
Solar Photooxygenations for the Manufacturing of Fine Chemicals—Technologies and Applications



Abstract
:
1. Introduction
2. Solar Reactors for Synthetic Applications
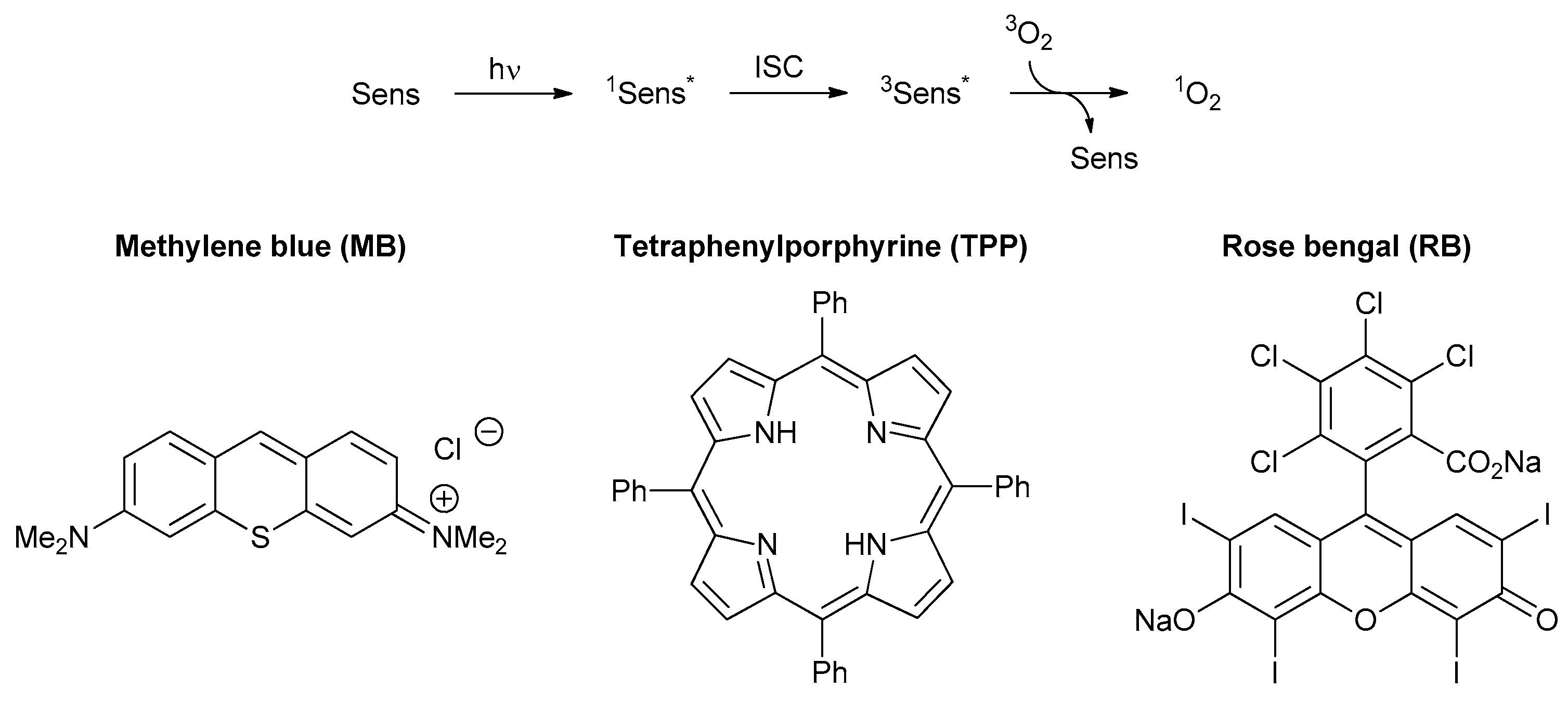
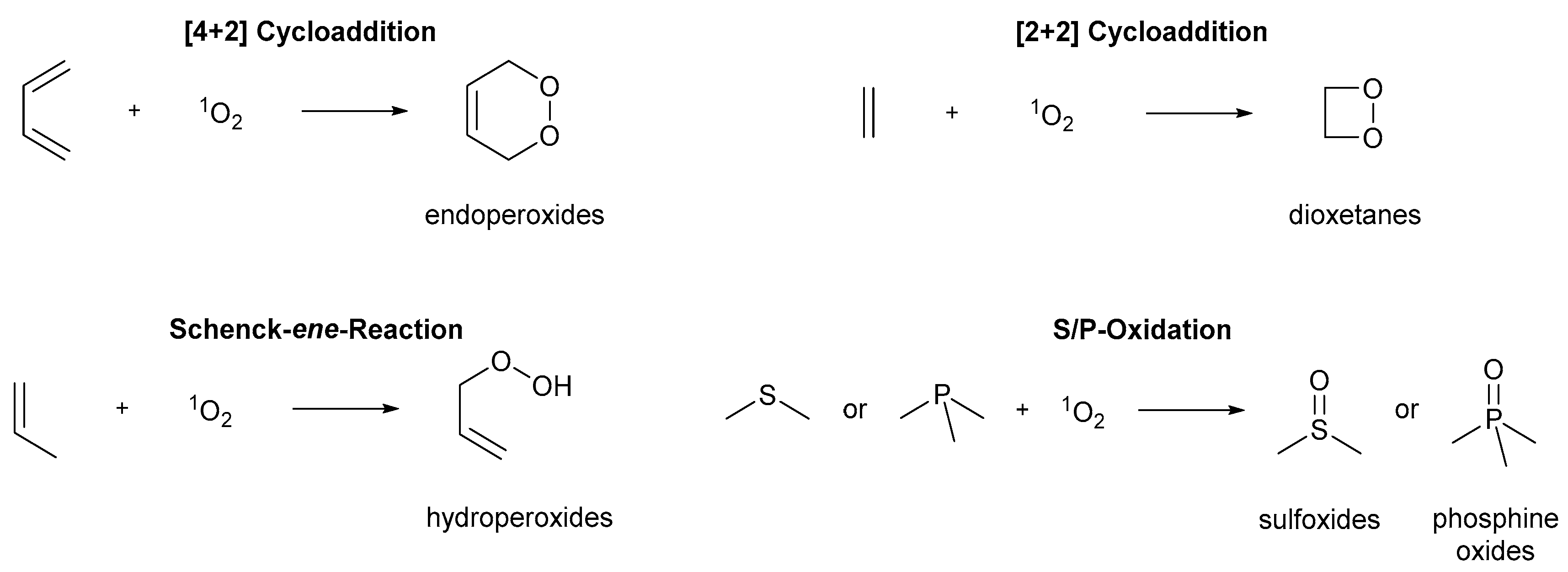
3. Photooxygenations in Organic Synthesis
4. Solar Preparative Photooxygenations
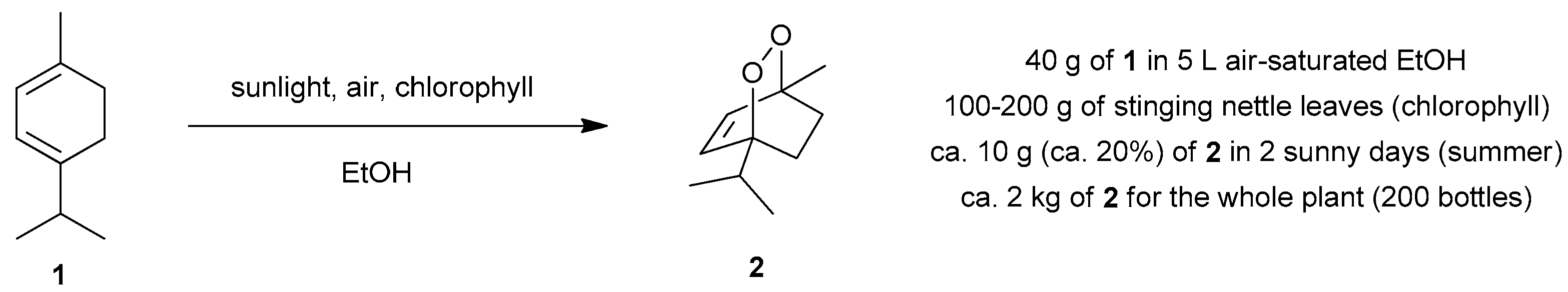
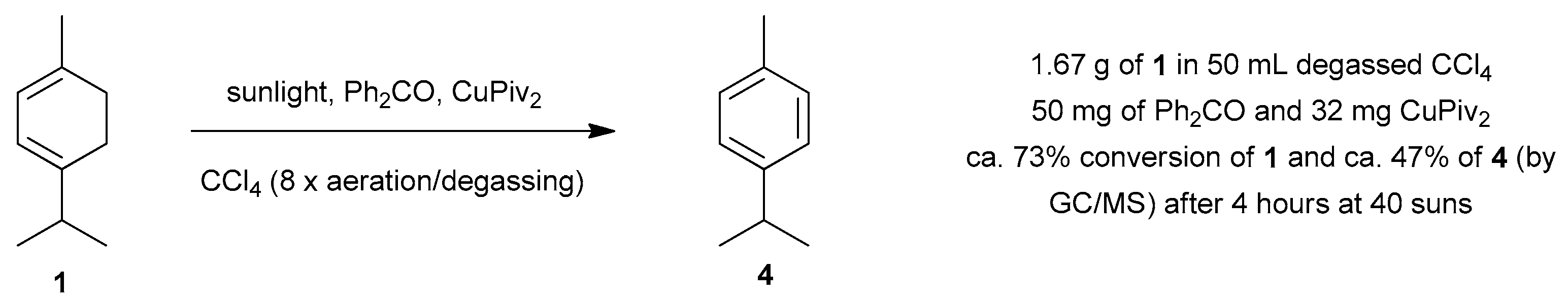
4.1. Photooxygenation of α-Terpinene and Related Reactions
4.2. Photooxygenation of Citronellol for the Production of Rose Oxide
4.3. Photooxygenations of β-Pinene for the Production of Myrtenol
4.4. Photooxygenations of α-Thujene for the Production of Trans-Sabinene Hydrate
4.5. Photooxygenations of Furfural and Furfuraldiethylacetal to 5-Hydroxy- and 5-Alkoxyfuranones
4.6. Photooxygenations of 1,5-Dihydroxynaphthalene to Juglone
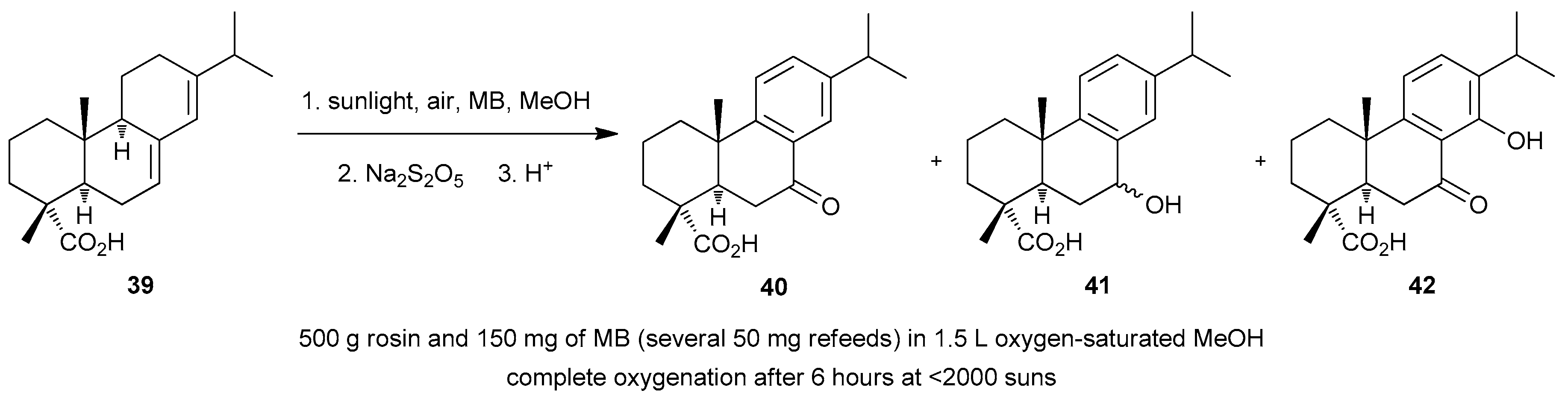
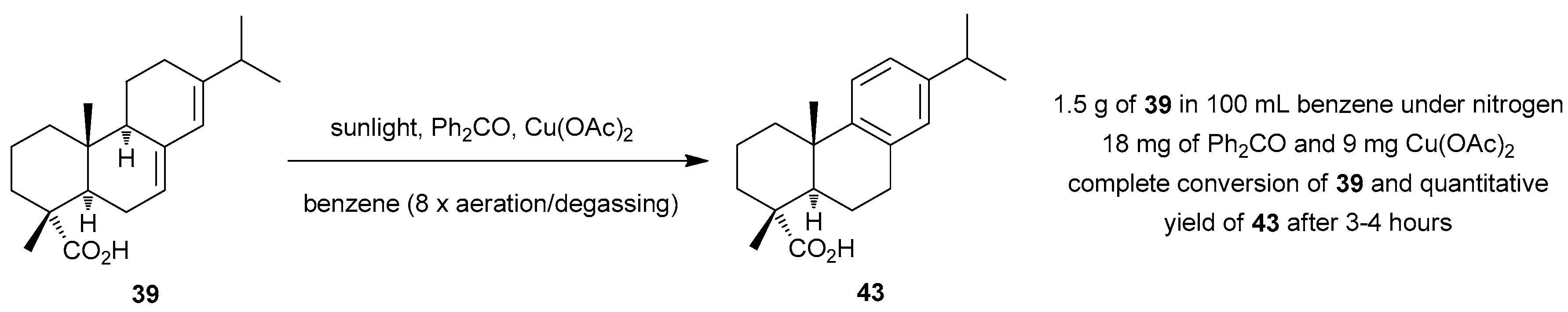
4.7. Miscellaneous Solar Photooxygenations and Photooxidations
5. Limitations, Challenges and Opportunities
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonfield, H.E.; Knauber, T.; Lévesque, F.; Moschetta, E.G.; Susanne, F.; Edwards, L.J. Photons as a 21st century reagent. Nat. Commun. 2020, 11, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, A.G. The Future of Photochemistry: Just Bright. ChemPhotoChem 2018, 3, 8–9. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, M. Making APIs and fine chemicals with light. Chim. Oggi—Chem. Today 2017, 35, 84–85. [Google Scholar]
- Bach, T. More Chemistry with light! More light in chemistry! Angew. Chem. Int. Ed. 2015, 54, 11294–11295. [Google Scholar] [CrossRef] [Green Version]
- Balzani, V.; Bergamini, G.; Ceroni, P. Photochemistry and photocatalysis. Rend. Lincei 2017, 28, 125–142. [Google Scholar] [CrossRef]
- Kärkäs, M.D.; Porco, J.A., Jr.; Stephenson, C.R.J. Photochemical approaches to complex chemotypes: Applications in natural product synthesis. Chem. Rev. 2016, 116, 9683–9747. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Hoffmann, N. Studies in organic and physical photochemistry—An interdisciplinary approach. Org. Biomol. Chem. 2016, 14, 7392–7442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, N. Photochemical Reactions as Key Steps in Organic Synthesis. Chem. Rev. 2008, 108, 1052–1103. [Google Scholar] [CrossRef]
- Rehm, T.H. Reactor Technology Concepts for Flow Photochemistry. ChemPhotoChem 2019, 4, 235–254. [Google Scholar] [CrossRef]
- Di Filippo, M.; Bracken, C.; Baumann, M. Continuous Flow Photochemistry for the Preparation of Bioactive Molecules. Molecules 2020, 25, 356. [Google Scholar] [CrossRef] [Green Version]
- Politano, F.; Oksdath-Mansilla, G. Light on the Horizon: Current Research and Future Perspectives in Flow Photochemistry. Org. Process. Res. Dev. 2018, 22, 1045–1062. [Google Scholar] [CrossRef] [Green Version]
- Oelgemöller, M.; Hoffmann, N.; Shvydkiv, O.; Oelgemoeller, M. From ‘Lab & Light on a Chip’ to Parallel Microflow Photochemistry. Aust. J. Chem. 2014, 67, 337–342. [Google Scholar] [CrossRef]
- Sender, M.; Ziegenbalg, D. Light Sources for Photochemical Processes—Estimation of Technological Potentials. Chem. Ing. Tech. 2017, 89, 1159–1173. [Google Scholar] [CrossRef]
- Braun, A.M.; Oller do Nascimento, C.A. Angewandte (präparative) Photochemie. Nachr. Chem. Tech. Lab. 1991, 39, 515–526. [Google Scholar]
- Braun, A.; Peschl, G.H.; Oliveros, E. Industrial photochemistry. In CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; Griesbeck, A., Oelgemöller, M., Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; Volume 1, Chapter 1; pp. 1–19. [Google Scholar]
- Michelin, C.; Lefebvre, C.; Hoffmann, N. Les réactions photochimiques à l’échelle industrielle. L’Act. Chim. 2019, 436, 19–27. [Google Scholar]
- Pfoertner, K.-H.; Oppenländer, T. Photochemistry. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2012; pp. 1–45. [Google Scholar]
- Braun, A.M.; Jakob, L.; Oliveros, E.; Nascimento, C.A.O.D. Up-Scaling Photochemical Reactions. Adv. Photochem. 2007, 18, 235–313. [Google Scholar] [CrossRef]
- Fischer, M. Industrial Applications of Photochemical Syntheses. Angew. Chem. Int. Ed. 1978, 17, 16–26. [Google Scholar] [CrossRef]
- Mattay, J. Von der Laborsynthese zur Solarchemie. Chem. unsere Zeit 2002, 36, 98–106. [Google Scholar] [CrossRef]
- Wilkins, F.W.; Blake, D.M. Use solar energy to drive chemical processes. Chem. Eng. Prog. 1994, 90, 41–49. [Google Scholar]
- Funken, K.-H. Solare Photoreaktionen für die chemische Technik. Nachr. Chem. Tech. Lab. 1992, 40, 793–800. [Google Scholar] [CrossRef]
- Mathur, V.; Wong, E. Production of fuels and chemicals using solar photothermochemistry. Energy 1987, 12, 311–318. [Google Scholar] [CrossRef]
- Albini, A.; Dichiarante, V. The ‘belle ’epoque’ of photochemistry. Photochem. Photobiol. Sci. 2009, 8, 248–254. [Google Scholar] [CrossRef]
- Roth, H.D. Twentieth century developments in photochemistry. Brief historical sketches. Pure Appl. Chem. 2001, 73, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Roth, H.D. The Beginnings of Organic Photochemistry. Angew. Chem. Int. Ed. 1989, 28, 1193–1207. [Google Scholar] [CrossRef]
- Ciamician, G. The photochemistry of the future. Science 1912, 36, 385–394. [Google Scholar] [CrossRef] [Green Version]
- Protti, S.; Fagnoni, M. The sunny side of chemistry: Green synthesis by solar light. Photochem. Photobiol. Sci. 2009, 8, 1499–1516. [Google Scholar] [CrossRef]
- Ravelli, D.; Protti, S.; Fagnoni, M. Application of Visible and Solar Light in Organic Synthesis. Lect. Notes Chem. 2016, 92, 281–342. [Google Scholar] [CrossRef]
- Fuqiang, W.; Ziming, C.; Jianyu, T.; Yuan, Y.; Yong, S.; Linhua, L. Progress in concentrated solar power technology with parabolic trough collector system: A comprehensive review. Renew. Sustain. Energy Rev. 2017, 79, 1314–1328. [Google Scholar] [CrossRef]
- Weinstein, L.A.; Loomis, J.; Bhatia, B.S.; Bierman, D.M.; Wang, E.N.; Chen, G. Concentrating Solar Power. Chem. Rev. 2015, 115, 12797–12838. [Google Scholar] [CrossRef]
- Fernández-García, A.; Zarza, E.; Valenzuela, L.; Pérez, M. Parabolic-trough solar collectors and their applications. Renew. Sustain. Energy Rev. 2010, 14, 1695–1721. [Google Scholar] [CrossRef]
- Roeb, M.; Neises, M.; Monnerie, N.; Sattler, C.; Pitz-Paal, R. Technologies and trends in solar power and fuels. Energy Environ. Sci. 2011, 4, 2503–2511. [Google Scholar] [CrossRef]
- Funken, K.-H.; Ortner, J. Technologies for the Solar Photochemical and Photocatalytic Manufacture of Specialities and Commodities: A Review. Z. Phys. Chem. 1999, 213, 99–105. [Google Scholar] [CrossRef]
- Fendrich, M.A.; Quaranta, A.; Orlandi, M.; Bettonte, M.; Miotello, A. Solar Concentration for Wastewaters Remediation: A Review of Materials and Technologies. Appl. Sci. 2018, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Malato, S.; Maldonado, M.I.; Fernández-Ibáñez, P.; Oller, I.; Polo, I.; Sánchez-Moreno, R. Decontamination and disinfection of water by solar photocatalysis: The pilot plants of the Plataforma Solar de Almeria. Mater. Sci. Semicond. Process. 2016, 42, 15–23. [Google Scholar] [CrossRef]
- Tanveer, M.; Guyer, G.T. Solar assisted photo degradation of wastewater by compound parabolic collectors: Review of design and operational parameters. Renew. Sustain. Energy Rev. 2013, 24, 534–543. [Google Scholar] [CrossRef]
- Bahnemann, D. Photocatalytic water treatment: Solar energy applications. Sol. Energy 2004, 77, 445–459. [Google Scholar] [CrossRef]
- Oelgemöller, M. Solar Photochemical Synthesis: From the Beginnings of Organic Photochemistry to the Solar Manufacturing of Commodity Chemicals. Chem. Rev. 2016, 116, 9664–9682. [Google Scholar] [CrossRef]
- Spasiano, D.; Marotta, R.; Malato, S.; Fernandez-Ibañez, P. Solar photocatalysis: Materials, reactors, some commercial, and pre-industrialized applications. A comprehensive approach. Appl. Cat. B Environ. 2015, 170, 90–123. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Jung, C.; Mattay, J.; Oelgemoeller, M. Green photochemistry: Production of fine chemicals with sunlight. Pure Appl. Chem. 2007, 79, 1939–1947. [Google Scholar] [CrossRef]
- Esser, P.; Pohlmann, B.; Scharf, H.-D. The Photochemical Synthesis of Fine Chemicals with Sunlight. Angew. Chem. Int. Ed. 1994, 33, 2009–2023. [Google Scholar] [CrossRef]
- Mumtaz, S.; Sattler, C.; Oelgemöller, M. Solar photochemical manufacturing of fine chemicals—historical background, modern solar technologies, recent applications and future challenges. In Chemical Processes for a Sustainable Future; Letcher, T.M., Scott, J.L., Patterson, D., Eds.; Royal Society of Chemistry: Cambridge, UK, 2015; Chapter 7; pp. 158–191. [Google Scholar]
- Coyle, E.E.; Oelgemöller, M. Solar photochemistry from the beginnings of organic photochemistry to the solar production of chem-icals. In CRC Handbook of Organic Photochemistry and Photobiology, 3rd ed.; Griesbeck, A., Oelgemöller, M., Ghetti, F., Eds.; CRC Press: Boca Raton, FL, USA, 2012; Volume 1, Chapter 10; pp. 237–247. [Google Scholar]
- Oelgemöller, M.; Jung, C.; Ortner, J.; Mattay, J.; Schiel, C.; Zimmermann, E. Green photochemistry with moderately concentrated sunlight. Spectrum 2005, 18, 28–33. [Google Scholar]
- Oelgemöller, M.; Mattay, J.; Jung, C.; Schiel, C.; Ortner, C.; Zimmermann, E. Back to the roofs—the solarchemical production of fine chemicals with sunlight. In Proceedings of the International Solar Energy Conference, ISEC 2004 Conference, Portland, OR, USA, 11–14 July 2004; American Society of Mechanical Engineers (ASME): New York, NY, USA, 2004; pp. 523–531. [Google Scholar]
- Pohlmann, B.; Scharf, H.-D.; Jarolimek, U.; Mauermann, P. Photochemical production of fine chemicals with concentrated sunlight. Sol. Energy 1997, 61, 159–168. [Google Scholar] [CrossRef]
- Fu, Q. Radiation (solar). In Encyclopedia of Atmospheric Sciences; Holton, J., Ed.; Academic Press: Amsterdam, The Netherlands, 2003; pp. 1859–1863. [Google Scholar]
- Iqbal, M. An Introduction to Solar Radiation; Academic Press: New York, NY, USA, 1983. [Google Scholar]
- Pibiri, I.B.; Piccionello, S.; Palumbo, A.; Pace, A. Photochemically produced singlet oxygen: Applications and perspectives. ChemPhotoChem 2018, 2, 535–547. [Google Scholar] [CrossRef]
- Ghogare, A.A.; Greer, A. Using Singlet Oxygen to Synthesize Natural Products and Drugs. Chem. Rev. 2016, 116, 9994–10034. [Google Scholar] [CrossRef]
- DeRosa, M.C.; Crutchley, R.J. Photosensitized singlet oxygen and its applications. Coord. Chem. Rev. 2002, 233–234, 351–371. [Google Scholar] [CrossRef]
- Turconi, J.; Griolet, F.; Guevel, R.; Oddon, G.; Villa, R.; Geatti, A.; Hvala, M.; Rossen, K.; Göller, R.; Burgard, A. Semisynthetic Artemisinin, the Chemical Path to Industrial Production. Org. Process. Res. Dev. 2014, 18, 417–422. [Google Scholar] [CrossRef]
- Gollnick, K. Photooxygenation and its application in industry. Chim. Ind. 1982, 64, 156–166. [Google Scholar] [CrossRef]
- Rojahn, W.; Warnecke, H.-U. Die Photosensibilisierte Sauerstoffübertragung—eine Methode zur Herstellung hochwertiger Riech-stoffe. Dragoc. Rep. 1980, 27, 159–164. [Google Scholar]
- Demuth, M. Chemie mit Sonnenlicht—auch bei Bewölkung. In Innovationspreis Ruhrgebiet; Kommunalverband Ruhrgebiet: Essen, Germany, 2000; pp. 91–94. [Google Scholar]
- Blanco, J.; Malato, S.; Fernández, P.; Vidal, A.; Morales, A.; Trincado, P.; Oliveira, J.; Minero, C.; Musci, M.; Casalle, C.; et al. Compound parabolic concentrator technology development to commercial solar detoxification applications. Sol. Energy 1999, 67, 317–330. [Google Scholar] [CrossRef]
- Ajona, J.; Vidal, A. The use of CPC collectors for detoxification of contaminated water: Design, construction and preliminary results. Sol. Energy 2000, 68, 109–120. [Google Scholar] [CrossRef]
- Jung, C.; Funken, K.-H.; Ortner, J. Prophis: Parabolic trough-facility for organic photochemical syntheses in sunlight. Photochem. Photobiol. Sci. 2005, 4, 409–411. [Google Scholar] [CrossRef]
- Funken, K.-H. Solar chemistry using concentrating solar facilities. In Proceedings of the Sixth International Summer School Solar Energy 2000, Klagenfurt, Austria, 24 July–4 August 2000; Faninger, G., Bucher, W., Wolfer, U., Eds.; Interuniversitäres Institut für interdisziplinäre Forschung und Fortbildung (IFF), Universität Klagenfurt: Klagenfurt, Austria, 2001; pp. 118–137. [Google Scholar]
- Neumann, A.; Groer, U. Experimenting with concentrated sunlight using the DLR solar furnace. Sol. Energy 1996, 58, 181–190. [Google Scholar] [CrossRef]
- Fields, C.L.; Pitts, J.R.; Hale, M.J.; Bingham, C.; Lewandowski, A.; King, D.E. Formation of fullerenes in highly concentrated solar flux. J. Phys. Chem. 1993, 97, 8701–8702. [Google Scholar] [CrossRef]
- Flamant, G.; Luxembourg, D.; Robert, J.; Laplaze, D. Optimizing fullerene synthesis in a 50 kW solar reactor. Sol. Energy 2004, 77, 73–80. [Google Scholar] [CrossRef]
- Foote, C.S. Definition of type I and type II photosensitized oxidation. Photochem. Photobiol. 1991, 54, 659. [Google Scholar] [CrossRef]
- Schmidt, R. Photosensitized Generation of Singlet Oxygen. Photochem. Photobiol. 2007, 82, 1161–1177. [Google Scholar] [CrossRef]
- Osterberg, P.M.; Niemeier, J.K.; Welch, C.J.; Hawkins, J.M.; Martinelli, J.R.; Johnson, T.E.; Root, T.W.; Stahl, S.S. Experimental lim-iting oxygen concentrations for nine organic solvents at temperatures and pressures relevant to aerobic oxidations in the pharma-ceutical industry. Org. Process. Res. Dev. 2015, 19, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2006; pp. 542–548. [Google Scholar]
- Clarke, C.J.; Tu, W.-C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef]
- Fresnadillo, D.G.; Lacombe, S. Reference photosensitizers for the production of singlet oxygen. In Singlet Oxygen: Applications in Biosciences and Nanosciences; Nonell, S., Flors, C., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; Volume 1, Chapter 6; pp. 105–143. [Google Scholar]
- Marin, M.L.; Santos-Juanes, L.; Arques, A.; Amat, A.M.; Miranda, M.A. Organic photocatalysts for the oxidation of pollutants and model compounds. Chem. Rev. 2012, 112, 1710–1750. [Google Scholar] [CrossRef]
- Lacombe, S.; Pigot, T. Materials for selective photo-oxygenation vs. photocatalysis: Preparation, properties and applications in en-vironmental and health fields. Catal. Sci. Technol. 2016, 6, 1571–1592. [Google Scholar] [CrossRef]
- Wahlen, J.; De Vos, D.E.; Jacobs, P.A.; Alsters, P.L. Solid Materials as Sources for Synthetically Useful Singlet Oxygen. Adv. Synth. Catal. 2004, 346, 152–164. [Google Scholar] [CrossRef]
- Malakar, P.; Deb, A.R.; Goodine, T.; Robertson, M.J.; Oelgemöller, M. Continuous-flow photooxygenations: an advantageous and sustainable oxidation methodology with a bright future. In Catalytic Aerobic Oxidations—Catalysis Series; Mejía, P., Ed.; Royal Society of Chemistry: Cambridge, UK, 2020; Chapter 7; pp. 181–251. [Google Scholar] [CrossRef]
- Nardello-Rataj, V.; Alsters, P.L.; Aubry, J.-M. Industrial Prospects for the Chemical and Photochemical Singlet Oxygenation of Organic Compounds. Liq. Phase Aerob. Oxid. Catal. Ind. Appl. Acad. Persp. 2016, 22, 369–395. [Google Scholar] [CrossRef]
- Alberti, M.N.; Orfanopoulos, M. Unravelling the mechanism of the singlet oxygen ene reaction: Recent computational and experi-mental approaches. Chem. Eur. J. 2010, 16, 9414–9421. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.; Pérez-Ruiz, R.; Von Wangelin, A.J. Stereoselective Photooxidations by the Schenck Ene Reaction. ChemPhotoChem 2018, 2, 559–570. [Google Scholar] [CrossRef]
- Khayyat, S.A.; Roselin, L.S. Recent progress in photochemical reaction on main components of some essential oils. J. Saudi Chem. Soc. 2018, 22, 855–875. [Google Scholar] [CrossRef]
- Goodine, T.; Oelgemöller, M. Corymbia citriodora: A Valuable Resource from Australian Flora for the Production of Fragrances, Repellents, and Bioactive Compounds. ChemBioEng Rev. 2020, 7, 170–192. [Google Scholar] [CrossRef]
- Schenck, O.; Ziegler, K. Die Synthese des Ascaridols. Naturwissenschaften 1944, 32, 157. [Google Scholar] [CrossRef]
- Schenck, G.O. Autoxydation von Furan und anderen Dienen (Die Synthese des Ascaridols). Ang. Chem. 1944, 57, 101–102. [Google Scholar] [CrossRef]
- Schenck, G.; Schulze-Buschoff, H. Synthetisches Askaridol, eine neue Möglichkeit der Spulwurmbehandlung. DMW—Dtsch. Med. Wochenschr. 1948, 73, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Karapire, C.; Kolancilar, H.; Oyman, Ü.; Icli, S. Fluorescence emission and photooxidation studies with 5,6- and 6,7-benzocoumarins and a 5,6-benzochromone under direct and concentrated sun light. J. Photochem. Photobiol. Chem. 2002, 153, 173–184. [Google Scholar] [CrossRef]
- Dindar, B.; Içli, S. Unusual photoreactivity of zinc oxide irradiated by concentrated sunlight. J. Photochem. Photobiol. Chem. 2001, 140, 263–268. [Google Scholar] [CrossRef]
- Karapire, C.; Kuś, M.; Turkmen, G.; Trevithick-Sutton, C.; Foote, C.; Icli, S. Photooxidation studies with perylenediimides in solution, PVC and sol–gel thin films under concentrated sun light. Sol. Energy 2005, 78, 5–17. [Google Scholar] [CrossRef]
- Avcibasi, N.; Icli, S.; Gilbert, A. Photochemical reactions of α-terpinene and acenaphthene under concentrated sunlight. Turk. J. Chem. 2003, 27, 1–7. [Google Scholar]
- Ohloff, G.; Klein, E.; Schenck, G.O. Darstellung von “Rosenoxyden” und anderen Hydropyran-Derivaten über Photohydroper-oxyde. Angew. Chem. 1961, 73, 578. [Google Scholar] [CrossRef]
- Bicas, J.L.; Dionísio, A.P.; Pastore, G.M. Bio-oxidation of Terpenes: An Approach for the Flavor Industry. Chem. Rev. 2009, 109, 4518–4531. [Google Scholar] [CrossRef]
- Pickenhagen, W.; Schatkowski, D. Verfahren zur Herstellung von Rosenoxid. DE19645922A1, 14 February 2002. [Google Scholar]
- Demuth, M.; Ritter, A. Photochemical and Thermochemical Solar Syntheses Using Flat-Bed Solar Collectors/Solar Reactors. WO99/54042A1, 28 October 1999. [Google Scholar]
- Ritterskamp, P.; Hülsdünker, A.; Heimann, F.; Goeller, F.; Uzun, D.; Ritter, A.; Demuth, M.; Kleinwächter, J.; Ortner, J.; Funken, K.-H.; et al. Synthesis of biologically active compounds with sunlight and technological developments. In Solare Chemie und Solare Materialforschung; AG-Solar NRW: Jülich, Germany, 2002; Chapter 2.2.1; pp. 1–6. ISBN 3893363068. [Google Scholar]
- Oelgemöller, M.; Jung, C.; Ortner, J.; Mattay, J.; Zimmermann, E.; Oelgemoeller, M. Green photochemistry: Solar photooxygenations with medium concentrated sunlight. Green Chem. 2004, 7, 35–38. [Google Scholar] [CrossRef]
- Ortner, J.; Faust, D.; Funken, K.-H.; Lindner, T.; Schulat, J.; Stojanoff, C.G.; Fröning, P. New developments using holographic concentration in solar photochemical reactors. J. Phys. Colloq. 1999, 9, Pr3-379–Pr3-383. [Google Scholar] [CrossRef]
- Stojanoff, C.G.; Schulat, J.; Fröning, P. Development and fabrication of holographic concentrators for solar chemistry. In Solare Chemie und Solare Materialforschung; AG-Solar NRW: Jülich, Germany, 2002; Chapter 6.4; pp. 1–10. ISBN 3893363068. [Google Scholar]
- Funken, K.-H.; Pohlmann, B.; Ortner, J.; Faust, D. Verfahren zur Oxidation ungesättigter Kohlenwasserstoffe. DE19923071A1, 21 August 2003. [Google Scholar]
- Monniere, N.; Ortner, J. Economic evaluation of the industrial photosynthesis of rose oxide via lamp or solar operated photooxidation of citronellol. J. Sol. Energy Eng. 2001, 123, 171–174. [Google Scholar] [CrossRef]
- Dincalp, H.; Icli, S. Photosynthesis of rose oxide by concentrated sunlight in the absence of singlet oxygen. J. Photochem. Photobiol. Chem. 2001, 141, 147–151. [Google Scholar] [CrossRef]
- Schenck, G.O.; Eggert, H.; Denk, W. Über die Bildung von Hydroperoxyden bei photosensibilisierten Reaktionen von O2 mit ge-eigneten Akzeptoren, insbesondere mit α- und β-Pinen. Liebigs Ann. Chem. 1953, 584, 177–198. [Google Scholar] [CrossRef]
- Bhatia, S.; McGinty, D.; Letizia, C.; Api, A. Fragrance material review on myrtenol. Food Chem. Toxicol. 2008, 46, S237–S240. [Google Scholar] [CrossRef] [PubMed]
- Mihelich, E.D.; Eickhoff, D.J. A one-pot conversion of olefins to. alpha.,. beta.-unsaturated carbonyl compounds. An easy synthesis of 2-cyclopentenone and related compounds. J. Org. Chem. 1983, 48, 4135–4137. [Google Scholar] [CrossRef]
- Jung, C.; Ortner, J.; Jarolimek, U.; Mauermann, P.; Leyen, B. Advances in photochemical production of fine chemicals with con-centrated sunlight. In Solare Chemie und Solare Materialforschung; AG-Solar NRW: Jülich, Germany, 2002; Chapter 2.2.1; pp. 1–10. ISBN 3893363068. [Google Scholar]
- Neckers, D.C. Rose Bengal. J. Photochem. Photobiol. Chem. 1989, 47, 1–29. [Google Scholar] [CrossRef]
- Esser, P. Die Anwendung von Sonnenlicht zur Photochemischen Producktion von Feinchemikalien. Ph.D. Thesis, Rheinisch-Westfälische Technische Hochschule, Aachen, Germany, 1994. [Google Scholar]
- Funken, K.-H.; Schneider, G.; Esser, E.P.; Scharf, H.-D.; Esser, P.; Wöhrle, I. The SOLARIS-experiment: Demonstration of solar-photochemical syntheses of fine chemicals. In Proceedings of the 6th International Symposium on Solar Thermal Concentrating Technologies, Mojacar, Spain, 28 September–2 October 1992; Centro de Investigaciones Energétical, Medioambientales y Tecnológias (CIEMAT): Madrid, Spain, 1992; Volume 2, pp. 1027–1037. [Google Scholar]
- Klein, E.; Rojahn, W. Die photosensibilisierte O2-Übertragung auf (+)-α-Thujen. Chem. Ber. 1965, 98, 3045–3049. [Google Scholar] [CrossRef]
- Baeckström, P.; Koutek, B.; Šaman, D.; Vrkoč, J. A convenient synthesis of trans-sabinene hydrate from (−)-3-thujol via a highly selective ene reaction of singlet oxygen. Bioorg. Med. Chem. 1996, 4, 419–421. [Google Scholar] [CrossRef]
- Scharf, H.-D.; Esser, P.; Kuhn, W.; Pelzer, R. Process for the Photooxidation of Terpene Olefins. US5620569A, 15 April 1997. [Google Scholar]
- Schenck, G.O. Über die unsensibilisierte und photosensibilisierte Autoxydation von Furanen. Liebigs Ann. Chem. 1953, 584, 156–176. [Google Scholar] [CrossRef]
- Schenck, G.O. Verfahren zur Herstellung von β-Acyl-acrylsäuren bzw. ihren Pseudoestern. DE875650C, 4 May 1953. [Google Scholar]
- Bolz, G.; Wiersdorff, W.-W. Verfahren zur Herstellung von 2-Hydroxy-2,5-dihydrofuranon-(5). DE2111119A1, 14 September 1972. [Google Scholar]
- Hoydonckx, H.E.; Van Rhijn, W.M.; De Vos, D.E.; A Jacobs, P. Furfural and Derivatives. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2007; Volume 16, pp. 285–313. [Google Scholar]
- Esser, P.; Pelzer, R.; Völkl, F. Verwendung substituierter Lactone als Riechstoffe. EP0761808A2, 19 May 1999. [Google Scholar]
- Strugstad, M.P.; Despotovski, S. A summary of extraction, synthesis, properties, and potential uses of juglone: A literature review. J. Ecosyst. Manag. 2012, 13, 1–16. [Google Scholar]
- Griffiths, J.; Chu, K.-Y.; Hawkins, C. Photosensitised oxidation of 1-naphthols. J. Chem. Soc. Chem. Commun. 1976, 676–677. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Healy, N.; De Oliveira, L.; Jung, C.; Mattay, J. Green photochemistry: Solar-chemical synthesis of Juglone with medium concentrated sunlight. Green Chem. 2006, 8, 831–834. [Google Scholar] [CrossRef]
- Suchard, O.; Kane, R.; Roe, B.J.; Zimmermann, E.; Jung, C.; Waske, P.A.; Mattay, J.; Oelgemöller, M. Photooxygenations of 1-naphthols: An environmentally friendly access to 1,4-naphthoquinones. Tetrahedron 2006, 62, 1467–1473. [Google Scholar] [CrossRef]
- Dincalp, H.; Icli, S. Photoinduced electron transfer-catalyzed processes of sulfoamine perylene diimide under concentrated sun light. Sol. Energy 2006, 80, 332–346. [Google Scholar] [CrossRef]
- Icli, S. Production of oxygenated rosin emulsifier by a solar photoorganic chemical method. For. Chem. Rev. 1999, 102, 7–9. [Google Scholar]
- Icli, S.; Bulut, A.; Gül, Y. Room temperature generation of dehydrogenated colophony: Solar chemical production. Turk. J. Chem. 1992, 16, 289–292. [Google Scholar]
- Erten, S.; Alp, S.; Icli, S. Photooxidation quantum yield efficiencies of naphthalene diimides under concentrated sun light in com-parisons with perylene diimides. J. Photochem. Photobiol. Chem. 2005, 175, 214–220. [Google Scholar] [CrossRef]
- Funken, K.-H.; Ortner, J.; Riffelmann, K.-J.; Sattler, C. New developments in solar photochemistry. J. Inf. Rec. 1998, 24, 61–68. [Google Scholar]
- Tirronen, E.; Salmi, T. Process development in the fine chemical industry. Chem. Eng. J. 2003, 91, 103–114. [Google Scholar] [CrossRef]
- Bruggink, A. Growth and efficiency in the (fine) chemical industry. Chim. Oggi 1998, 16, 44–47. [Google Scholar]
- Pollak, P.; Vouillamoz, R. Fine Chemicals. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Hoboken, NJ, USA, 2000; pp. 1–17. [Google Scholar]
- Landgraf, S. Application of semiconductor light sources for investigations of photochemical reactions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2001, 57, 2029–2048. [Google Scholar] [CrossRef]
- Kalyani, N.T.; Dhoble, S. Organic light emitting diodes: Energy saving lighting technology—A review. Renew. Sustain. Energy Rev. 2012, 16, 2696–2723. [Google Scholar] [CrossRef]
- Monteiro, R.A.R.; Rodrigues-Silva, C.; Lopes, F.V.S.; Silva, A.M.T.; Boaventura, R.A.R.; Vilar, V.J.P. Evaluation of a solar/UV an-nular pilot scale reactor for 24 h continuous photocatalytic oxidation of n-decane. Chem. Eng. J. 2015, 280, 409–416. [Google Scholar] [CrossRef]
- Navntoft, C.; Araujo, P.; Litter, M.I.; Apella, M.C.; Fernández, D.; Puchulu, M.E.; Hidalgo, M.D.V.; Blesa, M.A. Field Tests of the Solar Water Detoxification SOLWATER Reactor in Los Pereyra, Tucumán, Argentina. J. Sol. Energy Eng. 2006, 129, 127–134. [Google Scholar] [CrossRef]
- Bolte, M.; Klaeden, K.; Beqiraj, A.; Oelgemöller, M. Photochemistry Down under—Solar chemicals from and for the Tropics. Eur. Photochem. Assoc. Newslett. 2013, 83–84, 79–83. [Google Scholar]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef]
- Horikoshi, S.; Nishimura, T.; Tsutsumi, H.; Serpone, N. Microwave Discharge Electrodeless Lamps. Part VIII: Continuous On-Site Solar Energy Remediation of Contaminated Water. Chem. Eng. Technol. 2015, 39, 102–107. [Google Scholar] [CrossRef]
- Lui, G.Y.; Roser, D.; Corkish, R.; Ashbolt, N.; Jagals, P.; Stuetz, R. Photovoltaic powered ultraviolet and visible light-emitting diodes for sustainable point-of-use disinfection of drinking waters. Sci. Total Environ. 2014, 493, 185–196. [Google Scholar] [CrossRef]
- Vassilikogiannakis, G. Singlet Oxygen and Dyes: Synthesis with Visible Light is Where the Future Lies. ChemPhotoChem 2020, 4, 385–387. [Google Scholar] [CrossRef]
- Montagnon, T.; Kalaitzakis, D.; Triantafyllakis, M.; Stratakis, M.; Vassilikogiannakis, G. Furans and singlet oxygen—why there is more to come from this powerful partnership. Chem. Commun. 2014, 50, 15480–15498. [Google Scholar] [CrossRef] [Green Version]
- Dinda, M.; Maiti, S.; Samanta, S.; Ghosh, P.K. Illustrations of Efficient Solar Driven Organic Reactions. Curr. Org. Synth. 2016, 13, 372–384. [Google Scholar] [CrossRef]
- Li, P.; Terrett, J.A.; Zbieg, J.R. Visible-Light Photocatalysis as an Enabling Technology for Drug Discovery: A Paradigm Shift for Chemical Reactivity. ACS Med. Chem. Lett. 2020, 11, 2120–2130. [Google Scholar] [CrossRef]
- McAtee, R.C.; McClain, E.J.; Stephenson, C.R. Illuminating Photoredox Catalysis. Trends Chem. 2019, 1, 111–125. [Google Scholar] [CrossRef]
- Douglas, J.J.; Sevrin, M.J.; Stephenson, C.R.J. Visible Light Photocatalysis: Applications and New Disconnections in the Synthesis of Pharmaceutical Agents. Org. Process. Res. Dev. 2016, 20, 1134–1147. [Google Scholar] [CrossRef]
- Nauth, A.M.; Lipp, A.; Lipp, B.; Opatz, T. Sunflow: Sunlight drives fast and green photochemical flow reactions in simple micro-capillary reactors—application to photoredox and H-atom-transfer chemistry. Eur. J. Org. Chem. 2017, 2017, 2099–2103. [Google Scholar] [CrossRef] [Green Version]
- Yaseen, M.A.; Mumtaz, S.; Hunter, R.L.; Wall, D.; Robertson, M.J.; Oelgemöller, M. Continuous-flow photochemical transformations of 1,4-naphthoquinones and phthalimides in a concentrating solar trough reactor. Aust. J. Chem. 2020, 73, 1149–1157. [Google Scholar] [CrossRef]
- Yaseen, M.A.; Mumtaz, S.; Hunter, R.L.; Wall, D.; Robertson, M.J.; Oelgemöller, M. Corrigendum to: Continuous-flow photochemical transformations of 1,4-naphthoquinones and phthalimides in a concentrating solar trough reactor. Aust. J. Chem. 2020, 73, 1301. [Google Scholar] [CrossRef]
- Papakonstantinou, I.; Portnoi, M.; Debije, M.G. The hidden potential of luminescent solar concentrators. Adv. Energy Mater. 2020, 11. [Google Scholar] [CrossRef]
- Poliakoff, M.; George, M.W. Manufacturing chemicals with light: Any role in the circular economy? Phil. Trans. R. Soc. A 2020, 378, #20190260. [Google Scholar] [CrossRef] [PubMed]
- Bochet, C.G. On the Sustainability of Photochemical Reactions. Chim. Int. J. Chem. 2019, 73, 720–723. [Google Scholar] [CrossRef] [Green Version]
- Michelin, C.; Hoffmann, N. Photocatalysis applied to organic synthesis—A green chemistry approach. Curr. Opin. Green Sustain. Chem. 2018, 10, 40–45. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Oelgemoeller, M. Green Photochemical Processes and Technologies for Research & Development, Scale-up and Chemical Production. J. Chin. Chem. Soc. 2014, 61, 743–748. [Google Scholar] [CrossRef]
- Ciana, C.-L.; Bochet, C.G. Clean and Easy Photochemistry. Chim. Int. J. Chem. 2007, 61, 650–654. [Google Scholar] [CrossRef]
- Albini, A.; Fagnoni, M.; Mella, M. Environment-friendly organic synthesis. The photochemical approach. Pure Appl. Chem. 2000, 72, 1321–1326. [Google Scholar] [CrossRef]
- Ciriminna, R.; Delisi, R.; Xu, Y.-J.; Pagliaro, M. Toward the waste-free synthesis of fine chemicals with visible light. Org. Process Res. Dev. 2016, 20, 403–408. [Google Scholar] [CrossRef]
- Oelgemöller, M. The sunny side of chemistry at James Cook Uni. Chemistry in Australia, November 2014; 7. [Google Scholar]














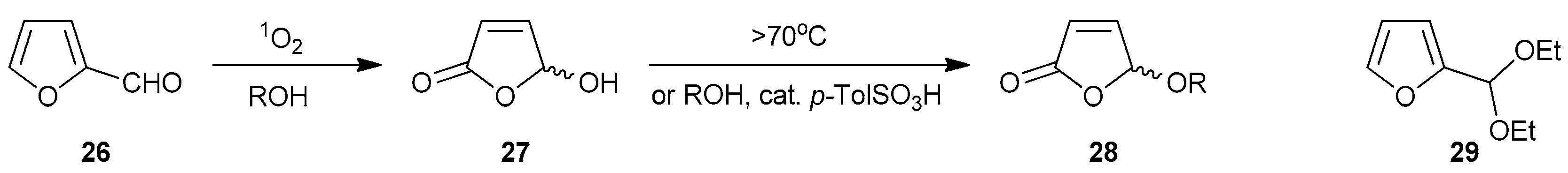


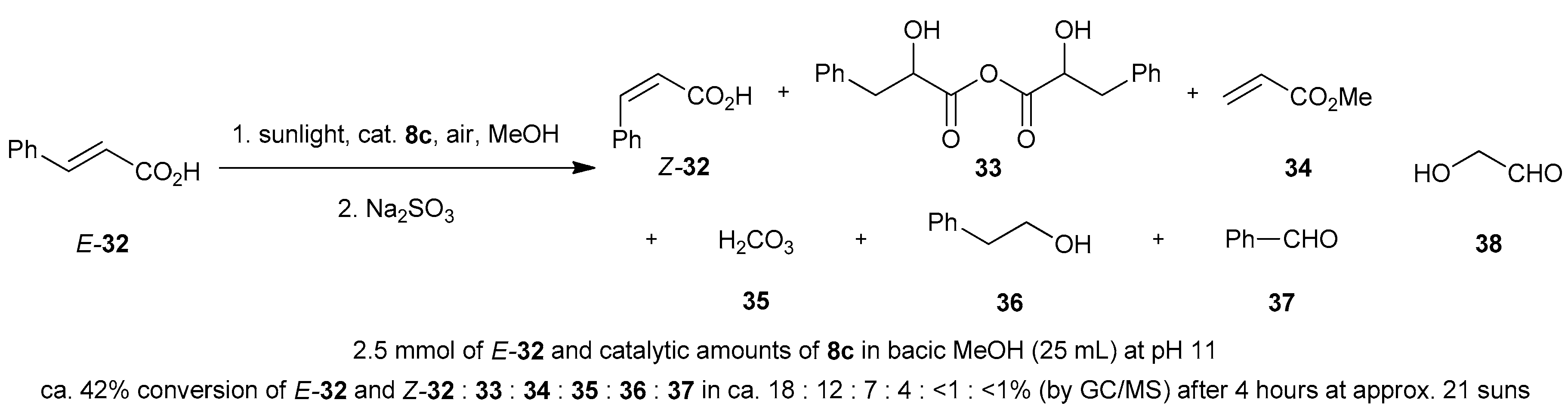



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction 1 [89] | Reaction 2 [89] | Reaction 3 [90] | Reaction 4 [90] | |
|---|---|---|---|---|
| citronellol (12) | 0.5 L (2.7 mol) | 0.2 L (1.1 mol) | 0.1 L (0.55 mol) | 0.1 L (0.55 mol) |
| methanol | 400 mL | 1 L | 2 L | 2 L |
| rose bengal 1 | 5 × 6 g | 3 g + 1 g | 5 g | 5 g |
| reactor | batch (1 m2) | in series (1 m2) | batch (1 m2) | batch (1 m2) |
| operation | static | flow (2 L/h) | static | static |
| time | 3 days | 11 h | 11 h | 7 h |
| solar conditions | 50% cloudy, 50% sunny | 70% cloudy; 30% sunny | 70% cloudy; 30% sunny | 100% sunny |
| yield of 15 2 | 50% (1.36 mol) | 52% (0.57 mol) | not reported 3 | not reported 3 |
| Reaction 5 [91] | Reaction 6 [91] | Reaction 7 [94] | Reaction 8 [94] | |
|---|---|---|---|---|
| citronellol (12) | 5.8 L (31.8 mol) | 8 L (43.9 mol) | 5.8 L (31.8 mol) | 5.8 L (31.8 mol) |
| isopropanol | 40 L | 72 L | 40 L | 40 L |
| rose bengal | 20 g 1 | 36 g 1 | 2 g | 2 g |
| O2-flow | 600 L/h | 200 L/h | not reported | not reported |
| # of troughs | 1 (8 m2) | 4 (32 m2) | 1 (8 m2) 2 | 1 (8 m2) |
| time 3 | 3 h | 2¼ h | 2⅓ h | 2¼ h |
| solar conditions | largely sunny | sunny | sunny | sunny |
| conversion of 12 4 | >95% | >95% (55% 5) | >95% | >95% |
| Reactions 9 | Reactions 10 | Reactions 11 | Reactions 12 | Reactions 13 | |
|---|---|---|---|---|---|
| reactor | PROPHIS | CPC | flatbed | horizontal tubes | vertical tubes |
| CF | 32 suns | 1 sun | 1 sun | 1 sun | 1 sun |
| aperture | 32 m2 | 3 m2 | 1.5 m2 | not given | not given |
| volume | 80 L (37 L 1) | 40 L (26 L 1) | 21 L 1 | 25 L (11 L 1) | 20 L (10 L 1) |
| operation | circulation | circulation | static | circulation | circulation |
| mixing | static mixer | 7 × 180° turns | air bubbling | 9 × 180° turns | air bubbling |
| citronellol (12) | 8 L (43.9 mol) | 4 L (22 mol) | 2.1 L (11.5 mol) | 2.5 L (13.7 mol) | 2 L (11 mol) |
| isopropanol | 72 L | 36 L | 18.9 L | 22.5 L | 18 L |
| rose bengal 2 | 40 g | 20 g | 10.5 g | 12.5 g | 10 g |
| O2 source | oxygen | air | air | air | air |
| time (sunny/cloudy) 4 | 2½ h 3/not operated | 7 h/30 h | 15 h/30 h | 12 h/33 h | 13 h/33 h |
| conversion of 12 5 | >95% 3/not operated | >95%/>95% | >95%/>95% | >95%/>65% | >95%/>65% |
| Reaction 14 | Reaction 15 | Reaction 16 | Reaction 17 | |
|---|---|---|---|---|
| sensitizer | ABIPER (8a) | BUNAP (16) | ABIPER (8a) | ABIPER (8a) |
| additive | none | none | CuPiv2 | FeMyr3 |
| exposure time 1 | 2 h | 1 h | 2 h | 2 h |
| residual 12 2 | 3% | 1% | 81% | 36% |
| 14a/14b2 | 6% | 20% | 2% | 5% |
| 15 (cis/trans 3) 2 | 76%/6% | 54%/23% | 11%/5% | 53%/6% |
| Reaction 18 | Reaction 19 [100] | Reaction 20 [102] | |
|---|---|---|---|
| reactor | CPC | DLR solar furnace | SOLARIS |
| CF | 1 sun | up to 4,800 suns | 20 suns (42 suns 1) |
| volume | 40 L (26 L 2) | 5.1 L (2.03 L 2) | 70 L (30 L 2) |
| β-pinene (17) | 2 L (12.8 mol) | 0.29 L (1.84 mol) | 6.2 L (37.7 mol) |
| solvent | 38 L (iPrOH) | 3.5 L (EtOH) | 35 L (iPrOH) |
| rose bengal | not reported | 40 g 3 | 21.77 g 3 |
| exposure time | 9 days | 14 h | 3 days (ca. 27 h) |
| conversion of 17 4 | 95% | 97% | ca. 56% |
| Reaction 21 | Reaction 22 | Reaction 23 | Reaction 24 | Reaction 25 | Reaction 26 | Reaction 27 | |
|---|---|---|---|---|---|---|---|
| α-thujene (21) 1 | 2.7 kg (17.2 mol) | 5.42 kg (34.5 mol) | 5.45 kg (34.6 mol) | ~3.3 kg (20.9 mol) | 5.63 kg (35.8 mol) | 5.64 kg (35.9 mol) | 5.63 kg (35.8 mol) |
| iPrOH | 44 L | 38 L | 35 L | ~32 L | 35 L | 35 L | 35 L |
| sensitizer 2 | 5.26 g (MB) | 9.61 g (MB) | 11.11 g (MB) | 12.29 g (MB) | 12.82 g (RB) | 9.02 g (RB) | 6.95 g (RB) |
| time 3 | ~7 h (1 day) | ~11 h (2 days) | 7 h (1 day) | ~7 h (1 day) | 6 h (1 day) | 5½ h (1 day) | 4½ h (1 day) |
| total radiation 4 | 16.5 kWh (110 mol) | 37.9 kWh (271 mol) | 40.8 kWh (287 mol) | 43.0 kWh (306 mol) | 29.9 kWh (123 mol) | 27.5 kWh (113 mol) | 24.2 kWh (106 mol) |
| ηs 5 | 14% | 12% | 12% | 7% | 29% | 31% | 33% |
| conversion 6 | 88% | 97% | 96% | 97% | ~100% | 99% | 98% |
| 217 | 12% | 2% | 3% | 2% | <1% | 1% | 1% |
| 23a7 | 61% | 56% | 63% | 66% | 65% | 67% | 61% |
| 23b7 | 7% | 11% | 11% | 10% | 10% | 11% | 13% |
| selectivity 8 | 82% | 67% | 75% | 78% | 75% | 78% | 72% |
| Reaction 28 | Reaction 29 | Reaction 30 | Reaction 31 | Reaction 32 | |
|---|---|---|---|---|---|
| furane | 2.06 kg 21.5 mol (26) | 2.14 kg 22.3 mol (26) | 4.32 kg 45.0 mol (26) | 3.61 kg 37.6 mol (26) | 3.61 kg 21.2 mol (29) |
| ethanol | 35 L | 35 | 30 | 35 | 35 |
| sensitizer 1 | 6.1 g (MB) | 11.2 g (RB) | 5.1 g (MB) | 9.0 g (MB) | 6.0 g (MB) |
| additive | none | none | none | c. HCl (5 mL) | none |
| temperature | 8 °C | 20 °C | 20 °C | 20 °C | 20 °C |
| fluid flow | 35 L/min | 55 L/min | 55 L/min | 45 L/min | 45 L/min |
| time 2 | 16 h (3 days) | ~11 h (2 days) | ~12½ h (2 days) | 5½ h (1 day) | ~3¼ h (1 day) |
| total radiation 3 | 22.2 kWh 140 mol | 54.3 kWh 413 mol | 61.9 kWh 436 mol | 32.4 kWh 226 mol | 16.9 kWh 114 mol |
| ηs 4 | 15% | 5% | 10% | 16% | 18% |
| conversion 5 | 98% | 99% | ~100% | 98% | 95% |
| 27:286 | 42:1 | 133:1 | 58:1 | 98:1 | 390:1 |
| Reaction 33 | Reaction 34 | Reaction 35 | Reaction 36 | Reaction 37 | |
|---|---|---|---|---|---|
| reactor | horizontal trough | horizontal trough | horizontal trough | horizontal trough | vertical trough |
| CF | 15 suns | 15 suns | 15 suns | 15 suns | 18 suns |
| reflector | holographic | holographic | aluminum | aluminum | aluminum |
| diol 30 | 2.0 g | 1.0 g | 2.0 g | 2.0 g | 0.5 g |
| solvent | 200 mL (iPrOH) | 200 mL (iPrOH) | 250 mL (iPrOH) | 250 mL (acetone) | 100 mL (iPrOH) |
| sensitizer | 0.1 g (RB) | 0.1 g (RB) | 0.1 g (RB) | 0.1 g (RB) | 0.05 g (RB) |
| time | 8 h 1 (2 days) | 3 h 1 (2 days) | 4 h 1 (1 day) | 4 h 1 (1 day) | 2/3 h 1 (1 day) |
| conversion 2 | 83% | >95% | 93% | 99% | >95% |
| yield of 31 3 | 54% | 79% | 75% | 79% | 71% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wau, J.S.; Robertson, M.J.; Oelgemöller, M. Solar Photooxygenations for the Manufacturing of Fine Chemicals—Technologies and Applications. Molecules 2021, 26, 1685. https://doi.org/10.3390/molecules26061685
Wau JS, Robertson MJ, Oelgemöller M. Solar Photooxygenations for the Manufacturing of Fine Chemicals—Technologies and Applications. Molecules. 2021; 26(6):1685. https://doi.org/10.3390/molecules26061685
Chicago/Turabian StyleWau, Jayson S., Mark J. Robertson, and Michael Oelgemöller. 2021. "Solar Photooxygenations for the Manufacturing of Fine Chemicals—Technologies and Applications" Molecules 26, no. 6: 1685. https://doi.org/10.3390/molecules26061685