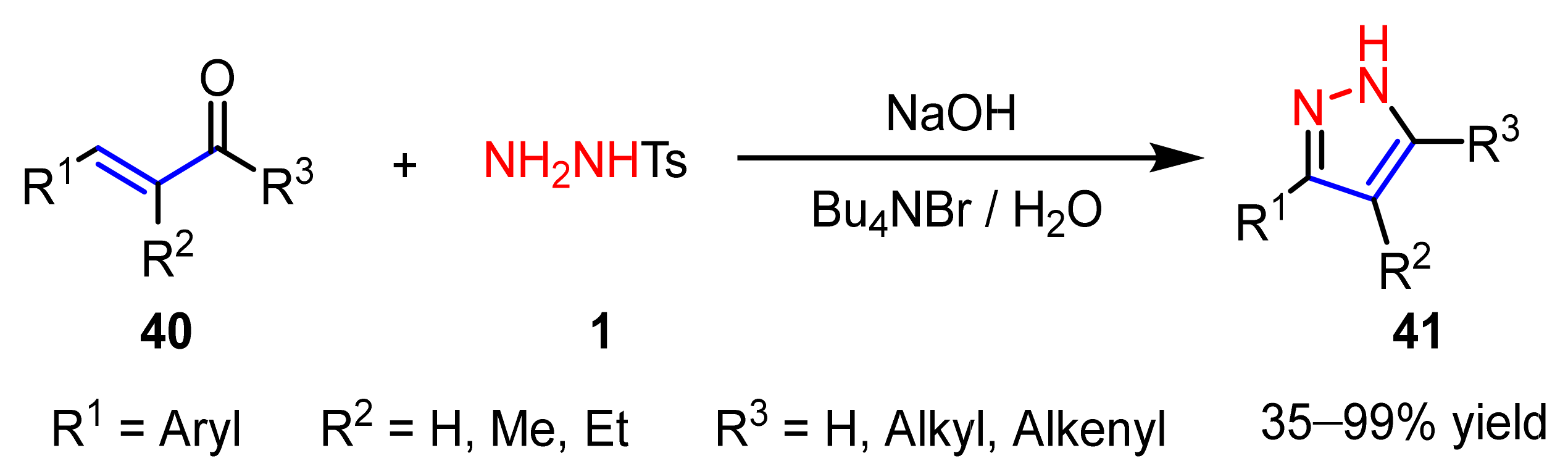
2.1.2. Cyclocondensation of Hydrazines with Enones and Related Compounds
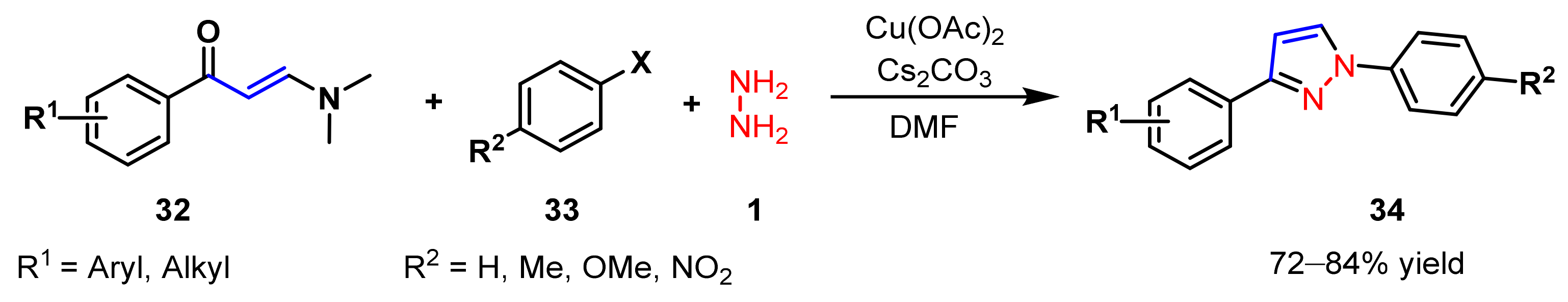
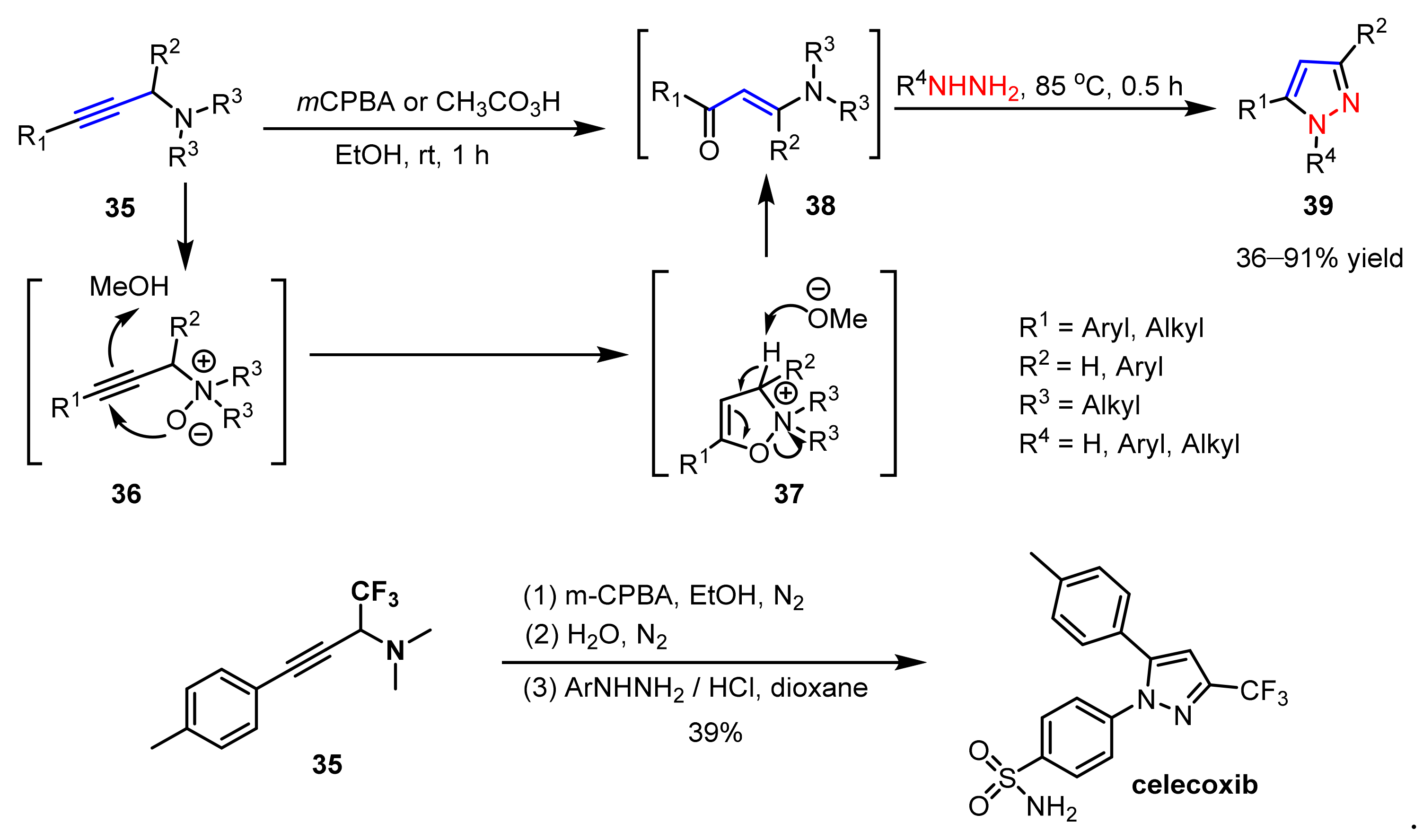
2.1.3. Cyclocondensation of Hydrazines with Ynones and Related Compounds
2.1.4. Cyclocondensation of Hydrazines with Vinyl Ketones Bearing a Leaving Group
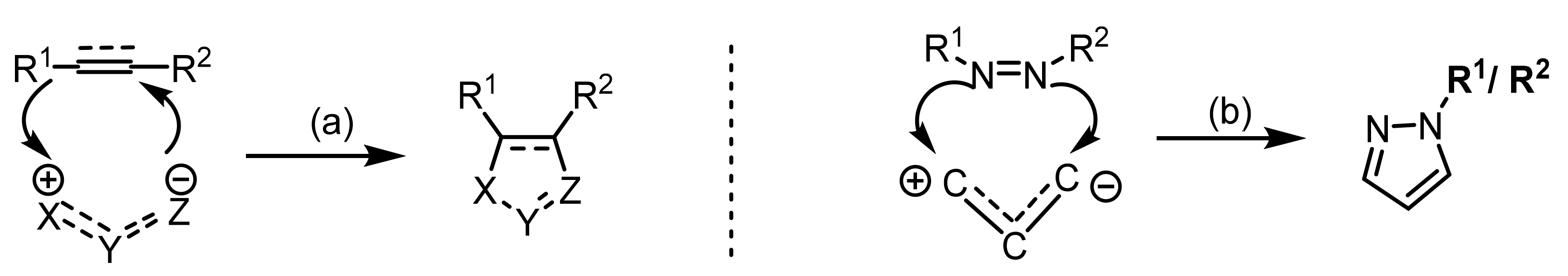
2.2. 1,3-Dipolar Cycloadditions
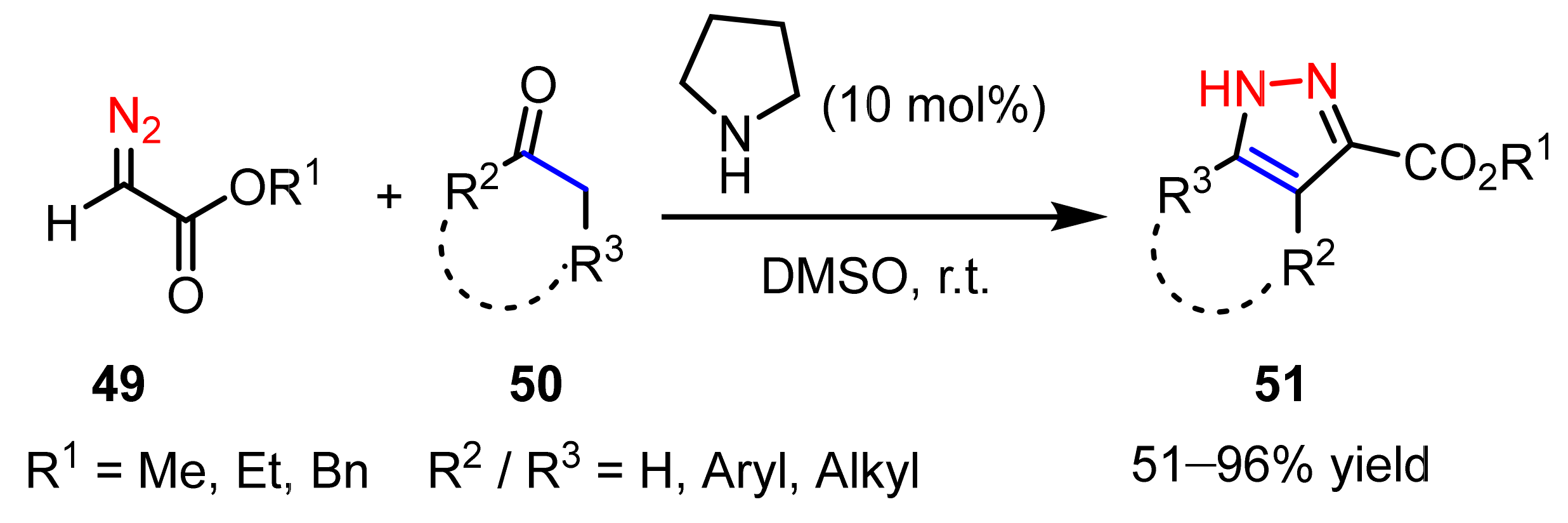
2.2.1. Diazoalkanes as 1,3-Dipoles
2.2.2. Nitrilimines as 1,3-Dipoles
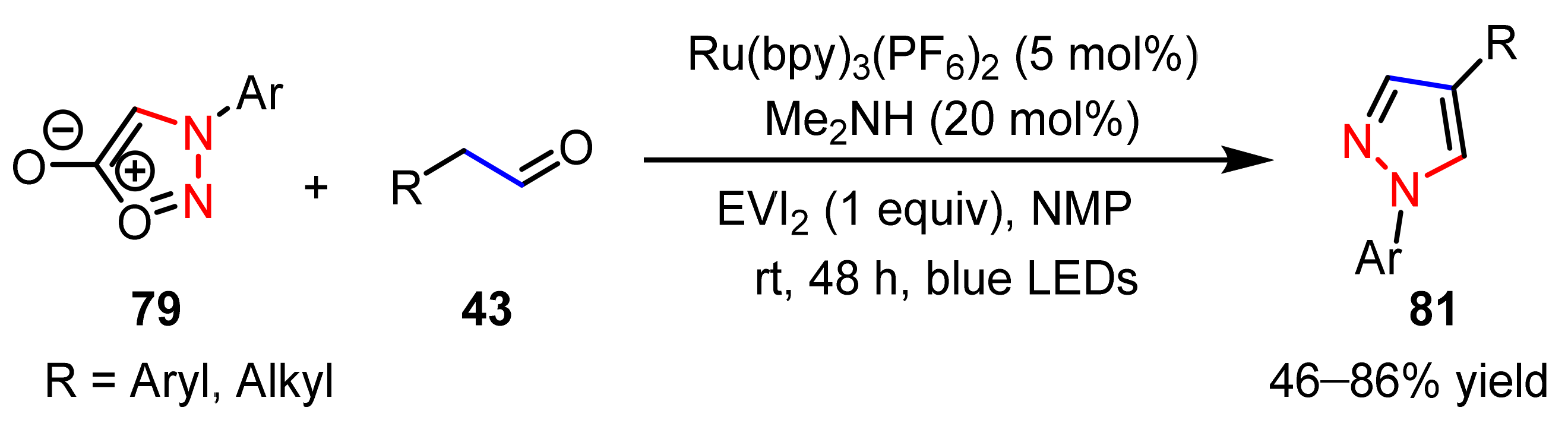
2.2.3. Sydnones as 1,3-Dipoles
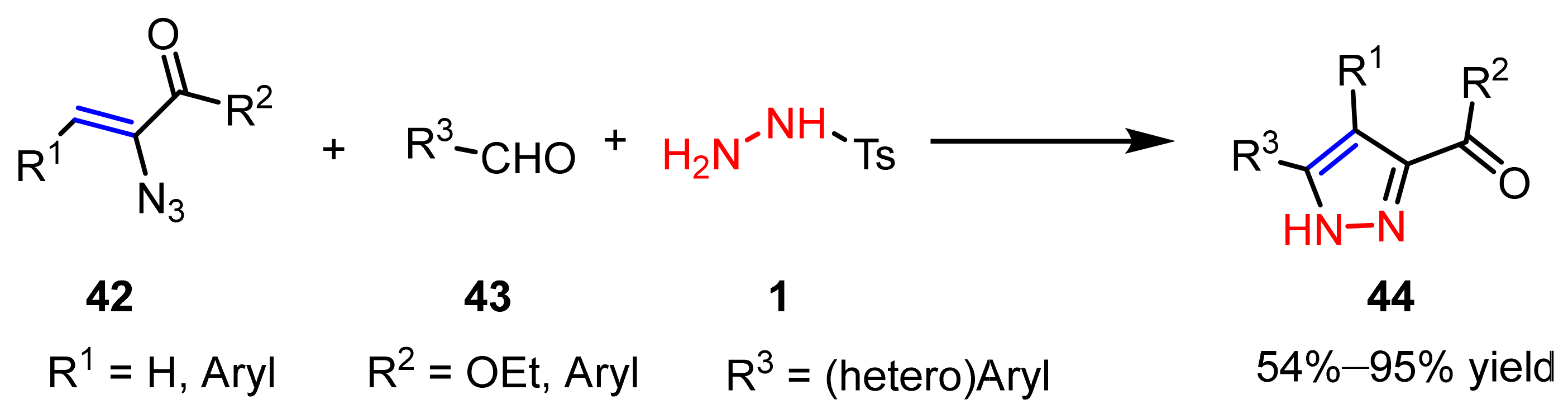
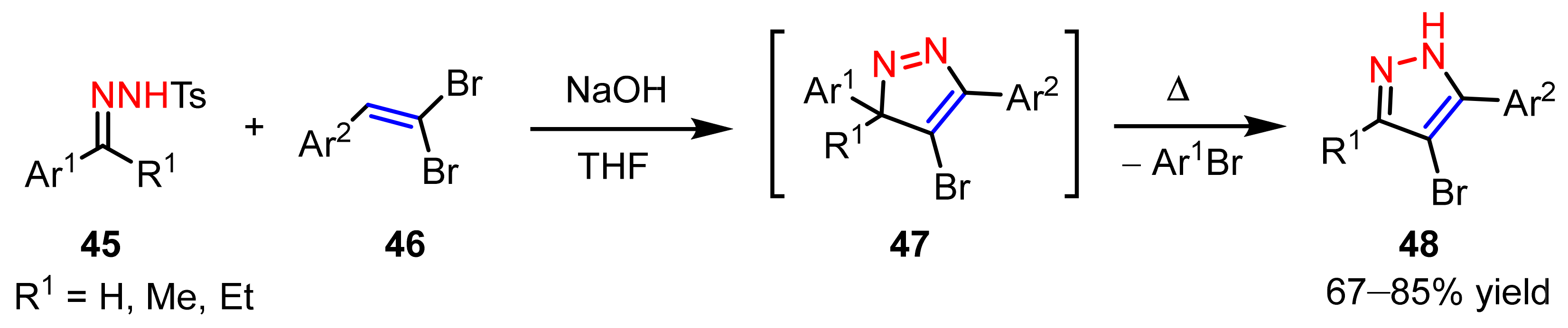
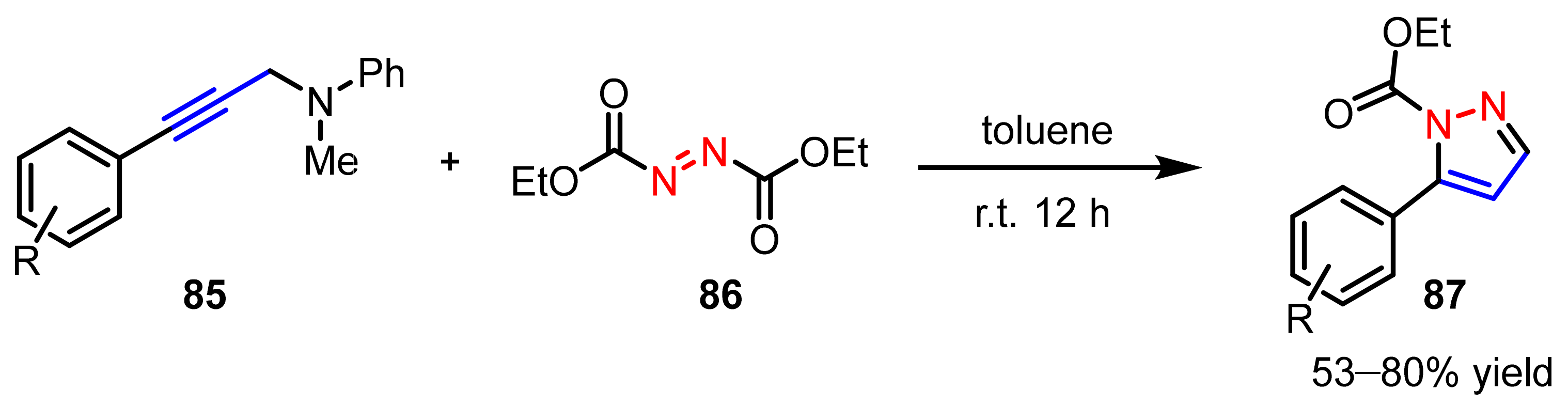
2.2.4. Azo Compounds as [NN] Fragments
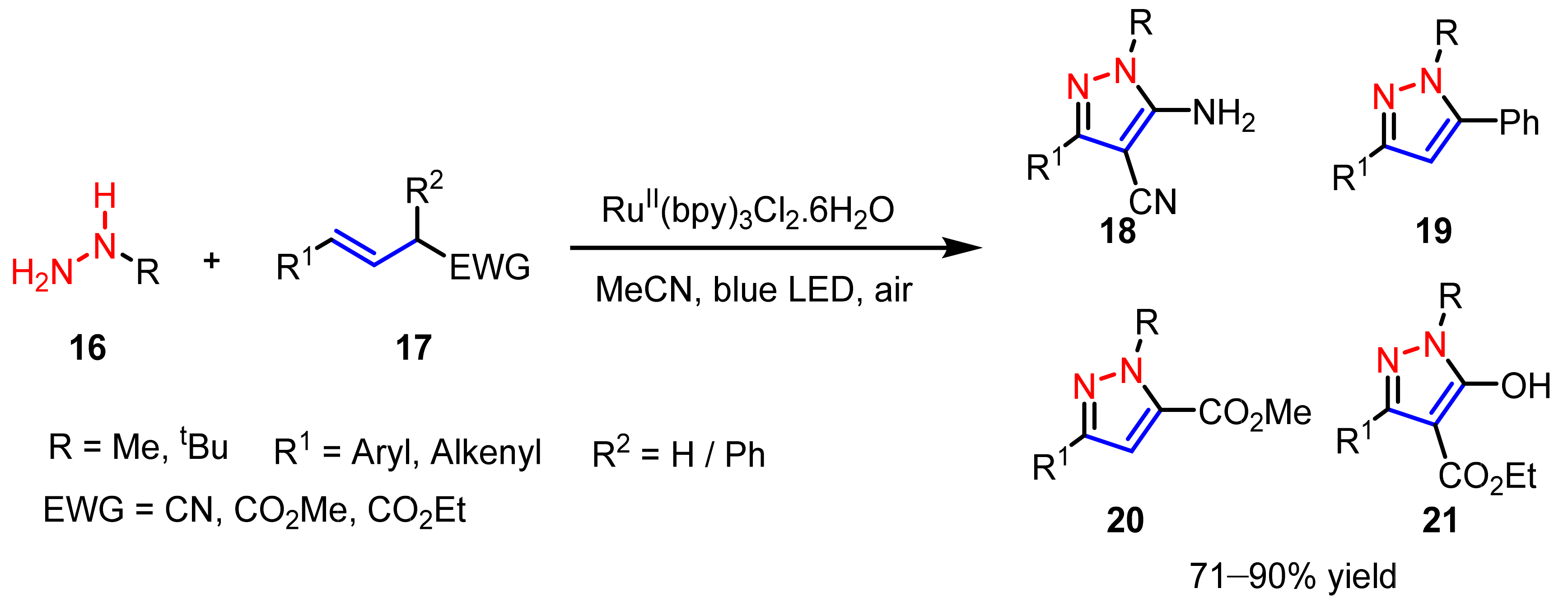
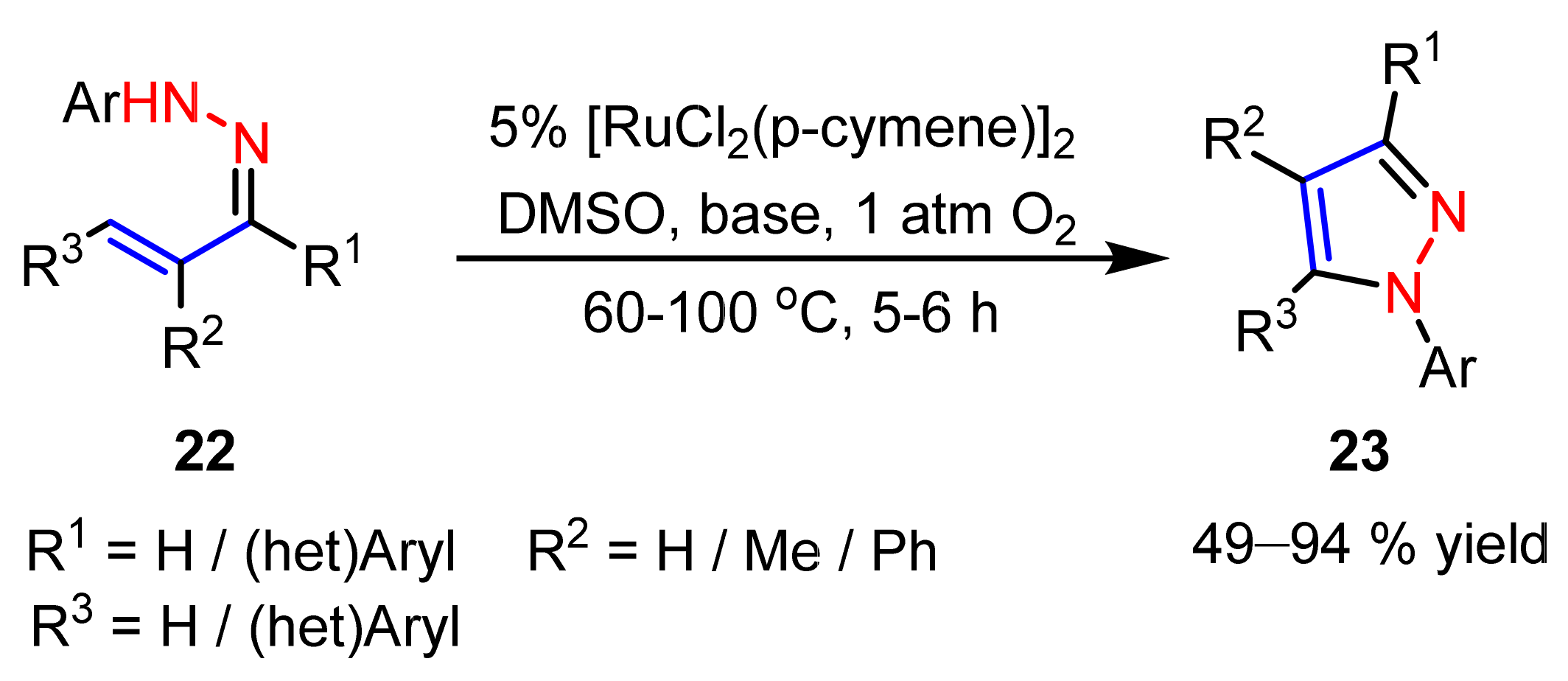
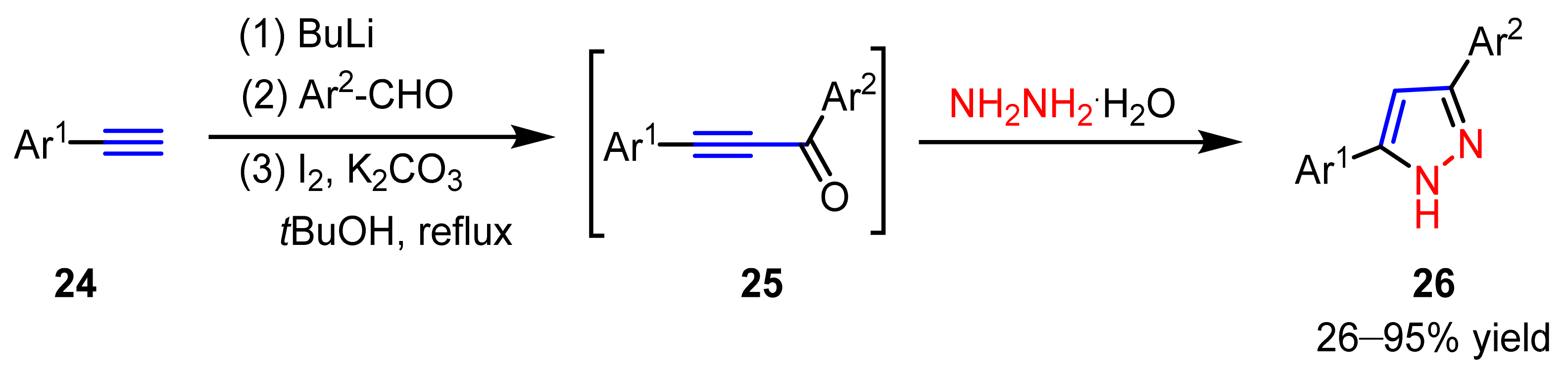
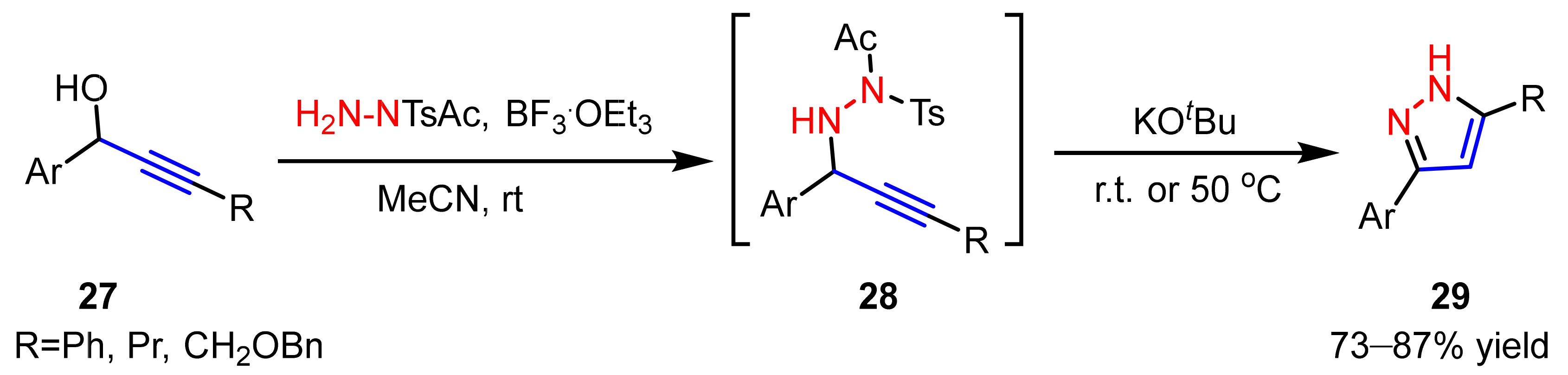
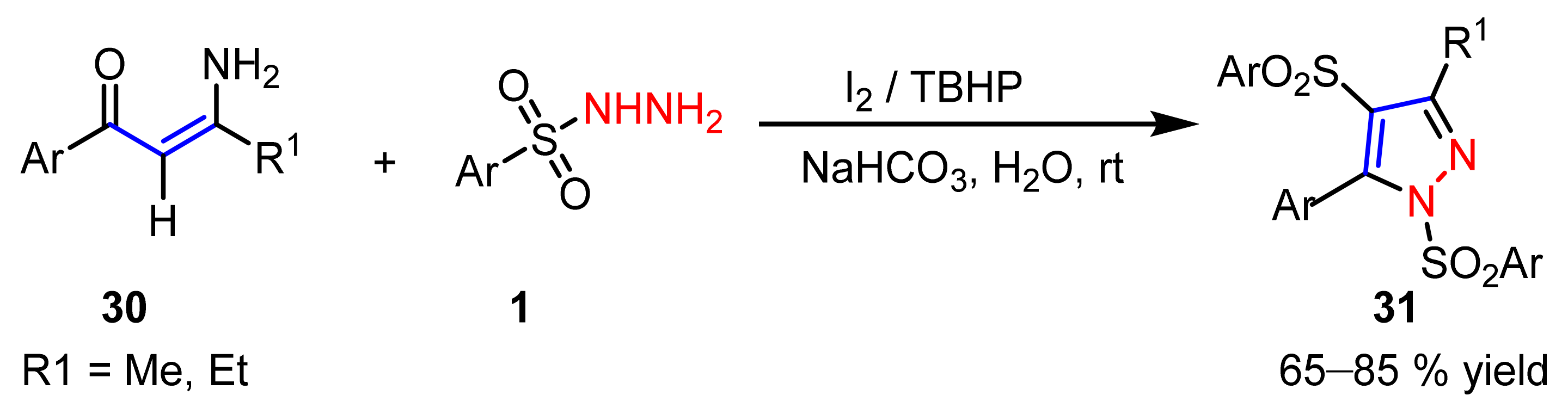
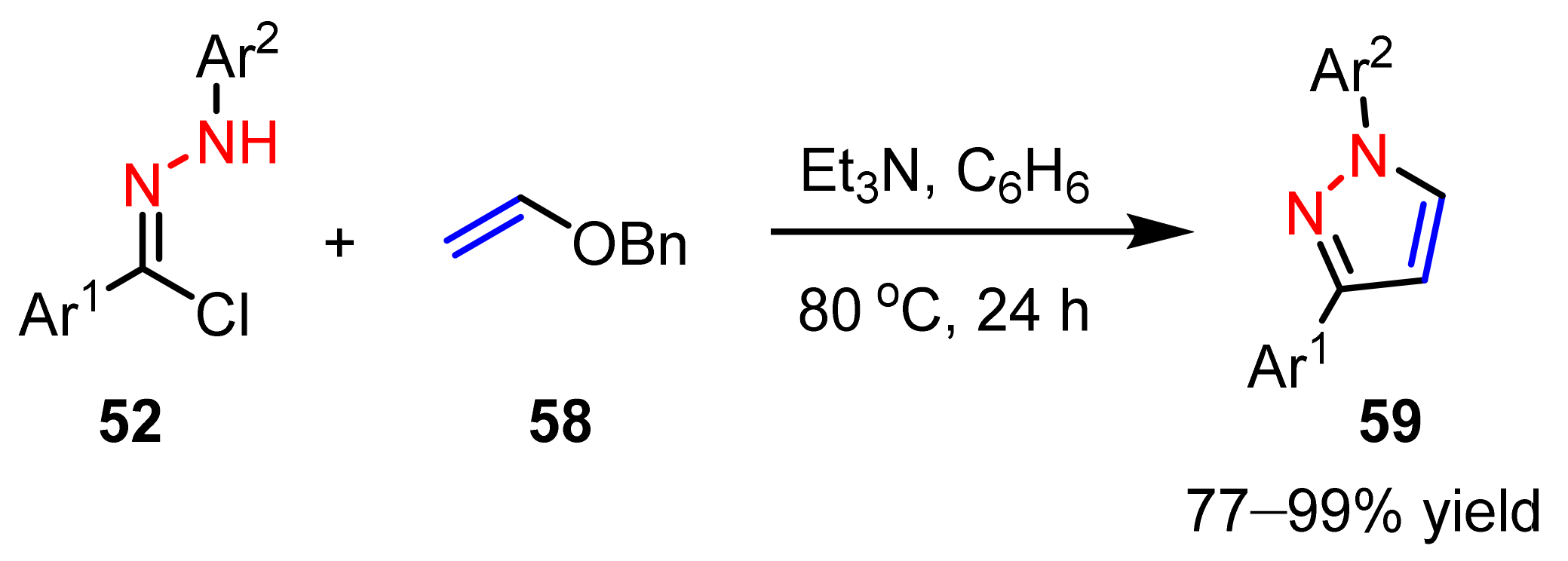
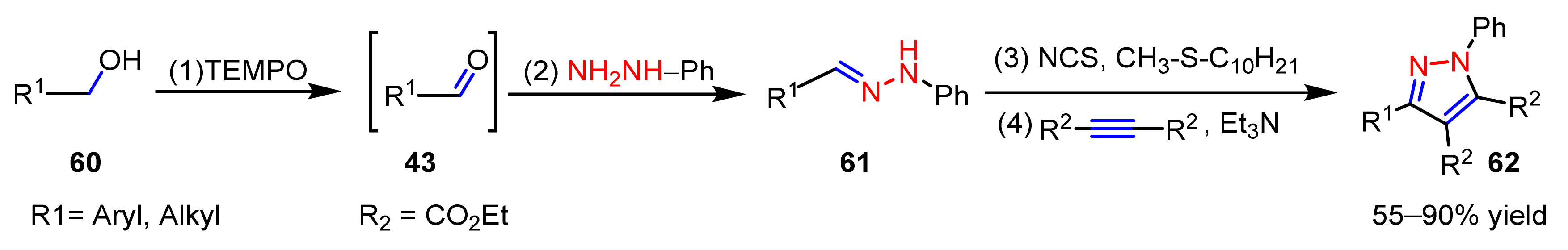
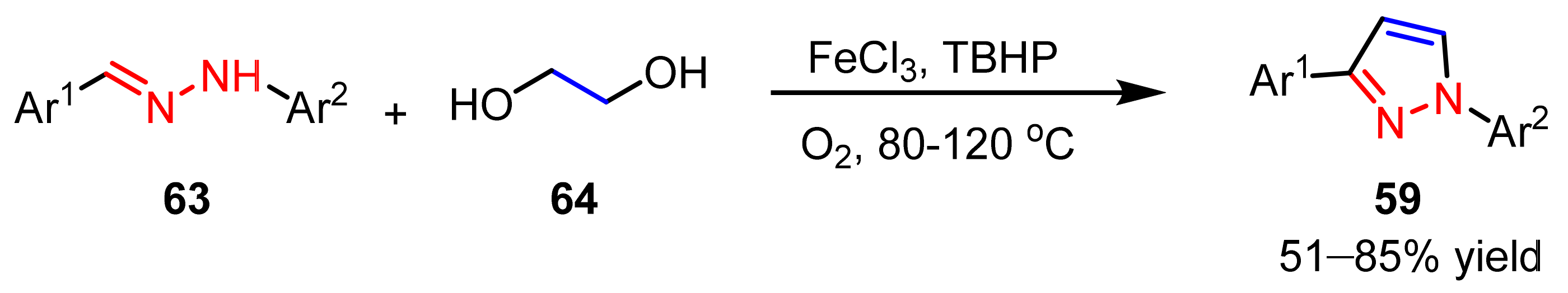
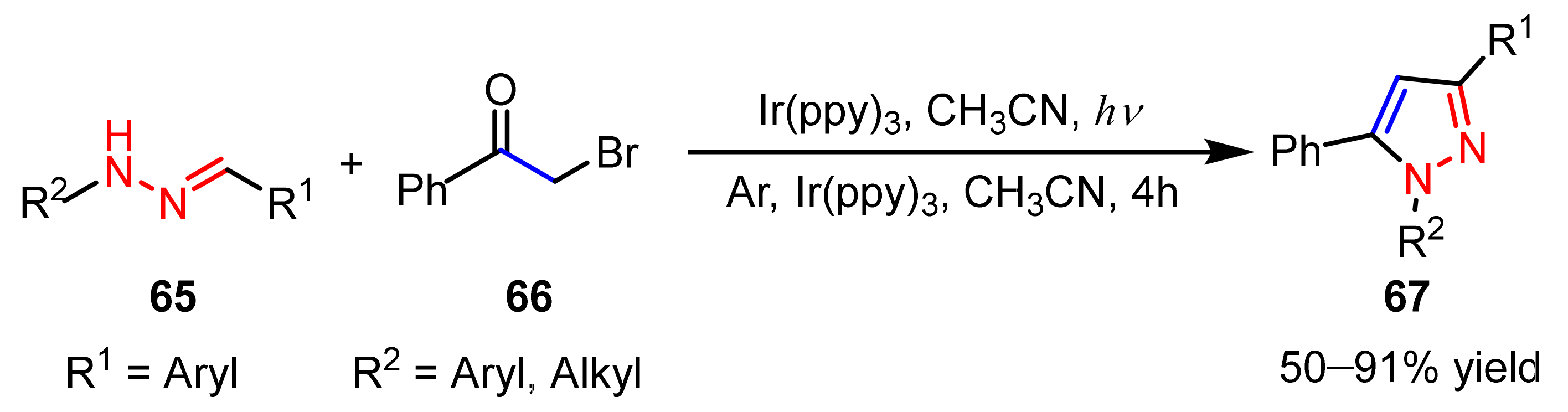
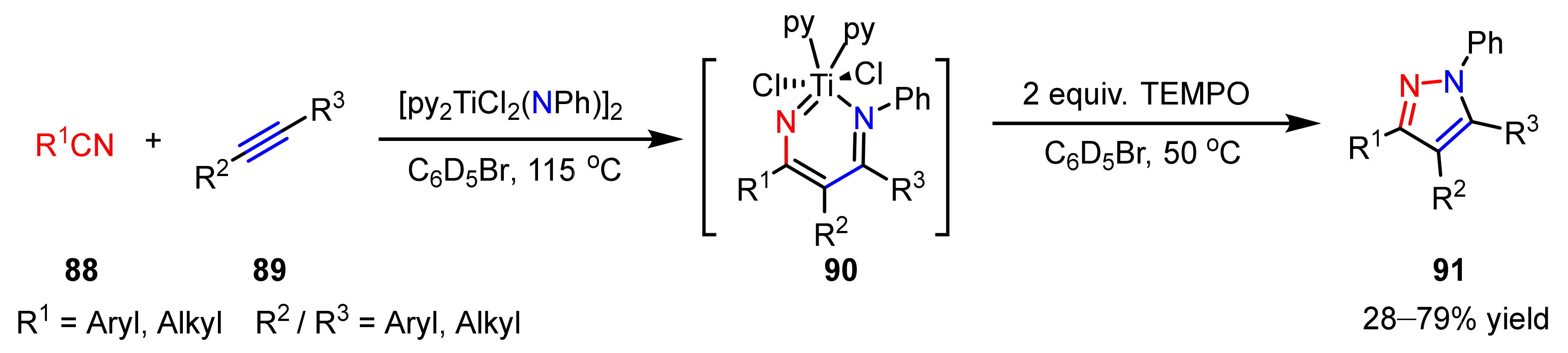
2.3. New Approaches to Reactions for Pyrazole Synthesis
3. The Functionalization of Pyrazoles
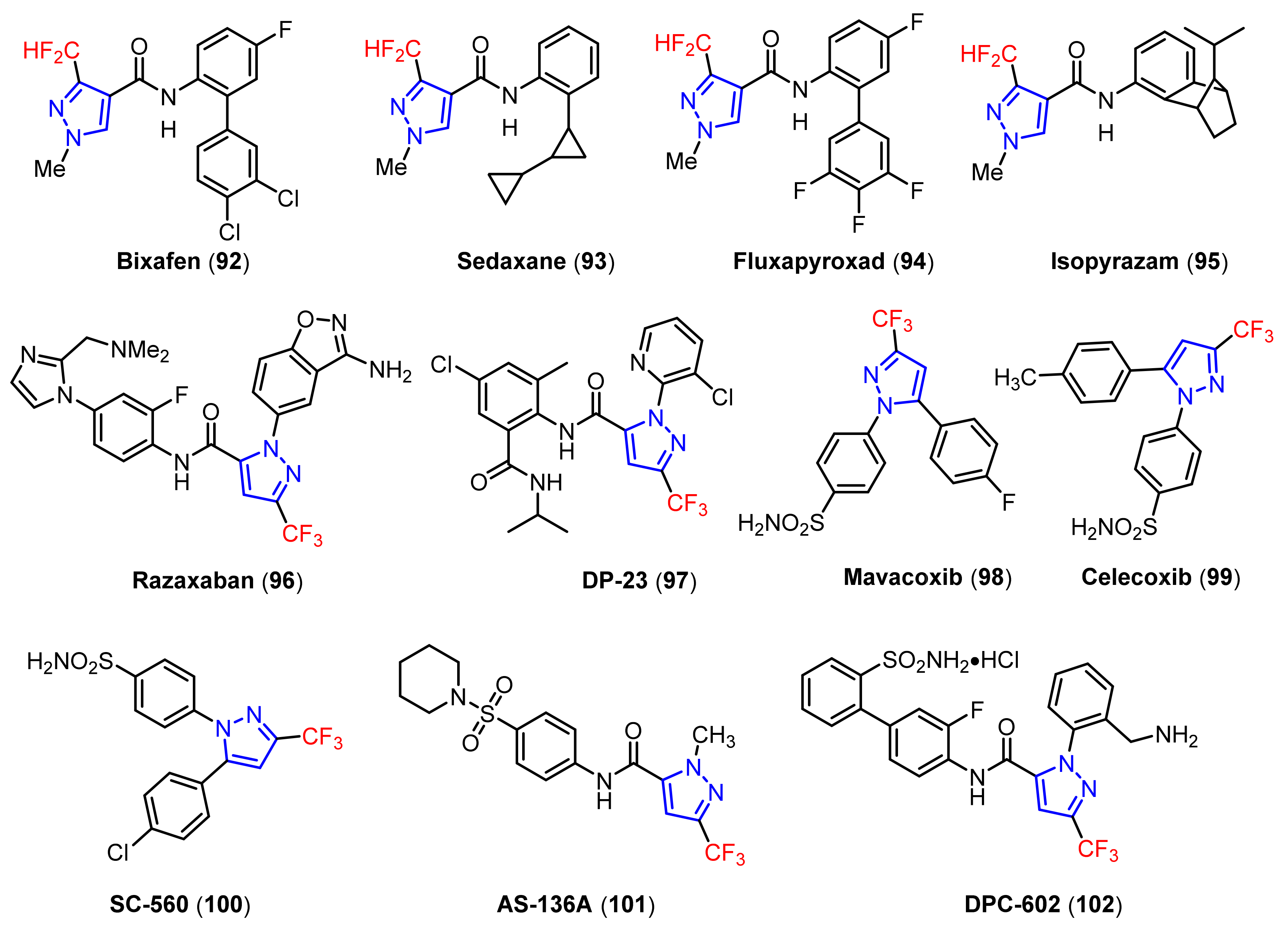
3.1. The Synthesis of Fluorine-Containing and Fluoroalkyl Substituted Pyrazoles
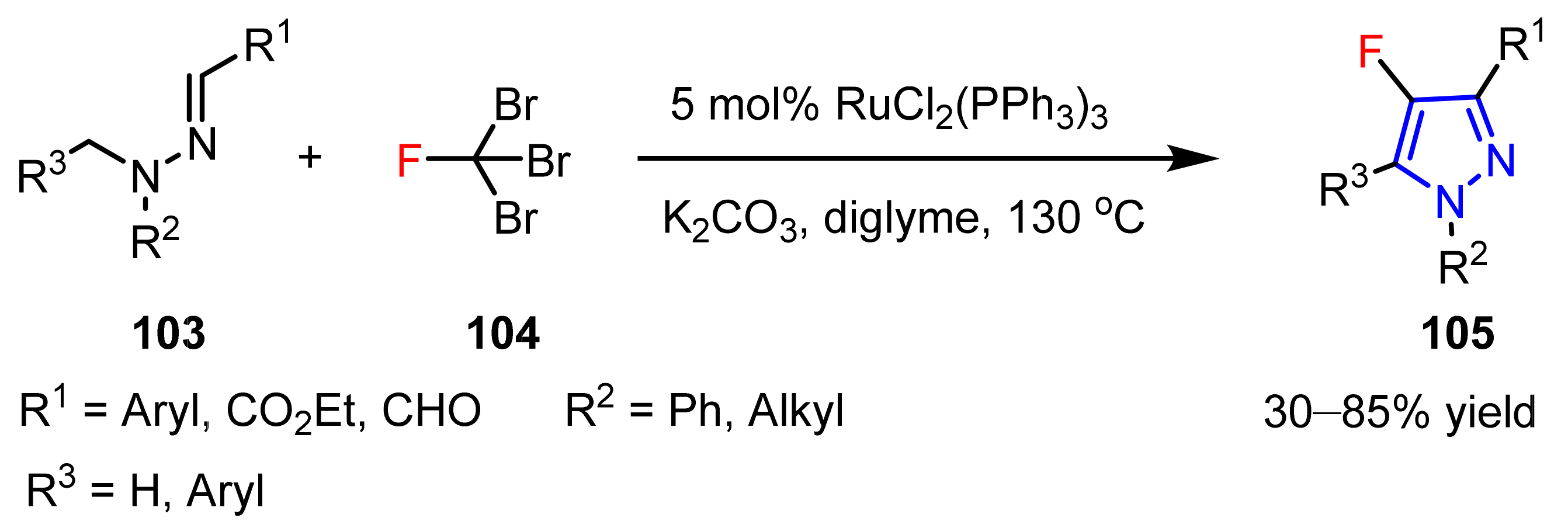
3.1.1. The Synthesis of Monofluorine-Substituted Pyrazoles
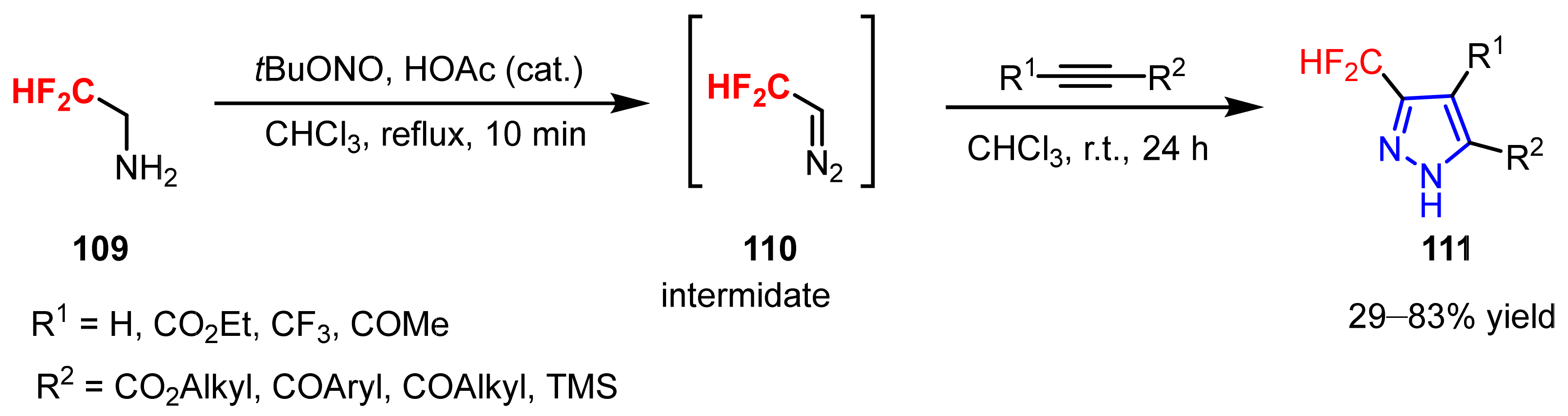
3.1.2. The Synthesis of Difluoromethylpyrazoles
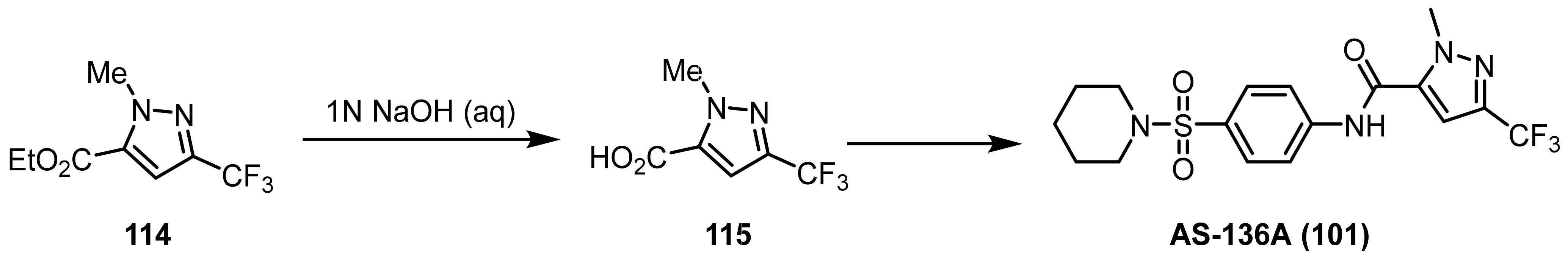
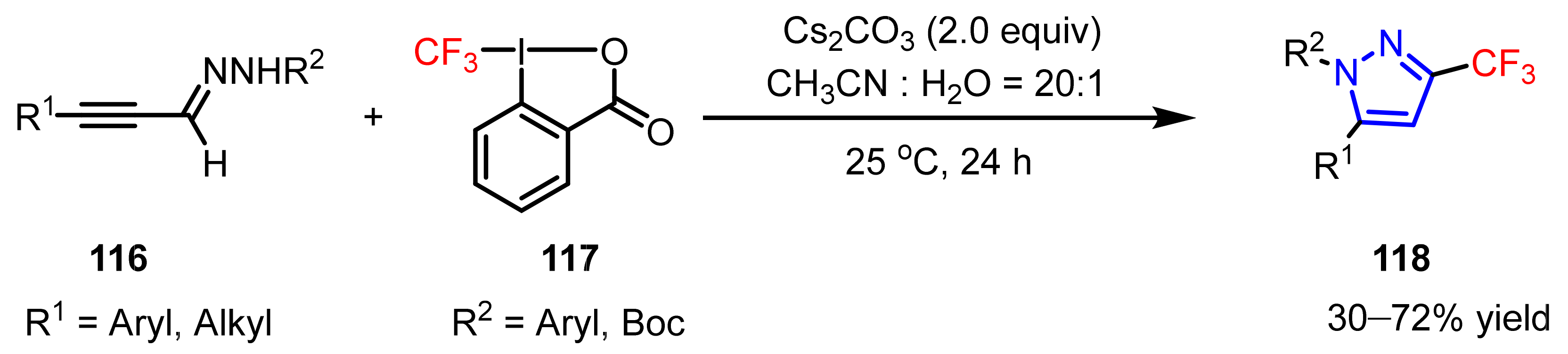
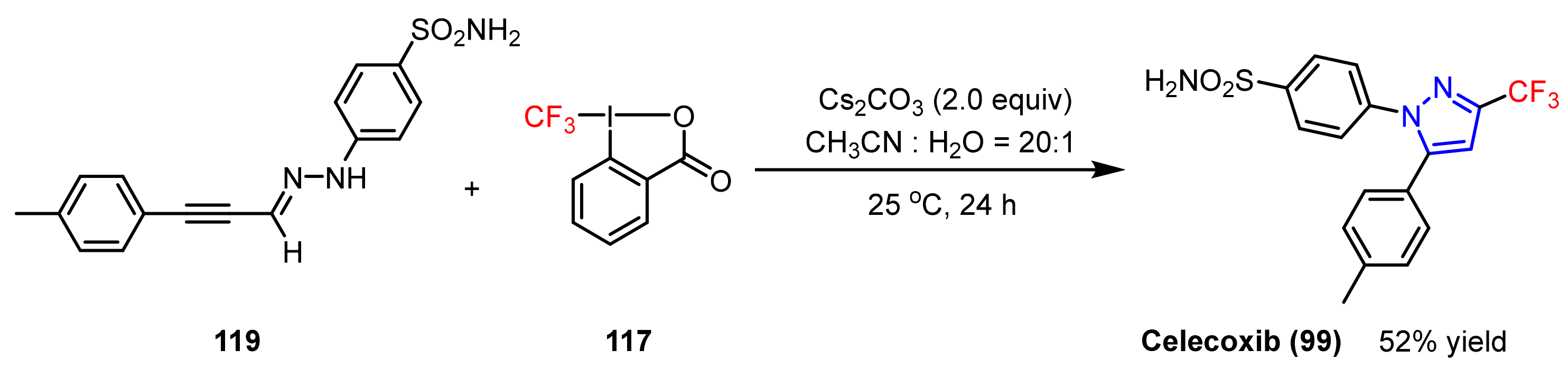
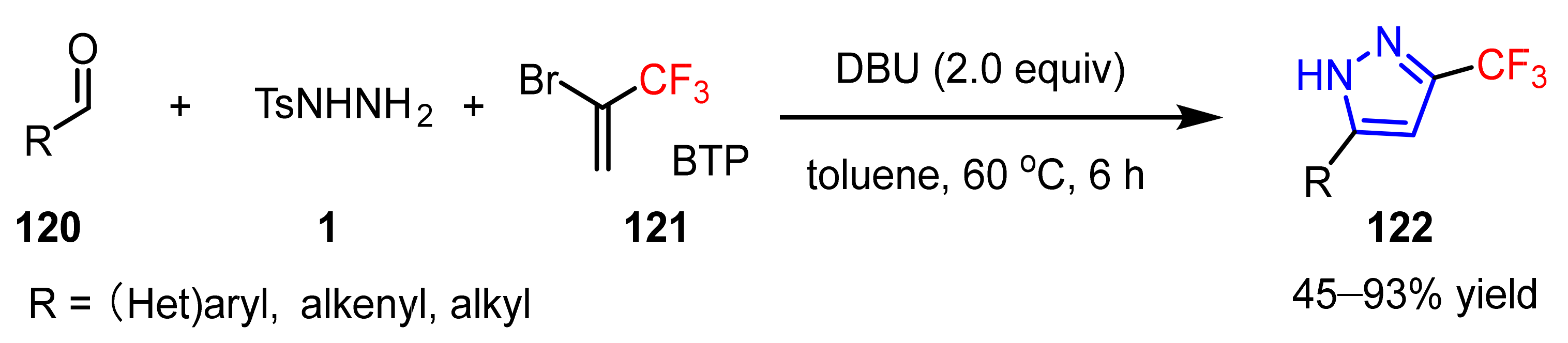
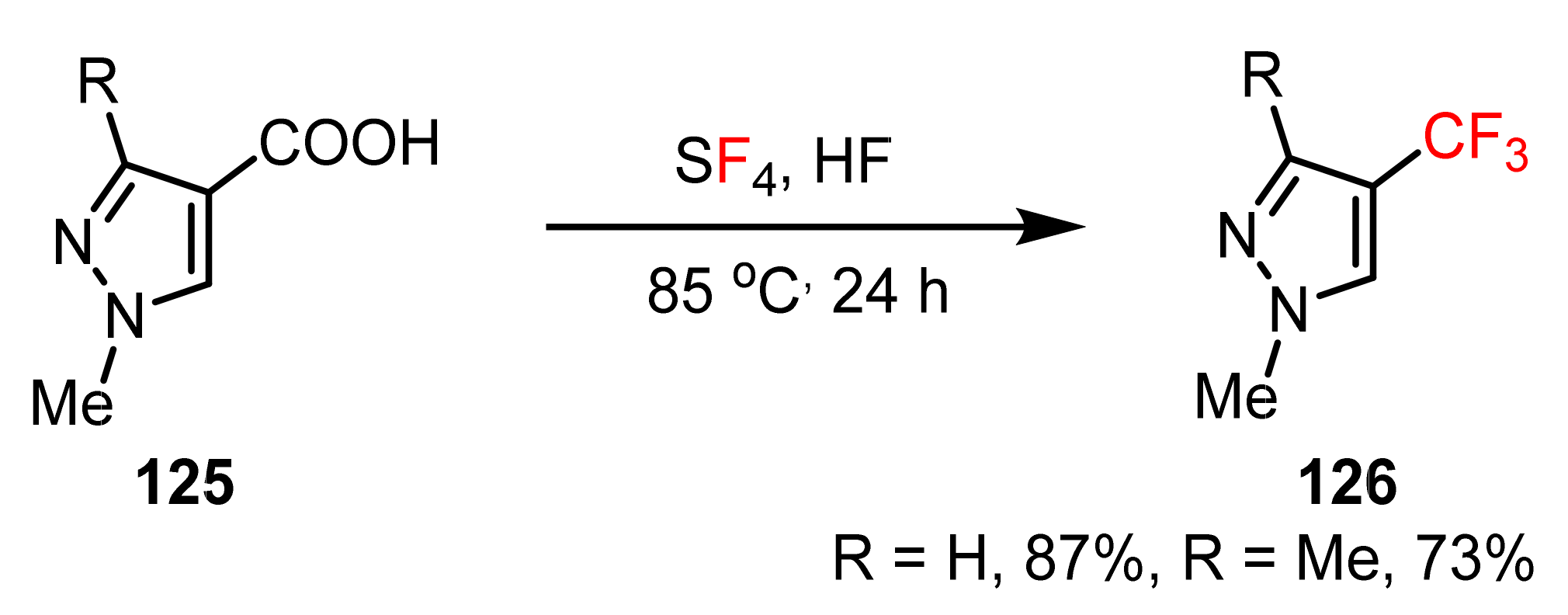
3.1.3. The Synthesis of Trifluoromethyl Pyrazoles
3.2. N-Alkylation, N-Arylation, and N-Alkenylation of Pyrazoles
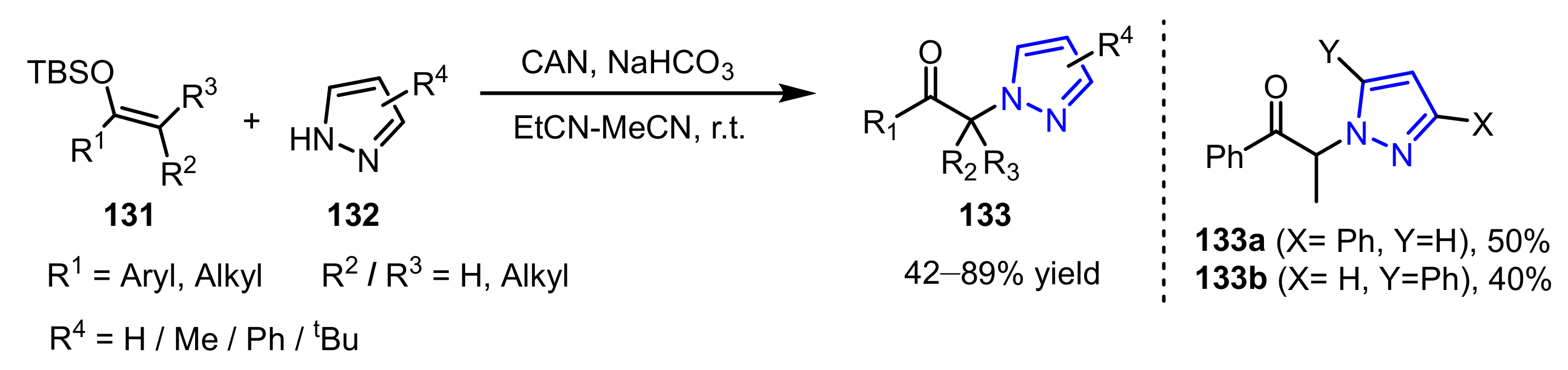
3.2.1. The Synthesis of N-Alkylated Pyrazoles
3.2.2. The Synthesis of N-Alkenylated Pyrazoles
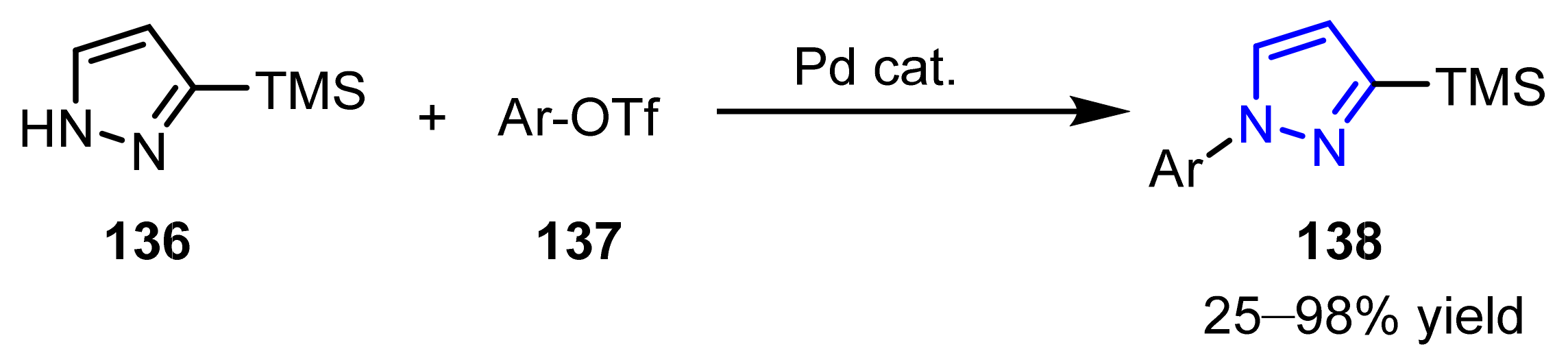
3.2.3. The Synthesis of N-Arylated Pyrazoles
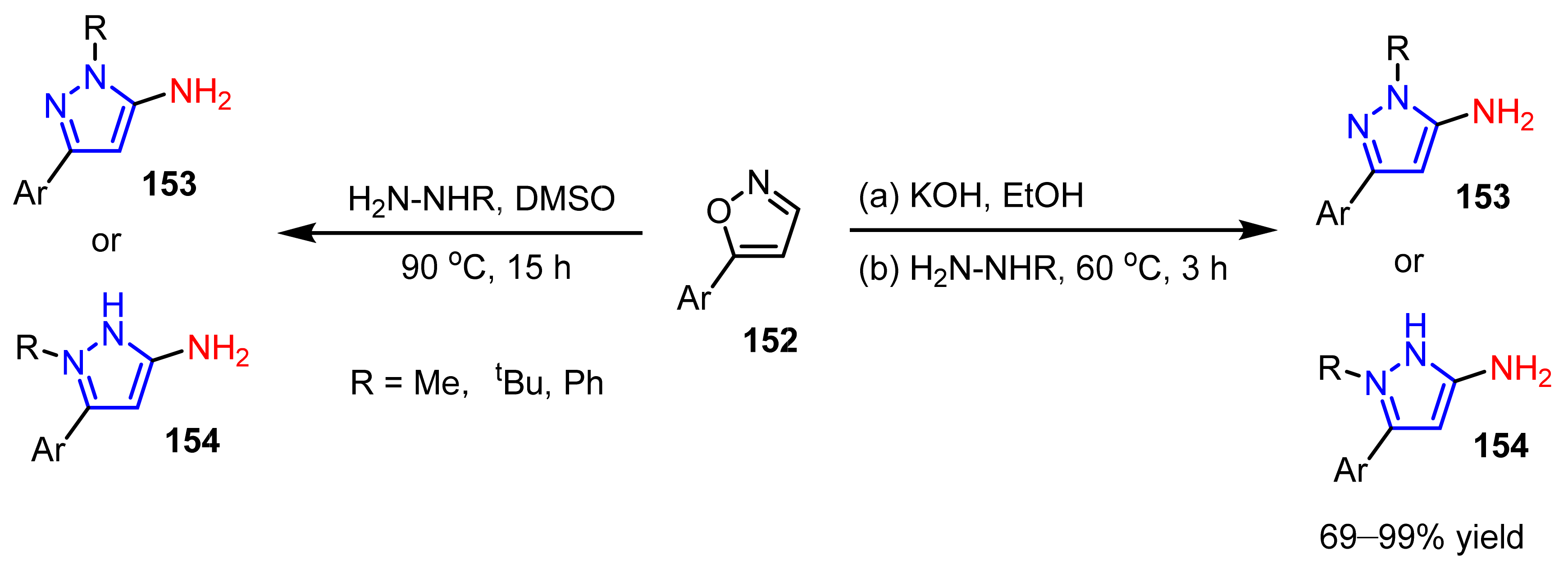
3.3. The Amination of Pyrazoles
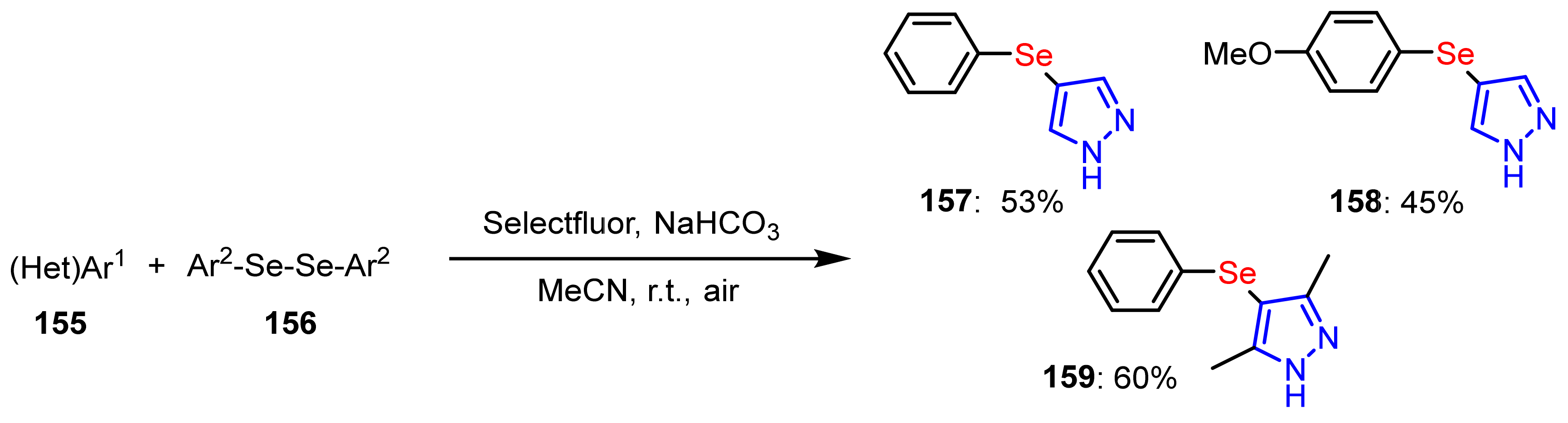
3.4. The Selanylation of Pyrazoles.
3.5. The Borylation of Pyrazoles
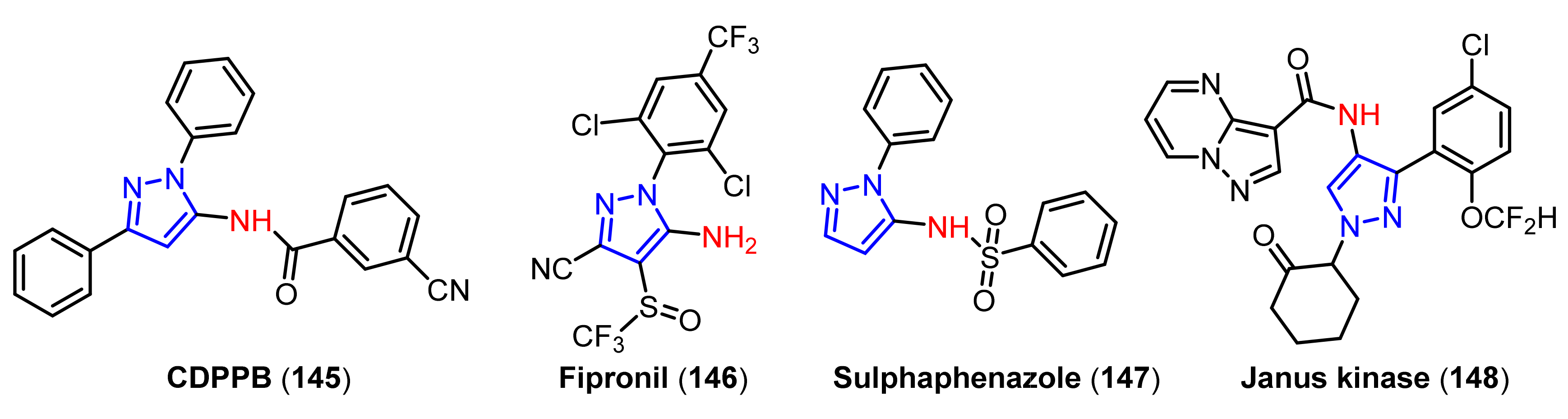
4. Applications of Pyrazoles in Neurodegenerative Diseases
4.1. Applications of Pyrazoles in Alzheimer’s Disease (AD) Treatment
4.1.1. Acetylcholinesterase (AChE) Inhibitors
4.1.2. Protein Aggregation Inhibitors
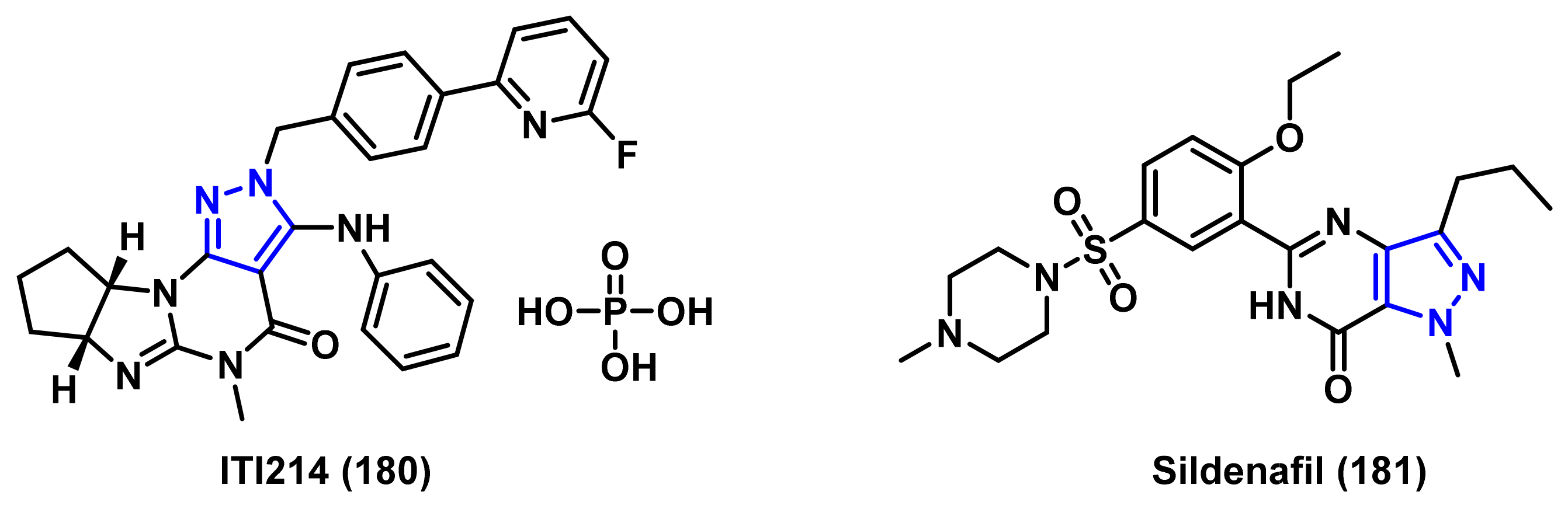
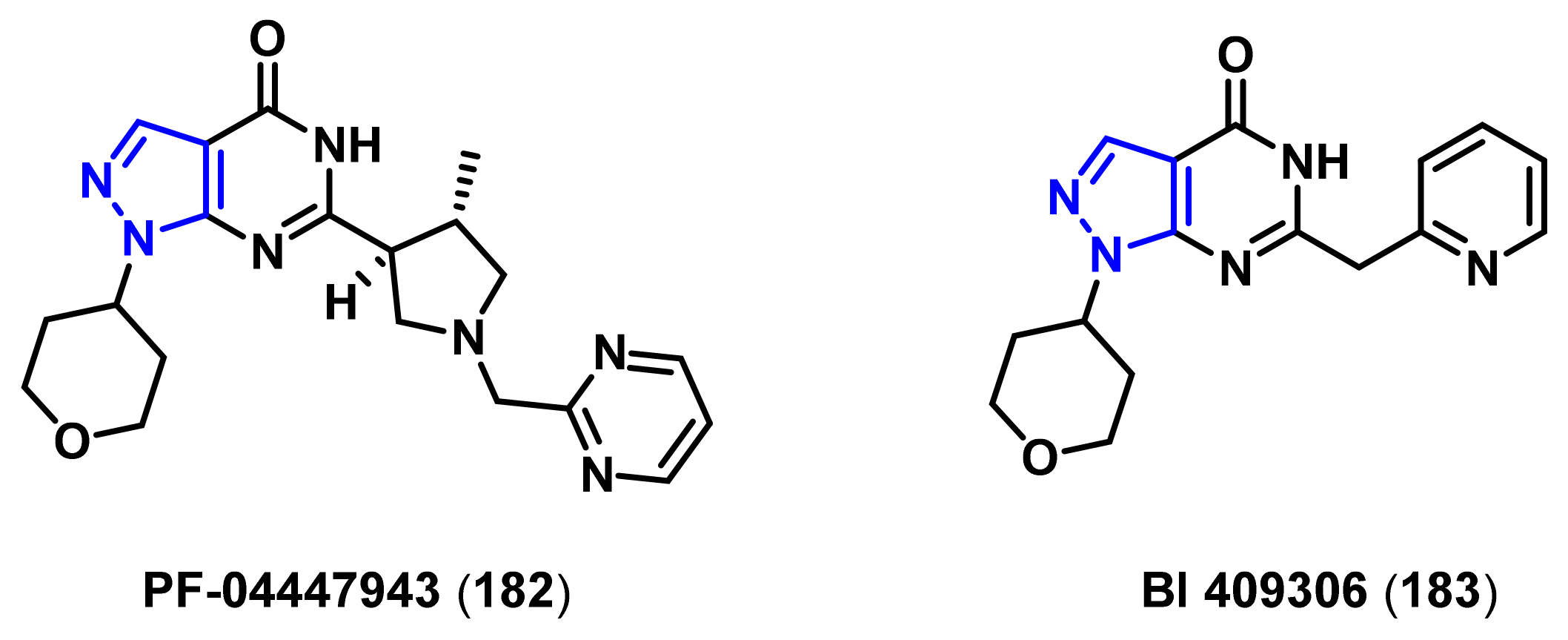
4.1.3. Phosphodiesterase (PDE) Inhibitors
4.1.4. Dual Leucine Zipper Kinase (DLK) Inhibitors
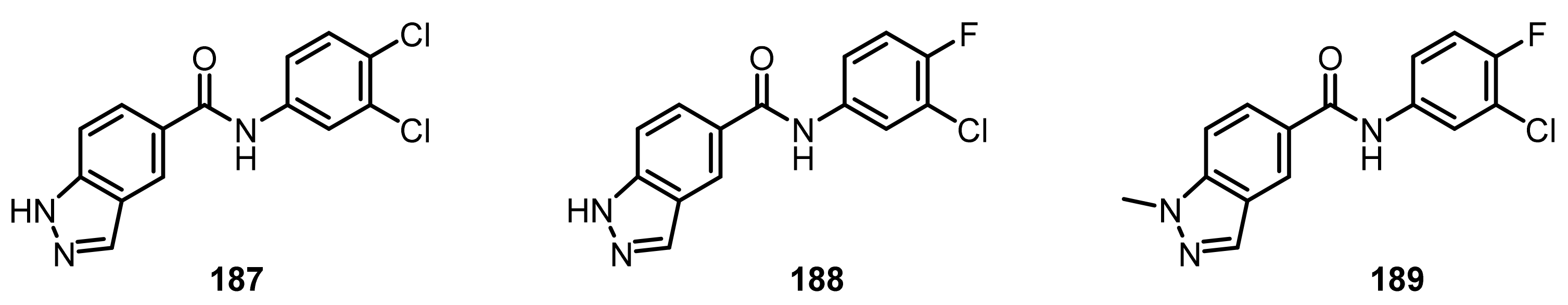
4.1.5. Monoamine Oxidases B (MAO-B) Inhibitors
4.2. Applications of Pyrazoles in Parkinson’s Disease (PD) Treatment
4.2.1. Antioxidants
4.2.2. Protein Aggregation Inhibitors
4.2.3. Adenosine Receptor A2A Receptor Antagonists
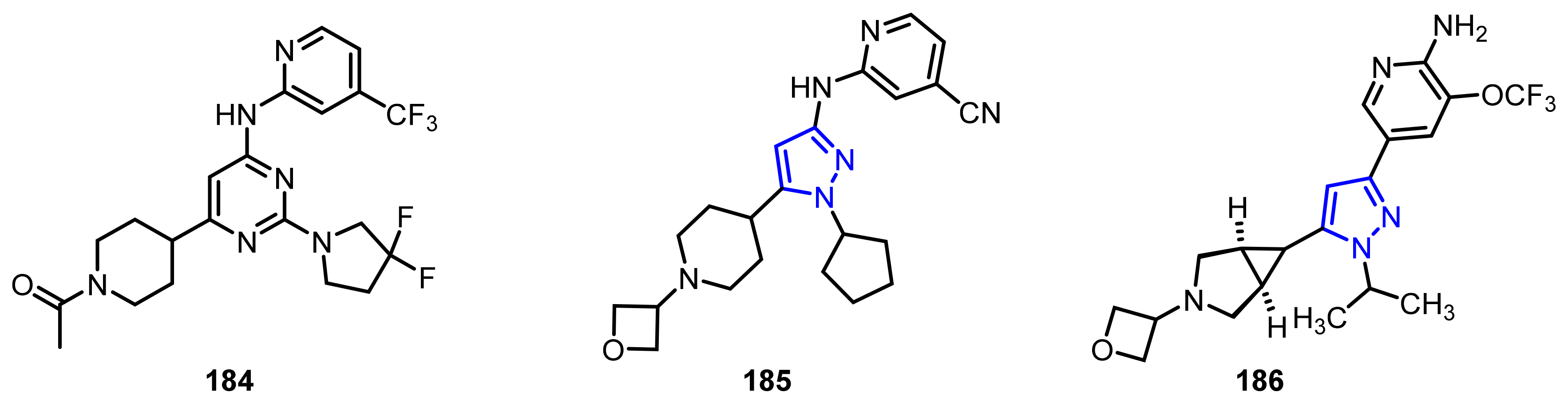
4.2.4. Phosphodiesterase-10A (PDE10A) Enzyme Inhibitors
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.P.; Leroux, F.R. Synthesis of diversely fluorinated pyrazoles as novel active agrochemical ingredients. J. Fluorine Chem. 2013, 152, 2–11. [Google Scholar] [CrossRef]
- Khan, M.F.; Alam, M.M.; Verma, G.; Akhtar, W.; Akhter, M.; Shaquiquzzaman, M. The therapeutic voyage of pyrazole and its analogs: A review. Eur. J. Med. Chem. 2016, 120, 170–201. [Google Scholar] [CrossRef] [PubMed]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and pharmacological activities of pyrazole derivatives: A review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A clinical pharmacokinetic and pharmacodynamic review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fustero, S.; Sanchez-Rosello, M.; Barrio, P.; Simon-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef]
- Gomes, P.M.O.; Silva, A.M.S.; Silva, V.L.M. Pyrazoles as key scaffolds for the development of fluorine-18-labeled radiotracers for positron emission tomography (PET). Molecules 2020, 25, 1722. [Google Scholar] [CrossRef]
- Turkan, F.; Cetin, A.; Taslimi, P.; Gulcin, I. Some pyrazoles derivatives: Potent carbonic anhydrase, α-glycosidase, and cholinesterase enzymes inhibitors. Arch. Pharm. 2018, 351, e1800200. [Google Scholar] [CrossRef]
- Hazarika, R.; Konwar, M.; Damarla, K.; Kumar, A.; Sarma, D. HBF4/ACN: A simple and efficient protocol for the synthesis of pyrazoles under ambient reaction conditions. Synth. Commun. 2019, 50, 329–337. [Google Scholar] [CrossRef]
- Wang, H.F.; Sun, X.L.; Zhang, S.L.; Liu, G.L.; Wang, C.J.; Zhu, L.L.; Zhang, H. Efficient copper-catalyzed synthesis of substituted pyrazoles at room temperature. Synlett 2018, 29, 2689–2692. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.V.; Yadav, S.K.; Raghava, B.; Saraiah, B.; Ila, H.; Rangappa, K.S.; Hazra, A. Cyclocondensation of arylhydrazines with 1,3-bis(het)arylmonothio-1,3-diketones and 1,3-bis(het)aryl-3-(methylthio)-2-propenones: Synthesis of 1-aryl-3,5-bis(het)arylpyrazoles with complementary regioselectivity. J. Org. Chem. 2013, 78, 4960–4973. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, D.C.; Taylor, A.P.; Flick, A.C.; Kyne, R.E., Jr. Synthesis of pyrazoles from 1,3-diols via hydrogen transfer catalysis. Org. Lett. 2015, 17, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kang, J.; Niu, P.; Wu, J.; Yu, W.; Chang, J. I2-mediated oxidative C-N bond formation for metal-free one-pot synthesis of di-, tri-, and tetrasubstituted pyrazoles from α,β-unsaturated aldehydes/ketones and hydrazines. J. Org. Chem. 2014, 79, 10170–10178. [Google Scholar] [CrossRef] [PubMed]
- Gutti, G.; Kumar, D.; Paliwal, P.; Ganeshpurkar, A.; Lahre, K.; Kumar, A.; Krishnamurthy, S.; Singh, S.K. Development of pyrazole and spiropyrazoline analogs as multifunctional agents for treatment of Alzheimer’s disease. Bioorg. Chem. 2019, 90, 103080. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Zhang, T.; Chen, Q.Y.; Zhu, C. Visible-light photocatalytic aerobic annulation for the green synthesis of pyrazoles. Org. Lett. 2016, 18, 4206–4209. [Google Scholar] [CrossRef]
- Hu, J.; Chen, S.; Sun, Y.; Yang, J.; Rao, Y. Synthesis of tri- and tetrasubstituted pyrazoles via Ru(II) catalysis: Intramolecular aerobic oxidative C-N coupling. Org. Lett. 2012, 14, 5030–5033. [Google Scholar] [CrossRef]
- Harigae, R.; Moriyama, K.; Togo, H. Preparation of 3,5-disubstituted pyrazoles and isoxazoles from terminal alkynes, aldehydes, hydrazines, and hydroxylamine. J. Org. Chem. 2014, 79, 2049–2058. [Google Scholar] [CrossRef] [PubMed]
- Raji Reddy, C.; Vijaykumar, J.; Grée, R. Facile one-pot synthesis of 3,5-disubstituted 1H-pyrazoles from propargylic alcohols via propargyl hydrazides. Synthesis 2013, 45, 830–836. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, G.; Wei, L.; Wan, J.P. Domino C-H sulfonylation and pyrazole annulation for fully substituted pyrazole synthesis in water using hydrophilic enaminones. J. Org. Chem. 2019, 84, 2984–2990. [Google Scholar] [CrossRef] [PubMed]
- Raghunadh, A.; Meruva, S.B.; Mekala, R.; Rao, K.R.; Krishna, T.; Chary, R.G.; Rao, L.V.; Kumar, U.K.S. An efficient regioselective copper catalyzed multi-component synthesis of 1,3-disubstituted pyrazoles. Tetrahedron Lett. 2014, 55, 2986–2990. [Google Scholar] [CrossRef]
- Chen, J.; Properzi, R.; Uccello, D.P.; Young, J.A.; Dushin, R.G.; Starr, J.T. One-pot oxidation and rearrangement of propargylamines and in situ pyrazole synthesis. Org. Lett. 2014, 16, 4146–4149. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Zhang, F.M. Efficient one-pot synthesis of substituted pyrazoles. Tetrahedron 2013, 69, 1427–1433. [Google Scholar] [CrossRef]
- Wen, J.; Fu, Y.; Zhang, R.Y.; Zhang, J.; Chen, S.Y.; Yu, X.Q. A simple and efficient synthesis of pyrazoles in water. Tetrahedron 2011, 67, 9618–9621. [Google Scholar] [CrossRef]
- Zhang, G.; Ni, H.; Chen, W.; Shao, J.; Liu, H.; Chen, B.; Yu, Y. One-pot three-component approach to the synthesis of polyfunctional pyrazoles. Org. Lett. 2013, 15, 5967–5969. [Google Scholar] [CrossRef]
- Sha, Q.; Wei, Y.Y. An efficient one-pot synthesis of 3,5-diaryl-4-bromopyrazoles by 1,3-dipolar cycloaddition of in situ generated diazo compounds and 1-bromoalk-1-ynes. Synthesis 2013, 45, 413–420. [Google Scholar] [CrossRef]
- Wang, L.; Huang, J.; Gong, X.; Wang, J. Highly regioselective organocatalyzed synthesis of pyrazoles from diazoacetates and carbonyl compounds. Chem. Eur. J. 2013, 19, 7555–7560. [Google Scholar] [CrossRef]
- Jackowski, O.; Lecourt, T.; Micouin, L. Direct synthesis of polysubstituted aluminoisoxazoles and pyrazoles by a metalative cyclization. Org. Lett. 2011, 13, 5664–5667. [Google Scholar] [CrossRef]
- Li, D.Y.; Mao, X.F.; Chen, H.J.; Chen, G.R.; Liu, P.N. Rhodium-catalyzed addition-cyclization of hydrazines with alkynes: Pyrazole synthesis via unexpected C-N bond cleavage. Org. Lett. 2014, 16, 3476–3479. [Google Scholar] [CrossRef]
- Ledovskaya, M.S.; Voronin, V.V.; Polynski, M.V.; Lebedev, A.N.; Ananikov, V.P. Primary vinyl ethers as acetylene surrogate: A flexible tool for deuterium-labeled pyrazole synthesis. Eur. J. Org. Chem. 2020, 2020, 4571–4580. [Google Scholar] [CrossRef]
- Kobayashi, E.; Togo, H. Facile one-pot transformation of primary alcohols into 3-aryl- and 3-alkyl-isoxazoles and -pyrazoles. Synthesis 2019, 51, 3723–3735. [Google Scholar] [CrossRef]
- Panda, N.; Jena, A.K. Fe-catalyzed one-pot synthesis of 1,3-di- and 1,3,5-trisubstituted pyrazoles from hydrazones and vicinal diols. J. Org. Chem. 2012, 77, 9401–9406. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.W.; Lei, T.; Zhou, C.; Meng, Q.Y.; Chen, B.; Tung, C.H.; Wu, L.Z. Radical addition of hydrazones by α-bromo ketones to prepare 1,3,5-trisubstituted pyrazoles via visible light catalysis. J. Org. Chem. 2016, 81, 7127–7133. [Google Scholar] [CrossRef]
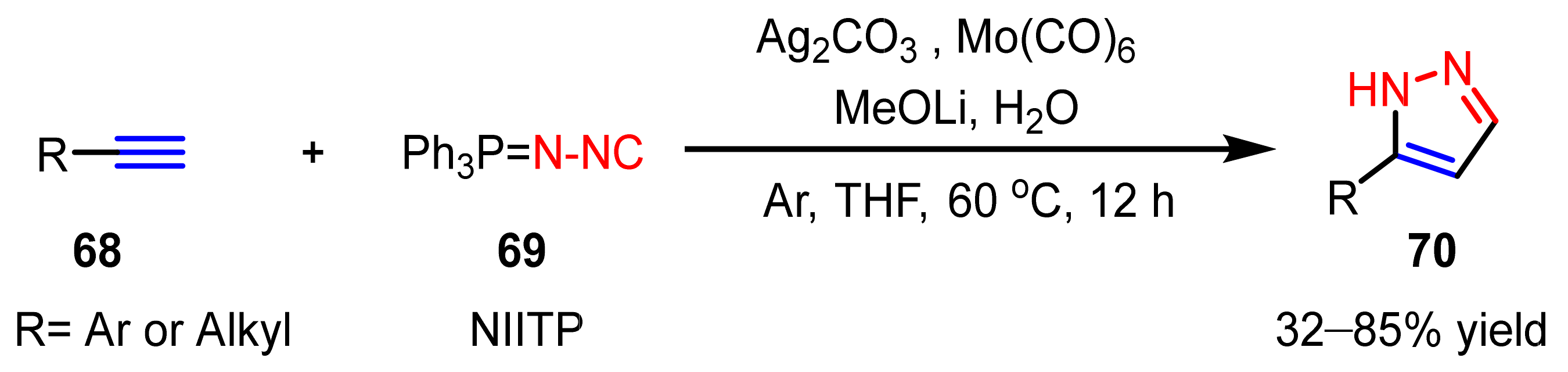
- Yi, F.; Zhao, W.; Wang, Z.; Bi, X. Silver-mediated [3+2] cycloaddition of alkynes and N-isocyanoiminotriphenylphosphorane: Access to monosubstituted pyrazoles. Org. Lett. 2019, 21, 3158–3161. [Google Scholar] [CrossRef]
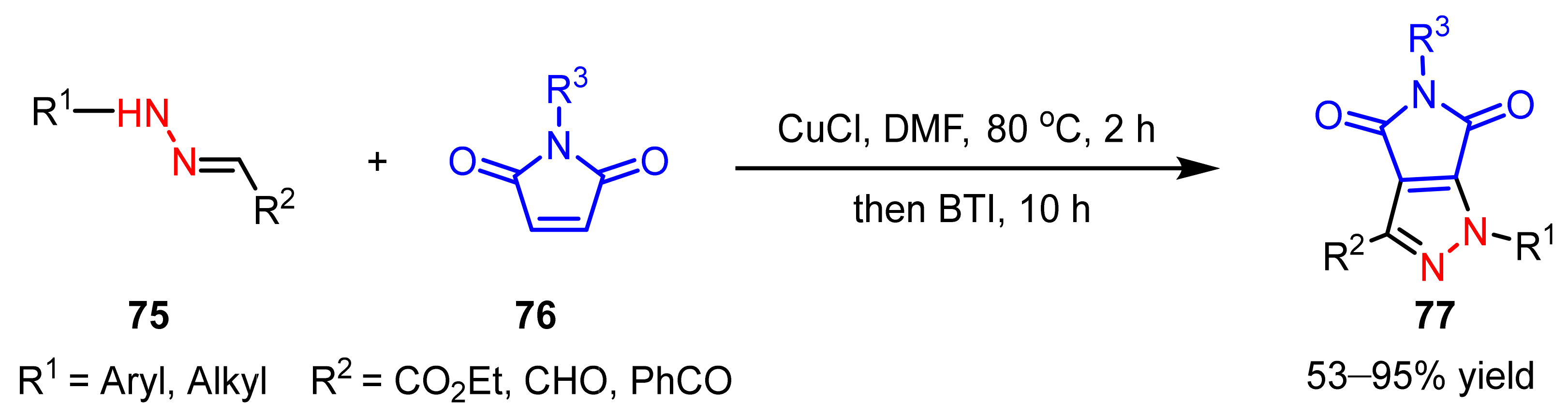
- Zhu, J.N.; Wang, W.K.; Jin, Z.H.; Wang, Q.K.; Zhao, S.Y. Pyrrolo[3,4-c]pyrazole synthesis via copper(I) chloride-catalyzed oxidative coupling of hydrazones to maleimides. Org. Lett. 2019, 21, 5046–5050. [Google Scholar] [CrossRef]
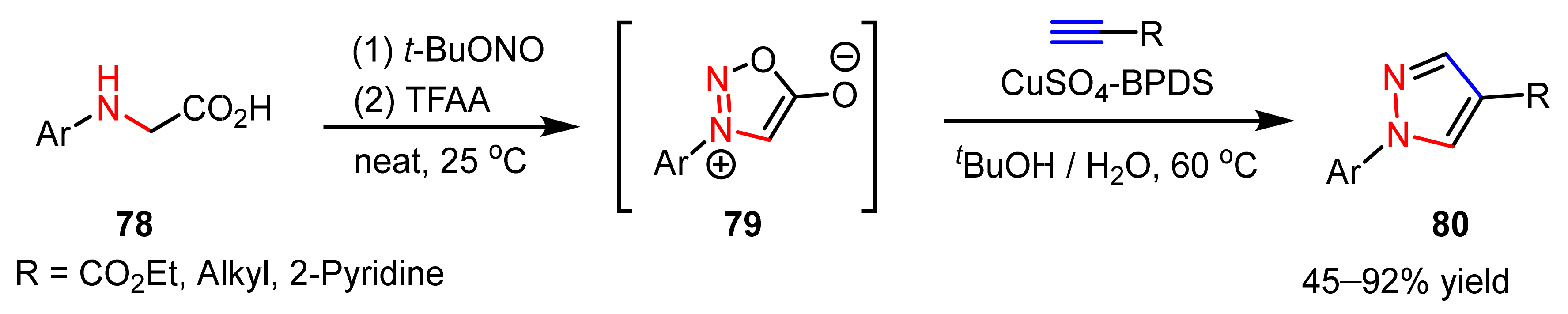
- Specklin, S.; Decuypere, E.; Plougastel, L.; Aliani, S.; Taran, F. One-pot synthesis of 1,4-disubstituted pyrazoles from arylglycines via copper-catalyzed sydnone-alkyne cycloaddition reaction. J. Org. Chem. 2014, 79, 7772–7777. [Google Scholar] [CrossRef]
- Lakeland, C.P.; Watson, D.W.; Harrity, J.P.A. Exploiting synergistic catalysis for an ambient temperature photocycloaddition to pyrazoles. Chem. Eur. J. 2020, 26, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Meng, L.G.; Wang, K.; Wang, L. nBu3P-catalyzed desulfonylative [3 + 2] cycloadditions of allylic carbonates with arylazosulfones to pyrazole derivatives. Org. Lett. 2015, 17, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.C.; Liu, J.; Jia, X.S. Phosphine-free [3+2] cycloaddition of propargylamines with dialkyl azodicarboxylates: An efficient access to pyrazole backbone. Synthesis 2018, 50, 3499–3505. [Google Scholar]
- Pearce, A.J.; Harkins, R.P.; Reiner, B.R.; Wotal, A.C.; Dunscomb, R.J.; Tonks, I.A. Multicomponent pyrazole synthesis from alkynes, nitriles, and titanium imido complexes via oxidatively induced N-N bond coupling. J. Am. Chem. Soc. 2020, 142, 4390–4399. [Google Scholar] [CrossRef]
- Sloop, J.C.; Holder, C.; Henary, M. Selective incorporation of fluorine in pyrazoles. Eur. J. Org. Chem. 2015, 2015, 3405–3422. [Google Scholar] [CrossRef]
- Prieto, A.; Bouyssi, D.; Monteiro, N. Ruthenium-catalyzed tandem C-H fluoromethylation/cyclization of N-alkylhydrazones with CBr3F: Access to 4-fluoropyrazoles. J. Org. Chem. 2017, 82, 3311–3316. [Google Scholar] [CrossRef]
- Schmitt, E.; Panossian, A.; Vors, J.P.; Funke, C.; Lui, N.; Pazenok, S.; Leroux, F.R. A major advance in the synthesis of fluoroalkyl pyrazoles: Tuneable regioselectivity and broad substitution patterns. Chem. Eur. J. 2016, 22, 11239–11244. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.; Bouvet, S.; Pegot, B.; Panossian, A.; Vors, J.P.; Pazenok, S.; Magnier, E.; Leroux, F.R. Fluoroalkyl amino reagents for the introduction of the fluoro(trifluoromethoxy)methyl group onto arenes and heterocycles. Org. Lett. 2017, 19, 4960–4963. [Google Scholar] [CrossRef] [PubMed]
- Mykhailiuk, P.K. In situ generation of difluoromethyl diazomethane for [3+2] cycloadditions with alkynes. Angew. Chem. Int. Ed. 2015, 54, 6558–6561. [Google Scholar] [CrossRef] [PubMed]
- Montoya, V.; Pons, J.; García-Antón, J.; Solans, X.; Font-Bardia, M.; Ros, J. Reaction of 2-hydroxyethylhydrazine with a trifluoromethyl-β-diketone: Study and structural characterization of a new 5-hydroxy-5-trifluoromethyl-4,5-dihydropyrazole intermediate. J. Fluorine Chem. 2007, 128, 1007–1011. [Google Scholar] [CrossRef]
- Li, F.; Nie, J.; Sun, L.; Zheng, Y.; Ma, J.A. Silver-mediated cycloaddition of alkynes with CF3CHN2: Highly regioselective synthesis of 3-trifluoromethylpyrazoles. Angew. Chem. Int. Ed. 2013, 52, 6255–6258. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Chandrakumar, N.; Yoon, J.-J.; Plemper, R.K.; Snyder, J.P. Non-nucleoside inhibitors of the measles virus RNA-dependent RNA polymerase complex activity: Synthesis and in vitro evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 5199–5203. [Google Scholar] [CrossRef] [Green Version]
- Ji, G.; Wang, X.; Zhang, S.; Xu, Y.; Ye, Y.; Li, M.; Zhang, Y.; Wang, J. Synthesis of 3-trifluoromethylpyrazoles via trifluoromethylation/cyclization of α,β-alkynic hydrazones using a hypervalent iodine reagent. Chem. Commun. 2014, 50, 4361–4363. [Google Scholar] [CrossRef]
- 50 Liu, H.; Laforest, R.; Gu, J.; Luo, Z.; Jones, L.A.; Gropler, R.J.; Benzinger, T.L.S.; Tu, Z. Acute rodent tolerability, toxicity, and radiation dosimetry estimates of the S1P1-specific radioligand [11C]CS1P1. Mol. Imaging. Biol. 2020, 22, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, L.; Li, K.K.; Tsui, G.C. Copper-mediated domino cyclization/trifluoromethylation/deprotection with TMSCF3: Synthesis of 4-(trifluoromethyl)pyrazoles. Org. Lett. 2017, 19, 658–661. [Google Scholar] [CrossRef]
- Trofymchuk, S.; Bugera, M.Y.; Klipkov, A.A.; Razhyk, B.; Semenov, S.; Tarasenko, K.; Starova, V.S.; Zaporozhets, O.A.; Tananaiko, O.Y.; Alekseenko, A.N.; et al. Deoxofluorination of (hetero)aromatic acids. J. Org. Chem. 2020, 85, 3110–3124. [Google Scholar] [CrossRef] [PubMed]
- Dhanju, S.; Caravana, A.C.; Thomson, R.J. Access to α-pyrazole and α-triazole derivatives of ketones from oxidative heteroarylation of silyl enolethers. Org. Lett. 2020, 22, 8055–8058. [Google Scholar] [CrossRef] [PubMed]
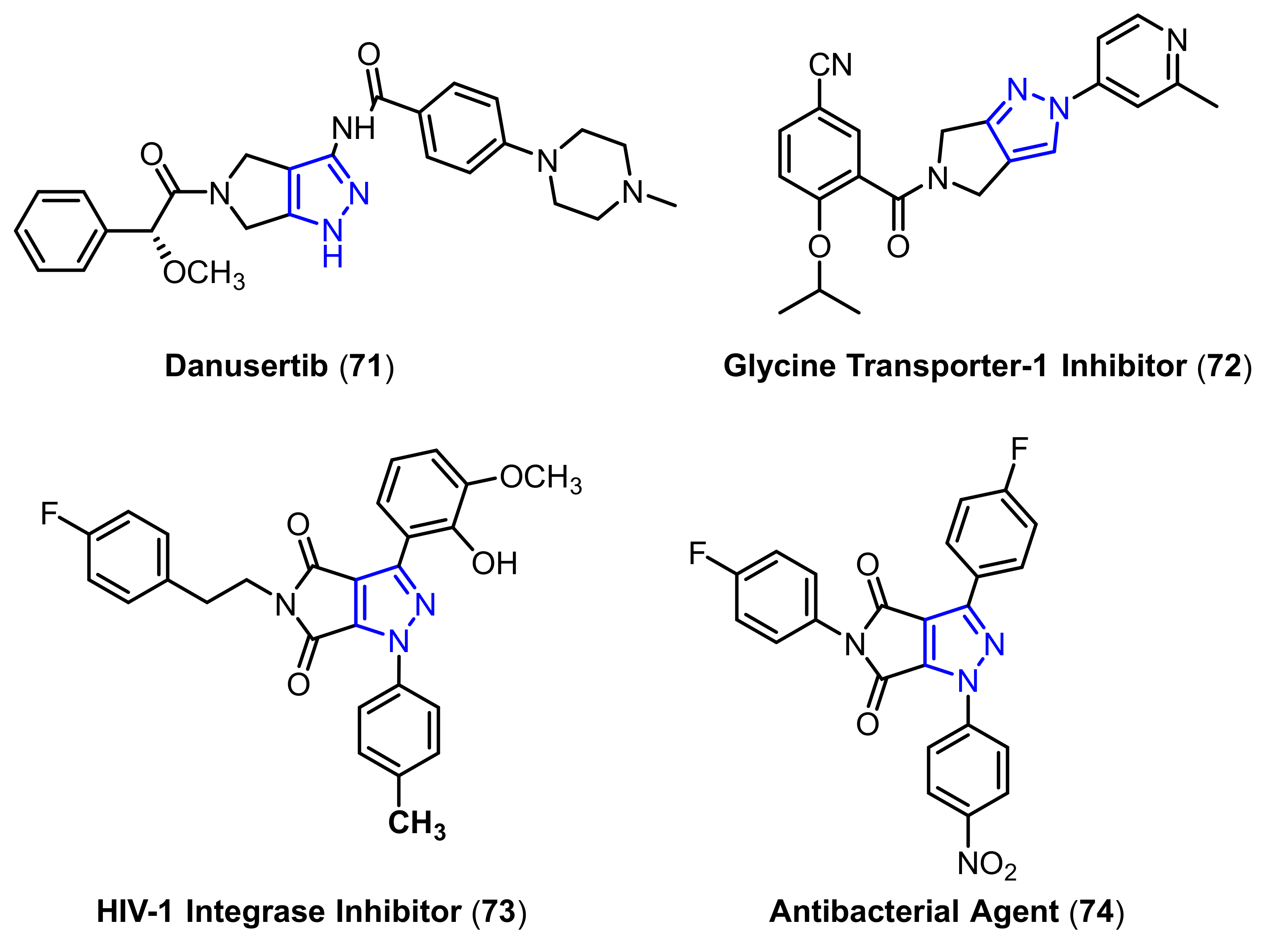
- Kumar, V.; Kaur, K.; Gupta, G.K.; Sharma, A.K. Pyrazole containing natural products: Synthetic preview and biological significance. Eur. J. Med. Chem. 2013, 69, 735–753. [Google Scholar] [CrossRef]
- Luo, G.; Chen, L. Gold-catalyzed stereoselective 1,4-conjugate addition of pyrazoles to propiolates and their hydrogenation to β-pyrazolyl acid esters. Tetrahedron Lett. 2015, 56, 6276–6278. [Google Scholar] [CrossRef]
- Roughley, S.D.; Jordan, A.M. The medicinal chemist’s toolbox: An analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Viciano-Chumillas, M.; Tanase, S.; de Jongh, L.J.; Reedijk, J. Coordination versatility of pyrazole-based ligands towards high-nuclearity transition-metal and rare-earth clusters. Eur. J. Inorg. Chem. 2010, 2010, 3403–3418. [Google Scholar] [CrossRef]
- Ruiz-Castillo, P.; Buchwald, S.L. Applications of palladium-catalyzed C-N cross-coupling reactions. Chem. Rev. 2016, 116, 12564–12649. [Google Scholar] [CrossRef]
- Ma, D.; Cai, Q.; Zhang, H. Mild method for Ullmann coupling reaction of amines and aryl halides. Org. Lett. 2003, 5, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Onodera, S.; Kochi, T.; Kakiuchi, F. Synthesis of N-arylpyrazoles by palladium-catalyzed coupling of aryl triflates with pyrazole derivatives. J. Org. Chem. 2019, 84, 6508–6515. [Google Scholar] [CrossRef]
- Zhou, Q.; Du, F.; Chen, Y.; Fu, Y.; Sun, W.; Wu, Y.; Chen, G. L-(-)-Quebrachitol as a ligand for selective copper(0)-catalyzed N-arylation of nitrogen-containing heterocycles. J. Org. Chem. 2019, 84, 8160–8167. [Google Scholar] [CrossRef] [PubMed]
- Cristau, H.-J.; Cellier, P.P.; Spindler, J.-F.; Taillefer, M. Mild conditions for copper-catalysed N-arylation of pyrazoles. Eur. J. Org. Chem. 2004, 2004, 695–709. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.H.; Guo, Y.; Dong, H.; Liu, Q.; Yu, X.Q. TEMPO-mediated C-H amination of benzoxazoles with N-heterocycles. J. Org. Chem. 2020, 85, 12797–12803. [Google Scholar] [CrossRef]
- Aggarwal, R.; Kumar, V.; Kumar, R.; Singh, S.P. Approaches towards the synthesis of 5-aminopyrazoles. Beilstein J. Org. Chem. 2011, 7, 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senadi, G.C.; Hu, W.P.; Lu, T.Y.; Garkhedkar, A.M.; Vandavasi, J.K.; Wang, J.J. I2-TBHP-catalyzed oxidative cross-coupling of N-sulfonyl hydrazones and isocyanides to 5-aminopyrazoles. Org. Lett. 2015, 17, 1521–1524. [Google Scholar] [CrossRef]
- Kallman, N.J.; Cole, K.P.; Koenig, T.M.; Buser, J.Y.; McFarland, A.D.; McNulty, L.M.; Mitchell, D. Synthesis of aminopyrazoles from isoxazoles: Comparison of preparative methods by in situ NMR analysis. Synthesis 2016, 48, 3537–3543. [Google Scholar]
- Soriano-Garcia, M. Organoselenium compounds as potential therapeutic and chemopreventive agents: A review. Curr. Med. Chem. 2004, 11, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Desai, D.; Kaushal, N.; Gandhi, U.H.; Arner, R.J.; D’Souza, C.; Chen, G.; Vunta, H.; El-Bayoumy, K.; Amin, S.; Prabhu, K.S. Synthesis and evaluation of the anti-inflammatory properties of selenium-derivatives of celecoxib. Chem. Biol. Interact. 2010, 188, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belladona, A.L.; Cervo, R.; Alves, D.; Barcellos, T.; Cargnelutti, R.; Schumacher, R.F. C-H functionalization of (hetero)arenes: Direct selanylation mediated by Selectfluor. Tetrahedron Lett. 2020, 61, 152035. [Google Scholar] [CrossRef]
- Wang, X.; Liu, W.G.; Tung, C.H.; Wu, L.Z.; Cong, H. A monophosphine ligand derived from anthracene photodimer: Synthetic applications for palladium-catalyzed coupling reactions. Org. Lett. 2019, 21, 8158–8163. [Google Scholar] [CrossRef]
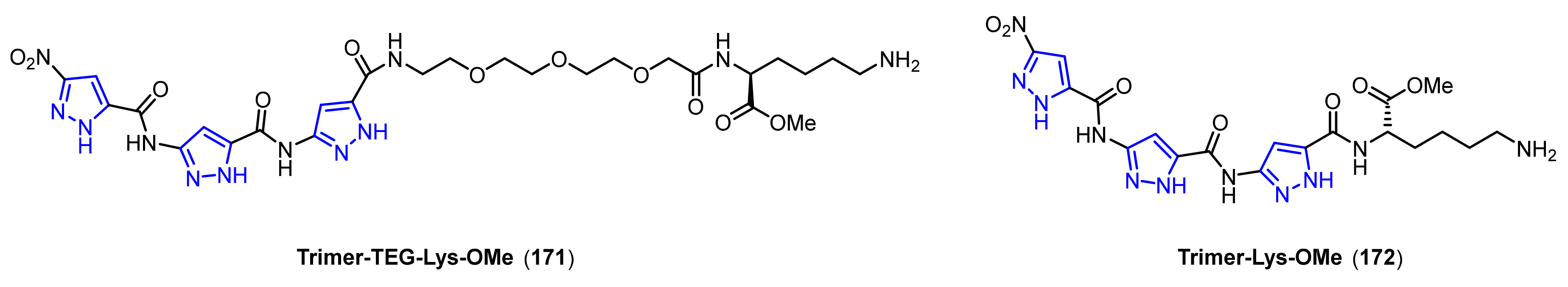
- Kirsten, C.N.; Schrader, T.H. Intermolecular β-sheet stabilization with aminopyrazoles. J. Am. Chem. Soc. 1997, 119, 12061–12068. [Google Scholar] [CrossRef]
- Rzepecki, P.; Schrader, T. β-Sheet ligands in action: KLVFF recognition by aminopyrazole hybrid receptors in water. J. Am. Chem. Soc. 2005, 127, 3016–3025. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Laakso, M.P.; Beltramello, A.; Geroldi, C.; Bianchetti, A.; Soininen, H.; Trabucchi, M. Hippocampal and entorhinal cortex atrophy in frontotemporal dementia and Alzheimer’s disease. Neurology 1999, 52, 91–100. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of multifunctional molecules as potential therapeutic candidates for Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis in the last decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Fonnum, F. Radiochemical micro assays for the determination of choline acetyltransferase and acetylcholinesterase activities. Biochem. J. 1969, 115, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Lockridge, O. Review of human butyrylcholinesterase structure, function, genetic variants, history of use in the clinic, and potential therapeutic uses. Pharmacol. Ther. 2015, 148, 34–46. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. β-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [Green Version]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care. Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
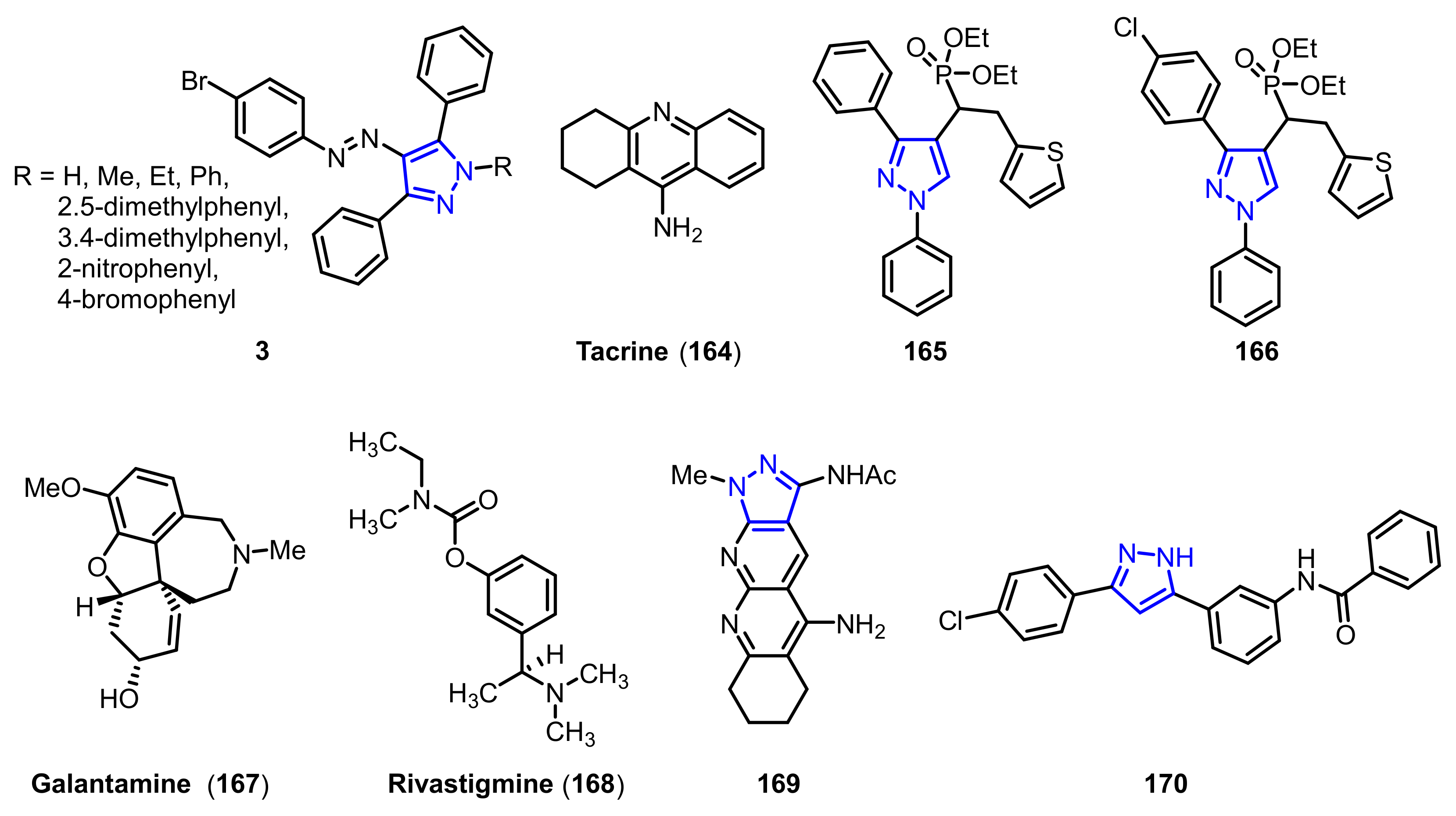
- Shaikh, S.; Dhavan, P.; Pavale, G.; Ramana, M.M.V.; Jadhav, B.L. Design, synthesis and evaluation of pyrazole bearing α-aminophosphonate derivatives as potential acetylcholinesterase inhibitors against Alzheimer’s disease. Bioorg. Chem. 2020, 96, 103589–103611. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.; Chioua, M.; Samadi, A.; Carmo Carreiras, M.; Jimeno, M.L.; Mendes, E.; Rios Cde, L.; Romero, A.; Villarroya, M.; Lopez, M.G.; et al. Synthesis and pharmacological assessment of diversely substituted pyrazolo[3,4-b]quinoline, and benzo[b]pyrazolo[4,3-g][1,8]naphthyridine derivatives. Eur. J. Med. Chem. 2011, 46, 4676–4681. [Google Scholar] [CrossRef] [Green Version]
- Esch, F.S.; Keim, P.S.; Beattie, E.C.; Blacher, R.W.; Culwell, A.R.; Oltersdorf, T.; McClure, D.; Ward, P.J. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science 1990, 248, 1122–1124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Nam, E.; Lee, H.J.; Savelieff, M.G.; Lim, M.H. Towards an understanding of amyloid-β oligomers: Characterization, toxicity mechanisms, and inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. [Google Scholar] [CrossRef]
- Kane, M.D.; Lipinski, W.J.; Callahan, M.J.; Bian, F.; Durham, R.A.; Schwarz, R.D.; Roher, A.E.; Walker, L.C. Evidence for seeding of β-amyloid by intracerebral infusion of Alzheimer brain extracts in β-amyloid precursor protein-transgenic mice. J. Neurosci. 2000, 20, 3606–3611. [Google Scholar] [CrossRef] [Green Version]
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polydoro, M.; Acker, C.M.; Duff, K.; Castillo, P.E.; Davies, P. Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J. Neurosci. 2009, 29, 10741–10749. [Google Scholar] [CrossRef] [Green Version]
- Ward, S.M.; Himmelstein, D.S.; Lancia, J.K.; Binder, L.I. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem. Soc. Trans. 2012, 40, 667–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochdorffer, K.; Marz-Berberich, J.; Nagel-Steger, L.; Epple, M.; Meyer-Zaika, W.; Horn, A.H.; Sticht, H.; Sinha, S.; Bitan, G.; Schrader, T. Rational design of β-sheet ligands against Aβ42-induced toxicity. J. Am. Chem. Soc. 2011, 133, 4348–4358. [Google Scholar] [CrossRef] [PubMed]
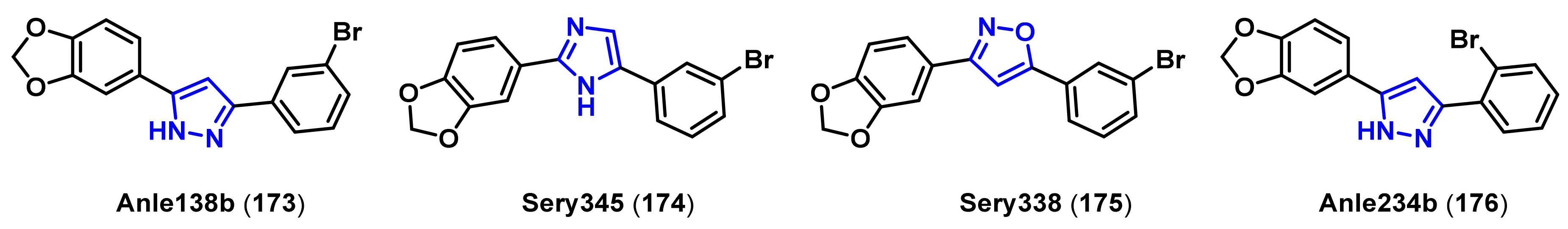
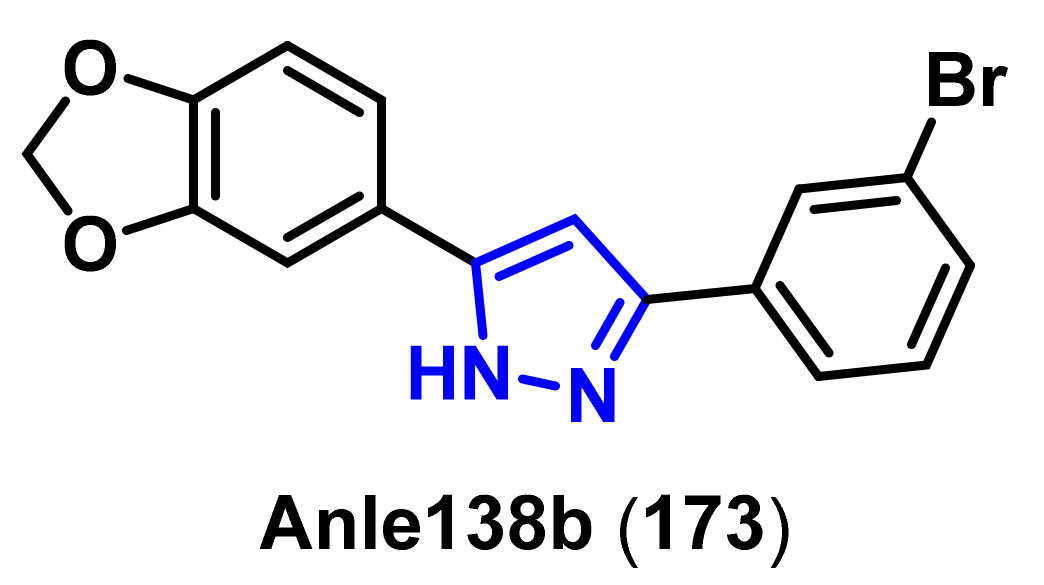
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta. Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.; Krauss, S.; Shi, S.; Ryazanov, S.; Steffen, J.; Miklitz, C.; Leonov, A.; Kleinknecht, A.; Goricke, B.; Weishaupt, J.H.; et al. Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta. Neuropathol. 2015, 130, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthes, D.; Gapsys, V.; Griesinger, C.; de Groot, B.L. Resolving the atomistic modes of anle138b inhibitory action on peptide oligomer formation. ACS Chem. Neurosci. 2017, 8, 2791–2808. [Google Scholar] [CrossRef] [Green Version]
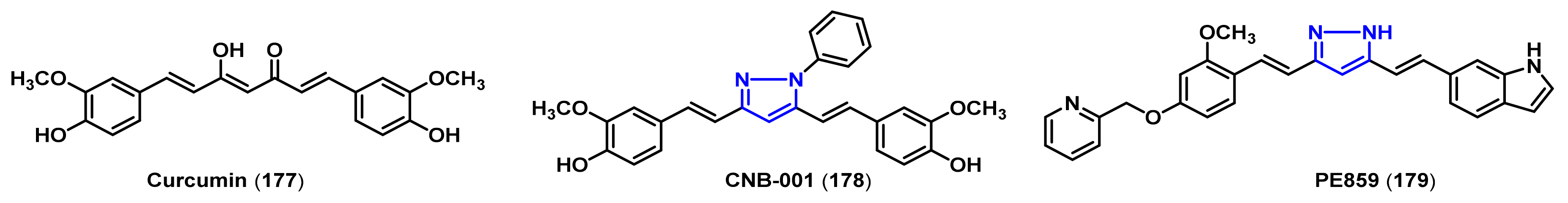
- Belkacemi, A.; Doggui, S.; Dao, L.; Ramassamy, C. Challenges associated with curcumin therapy in Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e34. [Google Scholar] [CrossRef]
- Mishra, S.; Patel, S.; Halpani, C.G. Recent updates in curcumin pyrazole and isoxazole derivatives: Synthesis and biological application. Chem. Biodivers. 2019, 16, e1800366. [Google Scholar] [CrossRef]
- Chainoglou, E.; Hadjipavlou-Litina, D. Curcumin in health and diseases: Alzheimer’s disease and curcumin analogues, derivatives, and hybrids. Int. J. Mol. Sci. 2020, 21, 1975. [Google Scholar] [CrossRef] [Green Version]
- Narlawar, R.; Pickhardt, M.; Leuchtenberger, S.; Baumann, K.; Krause, S.; Dyrks, T.; Weggen, S.; Mandelkow, E.; Schmidt, B. Curcumin-derived pyrazoles and isoxazoles: Swiss army knives or blunt tools for Alzheimer’s disease? ChemMedChem 2008, 3, 165–172. [Google Scholar] [CrossRef]
- Liu, Y.; Dargusch, R.; Maher, P.; Schubert, D. A broadly neuroprotective derivative of curcumin. J. Neurochem. 2008, 105, 1336–1345. [Google Scholar] [CrossRef]
- Valera, E.; Dargusch, R.; Maher, P.A.; Schubert, D. Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J. Neurosci. 2013, 33, 10512–10525. [Google Scholar] [CrossRef] [Green Version]
- Kotani, R.; Urano, Y.; Sugimoto, H.; Noguchi, N. Decrease of amyloid-β levels by curcumin derivative via modulation of amyloid-β protein precursor trafficking. J. Alzheimers Dis. 2017, 56, 529–542. [Google Scholar] [CrossRef]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Teruya, T.; Kawakami, H.; Takahashi, T.; Sugimoto, H. Design and synthesis of curcumin derivatives as tau and amyloid β dual aggregation inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5024–5028. [Google Scholar] [CrossRef]
- Okuda, M.; Fujita, Y.; Hijikuro, I.; Wada, M.; Uemura, T.; Kobayashi, Y.; Waku, T.; Tanaka, N.; Nishimoto, T.; Izumi, Y.; et al. PE859, a novel curcumin derivative, inhibits amyloid-β and tau aggregation, and ameliorates cognitive dysfunction in senescence-accelerated mouse prone 8. J. Alzheimers. Dis. 2017, 59, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Menniti, F.S.; Faraci, W.S.; Schmidt, C.J. Phosphodiesterases in the CNS: Targets for drug development. Nat. Rev. Drug. Discov. 2006, 5, 660–670. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Prickaerts, J.; Heckman, P.R.A.; Blokland, A. Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert. Opin. Investig. Drugs 2017, 26, 1033–1048. [Google Scholar] [CrossRef] [Green Version]
- Cuadrado-Tejedor, M.; Hervias, I.; Ricobaraza, A.; Puerta, E.; Perez-Roldan, J.M.; Garcia-Barroso, C.; Franco, R.; Aguirre, N.; Garcia-Osta, A. Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Heckman, P.R.; Wouters, C.; Prickaerts, J. Phosphodiesterase inhibitors as a target for cognition enhancement in aging and Alzheimer’s disease: A translational overview. Curr. Pharm. Des. 2015, 21, 317–331. [Google Scholar] [CrossRef]
- Li, P.; Zheng, H.; Zhao, J.; Zhang, L.; Yao, W.; Zhu, H.; Beard, J.D.; Ida, K.; Lane, W.; Snell, G.; et al. Discovery of potent and selective inhibitors of phosphodiesterase 1 for the treatment of cognitive impairment associated with neurodegenerative and neuropsychiatric diseases. J. Med. Chem. 2016, 59, 1149–1164. [Google Scholar] [CrossRef] [PubMed]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Shimizu-Albergine, M.; Rybalkin, S.D.; Rybalkina, I.G.; Feil, R.; Wolfsgruber, W.; Hofmann, F.; Beavo, J.A. Individual cerebellar Purkinje cells express different cGMP phosphodiesterases (PDEs): In vivo phosphorylation of cGMP-specific PDE (PDE5) as an indicator of cGMP-dependent protein kinase (PKG) activation. J. Neurosci. 2003, 23, 6452–6459. [Google Scholar] [CrossRef]
- Puzzo, D.; Staniszewski, A.; Deng, S.X.; Privitera, L.; Leznik, E.; Liu, S.; Zhang, H.; Feng, Y.; Palmeri, A.; Landry, D.W.; et al. Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J. Neurosci. 2009, 29, 8075–8086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Guo, J.; Zhao, X.; Chen, Z.; Wang, G.; Liu, A.; Wang, Q.; Zhou, W.; Xu, Y.; Wang, C. Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res. 2013, 250, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, D.; Muller, S.V.; Nager, W.; Stief, C.G.; Schlote, N.; Jonas, U.; Asvestis, C.; Johannes, S.; Munte, T.F. Central effects of sildenafil (Viagra) on auditory selective attention and verbal recognition memory in humans: A study with event-related brain potentials. World J. Urol. 2001, 19, 46–50. [Google Scholar] [CrossRef]
- Fisher, D.A.; Smith, J.F.; Pillar, J.S.; St Denis, S.H.; Cheng, J.B. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J. Biol. Chem. 1998, 273, 15559–15564. [Google Scholar] [CrossRef] [Green Version]
- Kleiman, R.J.; Chapin, D.S.; Christoffersen, C.; Freeman, J.; Fonseca, K.R.; Geoghegan, K.F.; Grimwood, S.; Guanowsky, V.; Hajos, M.; Harms, J.F.; et al. Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo. J. Pharmacol. Exp. Ther. 2012, 341, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Verhoest, P.R.; Fonseca, K.R.; Hou, X.; Proulx-Lafrance, C.; Corman, M.; Helal, C.J.; Claffey, M.M.; Tuttle, J.B.; Coffman, K.J.; Liu, S.; et al. Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyr an-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one (PF-04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J. Med. Chem. 2012, 55, 9045–9054. [Google Scholar]
- Schwam, E.M.; Nicholas, T.; Chew, R.; Billing, C.B.; Davidson, W.; Ambrose, D.; Altstiel, L.D. A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 413–421. [Google Scholar] [CrossRef]
- Moschetti, V.; Boland, K.; Feifel, U.; Hoch, A.; Zimdahl-Gelling, H.; Sand, M. First-in-human study assessing safety, tolerability and pharmacokinetics of BI 409306, a selective phosphodiesterase 9A inhibitor, in healthy males. Br. J. Clin. Pharmacol. 2016, 82, 1315–1324. [Google Scholar] [CrossRef]
- Rosenbrock, H.; Giovannini, R.; Schanzle, G.; Koros, E.; Runge, F.; Fuchs, H.; Marti, A.; Reymann, K.G.; Schroder, U.H.; Fedele, E.; et al. The novel phosphodiesterase 9A inhibitor BI 409306 increases cyclic guanosine monophosphate levels in the brain, promotes synaptic plasticity, and enhances memory function in rodents. J. Pharmacol. Exp. Ther. 2019, 371, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.S.; Wang, B.; Pozniak, C.D.; Chen, M.; Watts, R.J.; Lewcock, J.W. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell. Biol. 2011, 194, 751–764. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Sengupta Ghosh, A.; Gogineni, A.; Hanson, J.E.; Lee, S.H.; Larson, J.L.; Solanoy, H.; Bustos, D.; Li, H.; Ngu, H.; et al. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J. Exp. Med. 2013, 210, 2553–2567. [Google Scholar] [CrossRef]
- Le Pichon, C.E.; Meilandt, W.J.; Dominguez, S.; Solanoy, H.; Lin, H.; Ngu, H.; Gogineni, A.; Sengupta Ghosh, A.; Jiang, Z.; Lee, S.H.; et al. Loss of dual leucine zipper kinase signaling is protective in animal models of neurodegenerative disease. Sci. Transl. Med. 2017, 9, eaag0394. [Google Scholar] [CrossRef] [Green Version]
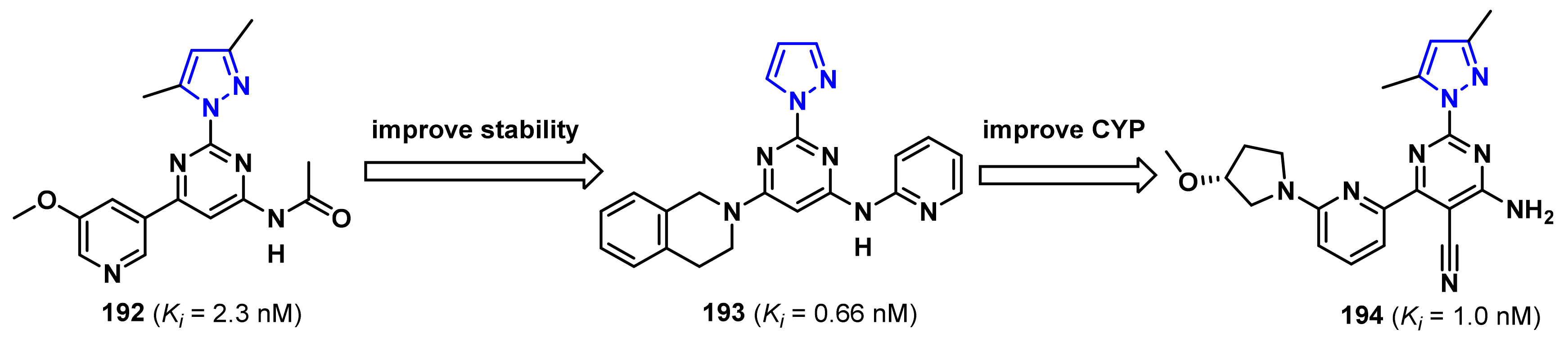
- Patel, S.; Cohen, F.; Dean, B.J.; De La Torre, K.; Deshmukh, G.; Estrada, A.A.; Ghosh, A.S.; Gibbons, P.; Gustafson, A.; Huestis, M.P.; et al. Discovery of dual leucine zipper kinase (DLK, MAP3K12) inhibitors with activity in neurodegeneration models. J. Med. Chem. 2015, 58, 401–418. [Google Scholar] [CrossRef]
- Patel, S.; Harris, S.F.; Gibbons, P.; Deshmukh, G.; Gustafson, A.; Kellar, T.; Lin, H.; Liu, X.; Liu, Y.; Liu, Y.; et al. Scaffold-hopping and structure-based discovery of potent, selective, and brain penetrant N-(1H-pyrazol-3-yl)pyridin-2-amine inhibitors of dual leucine zipper kinase (DLK, MAP3K12). J. Med. Chem. 2015, 58, 8182–8199. [Google Scholar] [CrossRef]
- Patel, S.; Meilandt, W.J.; Erickson, R.I.; Chen, J.; Deshmukh, G.; Estrada, A.A.; Fuji, R.N.; Gibbons, P.; Gustafson, A.; Harris, S.F.; et al. Selective inhibitors of dual leucine zipper kinase (DLK, MAP3K12) with activity in a model of Alzheimer’s disease. J. Med. Chem. 2017, 60, 8083–8102. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine oxidase: From genes to behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef] [Green Version]
- Sherif, F.; Gottfries, C.G.; Alafuzoff, I.; Oreland, L. Brain gamma-aminobutyrate aminotransferase (GABA-T) and monoamine oxidase (MAO) in patients with Alzheimer’s disease. J. Neural. Transm. 1992, 4, 227–240. [Google Scholar] [CrossRef]
- Kennedy, B.P.; Ziegler, M.G.; Alford, M.; Hansen, L.A.; Thal, L.J.; Masliah, E. Early and persistent alterations in prefrontal cortex MAO A and B in Alzheimer’s disease. J. Neural. Transm. 2003, 110, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Saura, J.; Luque, J.M.; Cesura, A.M.; Prada, M.D.; Chan-Palay, V.; Huber, G.; Löffler, J.; Richards, J.G. Increased monoamine oxidase b activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience 1994, 62, 15–30. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of alzheimer’s disease. Oxid. Med. Cell.Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Pisani, L.; Catto, M.; Leonetti, F.; Nicolotti, O.; Stefanachi, A.; Campagna, F.; Carotti, A. Targeting monoamine oxidases with multipotent ligands: An emerging strategy in the search of new drugs against neurodegenerative diseases. Cur. Med. Chem. 2011, 18, 4568–4587. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.; Herzig, Y.; Goren, T.; Finkelstein, N.; Lerner, D.; Goldenberg, W.; Miskolczi, I.; Molnar, S.; Rantal, F.; Tamas, T.; et al. Novel Dual Inhibitors of AChE and MAO Derived from hydroxy aminoindan and phenethylamine as potential treatment for Alzheimer’s disease. J. Med. Chem. 2002, 45, 5260–5279. [Google Scholar] [CrossRef]
- Huang, L.; Lu, C.; Sun, Y.; Mao, F.; Luo, Z.; Su, T.; Jiang, H.; Shan, W.; Li, X. Multitarget-directed benzylideneindanone derivatives: Anti-β-amyloid (Aβ) aggregation, antioxidant, metal chelation, and monoamine oxidase B (MAO-B) inhibition properties against Alzheimer’s disease. J. Med. Chem. 2012, 55, 8483–8492. [Google Scholar] [CrossRef]
- Tzvetkov, N.T.; Antonov, L. Subnanomolar indazole-5-carboxamide inhibitors of monoamine oxidase B (MAO-B) continued: Indications of iron binding, experimental evidence for optimised solubility and brain penetration. J. Enzyme Inhib. Med. Chem. 2017, 32, 960–967. [Google Scholar] [CrossRef] [Green Version]
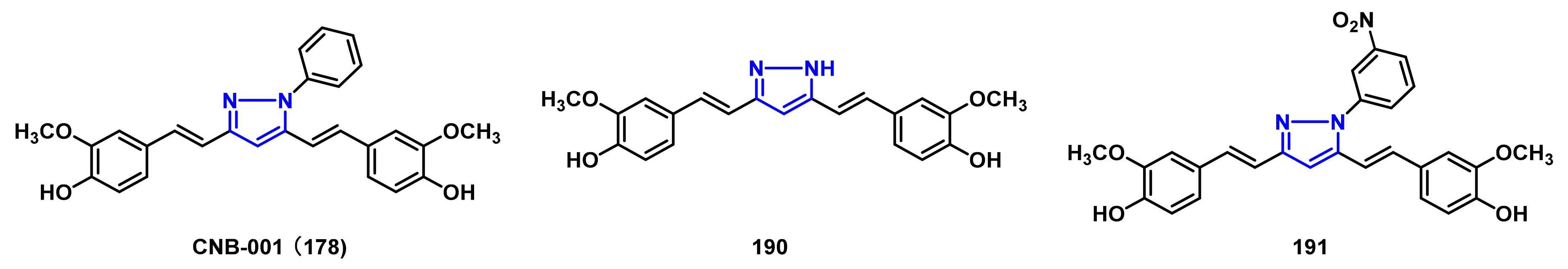
- Jayaraj, R.L.; Elangovan, N.; Dhanalakshmi, C.; Manivasagam, T.; Essa, M.M. CNB-001, a novel pyrazole derivative mitigates motor impairments associated with neurodegeneration via suppression of neuroinflammatory and apoptotic response in experimental Parkinson’s disease mice. Chem. Biol. Interact. 2014, 220, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, N.; Mishra, S.; Jain, M.K.; Surolia, A.; Gupta, S. Curcumin pyrazole and its derivative (N-(3-nitrophenylpyrazole) curcumin inhibit aggregation, disrupt fibrils and modulate toxicity of wild type and mutant α-synuclein. Sci. Rep. 2015, 5, 9862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeg, A.A.; Reiner, A.M.; Schmidt, F.; Schueder, F.; Ryazanov, S.; Ruf, V.C.; Giller, K.; Becker, S.; Leonov, A.; Griesinger, C.; et al. Anle138b and related compounds are aggregation specific fluorescence markers and reveal high affinity binding to α-synuclein aggregates. Biochim. Biophys. Acta. 2015, 1850, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Svenningsson, P.; Le Moine, C.; Fisone, G.; Fredholm, B.B. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog. Neurobiol. 1999, 59, 355–396. [Google Scholar] [CrossRef]
- Schwarzschild, M.A.; Xu, K.; Oztas, E.; Petzer, J.P.; Castagnoli, K.; Castagnoli, N., Jr.; Chen, J.F. Neuroprotection by caffeine and more specific A2A receptor antagonists in animal models of Parkinson’s disease. Neurology 2003, 61, S55–S61. [Google Scholar] [CrossRef]
- Bara-Jimenez, W.; Sherzai, A.; Dimitrova, T.; Favit, A.; Bibbiani, F.; Gillespie, M.; Morris, M.J.; Mouradian, M.M.; Chase, T.N. Adenosine A2A receptor antagonist treatment of Parkinson’s disease. Neurology 2003, 61, 293–296. [Google Scholar] [CrossRef]
- Armentero, M.T.; Pinna, A.; Ferre, S.; Lanciego, J.L.; Muller, C.E.; Franco, R. Past, present and future of A2A adenosine receptor antagonists in the therapy of Parkinson’s disease. Pharmacol. Ther. 2011, 132, 280–299. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Li, X.; Ma, H.; Zheng, J.; Zhen, X.; Zhang, X. Replacement of amide with bioisosteres led to a new series of potent adenosine A2A receptor antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 152–155. [Google Scholar] [CrossRef]
- Zheng, J.; Yang, Z.; Li, X.; Li, L.; Ma, H.; Wang, M.; Zhang, H.; Zhen, X.; Zhang, X. Optimization of 6-heterocyclic-2-(1H-pyrazol-1-yl)-N-(pyridin-2-yl)pyrimidin-4-amine as potent adenosine A2A receptor antagonists for the treatment of Parkinson’s disease. ACS Chem. Neurosci. 2014, 5, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Li, L.; Zheng, J.; Ma, H.; Tian, S.; Li, J.; Zhang, H.; Zhen, X.; Zhang, X. Identification of a new series of potent adenosine A2A receptor antagonists based on 4-amino-5-carbonitrile pyrimidine template for the treatment of Parkinson’s disease. ACS Chem. Neurosci. 2016, 7, 1575–1584. [Google Scholar] [CrossRef]
- Soderling, S.H.; Bayuga, S.J.; Beavo, J.A. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc. Natl. Acad. Sci. USA 1999, 96, 7071–7076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancesario, G.; Morrone, L.A.; D’Angelo, V.; Castelli, V.; Ferrazzoli, D.; Sica, F.; Martorana, A.; Sorge, R.; Cavaliere, F.; Bernardi, G.; et al. Levodopa-induced dyskinesias are associated with transient down-regulation of cAMP and cGMP in the caudate-putamen of hemiparkinsonian rats: Reduced synthesis or increased catabolism? Neurochem. Int. 2014, 79, 44–56. [Google Scholar] [CrossRef]
- Niccolini, F.; Foltynie, T.; Reis Marques, T.; Muhlert, N.; Tziortzi, A.C.; Searle, G.E.; Natesan, S.; Kapur, S.; Rabiner, E.A.; Gunn, R.N.; et al. Loss of phosphodiesterase 10A expression is associated with progression and severity in Parkinson’s disease. Brain 2015, 138, 3003–3015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movsesian, M.; Stehlik, J.; Vandeput, F.; Bristow, M.R. Phosphodiesterase inhibition in heart failure. Heart Fail. Rev. 2009, 14, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Heaslip, R.J.; Evans, D.Y. Emetic, central nervous system, and pulmonary activities of rolipram in the dog. Eur. J. Pharmacol. 1995, 286, 281–290. [Google Scholar] [CrossRef]
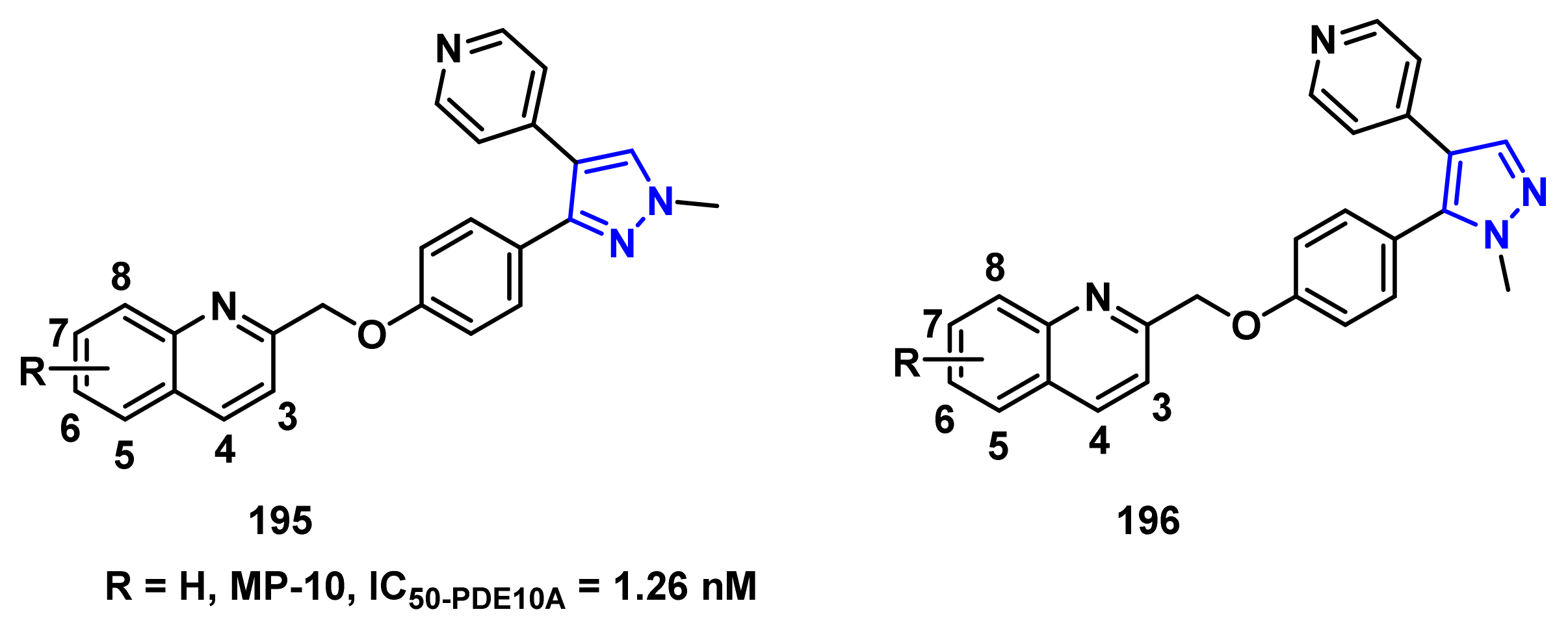
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O’Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jin, H.; Zhou, H.; Rothfuss, J.; Tu, Z. Synthesis and in vitro biological evaluation of pyrazole group-containing analogues for PDE10A. MedChemComm 2013, 4, 443–449. [Google Scholar] [CrossRef] [PubMed]
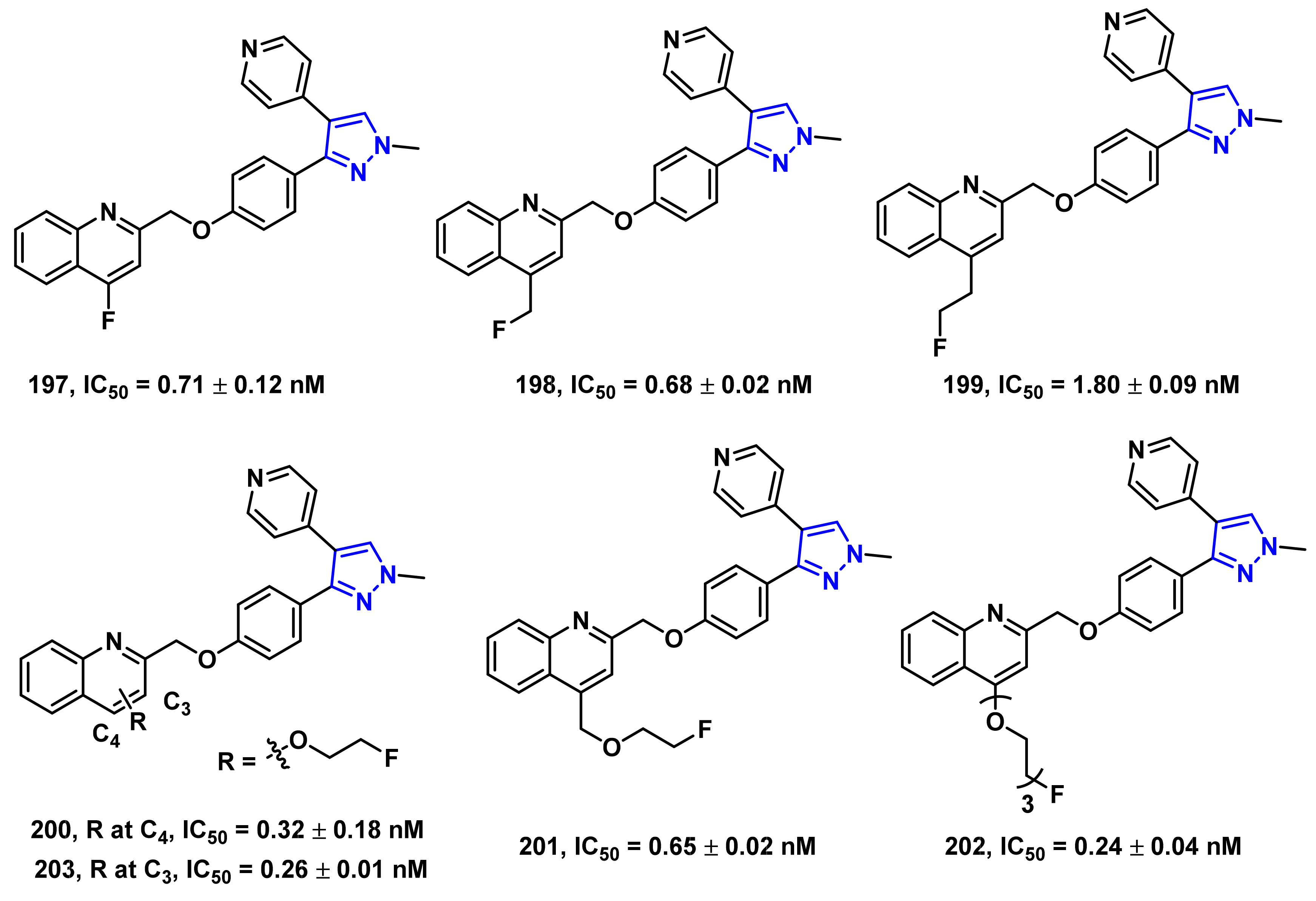
- Li, J.; Zhang, X.; Jin, H.; Fan, J.; Flores, H.; Perlmutter, J.S.; Tu, Z. Synthesis of fluorine-containing phosphodiesterase 10a (PDE10A) inhibitors and the in vivo evaluation of F-18 labeled PDE10A PET tracers in rodent and nonhuman primate. J. Med. Chem. 2015, 58, 8584–8600. [Google Scholar] [CrossRef] [PubMed] [Green Version]



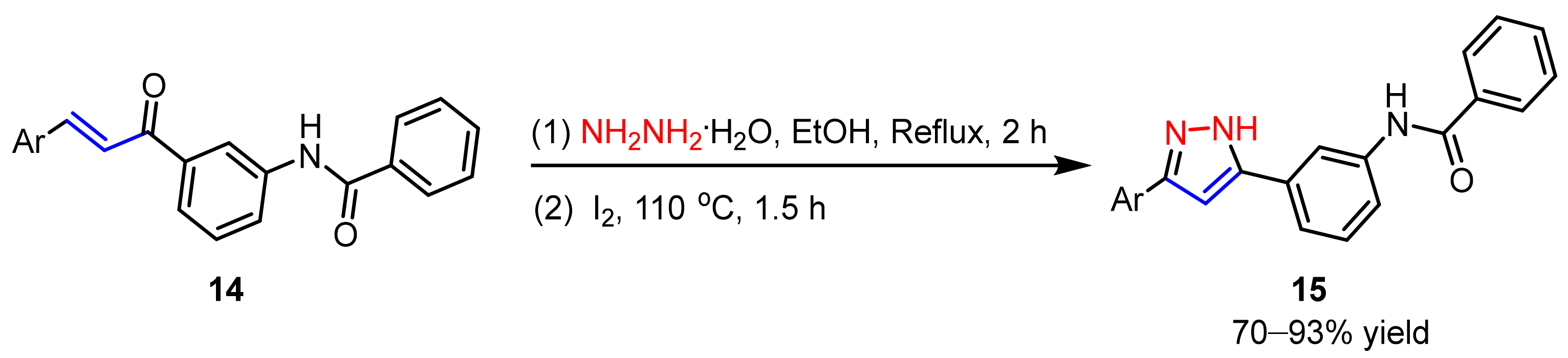

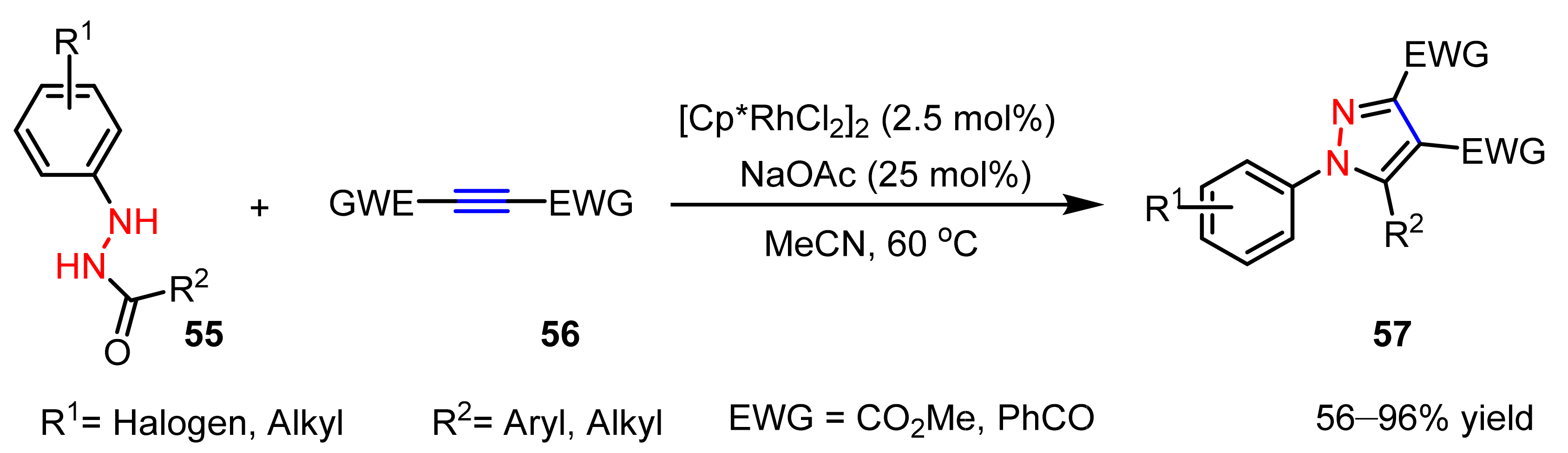











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ki1 or IC50 2 Values for Cholinesterase Inhibitors | ||||
|---|---|---|---|---|
| Compound | R | AChE | BuChE | AChE/BuChE |
| 3a 1 | H | 58.17 ± 4.84 nM | 74.82 ± 18.62 nM | 0.77 |
| 3b 1 | Me | 64.71 ± 13.04 nM | 80.36 ± 22.74 nM | 0.80 |
| 3c 1 | Et | 47.93 ± 4.92 nM | 58.12 ± 9.27 nM | 0.82 |
| 3d 1 | Ph | 44.66 ± 10.06 nM | 50.36 ± 13.88 nM | 0.88 |
| 3e 1 | 2, 5-dimethylphenyl | 78.34 ± 17.83 nM | 77.62 ± 18.32 nM | 1.00 |
| 3f 1 | 3, 4-dimethylphenyl | 48.16 ± 9.63 nM | 65.27 ± 12.73 nM | 0.73 |
| 3g 1 | 2-nitrophenyl | 56.23 ± 11.74 nM | 71.63 ± 8.93 nM | 0.78 |
| 3h 1 | 4-bromophenyl | 60.27 ± 15.67 nM | 88.36 ± 20.03 nM | 0.68 |
| 164 1 | -- | 126.13 ± 9.37 nM | 145.84 ± 13.44 nM | 0.86 |
| 165 2 | -- | 0.055 ± 0.143 µM | 8.863 ± 0.22 µM | 0.006 |
| 166 2 | -- | 0.017 ± 0.02 µM | 6.331 ±0.017 µM | 0.003 |
| 167 2 | -- | 3.148 ± 0.139 µM | 1.22 ± 0.05 µM | 2.58 |
| 168 2 | -- | 2.632 ± 0.021 µM | 6.901 ± 0.01 µM | 0.38 |
| 169 2 | -- | 0.069 ± 0.06 µM | 6.3 ± 0.6 µM | 0.01 |
| 170 2 | -- | 1937 ± 66 nM | 1166 ± 88 nM | 1.60 |
| IC50 Value (nM) | ||||
|---|---|---|---|---|
| Compound | hMAO-A | hMAO-B | hMAO-A/hMAO-B | Ki (nM) |
| 187 | >10,000 | 0.59 ± 0.09 | >16,959 | 0.26 ± 0.04 |
| 188 | >10,000 | 0.68 ± 0.04 | >14,706 | 0.30 ± 0.02 |
| 189 | ≥10,000 | 0.66 ± 0.06 | ≥15,151 | 0.29 ± 0.03 |
| IC50 Value | ||||||
|---|---|---|---|---|---|---|
| Compound | R | PDE10A (nM) | PDE3A (103 × nM) | PDE3B (103 × nM) | PDE4A (103 × nM) | PDE4B (103 × nM) |
| 195a | 3-OMe | 0.40 ± 0.02 | 123 ± 21 | 82.7 ± 10 | 3.85 ± 0.23 | 3.43 ± 0.21 |
| 195b | 4-OMe | 0.28 ± 0.06 | 27.5 ± 2.5 | 3.85 ± 0.95 | 2.56 ± 0.11 | 1.79 ± 0.10 |
| 195c | 6-OMe | 1.82 ± 0.25 | 78.0 ± 4.0 | 9.75 ± 1.25 | 3.37 ± 0.18 | 2.56 ± 0.09 |
| 196a | 3-OMe | 0.24 ± 0.05 | 18.7 ± 3.1 | 16.9 ± 2.1 | 199 ± 16.0 | 31.5 ± 3.60 |
| 196b | 4-OMe | 0.36 ± 0.03 | 29.0 ± 4.0 | 4.80 ± 1.20 | 4.18 ± 0.33 | 5.06 ± 0.40 |
| 196c | 6-OMe | 1.78 ± 0.03 | 1.5 ± 0.5 | 3.00 ± 0.14 | 5.60 ± 0.52 | 7.10 ± 0.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yu, Y.; Tu, Z. Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer’s Disease and Parkinson’s Disease Treatment (2011–2020). Molecules 2021, 26, 1202. https://doi.org/10.3390/molecules26051202
Li X, Yu Y, Tu Z. Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer’s Disease and Parkinson’s Disease Treatment (2011–2020). Molecules. 2021; 26(5):1202. https://doi.org/10.3390/molecules26051202
Chicago/Turabian StyleLi, Xuefei, Yanbo Yu, and Zhude Tu. 2021. "Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer’s Disease and Parkinson’s Disease Treatment (2011–2020)" Molecules 26, no. 5: 1202. https://doi.org/10.3390/molecules26051202