
The TDDFT Excitation Energies of the BODIPYs; The DFT and TDDFT Challenge Continues

 ,
,  ,
, 
Abstract
:1. Introduction
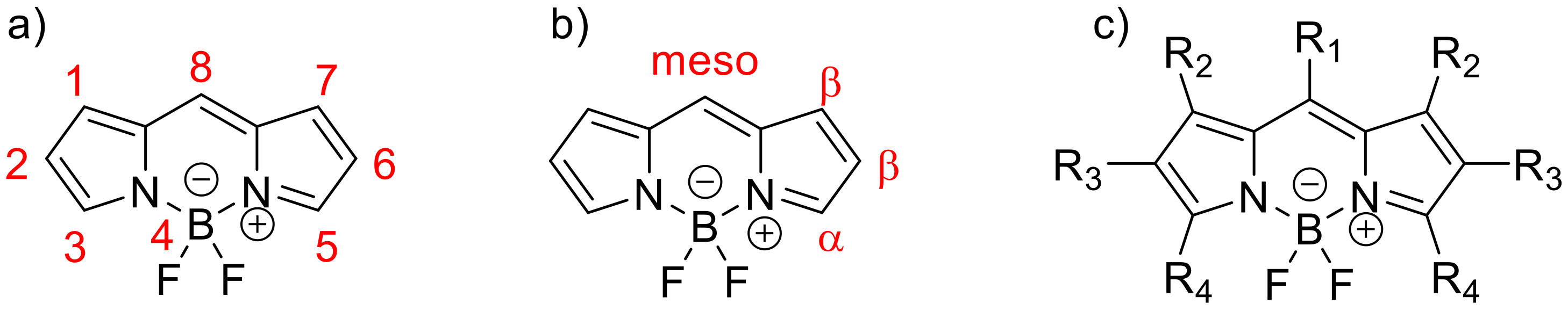
2. Investigated Structures
3. Results and Discussion
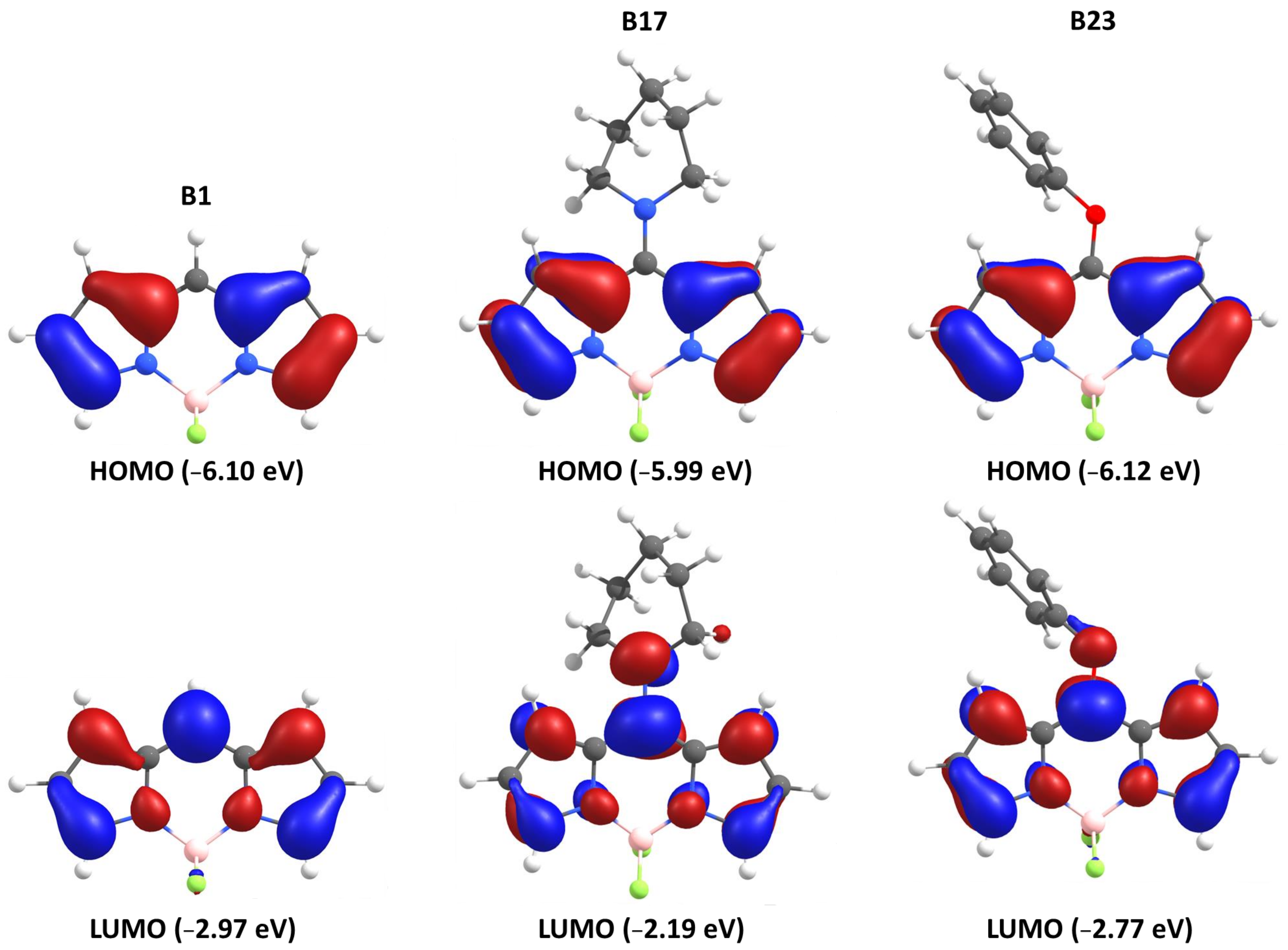
3.1. Optimisation of the BODIPYs Structures and Orbitals Parameters
3.2. TD-DFT Assessing the First Excited State
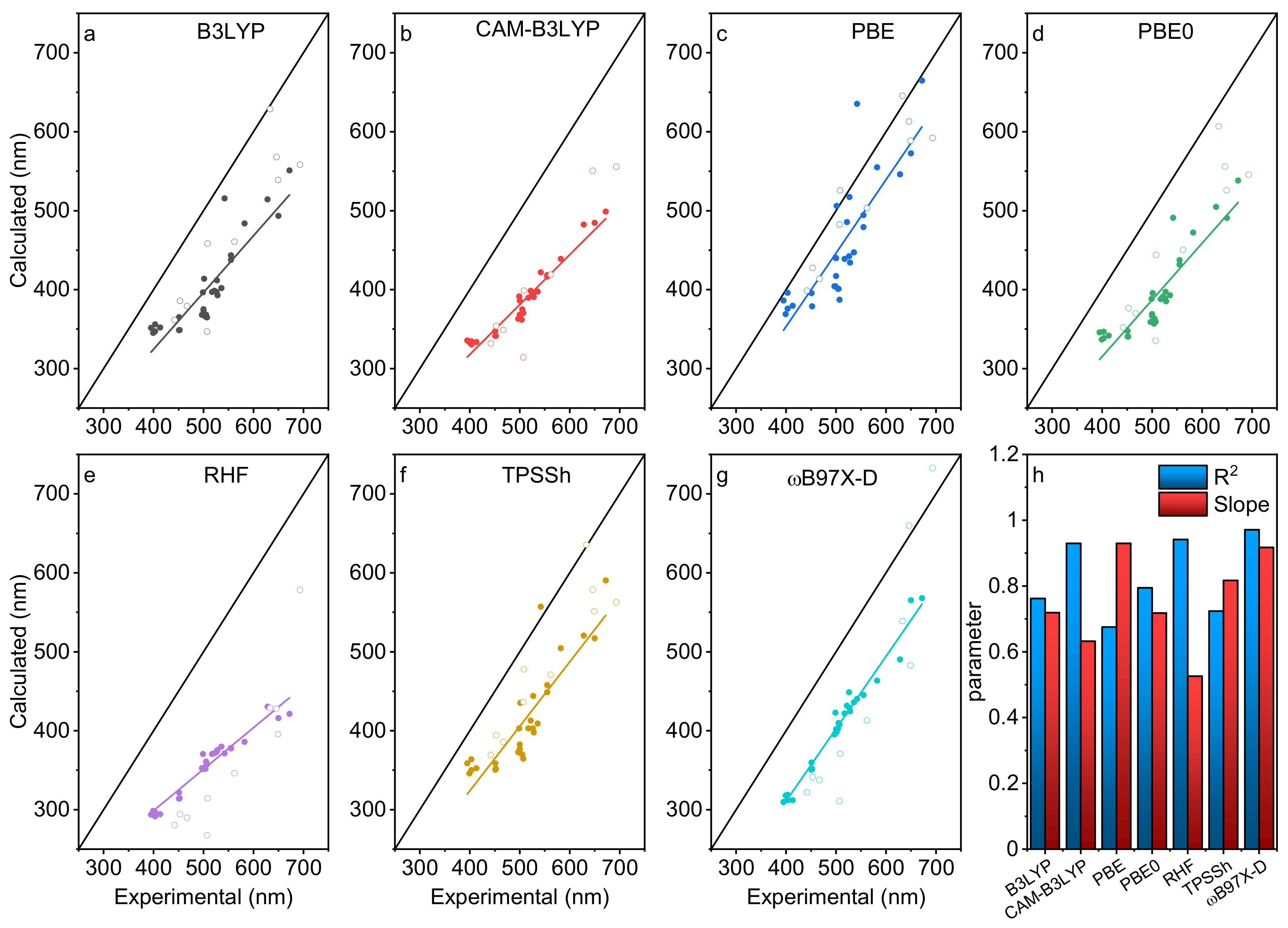
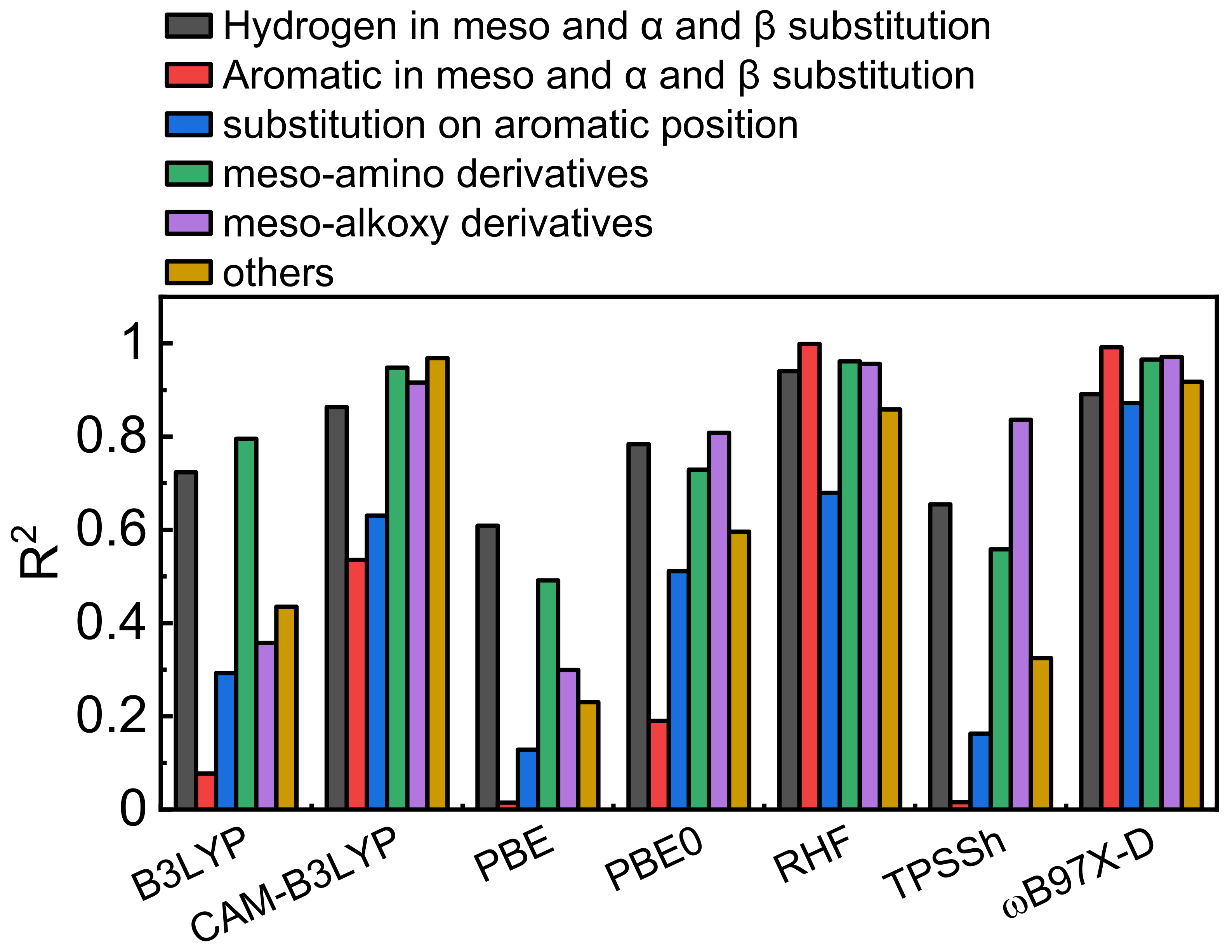
3.3. Model Validation
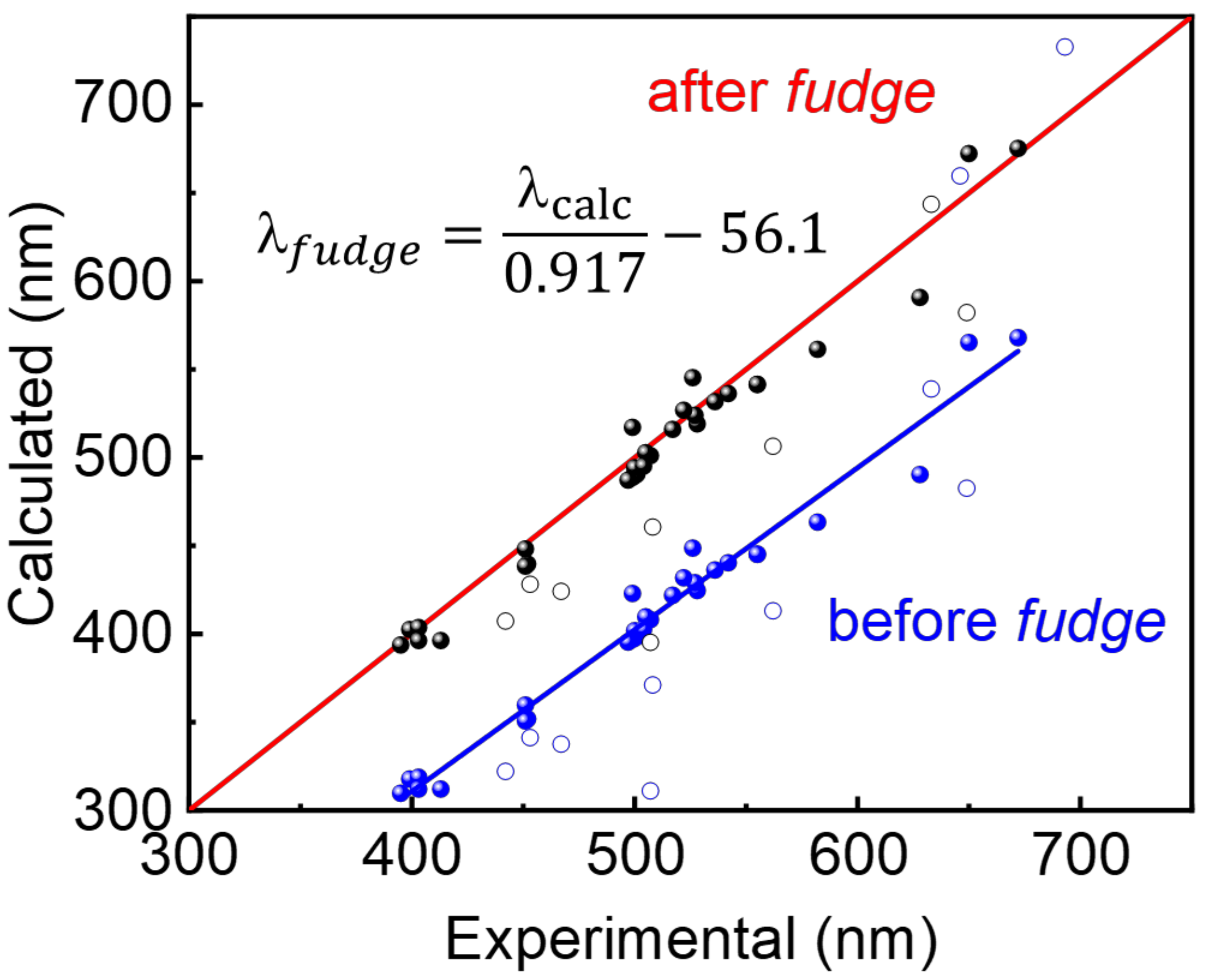
3.4. The “Fudge Factor” Approach
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A

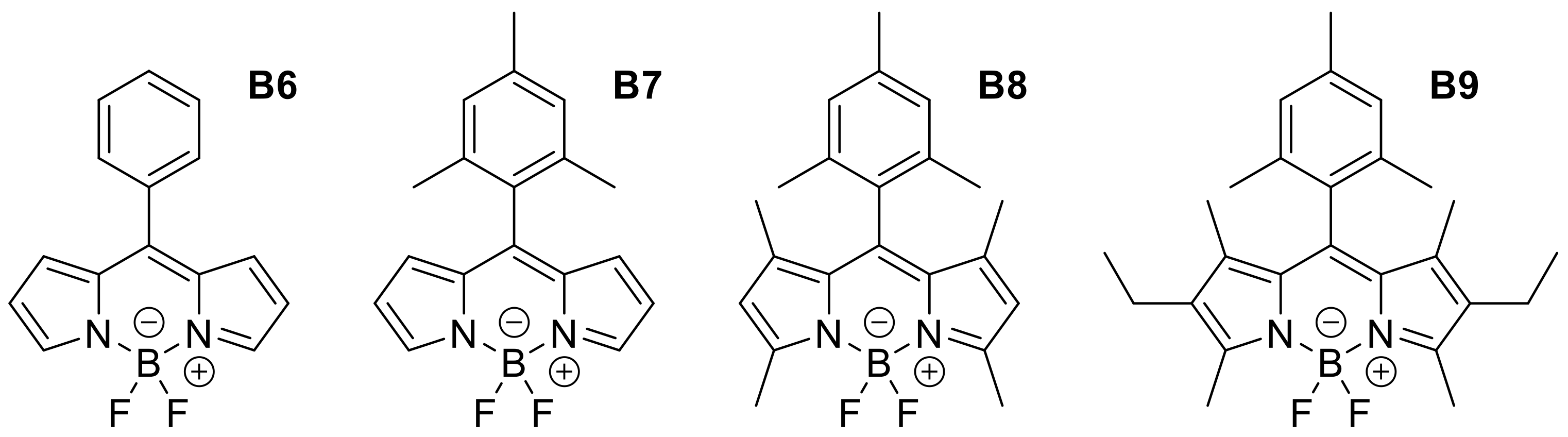
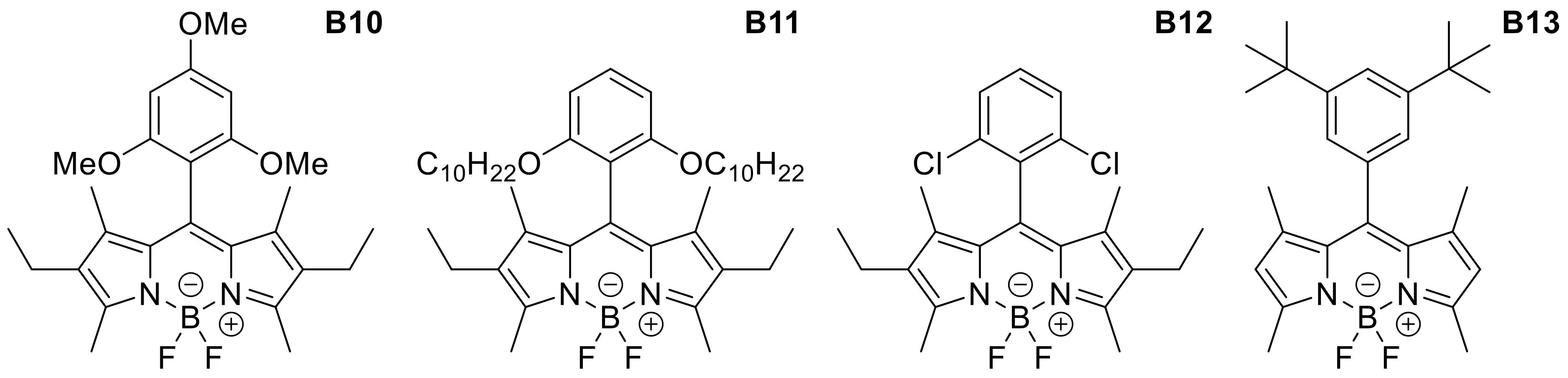
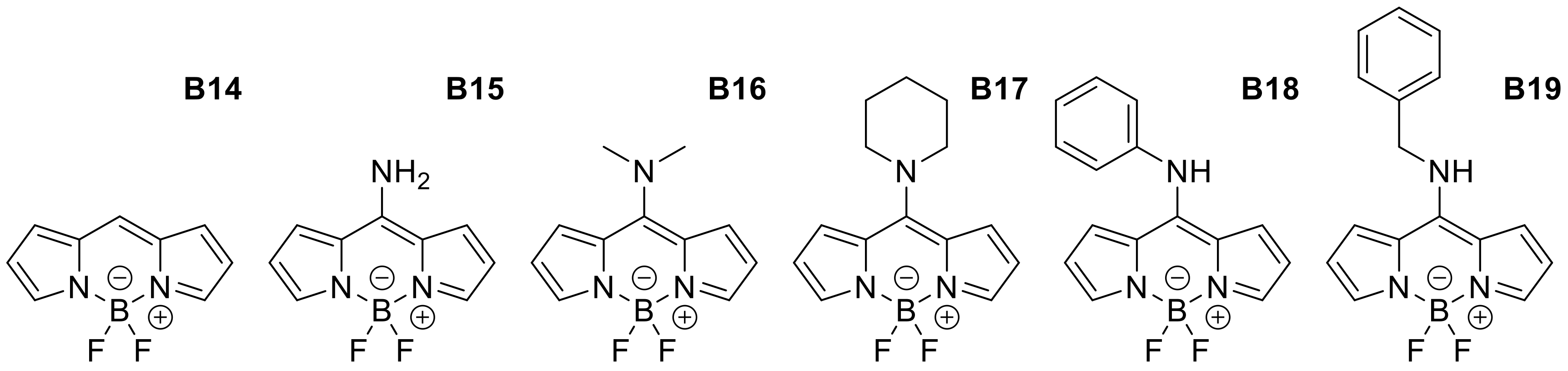
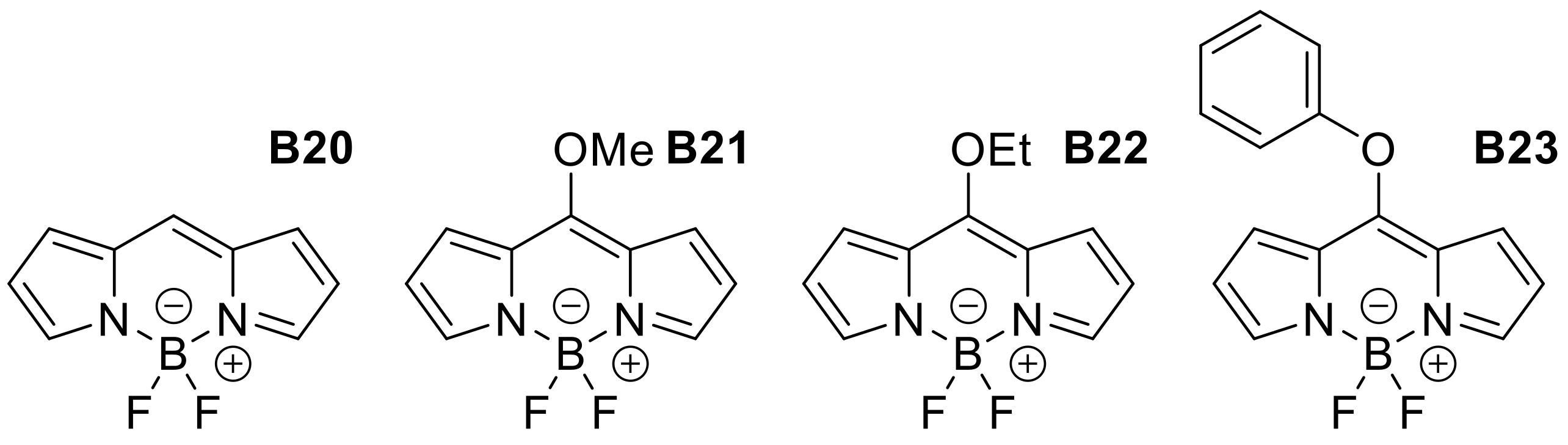
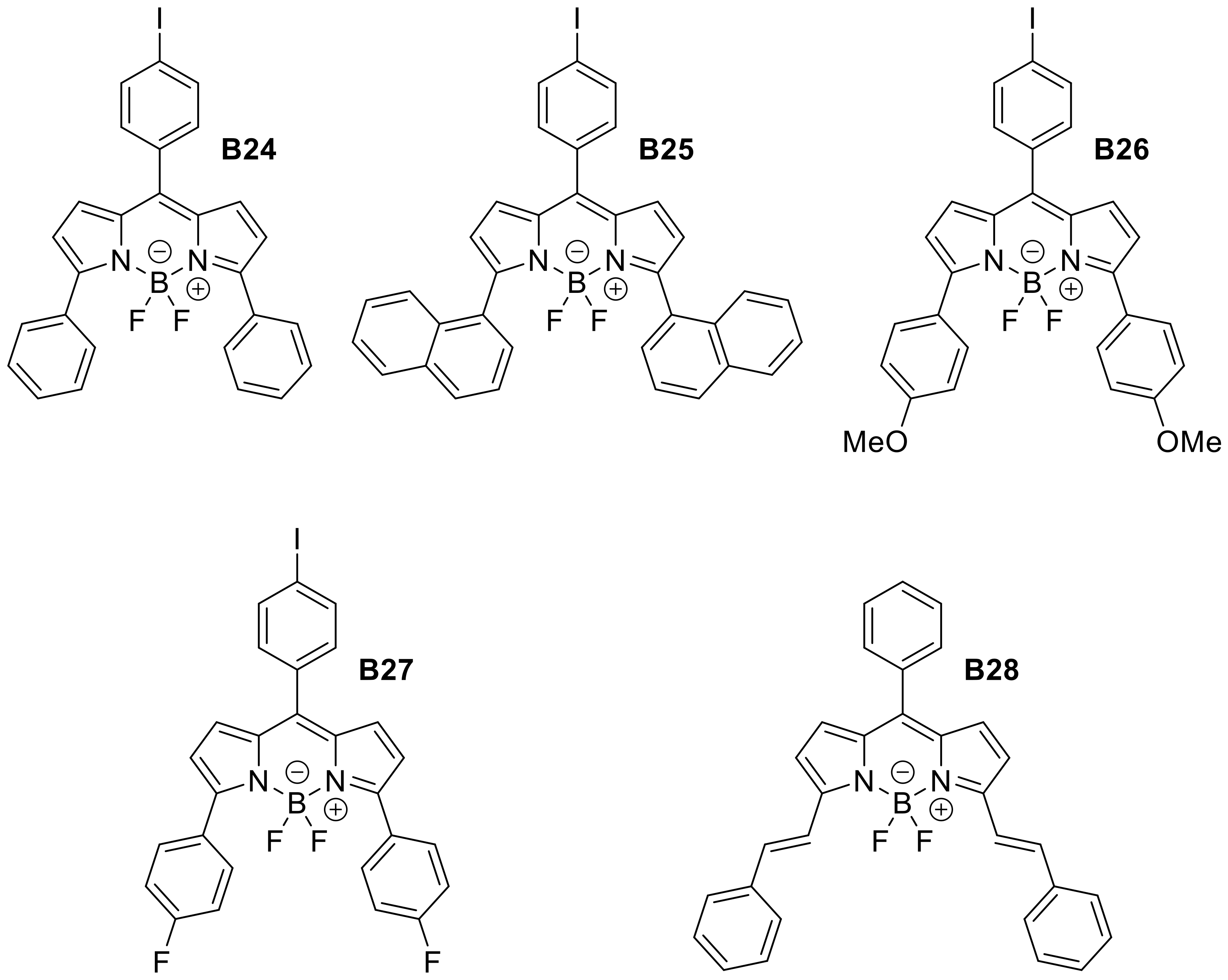

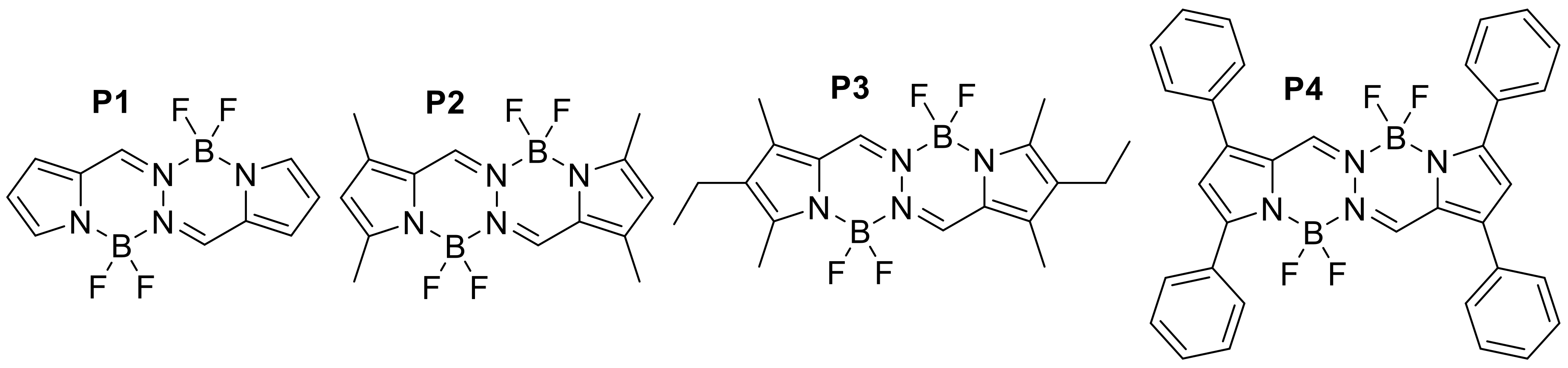
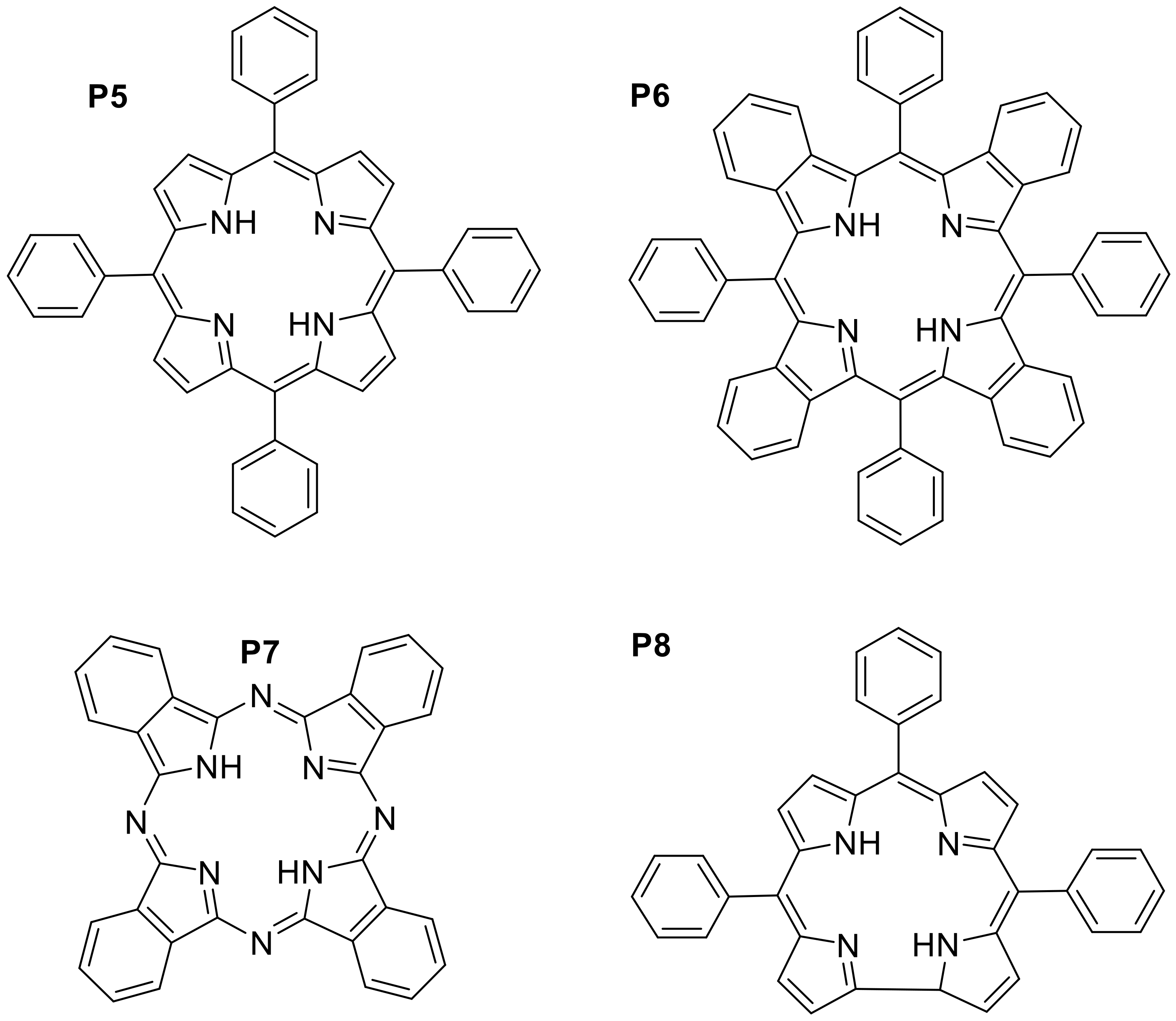
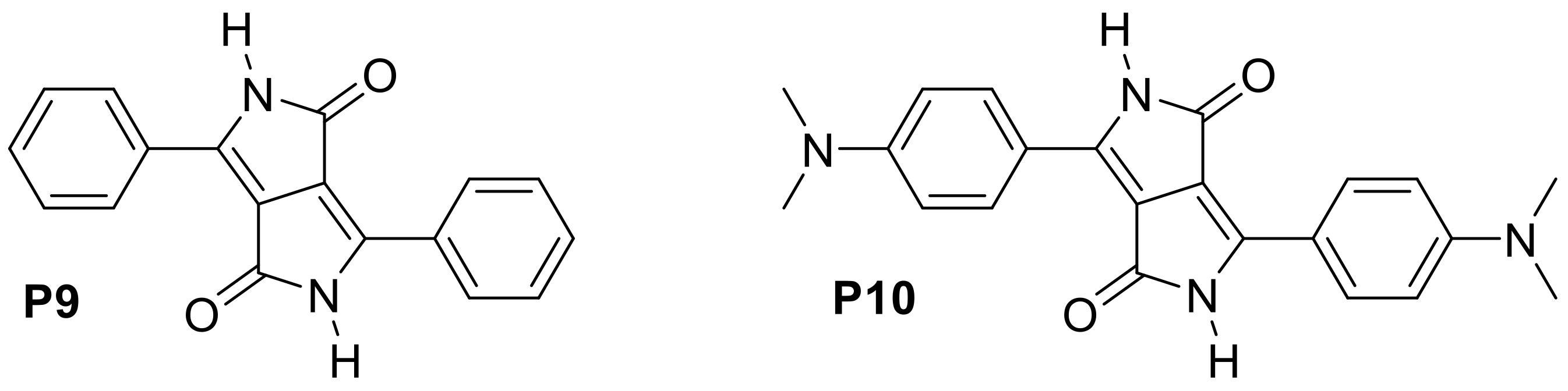
Appendix B

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λmax abs (nm) | B3LYP | CAM- B3LYP | PBE | PBE0 | RHF | TPSSh | ωB97X-D | ωB97X-D fudge |
|---|---|---|---|---|---|---|---|---|---|
| B1 | 499 | 368 | 363.1 | 404.2 | 358.9 | 352.5 | 372.9 | 396.9 | 488.81 |
| B2 | 507 | 364.9 | 370.4 | 387.3 | 359.5 | 357.5 | 364.6 | 408.12 | 501.05 |
| B3 | 505 | 368.3 | 375 | 400.9 | 363.1 | 360.9 | 369.7 | 409.75 | 502.82 |
| B4 | 528 | 393 | 390.8 | 434.2 | 385.3 | 375.7 | 397.7 | 424.65 | 519.07 |
| B5 | 517 | 397.1 | 389.8 | 438.8 | 388 | 370.6 | 403 | 421.87 | 516.04 |
| B6 | 500 | 375.3 | 368.2 | 417.3 | 366.9 | 351.9 | 382.5 | 397.65 | 489.63 |
| B7 | 501 | 413.9 | 366.8 | 506 | 395.5 | 353.4 | 435.3 | 398.58 | 490.65 |
| B8 | 500 | 374 | 385.9 | 439.9 | 369 | 351.8 | 377 | 401.89 | 494.25 |
| B9 | 526 | 397 | 393.1 | 442.4 | 388.6 | 373.2 | 403 | 448.72 | 545.31 |
| B10 | 527 | 411.8 | 394.9 | 517.4 | 397.4 | 373.9 | 444 | 429.12 | 523.94 |
| B11 | 522 | 398.6 | 398.6 | 485.7 | 391.2 | 371.6 | 412.8 | 431.8 | 526.86 |
| B12 | 536 | 401.9 | 397.5 | 447.3 | 393 | 379.9 | 409.2 | 436.21 | 531.67 |
| B13 | 499 | 396.6 | 391.3 | 440.6 | 388.2 | 370.4 | 403 | 422.89 | 517.15 |
| B14 | 497 | 368.1 | 363.2 | 404.2 | 359 | 352.6 | 372.8 | 395.37 | 487.15 |
| B15 | 399 | 345.1 | 333.3 | 369 | 336.7 | 298.4 | 345.9 | 317.78 | 402.56 |
| B16 | 395 | 351.6 | 335.5 | 386.2 | 345.8 | 293.9 | 358.8 | 309.67 | 393.72 |
| B17 | 413 | 352 | 333.6 | 379.7 | 341.7 | 294.3 | 352.5 | 312.05 | 396.31 |
| B18 | 403 | 355.9 | 334.6 | 396 | 346.7 | 296.7 | 363.8 | 318.81 | 403.68 |
| B19 | 403 | 347.2 | 330.7 | 375.9 | 338.5 | 291.5 | 350.2 | 312.05 | 396.31 |
| B20 | 504 | 365.8 | 362 | 400.9 | 357.1 | 351.7 | 370.3 | 402.67 | 495.11 |
| B21 | 452 | 348.9 | 341.6 | 378.8 | 340.6 | 314.5 | 351.7 | 351.78 | 439.63 |
| B22 | 451 | 348.7 | 341.5 | 395.7 | 340.5 | 313.9 | 350.5 | 350.67 | 438.42 |
| B23 | 451 | 365 | 347.3 | 395.7 | 347.7 | 321.8 | 358.7 | 359.66 | 448.22 |
| B24 | 555 | 437.6 | 416.5 | 479.2 | 431.7 | 378.2 | 448.6 | 445.17 | 541.44 |
| B25 | 542 | 515.5 | 421.9 | 635.3 | 491.1 | 371.3 | 557.3 | 440.39 | 536.23 |
| B26 | 582 | 483.9 | 438.8 | 554.9 | 472.4 | 385.9 | 504.6 | 463.41 | 561.32 |
| B27 | 555 | 443.5 | 418.5 | 494.5 | 437.5 | 377.6 | 457.7 | 445.2 | 541.47 |
| B28 | 628 | 514.3 | 482.3 | 546.1 | 504.7 | 430.4 | 520.5 | 490.36 | 590.70 |
| B29 | 650 | 493.5 | 484.7 | 572.7 | 490.7 | 416 | 517 | 565.21 | 672.30 |
| B30 | 664 | 551 | 498.9 | 664.8 | 538.3 | 421.3 | 590.5 | 567.93 | 675.27 |
| P1 | 442 | 361.6 | 332 | 398.7 | 352 | 280.4 | 369.2 | 322.12 | 407.29 |
| P2 | 467 | 379 | 348.8 | 413.8 | 370.1 | 289.9 | 385.9 | 337.65 | 424.22 |
| P3 | 453 | 385.7 | 353.8 | 427.6 | 376.4 | 294.2 | 394.3 | 341.29 | 428.19 |
| P4 | 508 | 458.2 | 398.8 | 525.6 | 444 | 314.5 | 477.7 | 371.07 | 460.66 |
| P5 | 646 | 567.9 | 550.6 | 613 | 556 | 427.8 | 578.7 | 659.64 | 775.25 |
| P6 | 633 | 628.8 | - | 645.4 | 606.9 | 428.9 | 635.7 | 538.87 | 643.59 |
| P7 | 693 | 558.3 | 555.6 | 592.2 | 545.6 | 578.4 | 562.6 | 732.8 | 855.01 |
| P8 | 649 | 538.9 | - | 588.6 | 525.9 | 395.5 | 551.2 | 482.58 | 582.22 |
| P9 | 507 | 346.8 | 314.1 | 482.4 | 335.4 | 267.5 | 436.5 | 311.06 | 395.23 |
| P10 | 562 | 460.7 | 419 | 503.2 | 450.4 | 346.1 | 470.8 | 413.11 | 506.49 |
Appendix C
| Removed Dye | B3LYP | CAM- B3LYP | PBE | PBE0 | RHF | TPSSh | ωB97X-D | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Slope | R2 | Slope | R2 | Slope | R2 | Slope | R2 | Slope | R2 | Slope | R2 | Slope | R2 | |
| None | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.79 | 0.53 | 0.94 | 0.82 | 0.72 | 0.92 | 0.97 |
| B1 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.53 | 0.94 | 0.81 | 0.73 | 0.92 | 0.97 |
| B2 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.69 | 0.72 | 0.80 | 0.53 | 0.94 | 0.82 | 0.74 | 0.92 | 0.97 |
| B3 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.69 | 0.72 | 0.80 | 0.53 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B4 | 0.72 | 0.77 | 0.63 | 0.93 | 0.94 | 0.68 | 0.72 | 0.80 | 0.52 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B5 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.52 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B6 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.53 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B7 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.69 | 0.72 | 0.79 | 0.53 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B8 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.53 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B9 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.52 | 0.94 | 0.82 | 0.73 | 0.91 | 0.98 |
| B10 | 0.72 | 0.76 | 0.63 | 0.93 | 0.92 | 0.68 | 0.72 | 0.79 | 0.52 | 0.94 | 0.81 | 0.72 | 0.92 | 0.97 |
| B11 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.52 | 0.94 | 0.82 | 0.72 | 0.92 | 0.97 |
| B12 | 0.72 | 0.77 | 0.63 | 0.93 | 0.94 | 0.68 | 0.72 | 0.80 | 0.52 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B13 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.67 | 0.72 | 0.79 | 0.53 | 0.95 | 0.82 | 0.72 | 0.92 | 0.98 |
| B14 | 0.72 | 0.77 | 0.63 | 0.93 | 0.93 | 0.68 | 0.72 | 0.80 | 0.53 | 0.94 | 0.81 | 0.73 | 0.92 | 0.97 |
| B15 | 0.74 | 0.76 | 0.65 | 0.93 | 0.94 | 0.66 | 0.74 | 0.79 | 0.53 | 0.94 | 0.84 | 0.72 | 0.92 | 0.97 |
| B16 | 0.75 | 0.77 | 0.65 | 0.94 | 0.96 | 0.68 | 0.75 | 0.80 | 0.52 | 0.94 | 0.85 | 0.73 | 0.92 | 0.97 |
| B17 | 0.73 | 0.76 | 0.64 | 0.93 | 0.94 | 0.67 | 0.73 | 0.79 | 0.52 | 0.94 | 0.83 | 0.72 | 0.91 | 0.97 |
| B18 | 0.74 | 0.77 | 0.64 | 0.93 | 0.96 | 0.68 | 0.74 | 0.80 | 0.52 | 0.94 | 0.85 | 0.73 | 0.92 | 0.97 |
| B19 | 0.74 | 0.76 | 0.64 | 0.93 | 0.95 | 0.67 | 0.74 | 0.79 | 0.52 | 0.94 | 0.84 | 0.72 | 0.92 | 0.97 |
| B20 | 0.72 | 0.77 | 0.63 | 0.94 | 0.93 | 0.68 | 0.72 | 0.80 | 0.53 | 0.94 | 0.82 | 0.73 | 0.92 | 0.97 |
| B21 | 0.71 | 0.76 | 0.63 | 0.93 | 0.92 | 0.67 | 0.71 | 0.79 | 0.52 | 0.94 | 0.81 | 0.72 | 0.91 | 0.97 |
| B22 | 0.71 | 0.76 | 0.63 | 0.93 | 0.93 | 0.67 | 0.71 | 0.79 | 0.52 | 0.94 | 0.81 | 0.72 | 0.91 | 0.97 |
| B23 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.67 | 0.72 | 0.79 | 0.52 | 0.94 | 0.81 | 0.72 | 0.92 | 0.97 |
| B24 | 0.72 | 0.76 | 0.63 | 0.93 | 0.94 | 0.68 | 0.72 | 0.79 | 0.53 | 0.94 | 0.82 | 0.72 | 0.92 | 0.97 |
| B25 | 0.70 | 0.83 | 0.63 | 0.93 | 0.89 | 0.77 | 0.70 | 0.84 | 0.53 | 0.94 | 0.79 | 0.81 | 0.92 | 0.97 |
| B26 | 0.70 | 0.75 | 0.63 | 0.93 | 0.91 | 0.66 | 0.70 | 0.79 | 0.53 | 0.94 | 0.80 | 0.71 | 0.93 | 0.97 |
| B27 | 0.72 | 0.76 | 0.63 | 0.93 | 0.93 | 0.67 | 0.71 | 0.79 | 0.53 | 0.94 | 0.81 | 0.72 | 0.92 | 0.97 |
| B28 | 0.69 | 0.73 | 0.61 | 0.93 | 0.95 | 0.66 | 0.69 | 0.77 | 0.51 | 0.94 | 0.81 | 0.70 | 0.95 | 0.98 |
| B29 | 0.73 | 0.74 | 0.62 | 0.92 | 0.95 | 0.65 | 0.72 | 0.77 | 0.54 | 0.94 | 0.83 | 0.70 | 0.89 | 0.97 |
| B30 | 0.67 | 0.70 | 0.62 | 0.91 | 0.84 | 0.59 | 0.68 | 0.74 | 0.56 | 0.95 | 0.75 | 0.65 | 0.91 | 0.96 |
References
- Chen, D.; Zhong, Z.; Ma, Q.; Shao, J.; Huang, W.; Dong, X.; Dong, X. Aza-BODIPY-Based Nanomedicines in Cancer Phototheranostics. ACS Appl. Mater. Interfaces 2020, 12, 26914–26925. [Google Scholar] [CrossRef]
- Liu, M.; Li, C. Recent Advances in Activatable Organic Photosensitizers for Specific Photodynamic Therapy. Chempluschem 2020, 85, 948–957. [Google Scholar] [CrossRef]
- Poddar, M.; Misra, R. Recent advances of BODIPY based derivatives for optoelectronic applications. Coord. Chem. Rev. 2020, 421, 213462. [Google Scholar] [CrossRef]
- Klfout, H.; Stewart, A.; Elkhalifa, M.; He, H. BODIPYs for Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2017, 9, 39873–39889. [Google Scholar] [CrossRef]
- Ho, D.; Ozdemir, R.; Kim, H.; Earmme, T.; Usta, H.; Kim, C. BODIPY-Based Semiconducting Materials for Organic Bulk Heterojunction Photovoltaics and Thin-Film Transistors. Chempluschem 2018, 84, cplu.201800543. [Google Scholar] [CrossRef]
- Bessette, A.; Hanan, G.S. Design, synthesis and photophysical studies of dipyrromethene-based materials: Insights into their applications in organic photovoltaic devices. Chem. Soc. Rev. 2014, 43, 3342–3405. [Google Scholar] [CrossRef] [PubMed]
- Squeo, B.M.; Pasini, M. BODIPY platform: A tunable tool for green to NIR OLEDs. Supramol. Chem. 2020, 32, 56–70. [Google Scholar] [CrossRef]
- Chiba, Y.; Nakamura, T.; Matsuoka, R.; Nabeshima, T. Synthesis and Functions of Oligomeric and Multidentate Dipyrrin Derivatives and their Complexes. Synlett 2020, 31, 1663–1681. [Google Scholar] [CrossRef]
- Praveen, V.K.; Vedhanarayanan, B.; Mal, A.; Mishra, R.K.; Ajayaghosh, A. Self-Assembled Extended π-Systems for Sensing and Security Applications. Acc. Chem. Res. 2020, 53, 507. [Google Scholar] [CrossRef]
- Liu, Z.; Jiang, Z.; Yan, M.; Wang, X. Recent Progress of BODIPY Dyes with Aggregation-Induced Emission. Front. Chem. 2019, 7, 712. [Google Scholar] [CrossRef] [PubMed]
- Toliautas, S.; Dodonova, J.; Žvirblis, A.; Čiplys, I.; Polita, A.; Devižis, A.; Tumkevičius, S.; Šulskus, J.; Vyšniauskas, A. Enhancing the Viscosity-Sensitive Range of a BODIPY Molecular Rotor by Two Orders of Magnitude. Chem. A Eur. J. 2019, 25, 10342–10349. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Kohn, A.W.; Van Voorhis, T. Toward Prediction of Nonradiative Decay Pathways in Organic Compounds II: Two Internal Conversion Channels in BODIPYs. J. Phys. Chem. C 2020, 124, 3925–3938. [Google Scholar] [CrossRef]
- Ou, Q.; Peng, Q.; Shuai, Z. Toward Quantitative Prediction of Fluorescence Quantum Efficiency by Combining Direct Vibrational Conversion and Surface Crossing: BODIPYs as an Example. J. Phys. Chem. Lett. 2020, 11, 7790–7797. [Google Scholar] [CrossRef]
- López Arbeloa, F.; Bañuelos, J.; Martínez, V.; Arbeloa, T.; López Arbeloa, I. Structural, photophysical and lasing properties of pyrromethene dyes. Int. Rev. Phys. Chem. 2005, 24, 339–374. [Google Scholar] [CrossRef]
- Wei, Y.; Zheng, M.; Zhou, Q.; Zhou, X.; Liu, S. Application of a bodipy-C70 dyad in triplet-triplet annihilation upconversion of perylene as a metal-free photosensitizer. Org. Biomol. Chem. 2018, 16, 5598–5608. [Google Scholar] [CrossRef]
- Spiegel, J.D.; Lyskov, I.; Kleinschmidt, M.; Marian, C.M. Charge-transfer contributions to the excitonic coupling matrix element in BODIPY-based energy transfer cassettes. Chem. Phys. 2017, 482, 265–276. [Google Scholar] [CrossRef]
- Thorat, K.G.; Kamble, P.; Ray, A.K.; Sekar, N. Novel pyrromethene dyes with N-ethyl carbazole at the meso position: A comprehensive photophysical, lasing, photostability and TD-DFT study. Phys. Chem. Chem. Phys. 2015, 17, 17221–17236. [Google Scholar] [CrossRef]
- Thorat, K.G.; Kamble, P.; Mallah, R.; Ray, A.K.; Sekar, N. Congeners of Pyrromethene-567 Dye: Perspectives from Synthesis, Photophysics, Photostability, Laser, and TD-DFT Theory. J. Org. Chem. 2015, 80, 6152–6164. [Google Scholar] [CrossRef] [PubMed]
- Ünal, H.; Gunceler, D.; Mete, E. A study of the density functional methods on the photoabsorption of Bodipy dyes. J. Photochem. Photobiol. A Chem. 2014, 278, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Shandura, M.P.; Yakubovskyi, V.P.; Gerasov, A.O.; Kachkovsky, O.D.; Poronik, Y.M.; Kovtun, Y.P. α-Polymethine-Substituted Boron Dipyrromethenes—BODIPY-Based NIR Cyanine-Like Dyes. Eur. J. Org. Chem. 2012, 2012, 1825–1834. [Google Scholar] [CrossRef]
- Irmler, P.; Winter, R.F. σ-Pt-BODIPY Complexes with Platinum Attachment to Carbon Atoms C2 or C3: Spectroscopic, Structural, and (Spectro)Electrochemical Studies and Photocatalysis. Organometallics 2018, 37, 235–253. [Google Scholar] [CrossRef]
- Dong, Y.; Iagatti, A.; Foggi, P.; Zhao, J.; Mazzone, G.; Xu, K.; Ji, W.; Di Donato, M.; Russo, N. Bodipy-squaraine triads: Preparation and study of the intramolecular energy transfer, charge separation and intersystem crossing. Dye. Pigment. 2017, 147, 560–572. [Google Scholar] [CrossRef]
- Shi, W.-J.; Kinoshita, T.; Ng, D.K.P. Ethynyl-Linked Donor-π-Acceptor Boron Dipyrromethenes for Panchromatic Dye-Sensitized Solar Cells. Asian J. Org. Chem. 2017, 6, 758–767. [Google Scholar] [CrossRef]
- Chong, H.; Fron, E.; Liu, Z.; Boodts, S.; Thomas, J.; Harvey, J.N.; Hofkens, J.; Dehaen, W.; Van der Auweraer, M.; Smet, M. Acid-Sensitive BODIPY Dyes: Synthesis through Pd-Catalyzed Direct C(sp 3)−H Arylation and Photophysics. Chem. A Eur. J. 2017, 23, 4687–4699. [Google Scholar] [CrossRef]
- Misra, R. Tuning of Second-Order Nonlinear Optical Response Properties of Aryl-Substituted Boron-Dipyrromethene Dyes: Unidirectional Charge Transfer Coupled with Structural Tailoring. J. Phys. Chem. C 2017, 121, 5731–5739. [Google Scholar] [CrossRef] [Green Version]
- Mao, M.; Li, Q.S.; Zhang, X.L.; Wu, G.H.; Dai, C.G.; Ding, Y.; Dai, S.Y.; Song, Q.H. Effects of donors of bodipy dyes on the performance of dye-sensitized solar cells. Dye. Pigment. 2017, 141, 148–160. [Google Scholar] [CrossRef]
- Orte, A.; Debroye, E.; Ruedas-Rama, M.J.; Garcia-Fernandez, E.; Robinson, D.; Crovetto, L.; Talavera, E.M.; Alvarez-Pez, J.M.; Leen, V.; Verbelen, B.; et al. Effect of the substitution position (2, 3 or 8) on the spectroscopic and photophysical properties of BODIPY dyes with a phenyl, styryl or phenylethynyl group. RSC Adv. 2016, 6, 102899–102913. [Google Scholar] [CrossRef] [Green Version]
- Zinna, F.; Bruhn, T.; Guido, C.A.; Ahrens, J.; Bröring, M.; Di Bari, L.; Pescitelli, G. Circularly Polarized Luminescence from Axially Chiral BODIPY DYEmers: An Experimental and Computational Study. Chem. A Eur. J. 2016, 22, 16089–16098. [Google Scholar] [CrossRef] [PubMed]
- Balsukuri, N.; Lone, M.Y.; Jha, P.C.; Mori, S.; Gupta, I. Synthesis, Structure, and Optical Studies of Donor-Acceptor-Type Near-Infrared (NIR) Aza-Boron-Dipyrromethene (BODIPY) Dyes. Chem. Asian J. 2016, 11, 1572–1587. [Google Scholar] [CrossRef]
- De Vetta, M.; Corral, I. Insight into the optical properties of meso-pentafluorophenyl(PFP)-BODIPY: An attractive platform for functionalization of BODIPY dyes. Comput. Theor. Chem. 2019, 1150, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.N.; Habib, M.; Pramanik, A.; Sarkar, P.; Pal, S. Tuning the BODIPY core for its potential use in DSSC: A quantum chemical approach. Bull. Mater. Sci. 2018, 41, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pirillo, J.; Mazzone, G.; Russo, N. Theoretical Insights into the Switching Off/On of 1 O 2 Photosensitization in Chemicontrolled Photodynamic Therapy. Chem. A Eur. J. 2018, 24, 3512–3519. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, G.; Quartarolo, A.D.; Russo, N. PDT-correlated photophysical properties of thienopyrrole BODIPY derivatives. Theoretical insights. Dye. Pigment. 2016, 130, 9–15. [Google Scholar] [CrossRef]
- Le Guennic, B.; Jacquemin, D. Taking Up the Cyanine Challenge with Quantum Tools. Acc. Chem. Res. 2015, 48, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Charaf-Eddin, A.; Le Guennic, B.; Jacquemin, D. Excited-states of BODIPY–cyanines: Ultimate TD-DFT challenges? RSC Adv. 2014, 4, 49449–49456. [Google Scholar] [CrossRef]
- Laurent, A.D.; Adamo, C.; Jacquemin, D. Dye chemistry with time-dependent density functional theory. Phys. Chem. Chem. Phys. 2014, 16, 14334–14356. [Google Scholar] [CrossRef] [PubMed]
- Egidi, F.; Trani, F.; Ballone, P.A.; Barone, V.; Andreoni, W. Low-lying electronic excitations of a water-soluble BODIPY: From the gas phase to the solvated molecule. Theor. Chem. Acc. 2016, 135, 264. [Google Scholar] [CrossRef]
- Rose, A.; Kumar, S.V.; Swavey, S.; Erb, J. A simple and efficient protocol for screening boron-dipyrromethene dyes using TD-DFT and an examination of the aryl-meso position. Comput. Theor. Chem. 2017, 1118, 107–114. [Google Scholar] [CrossRef]
- Momeni, M.R.; Brown, A. Why do TD-DFT excitation energies of BODIPY/aza-BODIPY families largely deviate from experiment? Answers from electron correlated and multireference methods. J. Chem. Theory Comput. 2015, 11, 2619–2632. [Google Scholar] [CrossRef] [PubMed]
- Chibani, S.; Laurent, A.D.; Le Guennic, B.; Jacquemin, D. Improving the accuracy of excited-state simulations of BODIPY and Aza-BODIPY dyes with a joint SOS-CIS(D) and TD-DFT approach. J. Chem. Theory Comput. 2014, 10, 4574–4582. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, M.; Kitagawa, Y.; Teramoto, R.; Miyagi, K.; Natori, Y.; Nakano, M. Origin of Solvent-independent Optical Property of Unsubstituted BODIPY Revisited. Chem. Lett. 2017, 46, 536–538. [Google Scholar] [CrossRef]
- Chibani, S.; Jacquemin, D.; Laurent, A.D. Modelling solvent effects on the absorption and emission spectra of constrained cyanines with both implicit and explicit QM/EFP models. Comput. Theor. Chem. 2014, 1040–1041, 321–327. [Google Scholar] [CrossRef]
- Filarowski, A.; Lopatkova, M.; Lipkowski, P.; Van Der Auweraer, M.; Leen, V.; Dehaen, W. Solvatochromism of BODIPY-Schiff dye. J. Phys. Chem. B 2015, 119, 2576–2584. [Google Scholar] [CrossRef] [PubMed]
- Momeni, M.R.; Brown, A. A Local CC2 and TDA-DFT Double Hybrid Study on BODIPY/aza-BODIPY Dimers as Heavy Atom Free Triplet Photosensitizers for Photodynamic Therapy Applications. J. Phys. Chem. A 2016, 120, 2550–2560. [Google Scholar] [CrossRef]
- Laurent, A.D.; Blondel, A.; Jacquemin, D. Choosing an atomic basis set for TD-DFT, SOPPA, ADC(2), CIS(D), CC2 and EOM-CCSD calculations of low-lying excited states of organic dyes. Theor. Chem. Acc. 2015, 134, 76. [Google Scholar] [CrossRef]
- Briggs, E.A.; Besley, N.A.; Robinson, D. QM/MM excited state molecular dynamics and fluorescence spectroscopy of BODIPY. J. Phys. Chem. A 2013, 117, 2644–2650. [Google Scholar] [CrossRef]
- Gawale, Y.; Sekar, N. Investigating the excited state optical properties and origin of large stokes shift in Benz[c,d]indole N-Heteroarene BF2 dyes with ab initio tools. J. Photochem. Photobiol. B Biol. 2018, 178, 472–480. [Google Scholar] [CrossRef]
- Chibani, S.; Le Guennic, B.; Charaf-Eddin, A.; Laurent, A.D.; Jacquemin, D. Revisiting the optical signatures of BODIPY with ab initio tools. Chem. Sci. 2013, 4, 1950–1963. [Google Scholar] [CrossRef]
- Machado, L.A.; de Souza, M.C.; da Silva, C.M.; Yoneda, J.; de Rezende, L.C.D.; Emery, F.S.; de Simone, C.A.; da Silva Júnior, E.N.; Pedrosa, L.F. On the synthesis, optical and computational studies of novel BODIPY-based phosphoramidate fluorescent dyes. J. Fluor. Chem. 2019, 220, 9–15. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef] [PubMed]
- Tamgho, I.-S.; Hasheminasab, A.; Engle, J.T.; Nemykin, V.N.; Ziegler, C.J. A New Highly Fluorescent and Symmetric Pyrrole–BF 2 Chromophore: BOPHY. J. Am. Chem. Soc. 2014, 136, 5623–5626. [Google Scholar] [CrossRef] [PubMed]
- Boodts, S.; Fron, E.; Hofkens, J.; Dehaen, W. The BOPHY fluorophore with double boron chelation: Synthesis and spectroscopy. Coord. Chem. Rev. 2018, 371, 1–10. [Google Scholar] [CrossRef]
- Taniguchi, M.; Du, H.; Lindsey, J.S. PhotochemCAD 3: Diverse Modules for Photophysical Calculations with Multiple Spectral Databases. Photochem. Photobiol. 2018, 94, 277–289. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Lindsey, J.S. Database of Absorption and Fluorescence Spectra of >300 Common Compounds for use in PhotochemCAD. Photochem. Photobiol. 2018, 94, 290–327. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Zhao, J.; Yang, P.; Sun, J. Zinc(ii) tetraphenyltetrabenzoporphyrin complex as triplet photosensitizer for triplet-triplet annihilation upconversion. Chem. Commun. 2013, 49, 10221–10223. [Google Scholar] [CrossRef] [PubMed]
- Freyer, W.; Mueller, S.; Teuchner, K. Photophysical properties of benzoannelated metal-free phthalocyanines. J. Photochem. Photobiol. A Chem. 2004, 163, 231–240. [Google Scholar] [CrossRef]
- Vala, M.; Vyňuchal, J.; Toman, P.; Weiter, M.; Luňák, S. Novel, soluble diphenyl-diketo-pyrrolopyrroles: Experimental and theoretical study. Dye. Pigment. 2010, 84, 176–182. [Google Scholar] [CrossRef]
- Jacquemin, D.; Wathelet, V.; Perpète, E.A.; Adamo, C. Extensive TD-DFT benchmark: Singlet-excited states of organic molecules. J. Chem. Theory Comput. 2009, 5, 2420–2435. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hočevar, T.; Demčar, J. Computation of Graphlet Orbits for Nodes and Edges in Sparse Graphs. J. Stat. Softw. 2016, 71. [Google Scholar] [CrossRef] [Green Version]
- Barone, V.; Cossi, M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable adiabatic connection models free from adjustable parameters. Chem. Phys. Lett. 1997, 274, 242–250. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, S.E.; Allen, W.D.; Schaefer, H.F. On the convergence of Z-averaged perturbation theory. J. Chem. Phys. 2008, 128, 074107. [Google Scholar] [CrossRef] [PubMed]
| Compound | R1 | R2 | R3 | R4 | Solvent | λmax abs (nm) | λmax em (nm) | Φ |
|---|---|---|---|---|---|---|---|---|
| Hydrogen in Meso and α and β Substitution | ||||||||
| B1 | H | H | H | H | EtOH | 499 | 535 | 0.93 |
| B2 | H | H | H | Me | EtOH | 507 | 520 | 0.81 |
| B3 | H | Me | H | Me | EtOH | 505 | 516 | 0.80 |
| B4 | H | Me | Me | Me | EtOH | 528 | 535 | 0.56 |
| B5 | H | Me | Et | Me | EtOH | 517 | 546 | 0.70 |
| Aromatic in Meso and α and β Substitution | ||||||||
| B6 | Phenyl | H | H | H | CH2Cl2 | 500 | 527 | 0.03 |
| B7 | 2,4,6-trimethylbenzene | H | H | H | CH2Cl2 | 501 | 521 | 0.84 |
| B8 | 2,4,6-trimethylbenzene | Me | H | Me | AcOEt | 500 | 508 | 0.92 |
| B9 | 2,4,6-trimethylbenzene | Me | Et | Me | CH2Cl2 | 526 | 535 | 0.72 |
| Substitution on Aromatic Position | ||||||||
| B10 | 2,4,6-trimethoxybenzene | Me | Et | Me | AcOEt | 527 | 535 | 0.86 |
| B11 | 2,6-didecyloxybenzene | Me | Et | Me | EtOH | 522 | 536 | 0.82 |
| B12 | 2,6-dichlorobenzene | Me | Et | Me | CH2Cl2 | 536 | 548 | 0.65 |
| B13 | 1,3-di-tert-butylbenzene | Me | H | Me | CH2Cl2 | 499 | 507 | 0.97 |
| Meso-Amino Derivatives | ||||||||
| B14 | H | H | H | H | MeOH | 497 | 507 | 0.87 |
| B15 | NH2 | H | H | H | MeOH | 399 | 437 | 0.92 |
| B16 | NMe2 | H | H | H | MeOH | 395 | 438 | 0.09 |
| B17 | Piperidine | H | H | H | MeOH | 413 | 537 | 0.001 |
| B18 | N-aniline | H | H | H | MeOH | 403 | 461 | 0.16 |
| B19 | N-phenylmethanamine | H | H | H | MeOH | 403 | 453 | 0.09 |
| Meso-alkoxy derivatives | ||||||||
| B20 | H | H | H | H | Cy | 504 | 511 | 0.96 |
| B21 | OMe | H | H | H | Cy | 452 | 487 | 0.84 |
| B22 | OEt | H | H | H | Cy | 451 | 487 | 0.96 |
| B23 | OPh | H | H | H | Cy | 451 | 486 | 0.88 |
| Others | ||||||||
| B24 | 4-iodobenzene | H | H | Ph | CHCl3 | 555 | 588 | 0.20 |
| B25 | 4-iodobenzene | H | H | 1-napthalene | CHCl3 | 542 | 607 | 0.38 |
| B26 | 4-iodobenzene | H | H | PhOMe | CHCl3 | 582 | 626 | 0.42 |
| B27 | 4-iodobenzene | H | H | 4-fluorobenzene | CHCl3 | 555 | 590 | 0.22 |
| B28 | Phenyl | H | H | CH=CH2Ph | CH3CN | 628 | 642 | 0.84 |
| B29 | - | Ph | H | Ph | CHCl3 | 650 | 672 | 0.34 |
| B30 | - | PhOMe | H | Ph | CHCl3 | 672 | 695 | 0.23 |
| Compound | Solvent | λmax abs (nm) | λmax em (nm) | Φ | Ref |
|---|---|---|---|---|---|
| P1 | CH2Cl2 | 442 | 465 | 0.95 | [51] |
| P2 | CH2Cl2 | 467 | 485 | 0.92 | [51] |
| P3 | CH2Cl2 | 453 | 497 | 1.00 | [52] |
| P4 | CHCl3 | 508 | 524 | 0.96 | [52] |
| P5 | Toluene | 419 | 649 | 0.11 | [53,54] |
| P6 | Toluene | 633 | 783 | 0.28 | [55] |
| P7 | Benzene | 693 | 698 | 0.43 | [56] |
| P8 | CH2Cl2 | 416 | 671 | 0.14 | [53,54] |
| P9 | DMSO | 507 | 519 | 0.74 | [57] |
| P10 | DMSO | 562 | 580 | 0.57 | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schlachter, A.; Fleury, A.; Tanner, K.; Soldera, A.; Habermeyer, B.; Guilard, R.; Harvey, P.D. The TDDFT Excitation Energies of the BODIPYs; The DFT and TDDFT Challenge Continues. Molecules 2021, 26, 1780. https://doi.org/10.3390/molecules26061780
Schlachter A, Fleury A, Tanner K, Soldera A, Habermeyer B, Guilard R, Harvey PD. The TDDFT Excitation Energies of the BODIPYs; The DFT and TDDFT Challenge Continues. Molecules. 2021; 26(6):1780. https://doi.org/10.3390/molecules26061780
Chicago/Turabian StyleSchlachter, Adrien, Alexandre Fleury, Kevin Tanner, Armand Soldera, Benoit Habermeyer, Roger Guilard, and Pierre D. Harvey. 2021. "The TDDFT Excitation Energies of the BODIPYs; The DFT and TDDFT Challenge Continues" Molecules 26, no. 6: 1780. https://doi.org/10.3390/molecules26061780
APA StyleSchlachter, A., Fleury, A., Tanner, K., Soldera, A., Habermeyer, B., Guilard, R., & Harvey, P. D. (2021). The TDDFT Excitation Energies of the BODIPYs; The DFT and TDDFT Challenge Continues. Molecules, 26(6), 1780. https://doi.org/10.3390/molecules26061780