
A Novel Validated Injectable Colistimethate Sodium Analysis Combining Advanced Chemometrics and Design of Experiments


Abstract
:1. Introduction
2. Results and Discussion
2.1. Design of Experiments
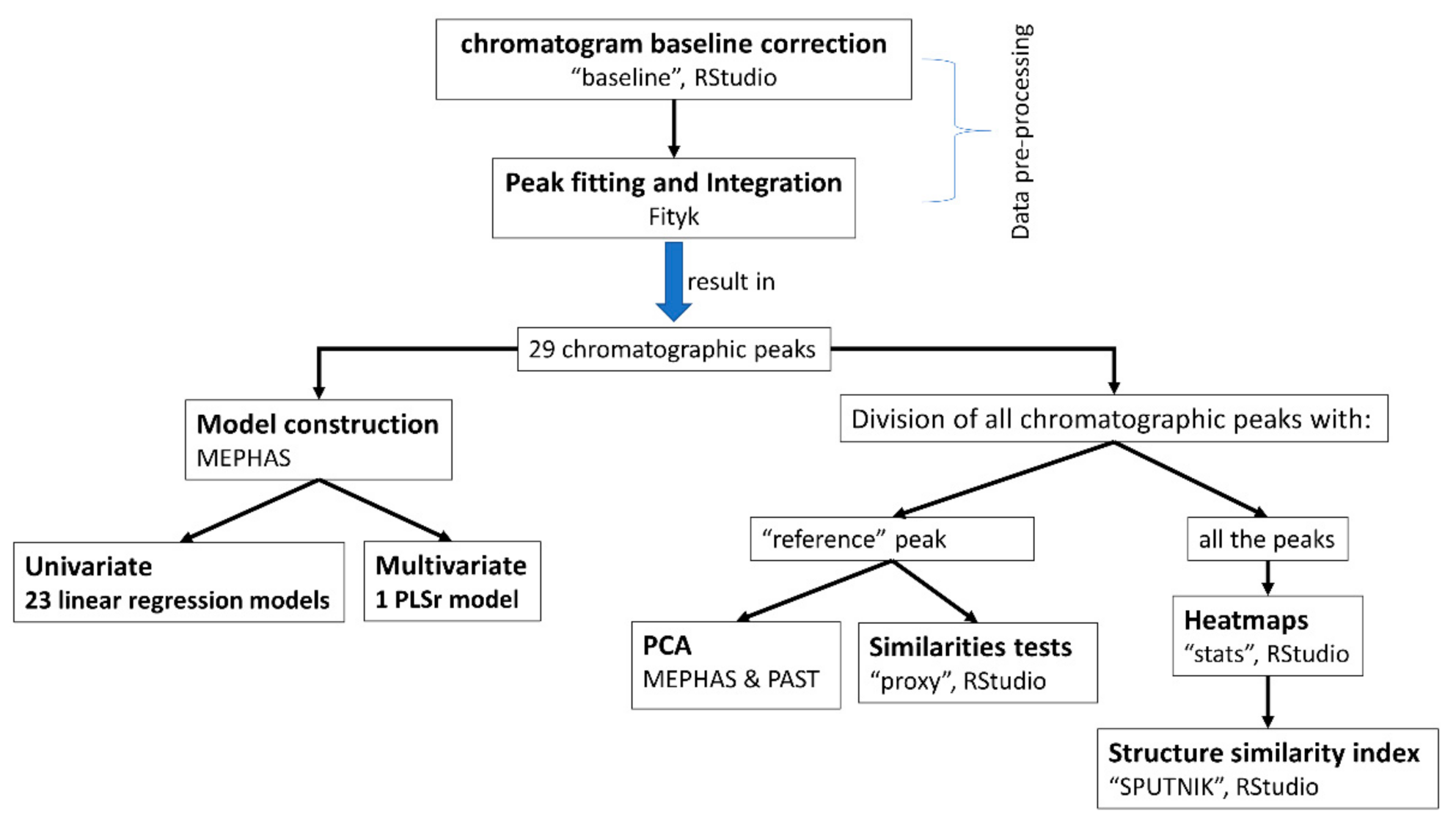
2.2. Data Processing
2.2.1. Data Pre-Processing
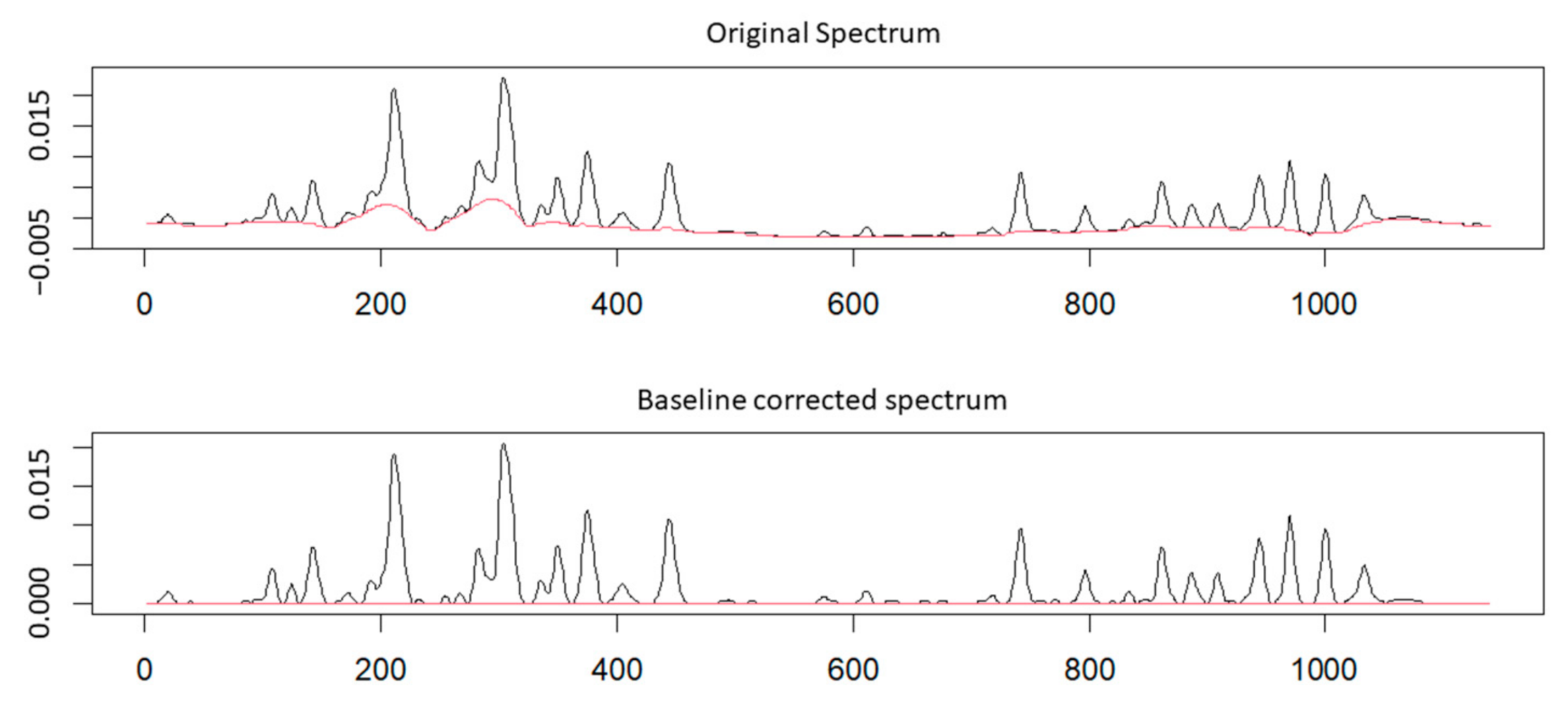
Baseline Correction
Peak Fitting and Integration
2.2.2. Data Analysis—Application to CMS Commercial Batches
Univariate Model Construction
Multivariate Model Construction
Validation of the Univariate and Multivariate Models
Principal Component Analysis
Similarity Tests
Heatmaps and Structure Similarity Index
3. Materials and Methods
3.1. Chemicals
3.2. Instrumentation
3.3. Sample Preparation
3.4. Design of Experiments
3.5. Data Processing
3.5.1. Baseline Correction
3.5.2. Peak Fitting and Integration
3.5.3. Univariate and Multivariate Data Analysis
3.5.4. Similarity Measures and Structure Similarity Index
3.6. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bergen, P.J.; Landersdorfer, C.B.; Lee, H.J.; Li, J.; Nation, R.L. “Old” antibiotics for emerging multidrug-resistant bacteria. Curr. Opin. Infect. Dis. 2012, 25, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Wilson, A.P.R.; Livermore, D.M.; Otter, J.A.; Warren, R.E.; Jenks, P.; Enoch, D.A.; Newsholme, W.; Oppenheim, B.; Leanord, A.; Mcnulty, C.; et al. Prevention and control of multi-drug-resistant Gram-negative bacteria: Recommendations from a Joint Working Party. J. Hosp. Infect. 2016, 92, 1–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, J.S.; Arora, S.; Khanna, S.P.; Kumar, K.V.S.H. Prevalence of Multidrug-resistant, extensively drug-resistant, and pandrug-resistant Pseudomonas aeruginosa from a tertiary level intensive care unit. J. Glob. Infect. Dis. 2016, 8, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.F.; Santiago, M. Microbial approaches for targeting antibiotic-resistant bacteria. Microb. Biotechnol. 2017, 10, 1047–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialvaei, A.Z.; Samadi Kafil, H. Colistin, mechanisms and prevalence of resistance. Curr. Med. Res. Opin. 2015, 31, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, I.; Souli, M.; Galani, I.; Giamarellou, H. Colistin: Still a lifesaver for the 21st century? Expert Opin. Drug Metab. Toxicol. 2017, 13, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Li, J.; Rayner, C.R.; Nation, R.L. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2006, 50, 1953–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, B.; Karvanen, M.; Cars, O.; Plachouras, D.; Friberg, L.E. Quantitative analysis of colistin A and colistin B in plasma and culture medium using a simple precipitation step followed by LC/MS/MS. J. Pharm. Biomed. Anal. 2009, 49, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Gobin, P.; Lemaître, F.; Marchand, S.; Couet, W.; Olivier, J.C. Assay of colistin and colistin methanesulfonate in plasma and urine by liquid chromatography-tandem mass spectrometry. Antimicrob. Agents Chemother. 2010, 54, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Gikas, E.; Bazoti, F.N.; Katsimardou, M.; Anagnostopoulos, D.; Papanikolaou, K.; Inglezos, I.; Skoutelis, A.; Daikos, G.L.; Tsarbopoulos, A. Determination of colistin A and colistin B in human plasma by UPLC-ESI high resolution tandem MS: Application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2013, 83, 228–236. [Google Scholar] [CrossRef]
- Mercier, T.; Tissot, F.; Gardiol, C.; Corti, N.; Wehrli, S.; Guidi, M.; Csajka, C.; Buclin, T.; Couet, W.; Marchetti, O.; et al. High-throughput hydrophilic interaction chromatography coupled to tandem mass spectrometry for the optimized quantification of the anti-Gram-negatives antibiotic colistin A/B and its pro-drug colistimethate. J. Chromatogr. A 2014, 1369, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Bihan, K.; Lu, Q.; Enjalbert, M.; Apparuit, M.; Langeron, O.; Rouby, J.-J.; Funck-Brentano, C.; Zahr, N. Determination of Colistin and Colistimethate levels in human plasma and urine by high-performance liquid chromatography–tandem mass spectrometry. Ther. Drug Monit. 2016, 38, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wu, X.J.; Fan, Y.X.; Guo, B.N.; Zhang, J. Development and validation of a UHPLC-MS/MS assay for colistin methanesulphonate (CMS) and colistin in human plasma and urine using weak-cation exchange solid-phase extraction. J. Pharm. Biomed. Anal. 2016, 124, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.; Bushby, S.R.M.; Wilkinson, S. Sodium sulphomethyl derivatives of polymyxins. J. Pharmacol. 1964, 23, 552–574. [Google Scholar] [CrossRef] [Green Version]
- European Medicines Agency. Assessment Report Polymyxin-Based Products; European Medicines Agency: London, UK, 2015. [Google Scholar]
- He, H.; Li, J.C.; Nation, R.L.; Jacob, J.; Chen, G.; Lee, H.J.; Tsuji, B.T.; Thompson, P.E.; Roberts, K.; Velkov, T.; et al. Pharmacokinetics of four different brands of colistimethate and formed colistin in rats. J. Antimicrob. Chemother. 2013, 68, 2311–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivanesan, S.; Roberts, K.; Wang, J.; Chea, S.-E.; Thompson, P.E.; Li, J.; Nation, R.L.; Velkov, T. Pharmacokinetics of the individual major components of polymyxin B and colistin in rats. J. Nat. Prod. 2017, 80, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.D.; Azad, M.A.K.; Wang, J.; Horne, A.S.; Thompson, P.E.; Nation, R.L.; Velkov, T.; Li, J. Antimicrobial activity and toxicity of the major lipopeptide components of polymyxin B and colistin: Last-line antibiotics against multidrug-resistant gram-negative bacteria HHS public access. ACS Infect. Dis. Novemb. 2015, 13, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagla, I.; Tsarbopoulos, A.; Gikas, E. Design of experiments guided multivariate calibration for the quantitation of injectable colistimethate sodium by ultra performance liquid chromatography–High resolution mass spectrometry. Talanta 2020, 220, 121406. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, A.P.; Hardaker, L.E.A.; Hatley, R.H.M. A simple method for assaying colistimethate sodium in pharmaceutical aerosol samples using high performance liquid chromatography. J. Pharm. Biomed. Anal. 2017, 142, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.L.C.; Bruns, R.E.; Ferreira, H.S.; Matos, G.D.; David, J.M.; Brandão, G.C.; da Silva, E.G.P.; Portugal, L.A.; dos Reis, P.S.; Souza, A.S.; et al. Box-Behnken design: An alternative for the optimization of analytical methods. Anal. Chim. Acta 2007, 597, 179–186. [Google Scholar] [CrossRef]
- Wang, Z.; Bovik, A.C.; Sheikh, H.R.; Simoncelli, E.P. Image Quality Assessment: From Error Visibility to Structural Similarity; IEEE: Piscataway, NJ, USA, 2004; Volume 13. [Google Scholar]
- Hawrył, A.M.; Ziobro, A.; Swieboda, R.S.; Hawrył, M.A.; Chernetskyy, M.; Waksmundzka-Hajnos, M. The HPLC fingerprint analysis of selected cirsium species with aid of chemometrics. Artic. J. Braz. Chem. Soc. 2016, 27, 1736–1743. [Google Scholar] [CrossRef]
- Zhou, Y.; Leung, S.W.; Mizutani, S.; Takagi, T.; Tian, Y.S. MEPHAS: An interactive graphical user interface for medical and pharmaceutical statistical analysis with R and shiny. BMC Bioinform. 2020, 21. [Google Scholar] [CrossRef]
- Hammer, D.A.T.; Ryan, P.D.; Hammer, Ø.; Harper, D.A.T. Past: Paleontological statistics software package for education and data analysis. Adv. Anthropol. 2001, 4, 9. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | Name | Best-Fit Values | Std. Error | R2 | Sy.× | ||
|---|---|---|---|---|---|---|---|
| B0 | B1 | B0 | B1 | ||||
| 1 | Peak_7.28 | 1.13 × 10−5 | 1.73 × 10−6 | 4.18 × 10−6 | 2.52 × 10−8 | 0.9994 | 2.39 × 10−6 |
| 2 | Peak_8.73 | −2.6 × 10−5 | 4.88 × 10−6 | 1.99 × 10−5 | 1.2 × 10−7 | 0.998 | 1.14 × 10−5 |
| 3 | Peak_9.02 | 4.36 × 10−5 | 1.73 × 10−6 | 1.55 × 10−5 | 9.33 × 10−8 | 0.991 | 8.85 × 10−6 |
| 4 | Peak_9.30 | 0.000105 | 8.27 × 10−6 | 2.82 × 10−5 | 1.7 × 10−7 | 0.998 | 1.61 × 10−5 |
| 5 | Peak_9.80 | 3.06 × 10−5 | 1.32 × 10−6 | 7.07 × 10−6 | 4.27 × 10−8 | 0.997 | 4.05 × 10−6 |
| 6 | Peak_10.43 | 0.000177 | 2.96 × 10−5 | 1.29 × 10−4 | 7.8 × 10−7 | 0.998 | 7.4 × 10−5 |
| 7 | Peak_11.99 | 3.45 × 10−5 | 3.62 × 10−5 | 1.45 × 10−4 | 8.79 × 10−7 | 0.998 | 8.34 × 10−5 |
| 8 | Peak_12.53 | 2.0 × 10−6 | 3.0 × 10−6 | 6.83 × 10−6 | 4.13 × 10−8 | 0.9994 | 3.92 × 10−6 |
| 9 | Peak_12.76 | 1.29 × 10−4 | 7.74 × 10−6 | 1.69 × 10−5 | 1.02 × 10−7 | 0.9995 | 9.71 × 10−6 |
| 10 | Peak_13.18 | 2.35 × 10−5 | 1.61 × 10−5 | 3.82 × 10−5 | 2.31 × 10−7 | 0.9994 | 2.19 × 10−5 |
| 11 | Peak_14.35 | −3 × 10−5 | 1.54 × 10−5 | 4.29 × 10−5 | 2.59 × 10−7 | 0.9996 | 2.46 × 10−5 |
| 12 | Peak_17.18 | 1.19 × 10−5 | 1.59 × 10−6 | 5.98 × 10−6 | 3.61 × 10−8 | 0.998 | 3.43 × 10−6 |
| 13 | Peak_18.95 | 1.11 × 10−5 | 1.18 × 10−6 | 5.73 × 10−6 | 3.46 × 10−8 | 0.997 | 3.28 × 10−6 |
| 14 | Peak_19.33 | 1.02 × 10−4 | 1.07 × 10−5 | 2.4 × 10−5 | 1.45 × 10−7 | 0.9994 | 1.38 × 10−5 |
| 15 | Peak_20.27 | 6.23 × 10−5 | 4.29 × 10−6 | 1.12 × 10−5 | 6.79 × 10−8 | 0.9993 | 6.44 × 10−6 |
| 16 | Peak_20.87 | 3.0 × 10−6 | 1.29 × 10−6 | 1.23 × 10−5 | 7.41 × 10−8 | 0.990 | 7.03 × 10−6 |
| 17 | Peak_21.33 | −3.8 × 10−5 | 8.46 × 10−6 | 2.03 × 10−5 | 1.23 × 10−7 | 0.9994 | 1.16 × 10−5 |
| 18 | Peak_21.75 | −3.2 × 10−5 | 4.77 × 10−6 | 2.36 × 10−5 | 1.43 × 10−7 | 0.997 | 1.35 × 10−5 |
| 19 | Peak_22.12 | 1.63 × 10−5 | 3.82 × 10−6 | 1.44 × 10−5 | 8.68 × 10−8 | 0.998 | 8.24 × 10−6 |
| 20 | Peak_22.70 | −9.4 × 10−5 | 1.13 × 10−5 | 2.47 × 10−5 | 1.49 × 10−7 | 0.9995 | 1.41 × 10−5 |
| 21 | Peak_23.13 | −4.7 × 10−7 | 1.24 × 10−5 | 3.11 × 10−5 | 1.88 × 10−7 | 0.9993 | 1.78 × 10−5 |
| 22 | Peak_23.65 | −6.4 × 10−5 | 1.08 × 10−5 | 3.77 × 10−5 | 2.28 × 10−7 | 0.998 | 2.16 × 10−5 |
| 23 | Peak_24.20 | 4.26 × 10−4 | 3.72 × 10−6 | 3.07 × 10−5 | 1.86 × 10−7 | 0.992 | 1.76 × 10−5 |
| Peaks | Batches | |||||||
|---|---|---|---|---|---|---|---|---|
| Reference | b1 | b2 | b3 | b4a | b4b | b4c | b5 | |
| Peak_7.28 | 161.38 | 172.37 | 160.18 | 177.90 | 197.40 | 203.45 | 183.76 | 215.34 |
| Peak_8.73 | 160.29 | 166.43 | 159.52 | 186.13 | 195.02 | 203.04 | 185.78 | 151.79 |
| Peak_9.02 | 163.82 | 155.89 | 159.66 | 163.45 | 159.12 | 164.16 | 152.44 | 151.37 |
| Peak_9.30 | 159.03 | 165.36 | 159.00 | 164.58 | 176.96 | 186.60 | 172.23 | 222.71 |
| Peak_9.80 | 157.88 | 123.53 | 158.68 | 216.73 | 174.85 | 183.56 | 170.11 | 219.82 |
| Peak_10.43 | 162.62 | 151.30 | 161.01 | 167.91 | 173.71 | 180.11 | 164.75 | 148.58 |
| Peak_11.99 | 162.82 | 167.38 | 161.37 | 175.33 | 179.80 | 187.03 | 171.63 | 206.85 |
| Peak_12.53 | 160.67 | 159.21 | 157.64 | 174.67 | 180.31 | 189.25 | 171.96 | 162.05 |
| Peak_12.76 | 160.98 | 153.46 | 156.65 | 162.76 | 169.68 | 176.89 | 159.61 | 200.00 |
| Peak_13.18 | 158.58 | 145.82 | 157.86 | 166.52 | 171.98 | 178.42 | 162.42 | 155.72 |
| Peak_14.35 | 159.22 | 154.64 | 158.66 | 169.20 | 176.24 | 182.93 | 166.23 | 173.91 |
| Peak_17.18 | 157.61 | 150.79 | 171.50 | 188.85 | 208.18 | 218.25 | 198.88 | 165.47 |
| Peak_18.95 | 160.51 | 129.14 | 168.93 | 173.40 | 178.19 | 182.98 | 189.46 | 143.35 |
| Peak_19.33 | 159.68 | 141.66 | 168.03 | 174.95 | 191.18 | 197.33 | 186.53 | 166.00 |
| Peak_20.27 | 159.49 | 141.25 | 165.80 | 174.65 | 182.64 | 191.58 | 179.32 | 234.93 |
| Peak_20.87 | 162.79 | 172.14 | 172.45 | 184.84 | 214.98 | 226.10 | 213.74 | 224.00 |
| Peak_21.33 | 159.86 | 158.55 | 169.28 | 178.59 | 196.83 | 204.21 | 192.78 | 200.75 |
| Peak_21.75 | 157.82 | 144.83 | 166.24 | 174.96 | 191.92 | 197.88 | 185.96 | 194.90 |
| Peak_22.12 | 159.21 | 140.39 | 166.33 | 166.10 | 179.57 | 186.70 | 175.73 | 239.08 |
| Peak_22.70 | 158.57 | 147.71 | 166.63 | 170.62 | 186.55 | 194.49 | 183.81 | 211.31 |
| Peak_23.13 | 157.88 | 133.52 | 165.97 | 173.85 | 183.17 | 188.17 | 178.13 | 134.49 |
| Peak_23.65 | 157.18 | 130.46 | 163.74 | 168.55 | 180.30 | 185.57 | 173.56 | 137.05 |
| Peak_24.20 | 167.26 | 166.95 | 176.09 | 179.92 | 198.30 | 206.27 | 193.16 | 234.58 |
| Batch | Interpolated Values CMS (μg mL−1) | Upper Limit | Lower Limit | % Error |
|---|---|---|---|---|
| b1 | 156.01 | 150.29 | 151.67 | −2.49 |
| b2 | 161.85 | 163.82 | 164.08 | 1.15 |
| b3 | 171.61 | 174.73 | 176.06 | 7.25 |
| b4a | 178.50 | 183.63 | 185.61 | 11.56 |
| b4b | 185.21 | 190.63 | 193.21 | 15.75 |
| b4c | 170.72 | 178.04 | 179.46 | 6.70 |
| b5 | 179.33 | 185.90 | 187.42 | 12.08 |
| 23 Univariate Linear Regression Models | ||||||
|---|---|---|---|---|---|---|
| Concentration Level (μg mL−1) | Accuracy (n = 3, %E) | Precision (n = 3, %RSD) | Stability (n = 3, %RSD) | Robustness (n = 3, %RSD) | ||
| Repeatability | Intermediate Precision | |||||
| 130 | <4.78 | <2.34 | <3.50 | <2.75 | ||
| 160 | <4.13 | <2.40 | <3.35 | <2.15 | <3.14 | |
| 190 | <3.35 | <2.28 | <2.95 | <2.37 | ||
| LOD (μg mL−1) | LOQ (μg mL−1) | |||||
| 4.12–17.98 | 12.48–54.49 | |||||
| Multivariate PLSr Model | ||||||
| Concentration Level (μg mL−1) | Accuracy (n = 3, %E) | Precision (n = 3, %RSD) | Stability (n = 3, %RSD) | Robustness (n = 3, %RSD) | ||
| Repeatability | Intermediate Precision | |||||
| 130 | 2.83 | 0.28 | 1.13 | 1.75 | ||
| 160 | 0.64 | 0.45 | 0.99 | 1.26 | <2.64 | |
| 190 | 1.17 | 0.57 | 1.24 | 1.38 | ||
| Tests | Batches | ||||||
|---|---|---|---|---|---|---|---|
| b1 | b2 | b3 | b4a | b4b | b4c | b5 | |
| Similarity measures | |||||||
| ejaccard | 0.995 | 0.999 | 0.999 | 0.999 | 0.999 | 0.997 | 0.972 |
| cosine | 0.997 | 1.000 | 1.000 | 0.999 | 0.999 | 0.999 | 0.986 |
| eDice | 0.997 | 1.000 | 1.000 | 0.999 | 0.999 | 0.999 | 0.986 |
| correlation | 0.996 | 1.000 | 0.999 | 0.999 | 0.999 | 0.998 | 0.974 |
| Gower | 0.693 | 0.855 | 0.795 | 0.707 | 0.695 | 0.612 | 0.236 |
| Distance measures | |||||||
| Bray | 0.967 | 0.987 | 0.985 | 0.980 | 0.980 | 0.973 | 0.923 |
| Canberra | 0.540 | 0.737 | 0.668 | 0.608 | 0.602 | 0.532 | 0.349 |
| Chord | 0.933 | 0.983 | 0.973 | 0.966 | 0.966 | 0.958 | 0.859 |
| divergence | 0.953 | 0.991 | 0.979 | 0.967 | 0.966 | 0.949 | 0.845 |
| Euclidean | 0.782 | 0.911 | 0.898 | 0.878 | 0.877 | 0.836 | 0.601 |
| Geodesic | 0.933 | 0.983 | 0.973 | 0.966 | 0.966 | 0.958 | 0.859 |
| Hellinger | 0.962 | 0.988 | 0.981 | 0.976 | 0.975 | 0.972 | 0.920 |
| Kullback | 0.997 | 1.000 | 0.999 | 0.999 | 0.999 | 0.998 | 0.985 |
| Manhattan | 0.544 | 0.748 | 0.724 | 0.663 | 0.662 | 0.581 | 0.319 |
| Podani | 0.975 | 0.983 | 0.967 | 0.943 | 0.943 | 0.943 | 0.878 |
| Soergel | 0.755 | 0.878 | 0.866 | 0.827 | 0.827 | 0.773 | 0.570 |
| supremum | 0.837 | 0.962 | 0.924 | 0.938 | 0.938 | 0.918 | 0.693 |
| Whittaker | 0.969 | 0.990 | 0.986 | 0.982 | 0.982 | 0.978 | 0.924 |
| Bhjattacharyya | 0.875 | 0.944 | 0.934 | 0.913 | 0.913 | 0.886 | 0.760 |
| Batch | SSIM |
|---|---|
| b1 | 0.961 |
| b2 | 0.998 |
| b3 | 0.994 |
| b4a | 0.960 |
| b4b | 0.961 |
| b4c | 0.958 |
| b5 | 0.918 |
| Software | Scope | Available on | |
|---|---|---|---|
| Rstudio | https://rstudio.com/ (accessed on 19 February 2021) | ||
| packages: | “baseline” | chromatogram baseline correction | https://cran.r-project.org/web/packages/baseline/index.html (accessed on 19 February 2021) |
| “proxy” | similarity and distance measures | https://cran.r-project.org/web/packages/proxy/index.html (accessed on 19 February 2021) | |
| “stats” | heatmaps | https://www.rdocumentation.org/packages/cran.stats/versions/0.1 (accessed on 19 February 2021) | |
| “SPUTNIK” | structure similarity index | https://cran.r-project.org/web/packages/SPUTNIK/index.html (accessed on 19 February 2021) | |
| Fityk | Peak fitting and integration | https://fityk.nieto.pl/ (accessed on 19 February 2021) | |
| MEPHAS | linear regression model, PLSr, PCA | https://alain003.phs.osaka-u.ac.jp/mephas/ (accessed on 19 February 2021) | |
| PAST | PCA | https://past.en.lo4d.com/windows (accessed on 19 February 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dagla, I.; Tsarbopoulos, A.; Gikas, E. A Novel Validated Injectable Colistimethate Sodium Analysis Combining Advanced Chemometrics and Design of Experiments. Molecules 2021, 26, 1546. https://doi.org/10.3390/molecules26061546
Dagla I, Tsarbopoulos A, Gikas E. A Novel Validated Injectable Colistimethate Sodium Analysis Combining Advanced Chemometrics and Design of Experiments. Molecules. 2021; 26(6):1546. https://doi.org/10.3390/molecules26061546
Chicago/Turabian StyleDagla, Ioanna, Anthony Tsarbopoulos, and Evagelos Gikas. 2021. "A Novel Validated Injectable Colistimethate Sodium Analysis Combining Advanced Chemometrics and Design of Experiments" Molecules 26, no. 6: 1546. https://doi.org/10.3390/molecules26061546