
Conventional vs. Microwave- or Mechanically-Assisted Synthesis of Dihomooxacalix[4]arene Phthalimides: NMR, X-ray and Photophysical Analysis §

 ,
,  , , ,
, , ,  ,
,  , and
, and 
Abstract
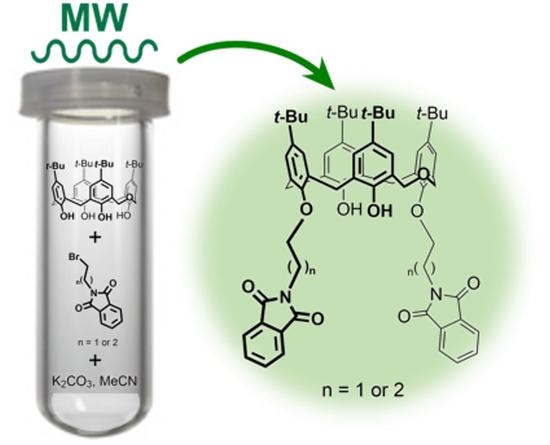
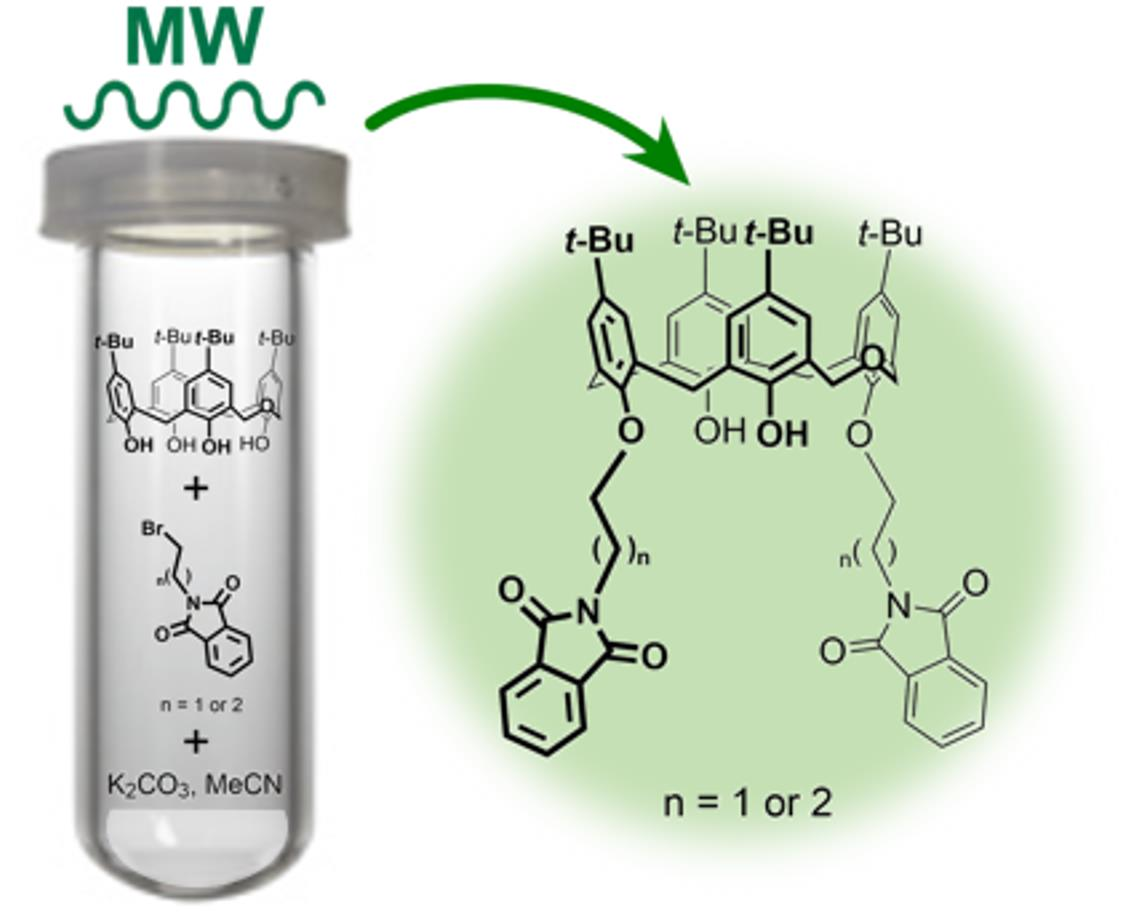
1. Introduction
2. Results and Discussion
2.1. Conventional vs. MW- and Mechanically-Assisted Synthesis
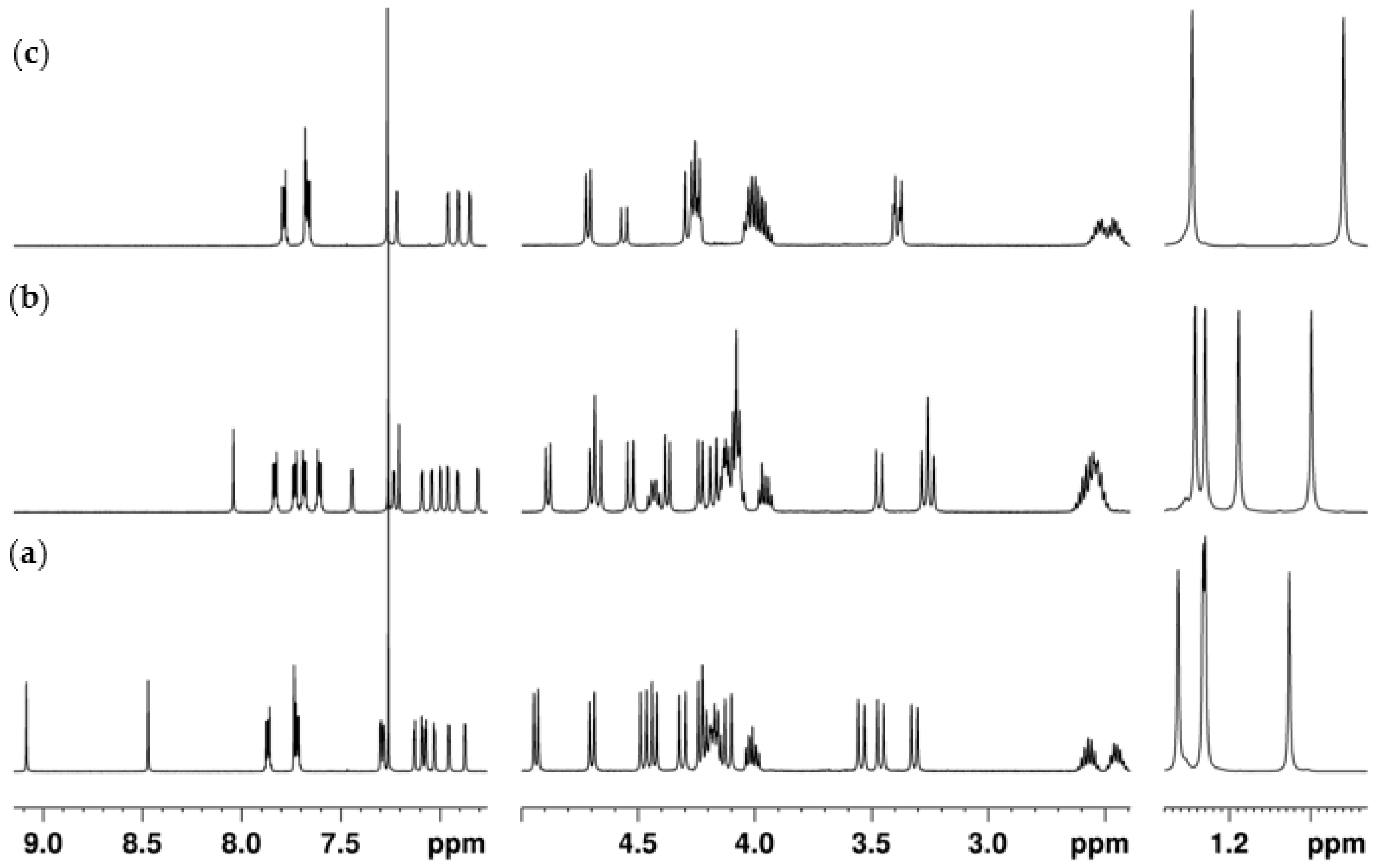
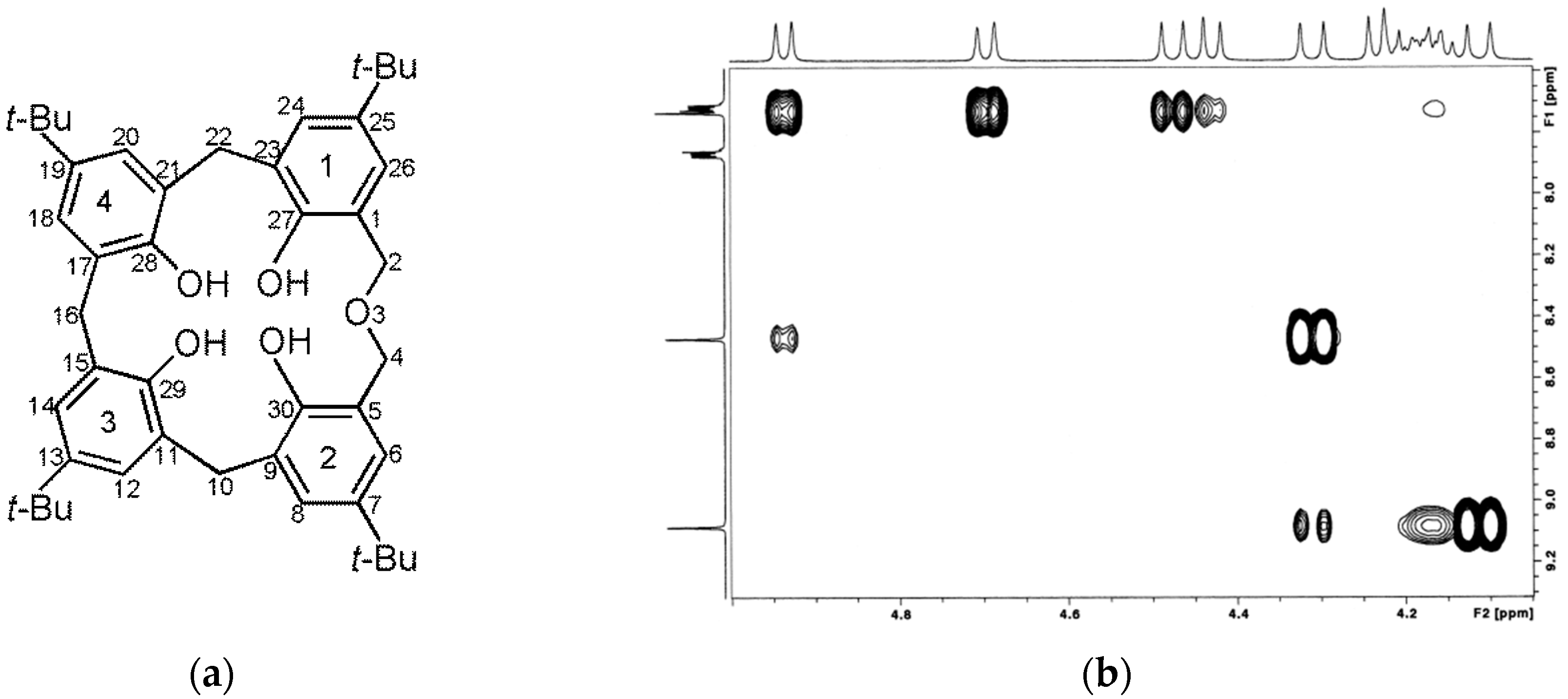
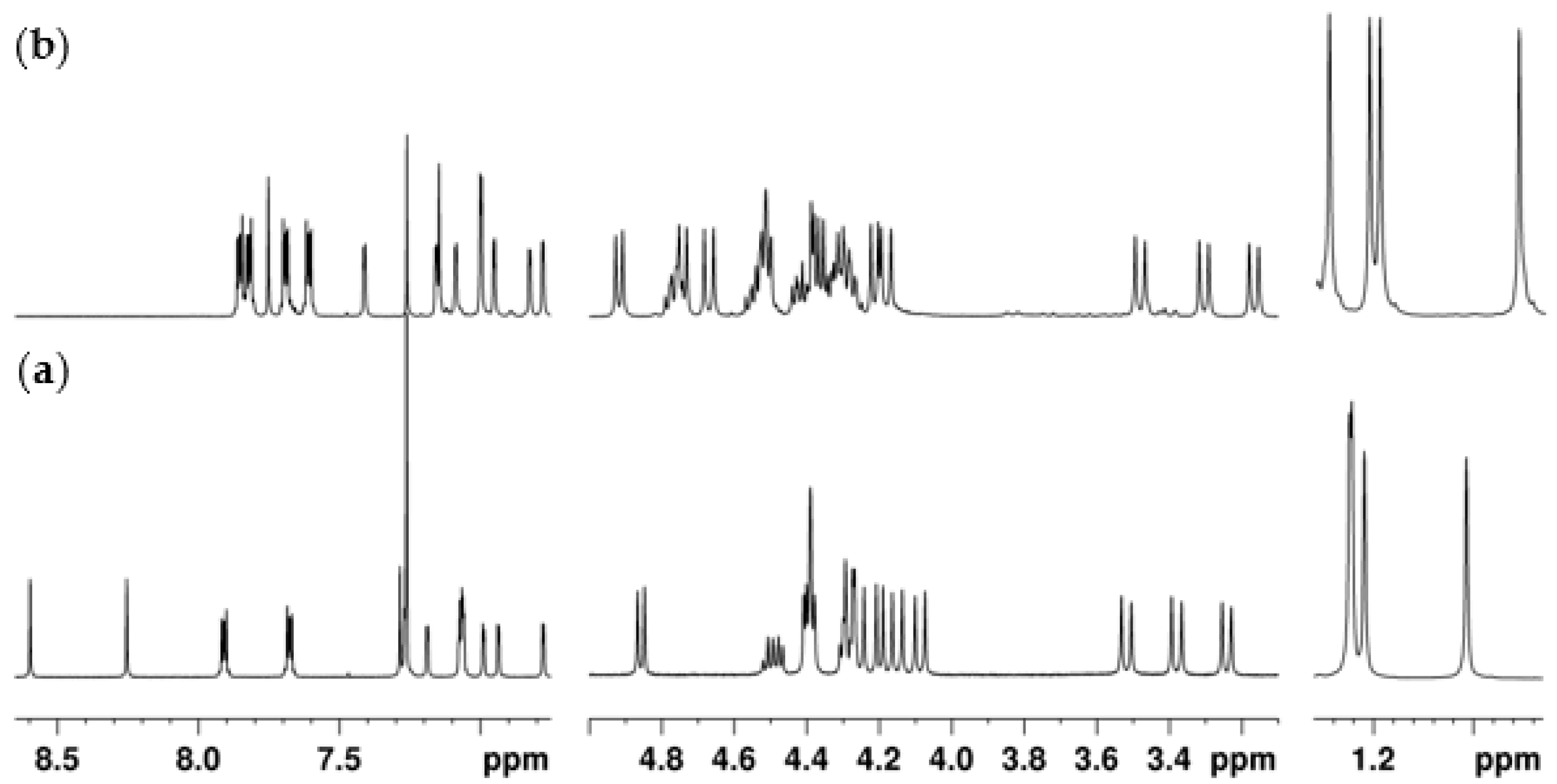
2.2. NMR Conformational Analysis
2.2.1. Mono-Propylphthalimide 2a
2.2.2. 1,3-Dipropylphthalimide 3a
2.2.3. 3,4-Dipropylphthalimide 3b
2.2.4. Mono-Ethylphthalimide 5a
2.2.5. 1,3- and 3,4-Diethylphthalimides 6a and 6b
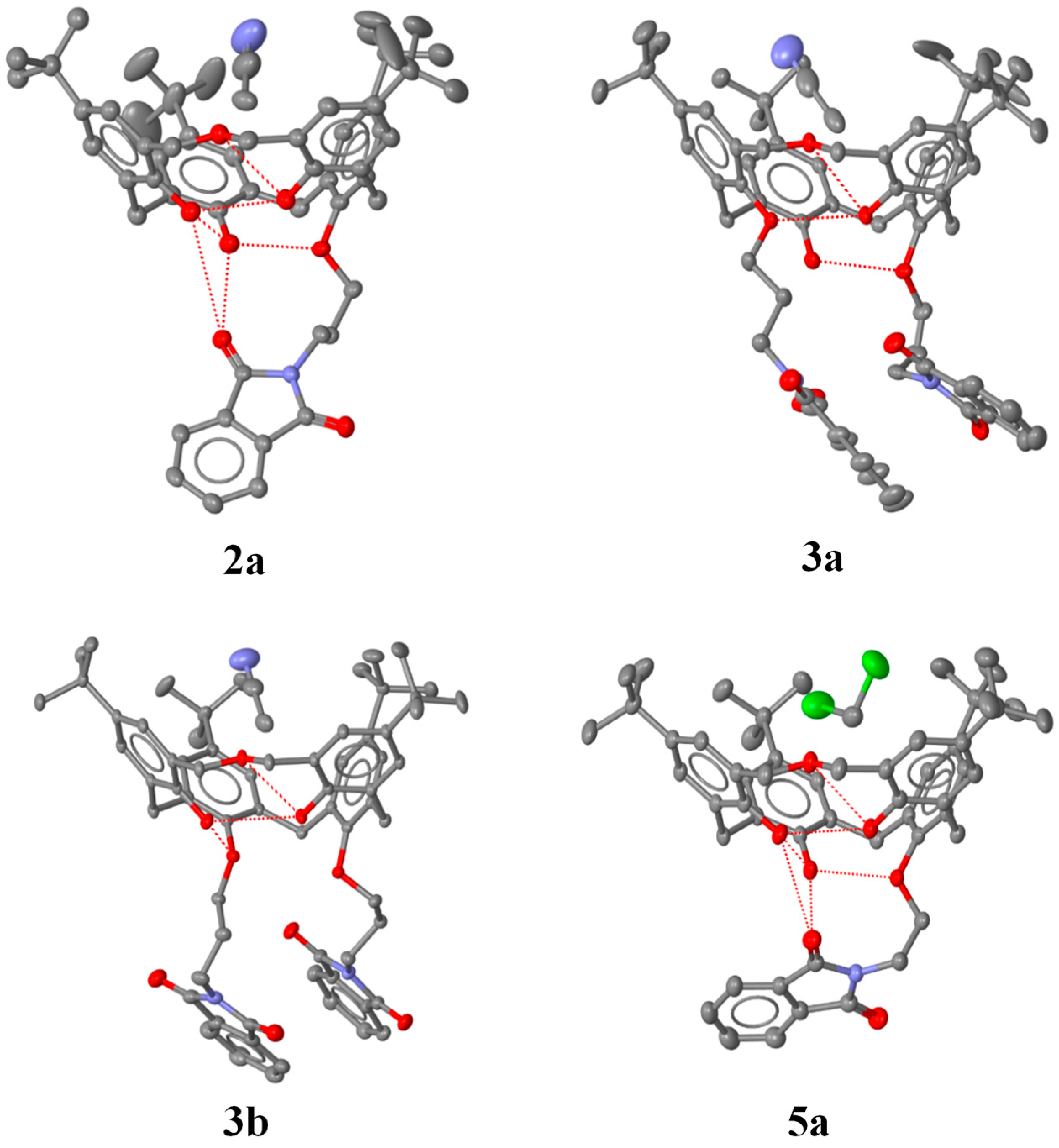
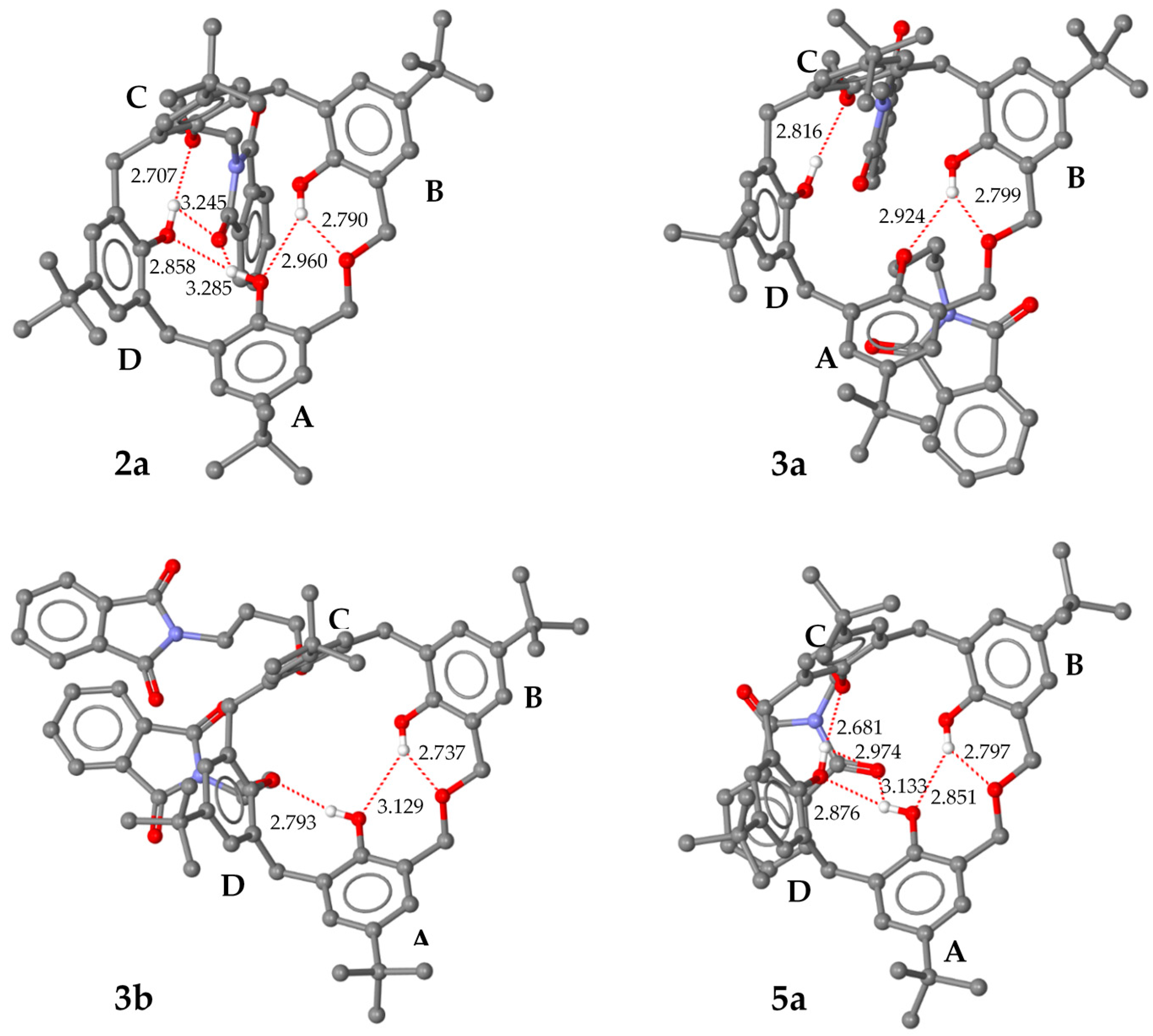
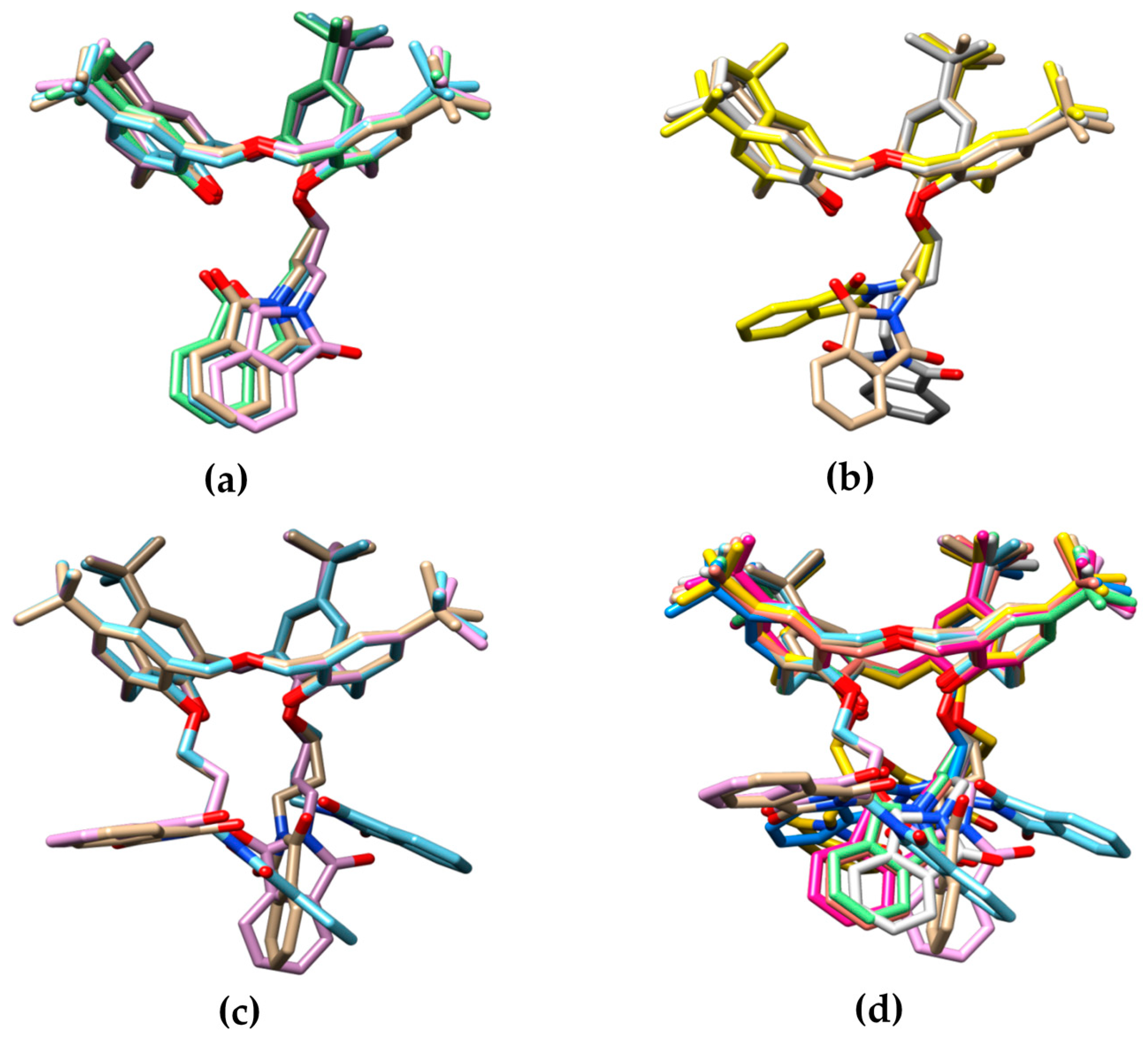
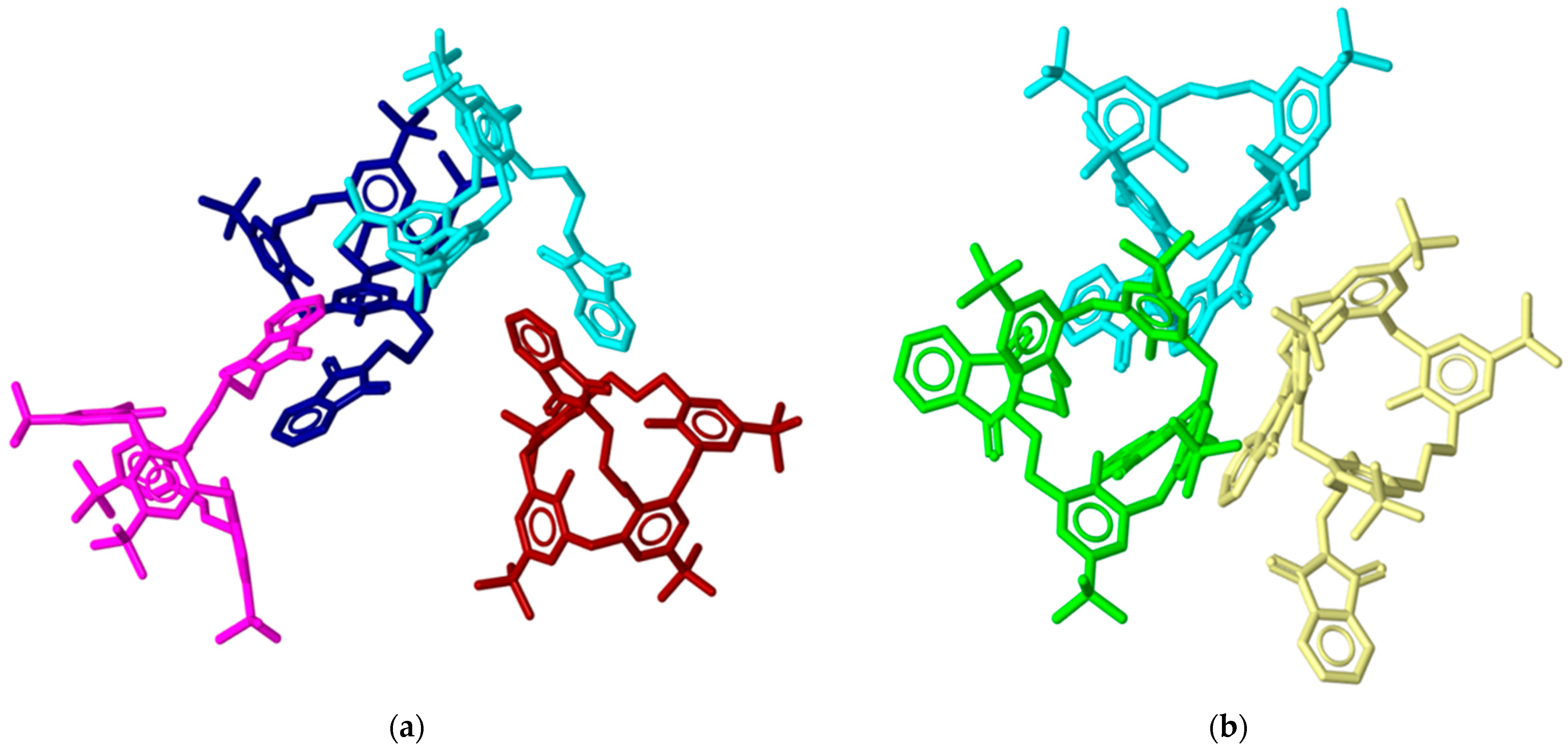
2.3. X-ray Structural Analysis
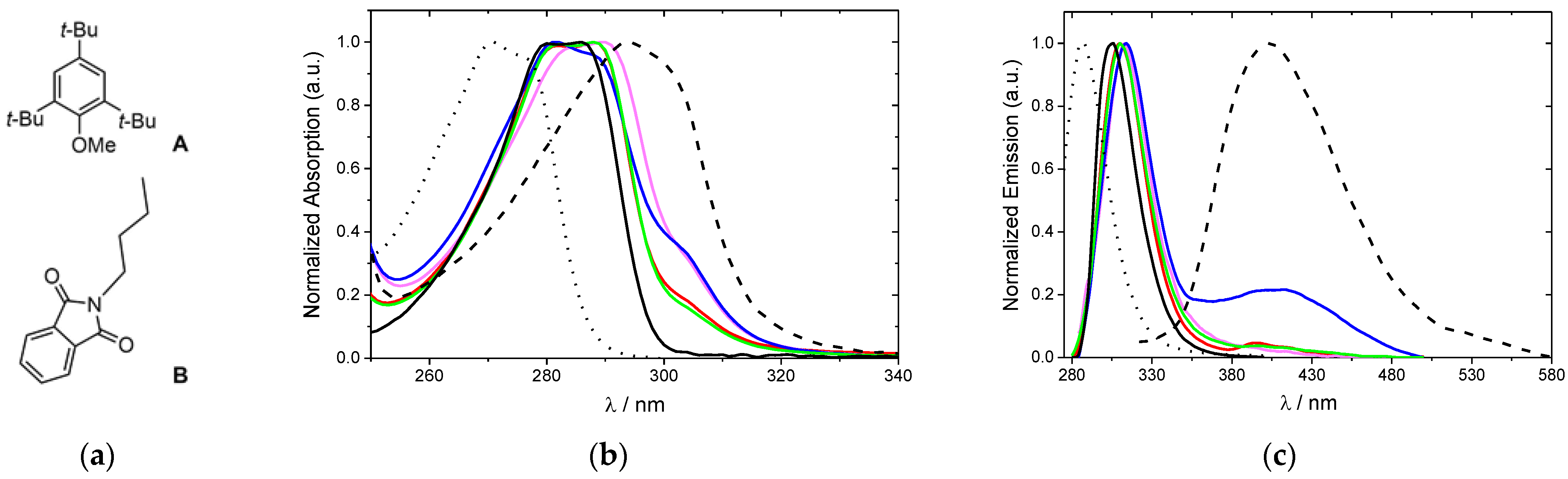
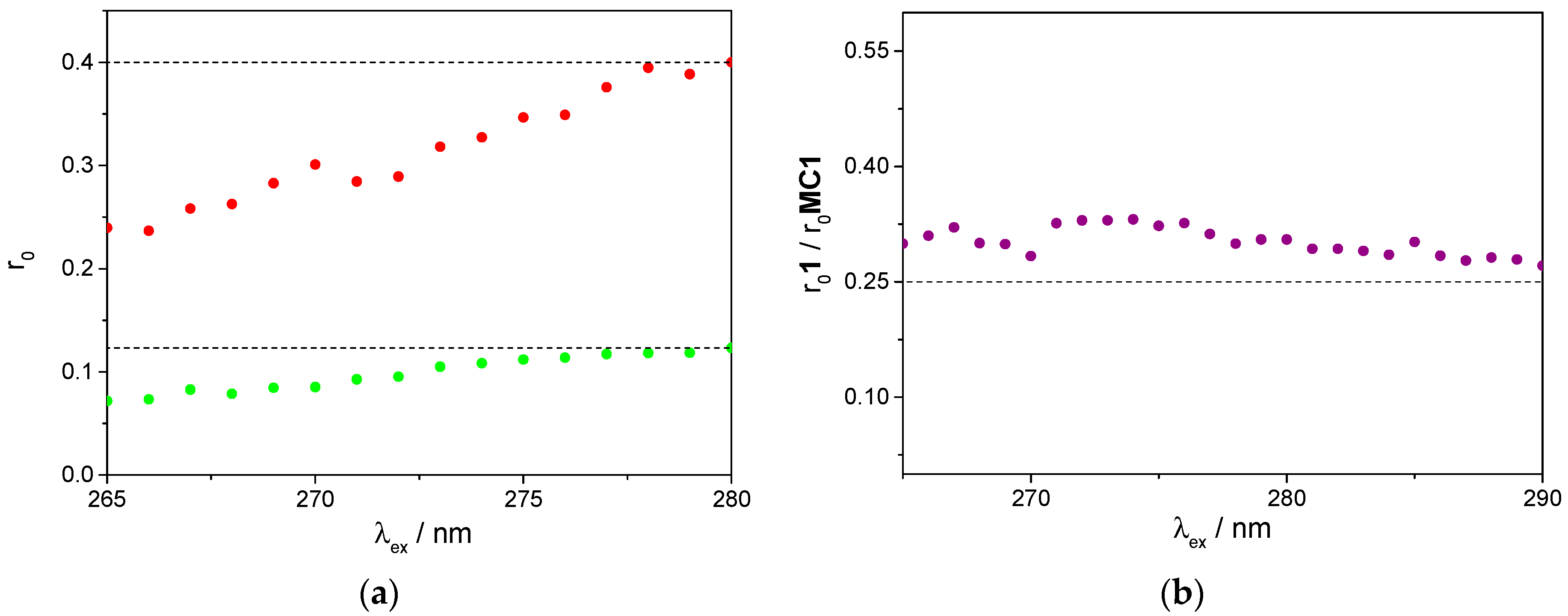
2.4. Photophysical Properties
3. Material and Methods
3.1. Synthesis
3.1.1. Conventional Procedure for the Synthesis of 2a, 3a and 3b
3.1.2. Conventional Procedure for the Synthesis of 5a and 6a
3.1.3. Microwave- and Mechanically-Assisted Syntheses
3.2. Determination of the Crystallographic Structures of 2a, 3a, 3b and 5a
3.3. Fluorescence Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gutsche, C.D. Calixarenes, an Introduction, 2nd ed.; Monographs in Supramolecular Chemistry; The Royal Society of Chemistry: Cambridge, UK, 2008. [Google Scholar]
- Neri, P.; Sessler, J.L.; Wang, M.-X. (Eds.) Calixarenes and Beyond; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Busschaert, N.; Caltagirone, C.; Van Rossom, W.; Gale, P.A. Applications of supramolecular anion recognition. Chem. Rev. 2015, 115, 8038–8155. [Google Scholar] [CrossRef]
- Gale, P.A.; Howe, E.N.W.; Wu, X. Anion receptor chemistry. Chem 2016, 1, 351–422. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M. Application of commercial microwave ovens to organic synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Strauss, C.R. A strategic, “green” approach to organic chemistry with microwave assistance and predictive yield optimization as core, enabling technologies. Aust. J. Chem. 2009, 62, 3–15. [Google Scholar] [CrossRef]
- Caddick, S.; Fitzmaurice, R. Microwave enhanced synthesis. Tetrahedron 2009, 65, 3325–3355. [Google Scholar] [CrossRef]
- Gawande, M.B.; Shelke, S.N.; Zboril, R.; Varma, R.S. Microwave-assisted chemistry: Synthetic applications for rapid assembly of nanomaterials and organics. Acc. Chem. Res. 2014, 47, 1338–1348. [Google Scholar] [CrossRef]
- Kumar, A.; Kuang, Y.; Liang, Z.; Sun, X. Microwave chemistry, recent advancements, and eco-friendly microwave-assisted synthesis of nanoarchitectures and their applications: A review. Mater. Today Nano 2020, 11, 100076. [Google Scholar] [CrossRef]
- Stolle, A.; Szuppa, T.; Leonhardt, S.E.S.; Ondruschka, B. Ball milling in organic synthesis: Solutions and challenges. Chem. Soc. Rev. 2011, 40, 2317–2329. [Google Scholar] [CrossRef]
- James, S.L.; Adams, C.J.; Bolm, C.; Braga, D.; Collier, P.; Friscie, T.; Grepioni, F.; Harris, K.D.M.; Hyett, G.; Jones, W.; et al. Mechanochemistry: Opportunities for new and cleaner synthesis. Chem. Soc. Rev. 2012, 41, 413–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.W. Mechanochemical organic synthesis. Chem. Soc. Rev. 2013, 42, 7668–7700. [Google Scholar] [CrossRef]
- Rahman, M.; Santra, S.; Kovalev, I.S.; Kopchuk, D.S.; Zyryanov, G.V.; Majee, A.; Chupakhin, O.N. Gree synthetic approaches for practically relevant (hetero)macrocycles: An overview. AIP Conf. Proc. 2020, 2280, 030014. [Google Scholar]
- Baozhi, L.; Gengliang, Y.; Jinsong, Z.; Kefang, D. Microwave-assisted synthesis of p-alkylcalix[n]arene catalysed by KOH. Eur. J. Chem. 2005, 2, 70–74. [Google Scholar]
- Takagaki, M.; Hosoda, A.; Mori, H.; Miyake, Y.; Kimura, K.; Taniguchi, H.; Nomura, E. Rapid and convenient laboratory method for the preparation of p-tert-butylcalix[4]arene using microwave irradiation. Green Chem. 2008, 10, 978–981. [Google Scholar] [CrossRef]
- Agrawal, Y.K.; Desai, N.C.; Mehta, N.D. Microwave-assisted synthesis of azocalixarenes. Synth. Commun. 2007, 37, 2243–2252. [Google Scholar] [CrossRef]
- Cecioni, S.; Lalor, R.; Blanchard, B.; Praly, J.P.; Imberty, A.; Matthews, S.E.; Vidal, S. Achieving high affinity towards a bacterial lectin through multivalent topological isomers of calix[4]arene glycoconjugates. Chem. Eur. J. 2009, 15, 13232–13240. [Google Scholar] [CrossRef] [PubMed]
- Fatykhova, G.A.; Burilov, V.A.; Dokuchaeva, M.N.; Solov’eva, S.E.; Antipin, I.S. Synthesis of tetraazide derivatives of p-tert-butylcalix[4]arene using copper-catalyzed nucleophilic aromatic substitution. Dokl. Chem. 2018, 479, 64–67. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, Z.; Chen, B.; Li, X.; Liu, M. Microwave-assisted synthesis of bidentate chiral unsymmetrical urea derivatives of p-tert-butylcalix[4]arene and their anion recognition properties. J. Chem. Res. 2015, 39, 303–306. [Google Scholar] [CrossRef]
- Nayak, S.K.; Choudhary, M.K. Microwave-assisted synthesis of 1,3-dialkyl ethers of calix[4]arenes: Application to the synthesis of cesium selective calix[4]crown-6 ionophores. Tetrahedron Lett. 2012, 53, 141–144. [Google Scholar] [CrossRef]
- Burilov, V.A.; Nugmanov, R.I.; Ibragimova, R.R.; Solovieva, S.E.; Antipin, I.S.; Konovalov, A.I. Microwave-assisted alkylation of p-tert-butylcalix[4]arene lower rim: The effect of alkyl halides. Mendeleev Commun. 2013, 23, 113–115. [Google Scholar] [CrossRef]
- Bakic, M.T.; Klaric, D.; Espinosa, M.S.; Kazazic, S.; Frkanec, L.; Babay, P.A.; Galic, N. Syntheses of ester and amide derivatives of calix[6]arene and their complexation affinities towards La3+, Eu3+, and Yb3+. Supramol. Chem. 2019, 31, 723–731. [Google Scholar] [CrossRef]
- Galán, H.; de Mendoza, J.; Prados, P. Microwave-assisted synthesis of a nitro-m-xylylenedioxycalix[6]arene building block functionalized at the upper rim. Eur. J. Org. Chem. 2010, 7005–7011. [Google Scholar] [CrossRef]
- Atwood, J.L.; Hardie, M.J.; Raston, C.L.; Sandoval, C.A. Convergent synthesis of p-benzylcalix[7]arene: Condensation and UHIG of p-benzylcalix[6 or 8]arenes. Org. Lett. 1999, 1, 1523–1526. [Google Scholar] [CrossRef]
- Chennakesavulu, K.; Raju, G.B. Mechanochemical synthesis of p-nitro calix[6]arene. Asian J. Chem. 2010, 22, 4947–4949. [Google Scholar]
- Marcos, P.M.; Teixeira, F.A.; Segurado, M.A.P.; Ascenso, J.R.; Bernardino, R.J.; Michel, S.; Hubscher-Bruder, V. Bidentate urea derivatives of p-tert-butyldihomooxacalix[4]arene: Neutral receptors for anion complexation. J. Org. Chem. 2014, 79, 742–751. [Google Scholar] [CrossRef]
- Marcos, P.M.; Teixeira, F.A.; Segurado, M.A.P.; Ascenso, J.R.; Bernardino, R.J.; Brancatelli, G.; Geremia, S. Synthesis and anion binding properties of new dihomooxacalix[4]arene diurea and dithiourea receptors. Tetrahedron 2014, 70, 6497–6505. [Google Scholar] [CrossRef]
- Augusto, A.S.; Miranda, A.S.; Ascenso, J.R.; Miranda, M.Q.; Félix, V.; Brancatelli, G.; Hickey, N.; Geremia, S.; Marcos, P.M. Anion recognition by partial cone dihomooxacalix[4]arene-based receptors bearing urea groups: Remarkable affinity for benzoate ion. Eur. J. Org. Chem. 2018, 5657–5667. [Google Scholar] [CrossRef]
- Miranda, A.S.; Serbetci, D.; Marcos, P.M.; Ascenso, J.R.; Berberan-Santos, M.N.; Hickey, N.; Geremia, S. Ditopic receptors based on dihomooxacalix[4]arenes bearing phenylurea moieties with electron-withdrawing groups for anions and organic ion pairs. Front. Chem. 2019, 7, 758. [Google Scholar] [CrossRef]
- Miranda, A.S.; Marcos, P.M.; Ascenso, J.R.; Berberan-Santos, M.N.; Schurhammer, R.; Hickey, N.; Geremia, S. Dihomooxacalix[4]arene-based fluorescent receptors for anion and organic ion pair recognition. Molecules 2020, 25, 4708. [Google Scholar] [CrossRef] [PubMed]
- Barboso, S.; Carrera, A.G.; Matthews, S.E.; Arnaud-Neu, F.; Bohmer, V.; Dozol, J.F.; Rouquette, H.; Schiwing-Weill, M.J. Calix[4]arenes with CMPO functions at the lower rim. Synthesis and extraction properties. J. Chem. Soc. Perkin Trans. 2 1999, 719–723. [Google Scholar] [CrossRef]
- Marcos, P.M.; Ascenso, J.R.; Pereira, J.L.C. Synthesis and NMR conformational studies of p-tert-butyldihomooxacalix[4]arene derivatives bearing pyridyl pendant groups at the lower rim. Eur. J. Org. Chem. 2002, 3034–3041. [Google Scholar] [CrossRef]
- Jaime, C.; de Mendoza, J.; Prados, P.; Nieto, P.; Sanchez, C. Carbon-13 NMR chemical shifts. A single rule to determine the conformation of calix[4]arenes. J. Org. Chem. 1991, 56, 3372–3376. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.; Yan, C.G. Synthesis and crystal structures of p-tert-butyldihomooxa-calix[4]arene mono-Schiff bases. J. Incl. Phenom. Macrocycl. Chem. 2017, 87, 157–166. [Google Scholar] [CrossRef]
- Miranda, A.S.; Martelo, L.M.; Fedorov, A.A.; Berberan-Santos, M.N.; Marcos, P.M. Fluorescence properties of p-tert-butyldihomooxacalix[4]arene derivatives and the effect of anion complexation. New J. Chem. 2017, 41, 5967–5973. [Google Scholar] [CrossRef]
- Griesbeck, A.G.; Görner, H. Laser flash photolysis study of N-alkylated phathalimides. J. Photochem. Photobiol. 1999, 129, 111–119. [Google Scholar] [CrossRef]
- Valeur, B.; Berberan-Santos, M.N. Molecular Fluorescence. Principles and Applications, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2012; p. 195. [Google Scholar]
- Weber, G. Fluorescence-polarization spectrum and electronic-energy transfer in tyrosine, tryptophan and related compounds. Biochem. J. 1960, 75, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Fornander, L.H.; Feng, B.; Beke-Somfai, T.; Nordén, B. UV transition moments of tyrosine. J. Phys. Chem. B 2014, 118, 9247–9257. [Google Scholar] [CrossRef]
- Berberan-Santos, M.N.; Canceill, J.; Brochon, J.C.; Jullien, L.; Lehn, J.-M.; Pouget, J.; Tauc, P.; Valeur, B. Multichromophoric cyclodextrins. 1. Synthesis of O-naphthoyl beta cyclodextrins and investigation of excimer formation and energy hopping. J. Am. Chem. Soc. 1992, 114, 6427–6436. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–integrated space-group and crystal-structure determination. Acta Crystallog. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallog. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for windows: An update. J. Appl. Crystallog. 2012, 45, 849–854. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
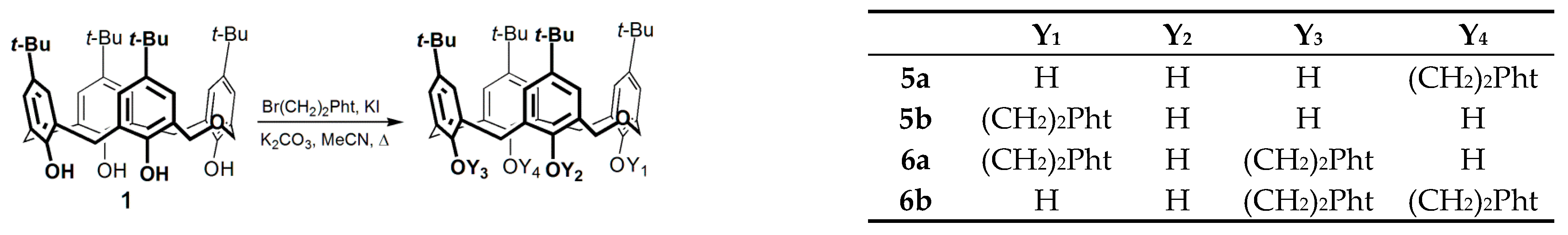{kind=link}
| Entry | Method | Reaction Time | 1:RX:Base Ratio | Reaction Mixture (%) 1 | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2a | 3a | 3b | 4 | ||||
| 1 | Reflux | 4 days | 1:2:2 | 6 | 13 | 65 | 3 | 12 |
| 2 | Reflux | 5 days | 1:2:2 | — | 4 | 76 | 15 | 5 |
| 3 | MW | 20 min | 1:2:2 | — | 10 | 86 | 2 | 2 |
| 4 | MW | 30 min | 1:2:2 | — | 3 | 93 | 2 | 2 |
| 5 | BM | 2 h | 1:2:2 | 70 | 30 | — | — | — |
| 6 | BM | 12 h | 1:4:4 | 27 | 46 | 19 | 4 | 4 |
| Entry | Method | Reaction Time | 1:RX:Base:KI Ratio | Reaction Mixture (%) 1 | ||||
|---|---|---|---|---|---|---|---|---|
| 1 | 5a | 5b | 6a | 6b | ||||
| 1 | Reflux | 3 days | 1:2:2:0 | 61 | 21 | 6 | 11 | 1 |
| 2 | Reflux | 3 days | 1:2.5:2:2 | 30 | 45 | 7 | 16 | 2 |
| 3 | Reflux | 3 days | 1:4:4:2 | 19 | 44 | 4 | 30 | 3 |
| 4 | Reflux | 7 days | 1:6:6:4 | 18 | 37 | 12 | 30 | 3 |
| 5 | MW | 6 + 12 min | 1:4:2:2 | 16 | 58 | — | 26 | — |
| 6 | MW | 6 + 36 min | 1:4:2:2 | — | 59 | — | 41 | — |
| 7 | MW | 12 + 36 min | 1:4:2:2 | — | 38 | — | 62 | — |
| 8 | MW | 12 + 36 min | 1:4:2:0 | — | 43 | — | 57 | — |
| 9 | MW | 18 + 36 min | 1:4:4:0 | — | — | — | 100 | — |
| 10 | MW | 6 + 36 min | 1:4:4:0 | — | — | — | 100 | — |
| 11 | BM | 12 h | 1:4:4:0 | 82 | 18 | — | — | — |
| Molecule | A | B | C | D | |
|---|---|---|---|---|---|
| 2a * | (I) | 138.4 | 154.7 | 105.0 | 126.1 |
| (II) | 141.0 | 146.1 | 107.9 | 132.5 | |
| (III) | 138.6 | 149.5 | 102.3 | 122.4 | |
| (IV) | 128.0 | 148.8 | 98.5 | 129.9 | |
| 3a ** | (I) | 137.02 | 145.72 | 100.30 | 118.38 |
| (II) | 134.54 | 146.88 | 100.23 | 118.73 | |
| (III) | 135.17 | 147.47 | 101.34 | 116.39 | |
| 3b | 140.96 | 146.90 | 94.82 | 120.85 | |
| 5a | 144.25 | 149.98 | 106.82 | 118.39 | |
| λmax,abs (nm) | ε (M–1 cm–1) | λmax,em (nm) | Stokes Shift a (nm) | τf (ns) | ΦFb | |
|---|---|---|---|---|---|---|
| A | 271 | 5.0 × 102 | 288 | 17 | 4.50 c | 7.5 × 10−3 |
| B | 294 | 8.2 × 102 | 403 | 109 | 0.26 d | 1.8 × 10−3 |
| 1 | 286 | 3.6 × 104 | 306 | 20 | 1.50 c | 1.3 × 10−1 |
| 2a | 288 | 2.0 × 104 | 310 | 22 | — | 1.1 × 10−2 |
| 3a | 290 | 1.9 × 104 | 314 | 22 | 1.32 d | 1.7 × 10−3 |
| 3b | 282 | 1.7 × 104 | 314 | 30 | 3.57 d | 2.5 × 10−3 e |
| 5a | 288 | 2.9 × 104 | 310 | 22 | — | 1.4 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda, A.S.; Marcos, P.M.; Ascenso, J.R.; Robalo, M.P.; Bonifácio, V.D.B.; Berberan-Santos, M.N.; Hickey, N.; Geremia, S. Conventional vs. Microwave- or Mechanically-Assisted Synthesis of Dihomooxacalix[4]arene Phthalimides: NMR, X-ray and Photophysical Analysis. Molecules 2021, 26, 1503. https://doi.org/10.3390/molecules26061503
Miranda AS, Marcos PM, Ascenso JR, Robalo MP, Bonifácio VDB, Berberan-Santos MN, Hickey N, Geremia S. Conventional vs. Microwave- or Mechanically-Assisted Synthesis of Dihomooxacalix[4]arene Phthalimides: NMR, X-ray and Photophysical Analysis. Molecules. 2021; 26(6):1503. https://doi.org/10.3390/molecules26061503
Chicago/Turabian StyleMiranda, Alexandre S., Paula M. Marcos, José R. Ascenso, M. Paula Robalo, Vasco D. B. Bonifácio, Mário N. Berberan-Santos, Neal Hickey, and Silvano Geremia. 2021. "Conventional vs. Microwave- or Mechanically-Assisted Synthesis of Dihomooxacalix[4]arene Phthalimides: NMR, X-ray and Photophysical Analysis" Molecules 26, no. 6: 1503. https://doi.org/10.3390/molecules26061503
APA StyleMiranda, A. S., Marcos, P. M., Ascenso, J. R., Robalo, M. P., Bonifácio, V. D. B., Berberan-Santos, M. N., Hickey, N., & Geremia, S. (2021). Conventional vs. Microwave- or Mechanically-Assisted Synthesis of Dihomooxacalix[4]arene Phthalimides: NMR, X-ray and Photophysical Analysis. Molecules, 26(6), 1503. https://doi.org/10.3390/molecules26061503