
Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro



Abstract
1. Introduction
2. Results
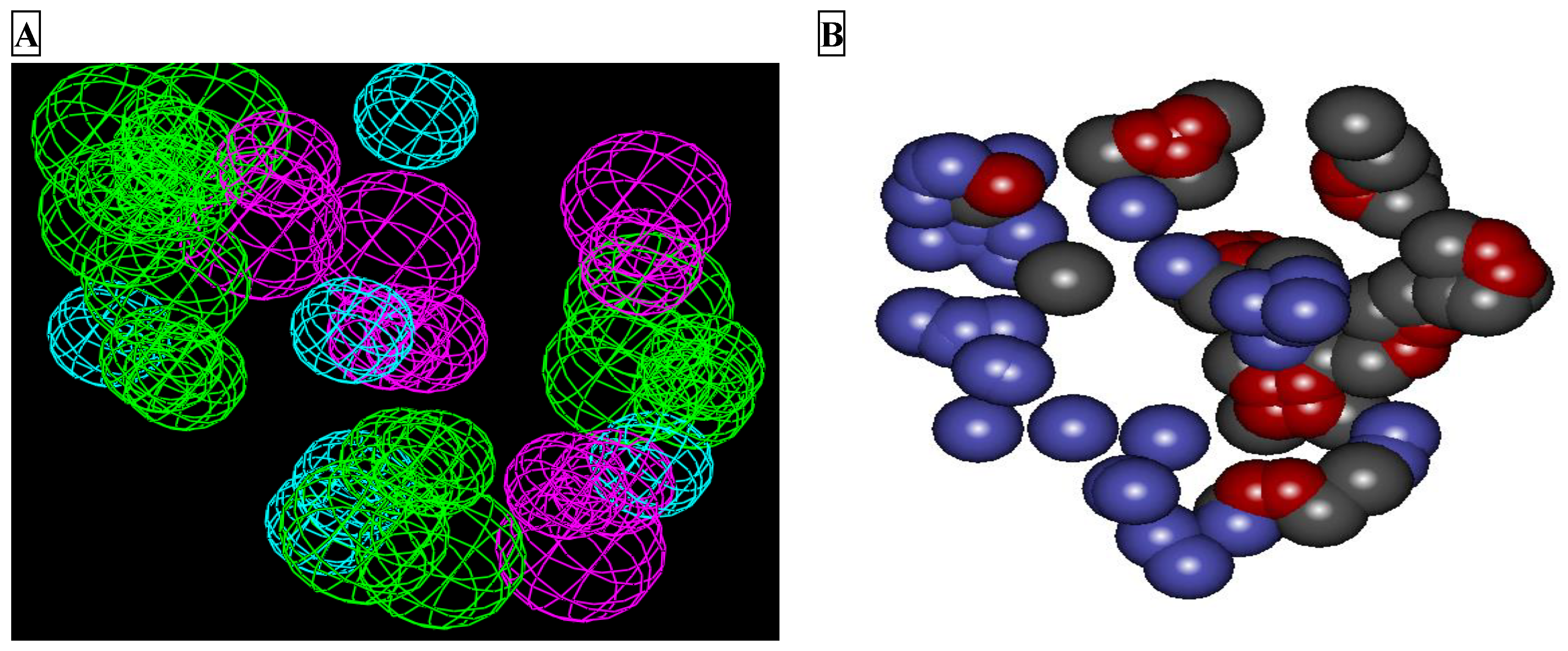
2.1. LUDI Receptor-Based Pharmacophore Generation
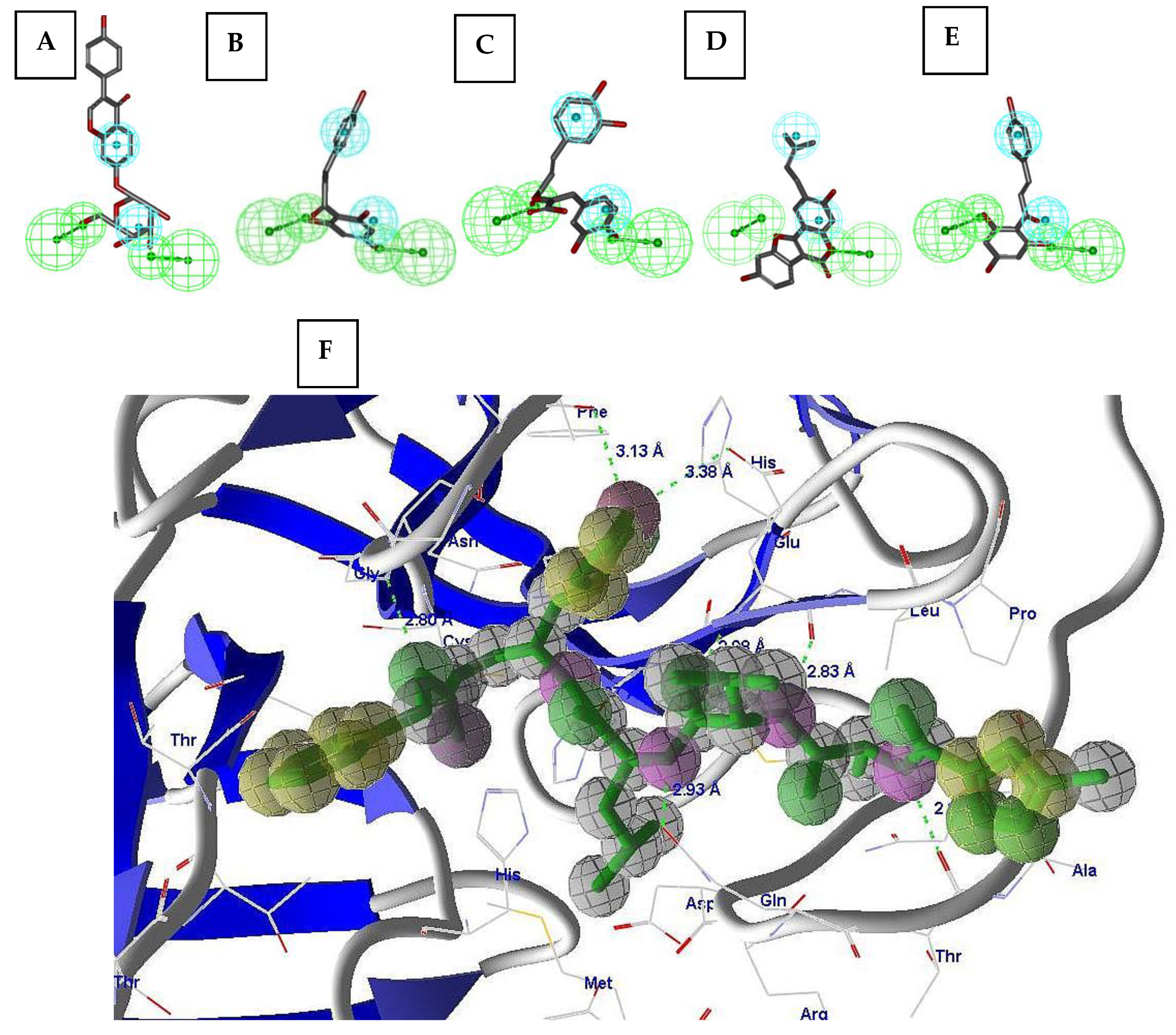
2.2. Pharmacophore Validation
2.3. Virtual Screening
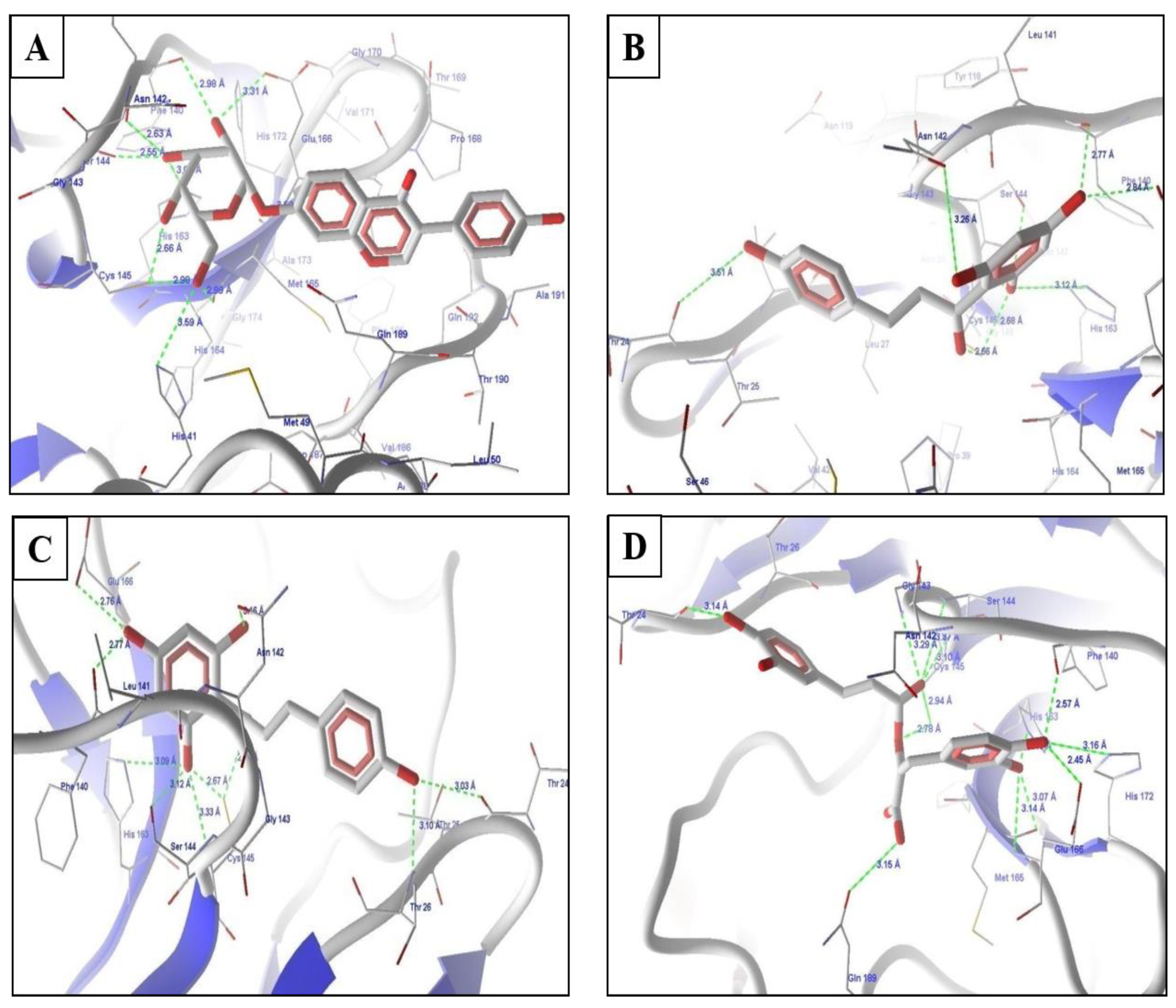
2.4. Molecular Docking Experiments
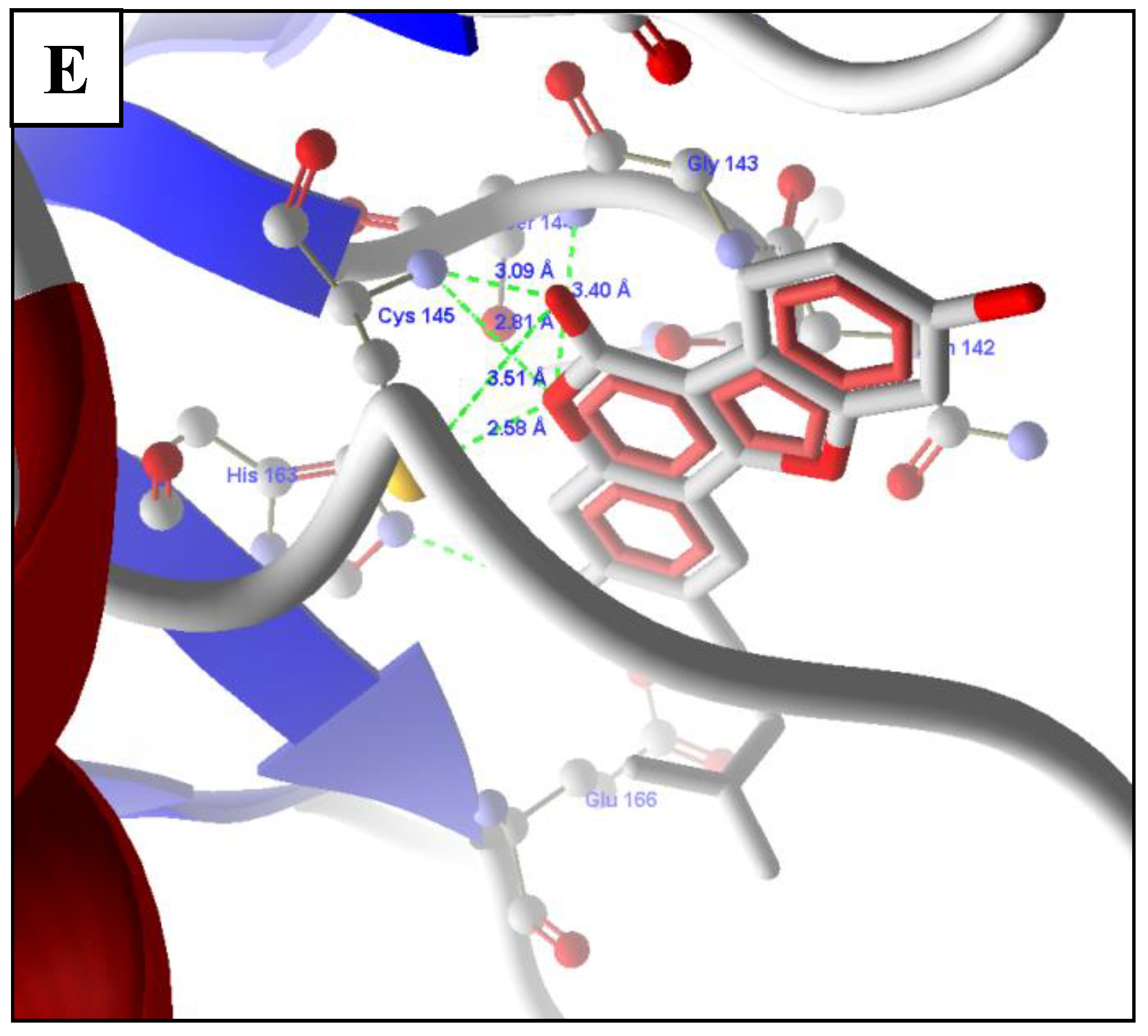
2.5. Molecular Docking of Prioritized Drugs
2.6. Revalidation of Docking Using Autodock
2.7. Physicochemical Parameters for the Selected Ligands
3. Discussion
4. Material and Methods
4.1. LUDI-Based Pharmacophore Model
4.2. Common Feature Pharmacophore Generation
4.3. Pharmacophore-Based Virtual Screening
4.4. Molecular Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kandimalla, R.; John, A.; Abburi, C.; Vallamkondu, J.; Reddy, P.H. Current status of multiple drug molecules, and vaccines: An update in SARS-CoV-2 therapeutics. Mol. Neurobiol. 2020, 57, 1–11. [Google Scholar] [CrossRef]
- Yi, Y.; Lagniton, P.N.; Ye, S.; Li, E.; Xu, R.-H. COVID-19: What has been learned and to be learned about the novel coronavirus disease. Int. J. Biol. Sci. 2020, 16, 1753. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferl, H.; Wenisch, C. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E. COVID-19: Drug targets and potential treatments. J. Med. Chem. 2020, 63, 12359–12386. [Google Scholar] [CrossRef] [PubMed]
- Prajapat, M.; Sarma, P.; Shekhar, N.; Avti, P.; Sinha, S.; Kaur, H.; Kumar, S.; Bhattacharyya, A.; Kumar, H.; Bansal, S. Drug targets for corona virus: A systematic review. Indian J. Pharmacol. 2020, 52, 56. [Google Scholar] [PubMed]
- Abd El-Aziz, T.M.; Al-Sabi, A.; Stockand, J.D. Human recombinant soluble ACE2 (hrsACE2) shows promise for treating severe COVID19. Signal. Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef] [PubMed]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Genet. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A. Drug targets for COVID-19 therapeutics: Ongoing global efforts. J. Biosci. 2020, 45, 1–24. [Google Scholar] [CrossRef]
- Balaramnavar, V.M.; Ahmad, K.; Saeed, M.; Ahmad, I.; Kamal, M.; Jawed, T. Pharmacophore-based approaches in the rational repurposing technique for FDA approved drugs targeting SARS-CoV-2 Mpro. RSC Adv. 2020, 10, 40264–40275. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Wang, X.; Jochmans, D.; Neyts, J.; Młynarski, W. SARS-CoV-2 M pro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2020, 17, 1–7. [Google Scholar]
- Saeed, M.; Saeed, A.; Alam, M.J.; Alreshidi, M. Identification of Persuasive Antiviral Natural Compounds for COVID-19 by Targeting Endoribonuclease NSP15: A Structural-Bioinformatics Approach. Molecules 2020, 25, 5657. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Pirhadi, S.; Shiri, F.; Ghasemi, J.B. Methods and applications of structure based pharmacophores in drug discovery. Curr. Top. Med. Chem. 2013, 13, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J. LUDI: Rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput. Aided Mol. Des. 1992, 6, 593–606. [Google Scholar] [CrossRef]
- Barillari, C.; Marcou, G.; Rognan, D. Hot-spots-guided receptor-based pharmacophores (HS-Pharm): A knowledge-based approach to identify ligand-anchoring atoms in protein cavities and prioritize structure-based pharmacophores. J. Chem. Inf. Model. 2008, 48, 1396–1410. [Google Scholar] [CrossRef]
- Puratchikody, A.; Irfan, N.; Balasubramaniyan, S. Conceptual design of hybrid PCSK9 lead inhibitors against coronary artery disease. Biocatal. Agric. Biotechnol. 2019, 17, 427–440. [Google Scholar] [CrossRef]
- Muthusamy, K.; Singh, K.D.; Chinnasamy, S.; Nagamani, S.; Krishnasamy, G.; Thiyagarajan, C.; Premkumar, P.; Anusuyadevi, M. High throughput virtual screening and E-pharmacophore filtering in the discovery of new BACE-1 inhibitors. Interdiscip. Sci. Comput. Life Sci. 2013, 5, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Chaudhary, M.; Qamar, I.; Singh, N.; Nain, V. In silico identification and validation of natural antiviral compounds as potential inhibitors of SARS-CoV-2 methyltransferase. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef]
- Rafii, F. The role of colonic bacteria in the metabolism of the natural isoflavone daidzin to equol. Metabolites 2015, 5, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: An in silico perspective. J. Biomol. Struct. Dyn. 2020, 1–9. [Google Scholar] [CrossRef]
- Lin, S.-C.; Chen, M.-C.; Liu, S.; Callahan, V.M.; Bracci, N.R.; Lehman, C.W.; Dahal, B.; Cynthia, L.; Lin, C.-C.; Wang, T.T. Phloretin inhibits Zika virus infection by interfering with cellular glucose utilisation. Int. J. Antimicrob. Agents 2019, 54, 80–84. [Google Scholar] [CrossRef]
- Behzad, S.; Sureda, A.; Barreca, D.; Nabavi, S.F.; Rastrelli, L.; Nabavi, S.M. Health effects of phloretin: From chemistry to medicine. Phytochem. Rev. 2017, 16, 527–533. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, Y.-I.; Lee, T.-H.; Jung, I.D.; Lee, J.S.; Lee, C.-M.; Kim, J.-I.; Joo, H.; Lee, J.-D.; Park, Y.-M. Rosmarinic acid inhibits indoleamine 2, 3-dioxygenase expression in murine dendritic cells. Biochem. Pharmacol. 2007, 73, 1412–1421. [Google Scholar] [CrossRef]
- Sharifi-Rad, J.; Kamiloglu, S.; Yeskaliyeva, B.; Beyatli, A.; Alfred, M.A.; Salehi, B.; Calina, D.; Docea, A.O.; Imran, M.; Kumar, N.V.A. Pharmacological activities of Psoralidin: A comprehensive review of the molecular mechanisms of action. Front. Pharmacol. 2020, 11, 571459. [Google Scholar] [CrossRef]
- Ahmad, K.; Balaramnavar, V.M.; Baig, M.H.; Srivastava, A.K.; Khan, S.; Kamal, M.A. Identification of potent caspase-3 inhibitors for treatment of multi-neurodegenerative diseases using pharmacophore modeling and docking approaches. CNS Neurol. Disord. Drug Targets 2014, 13, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Sprague, P.W. Automated chemical hypothesis generation and database searching with Catalyst®. Perspect. Drug Discov. Des. 1995, 3, 1–20. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Aided Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Milne, G.W.; Nicklaus, M.C.; Driscoll, J.S.; Wang, S.; Zaharevitz, D. National Cancer Institute drug information system 3D database. J. Chem. Inf. Comput. Sci. 1994, 34, 1219–1224. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Gehlhaar, D.K.; Bouzida, D.; Rejto, P.A. Fully automated and rapid flexible docking of inhibitors covalently bound to serine proteases. In Proceedings of the International Conference on Evolutionary Programming, San Diego, CA, USA, 25–27 March 1998; Springer: Berlin/Heidelberg, Germany, 1998; pp. 449–461. [Google Scholar]
- John, S.E.S.; Tomar, S.; Stauffer, S.R.; Mesecar, A.D. Targeting zoonotic viruses: Structure-based inhibition of the 3C-like protease from bat coronavirus HKU4—The likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS). Bioorg. Med. Chem. 2015, 23, 6036–6048. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Natural Product | MolDock Score | Rerank Score | Docking Score | Similarity Score | Fit Value a |
|---|---|---|---|---|---|
| Daidzin | −115.112 | −111.544 | −362.165 | −248.271 | 2.94536 |
| Phloretin | −113.173 | −90.7301 | −286.075 | −170.686 | 3.1189 |
| Rosmarinic acid | −130.853 | −103.821 | −361.153 | −225.21 | 3.08619 |
| Higenamine hydrochloride | −115.15 | −98.3765 | −257.79 | −142.498 | 3.16578 |
| Psoralidin | −115.765 | −92.1257 | −316.84 | −196.709 | 3.122 |
| Naringenin chalcone | −118.833 | −95.914 | −270.535 | −137.061 | 2.96379 |
| Pinoresinol dimethyl ether | −118.818 | −85.9625 | −312.815 | −195.726 | 3.16578 |
| AMPHICOL (chloramphenicol) | −77.3877 | −62.1465 | −218.843 | −141.977 | 2.90662 |
| Aloe-emodin | −97.7624 | −87.7886 | −234.279 | −135.969 | 3.2435 |
| Caffeic acid phenethyl ester (CAPE) | −101.871 | −85.2197 | −312.47 | −211.714 | 2.95378 |
| Dopamine hydrochloride | −69.3166 | −58.7612 | −176.013 | −107.296 | 3.16578 |
| Epinephrine bitartrate | −77.6416 | −64.9263 | −205.116 | −126.866 | 2.96379 |
| Genipin | −102.454 | −82.0033 | −221.112 | −114.184 | 3.26523 |
| Morin | −105.509 | −92.3222 | −278.217 | −165.081 | 3.00264 |
| Noradrenaline bitartrate | −74.4106 | −61.1462 | −192.769 | −119.856 | 3.45066 |
| Scopoletin | −88.1501 | −74.2017 | −195.512 | −102.584 | 2.95378 |
| N-Sulfo-glucosamine | −92.1255 | −68.4776 | −214.491 | −97.6027 | 2.94536 |
| Nordihydroguaiaretic acid | −100.616 | −71.7295 | −297.163 | −196.406 | 2.96379 |
| DL-Panthenol | −79.5991 | −68.3015 | −158.414 | −72.983 | 3.26523 |
| Oxyresveratrol | −94.9673 | −79.3005 | −259.463 | −161.834 | 3.00264 |
| Danshensu | −70.6003 | −62.9828 | −199.499 | −130.037 | 3.45066 |
| 3-Indolepropionic acid | −91.4718 | −71.9077 | −215.996 | −123.141 | 3.1189 |
| Pyridoxal 5-phosphate | −78.5887 | −65.6728 | −219.155 | −129.277 | 3.08619 |
| 2′-deoxyuridine | −88.4843 | −75.9807 | −225.239 | −131.446 | 2.90662 |
| D-Glucose 6-phosphate | −76.5959 | −58.835 | −191.605 | −110.676 | 3.2435 |
| Cadaverine | −55.1465 | −45.497 | −95.4426 | −40.6983 | 2.95378 |
| Ethyl caffeate | −95.3421 | −80.9775 | −209.814 | −115.739 | 2.94536 |
| Sesamin | −102.973 | −72.0989 | −337.484 | −235.388 | 3.26523 |
| XYLO-PFAN (xylose) | −70.0215 | −55.1009 | −138.678 | −55.264 | 3.00264 |
| Molecule | Daidzin | Phloretin | Rosmarinic Acid | Higenamine Hydrochloride | Naringenin Chalcone | |
|---|---|---|---|---|---|---|
| Physicochemical Properties | Formula | C21H20O9 | C15H14O5 | C18H16O8 | C16H18ClNO3 | C15H12O5 |
| MW | 416.38 | 274.27 | 360.31 | 307.77 | 272.25 | |
| Heavy atoms | 30 | 20 | 26 | 21 | 20 | |
| Aromatic heavy atoms | 16 | 12 | 12 | 12 | 12 | |
| Fraction Csp3 | 0.29 | 0.13 | 0.11 | 0.25 | 0 | |
| Rotatable bonds | 4 | 4 | 7 | 2 | 3 | |
| H-bond acceptors | 9 | 5 | 8 | 4 | 5 | |
| H-bond donors | 5 | 4 | 5 | 4 | 4 | |
| MR | 104.09 | 74.02 | 91.4 | 88.11 | 74.34 | |
| TPSA | 149.82 | 97.99 | 144.52 | 72.72 | 97.99 | |
| Pharmacokinetics | GI absorption | Low | High | Low | High | High |
| BBB permeant | No | No | No | Yes | No | |
| Pgp substrate | No | No | No | Yes | No | |
| CYP1A2 inhibitor | No | Yes | No | No | Yes | |
| CYP2C19 inhibitor | No | No | No | No | No | |
| CYP2C9 inhibitor | No | Yes | No | No | Yes | |
| CYP2D6 inhibitor | No | No | No | Yes | No | |
| CYP3A4 inhibitor | No | Yes | No | Yes | Yes | |
| log Kp (cm/s) | −8.36 | −6.11 | −6.82 | −6.01 | −5.96 | |
| Druglikeness | Lipinski violations | 0 | 0 | 0 | 0 | 0 |
| Ghose violations | 0 | 0 | 0 | 0 | 0 | |
| Veber violations | 1 | 0 | 1 | 0 | 0 | |
| Egan violations | 1 | 0 | 1 | 0 | 0 | |
| Muegge violations | 0 | 0 | 0 | 0 | 0 | |
| Bioavailability score | 0.55 | 0.55 | 0.56 | 0.55 | 0.55 | |
| Medicinal Chemistry | PAINS alerts | 0 | 0 | 1 | 1 | 0 |
| Brenk alerts | 0 | 0 | 2 | 1 | 1 | |
| Leadlikeness violations | 1 | 0 | 1 | 0 | 0 | |
| Synthetic accessibility | 5.01 | 1.88 | 3.38 | 2.7 | 2.56 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, M.; Saeed, A.; Alam, M.J.; Alreshidi, M. Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro. Molecules 2021, 26, 1549. https://doi.org/10.3390/molecules26061549
Saeed M, Saeed A, Alam MJ, Alreshidi M. Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro. Molecules. 2021; 26(6):1549. https://doi.org/10.3390/molecules26061549
Chicago/Turabian StyleSaeed, Mohd, Amir Saeed, Md Jahoor Alam, and Mousa Alreshidi. 2021. "Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro" Molecules 26, no. 6: 1549. https://doi.org/10.3390/molecules26061549
APA StyleSaeed, M., Saeed, A., Alam, M. J., & Alreshidi, M. (2021). Receptor-Based Pharmacophore Modeling in the Search for Natural Products for COVID-19 Mpro. Molecules, 26(6), 1549. https://doi.org/10.3390/molecules26061549