
Involvement of NRF2 in Breast Cancer and Possible Therapeutical Role of Polyphenols and Melatonin
 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mechanisms Underlying Breast Cancer
2.1. Oxidative Stress-Independent Mechanisms
2.2. Oxidative Stress-Dependent Mechanisms
ROS Formation via Estrogen Metabolism
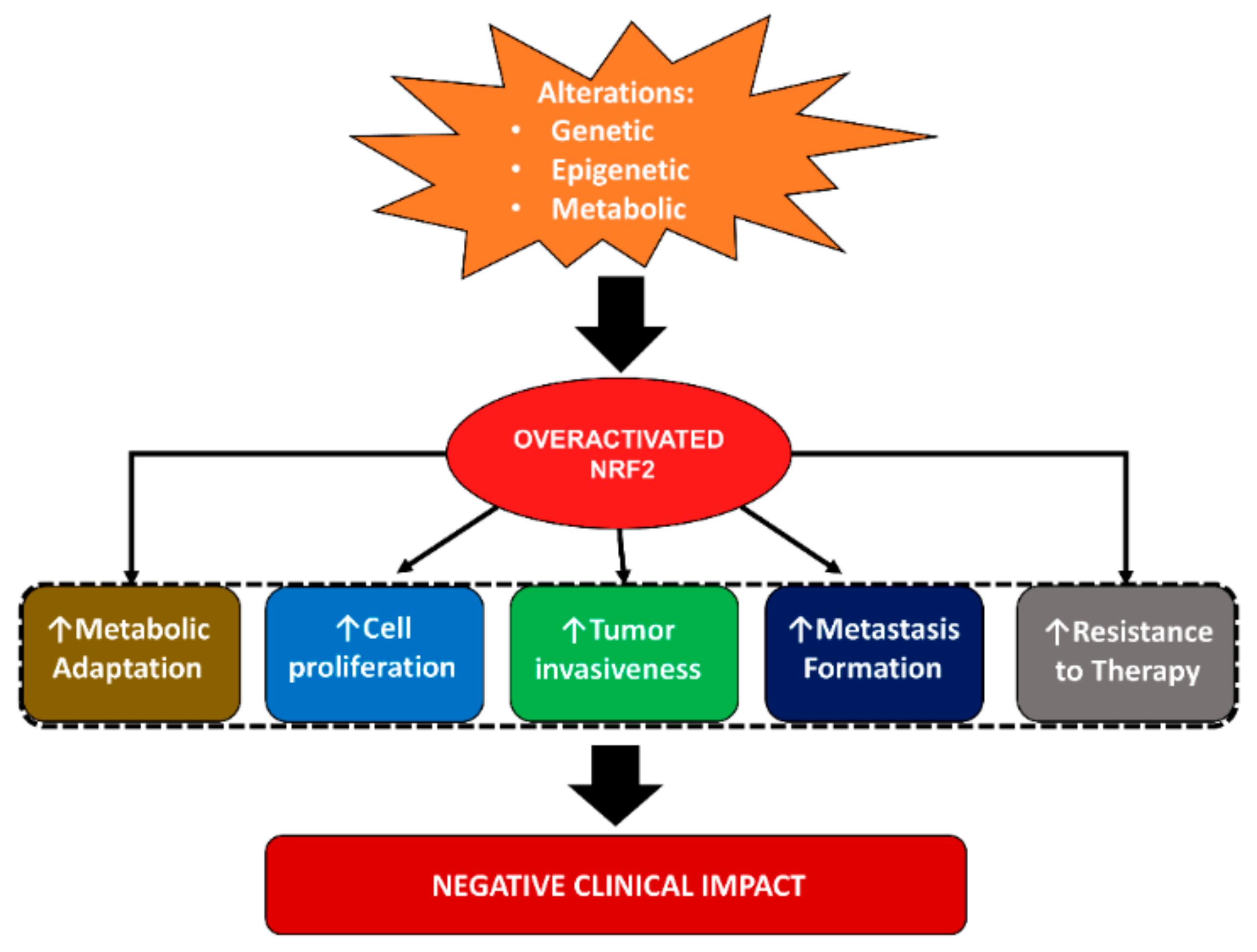
2.3. Role of Nuclear Factor Erythroid 2-Related Factor 2 (NRF2) in Breast Cancer
2.3.1. NRF2 in Breast Cancer Cell Proliferation, Growth, Invasion and Metastasis
2.3.2. NRF2 in the Regulation of Breast Cancer Cell Stemness and Therapy Resistance
2.3.3. NRF2 in Metabolic Adaptation of Breast Cancer Cells
2.3.4. NRF2 in Breast Cancer Prognosis
3. NRF2-Related Mechanisms as a Target in Breast Cancer
3.1. Therapeutical Role of Polyphenols
3.2. Therapeutical Role of Melatonin
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. GLOBOCAN 2018 Database. Issued by World Health Organization (WHO). Available online: http://gco.iarc.fr/today (accessed on 26 February 2021).
- Sims, A.H.; Howell, A.; Howell, S.J.; Clarke, R.B. Origins of breast cancer subtypes and therapeutic implications. Nat. Clin. Pr. Oncol. 2007, 4, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Planes-Laine, G.; Rochigneux, P.; Bertucci, F.; Chrétien, A.-S.; Viens, P.; Sabatier, R.; Gonçalves, A. PD-1/PD-L1 Targeting in Breast Cancer: The First Clinical Evidences Are Emerging. A Literature Review. Cancers 2019, 11, 1033. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Robert, A.W. Biological Hallmarks of cancer. Holl. Frei Cancer Med. 2017, 1, 1–10. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Klaunig, J.E. Chemical Carcinogenesis. In Casarette and Doull’s Toxicology; Klaassen, C.D., Ed.; Mc Graw Hill Educations: New York, NY, USA, 2013; pp. 393–444. [Google Scholar]
- Gurer-Orhan, H.; Ince, E.; Konyar, D.; Saso, L.; Suzen, S. The Role of Oxidative Stress Modulators in Breast Cancer. Curr. Med. Chem. 2018, 25, 4084–4101. [Google Scholar] [CrossRef]
- Thorlacius, S.; Struewing, J.P.; Hartage, P.; Olafsdottir, G.H.; Sigvaldason, H.; Tryggvadottir, L.; Wacholder, S.; Tulinius, H.; Eyfjörd, J.E. Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 1998, 352, 1337–1339. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleibl, Z.; Kristensen, V.N. Women at high risk of breast cancer: Molecular characteristics, clinical presentation and management. Breast 2016, 28, 136–144. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen Carcinogenesis in Breast Cancer. N. Engl. J. Med. 2006, 354, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilton, H.N.; Clarke, C.L.; Graham, J.D. Estrogen and progesterone signalling in the normal breast and its implications for cancer development. Mol. Cell. Endocrinol. 2018, 466, 2–14. [Google Scholar] [CrossRef]
- Barrios-Rodríguez, R.; Toledo, E.; Martinez-Gonzalez, M.A.; Aguilera-Buenosvinos, I.; Romanos-Nanclares, A.; Jiménez-Moleón, J.J. Adherence to the 2018 World Cancer Research Fund/American Institute for Cancer Research Recommendations and Breast Cancer in the SUN Project. Nutrients 2020, 12, 2076. [Google Scholar] [CrossRef]
- Freudenheim, J.L. Alcohol’s Effects on Breast Cancer in Women. Alcohol Res. Curr. Rev. 2020, 40, 11. [Google Scholar] [CrossRef]
- Rock, C.L.; Thomson, C.; Gansler, T.; Gapstur, S.M.; McCullough, M.L.; Patel, A.V.; Ba, K.S.A.; Bandera, E.V.; Spees, C.K.; Robien, K.; et al. American Cancer Society guideline for diet and physical activity for cancer prevention. CA Cancer J. Clin. 2020, 70, 245–271. [Google Scholar] [CrossRef]
- Jardé, T.; Caldefie-Chézet, F.; Damez, M.; Mishellany, F.; Perrone, D.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Adi-ponectin and leptin expression in primary ductal breast cancer and in adjacent healthy epithelial and myoepithelial tissue. Histopathology 2008, 53, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Sabol, R.A.; Beighley, A.; Giacomelli, P.; Wise, R.M.; Harrison, M.A.A.; O’Donnnell, B.A.; Sullivan, B.N.; Lampenfeld, J.D.; Matossian, M.D.; Bratton, M.R.; et al. Obesity-Altered Adipose Stem Cells Promote ER⁺ Breast Cancer Metastasis through Estrogen Independent Pathways. Int. J. Mol. Sci. 2019, 20, 1419. [Google Scholar] [CrossRef] [Green Version]
- La Barge, M.A.; Mora-Blanco, E.L.; Samson, S.; Miyano, M. Breast Cancer beyond the Age of Mutation. Gerontology 2016, 62, 434–442. [Google Scholar] [CrossRef]
- Sies, H. Oxidative Stress: Introductory Remarks. In Oxidative Stress; Academic Press: Cambridge, MA, USA, 1985; pp. 1–5. [Google Scholar]
- Jones, D.P. Redefining Oxidative Stress. Antioxid. Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, C. 8-hydroxy-2′ -deoxyguanosine (8-OHdG): A Critical Biomarker of Oxidative Stress and Carcinogenesis. J. Environ. Sci. Heal. Part C 2009, 27, 120–139. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eldin, E.E.M.N.; El-Readi, M.Z.; Eldein, M.M.N.; Alfalki, A.A.; Althubiti, M.A.; Kamel, H.F.M.; Eid, S.Y.; Al-Amodi, H.S.; Mirza, A.A. 8-Hydroxy-2′-deoxyguanosine as a Discriminatory Biomarker for Early Detection of Breast Cancer. Clin. Breast Cancer 2019, 19, e385–e393. [Google Scholar] [CrossRef] [PubMed]
- Sova, H.; Jukkolavuorinen, A.; Puistola, U.; Kauppila, S.; Karihtala, P. 8-Hydroxydeoxyguanosine: A new potential independent prognostic factor in breast cancer. Br. J. Cancer 2010, 102, 1018–1023. [Google Scholar] [CrossRef] [Green Version]
- Chuffa, L.G.D.A.; Lupi-Júnior, L.A.; Costa, A.B.; Amorim, J.P.D.A.; Seiva, F.R.F. The role of sex hormones and steroid receptors on female reproductive cancers. Steroids 2017, 118, 93–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalieri, E.; Frenkel, K.; Liehr, J.G.; Rogan, E.; Roy, D. Chapter 4: Estrogens as Endogenous Genotoxic Agents—DNA Adducts and Mutations. J. Natl. Cancer Inst. Monogr. 2000, 2000, 75–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.; Soares, M.; Ramalhinho, A.C.; Moutinho, J.F.; Breitenfeld, L.; Pereira, L. The prognostic value of NRF2 in breast cancer patients: A systematic review with meta-analysis. Breast Cancer Res. Treat. 2019, 179, 523–532. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, H.-J.; Bao, Q.-C.; Wang, L.; Guo, T.-K.; Chen, W.-L.; Xu, L.-L.; Zhou, H.-S.; Bian, J.-L.; Yang, Y.-R.; et al. NRF2 promotes breast cancer cell proliferation and metastasis by increasing RhoA/ROCK pathway signal transduction. Oncotarget 2016, 7, 73593–73606. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Huang, W.-Z.; Wen, Y.-Q.; Yi, Y.-C. Effect of miR-101 on proliferation and oxidative stress-induced apoptosis of breast cancer cells via Nrf2 signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8931–8939. [Google Scholar] [PubMed]
- De Blasio, A.; di Fiore, R.; Pratelli, G.; Drago-Ferrante, R.; Saliba, C.; Baldacchino, S.; Grech, G.; Scerri, C.; Vento, R.; Tesoriere, G. A loop involving NRF2, miR-29b-1-5p and AKT, regulates cell fate of MDA-MB-231 triple-negative breast cancer cells. J. Cell. Physiol. 2020, 235, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wu, X.; Li, F.; Huang, D.; Zhu, W. CDCA4, a downstream gene of the Nrf2 signaling pathway, regulates cell proliferation and apoptosis in the MCF-7/ADM human breast cancer cell line. Mol. Med. Rep. 2017, 17, 1507–1512. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, S.; Wang, X.; Guo, W.; Wang, L.; Zhang, D.; Yuan, L.; Zhang, Z.; Xu, Y.; Liu, S. Disordered signaling governing ferroportin transcription favors breast cancer growth. Cell. Signal. 2015, 27, 168–176. [Google Scholar] [CrossRef]
- Thangasamy, A.; Rogge, J.; Krishnegowda, N.K.; Freeman, J.W.; Ammanamanchi, S. Novel Function of Transcription Factor Nrf2 as an Inhibitor of RON Tyrosine Kinase Receptor-mediated Cancer Cell Invasion. J. Biol. Chem. 2011, 286, 32115–32122. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-F.; Chen, J.-H.; Chang, C.-N.; Lu, D.-Y.; Chang, P.-C.; Wang, S.-L.; Yeh, W.-L. Fisetin inhibits cell migration via inducing HO-1 and reducing MMPs expression in breast cancer cell lines. Food Chem. Toxicol. 2018, 120, 528–535. [Google Scholar] [CrossRef]
- Tertil, M.; Golda, S.; Skrzypek, K.; Florczyk, U.; Weglarczyk, K.; Kotlinowski, J.; Maleszewska, M.; Czauderna, S.; Pichon, C.; Kieda, C.; et al. Nrf2-heme oxygenase-1 axis in mucoepidermoid carcinoma of the lung: Antitumoral effects associated with down-regulation of matrix metalloproteinases. Free. Radic. Biol. Med. 2015, 89, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Chowdhury, S.R.; Mandal, G.; Purohit, S.; Gupta, A.; Bhattacharyya, A. RelA driven co-expression of CXCL13 and CXCR5 is governed by a multifaceted transcriptional program regulating breast cancer progression. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-L.; Zhu, C.-Y.; Wu, Z.-G.; Guo, X.; Zou, W. The oncoprotein HBXIP competitively binds KEAP1 to activate NRF2 and enhance breast cancer cell growth and metastasis. Oncogene 2019, 38, 4028–4046. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.-G.; Choi, B.-H.; Ku, S.-K.; Kwak, M.-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Harder, B.G.; Wong, P.K.; Lang, J.E.; Zhang, D.D. Oxidative stress, mammospheres and Nrf2-new implication for breast cancer therapy? Mol. Carcinog. 2014, 54, 1494–1502. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Liu, D.; Jin, X.; Gao, P.; Wang, Q.; Zhang, J.; Zhang, N. PA-MSHA inhibits the growth of doxorubicin-resistant MCF-7/ADR human breast cancer cells by downregulating Nrf2/p62. Cancer Med. 2016, 5, 3520–3531. [Google Scholar] [CrossRef]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-Differentiation Confers Multidrug Resistance Via Noncanonical PERK-Nrf2 Signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, S.; Ghosh, S.; Mandal, S.; Sau, S.; Pal, M. NRF2 transcriptionally activates the heat shock factor 1 promoter under oxidative stress and affects survival and migration potential of MCF7 cells. J. Biol. Chem. 2018, 293, 19303–19316. [Google Scholar] [CrossRef] [Green Version]
- Syu, J.-P.; Chi, J.-T.; Kung, H.-N. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget 2016, 7, 14659–14672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisi, D.; de Blasio, A.; Drago-Ferrante, R.; di Fiore, R.; Buttitta, G.; Morreale, M.; Scerri, C.; Vento, R.; Tesoriere, G. Parthenolide prevents resistance of MDA-MB231 cells to doxorubicin and mitoxantrone: The role of Nrf2. Cell Death Discov. 2017, 3, 17078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-Y.; Chen, J.; Liu, X.-M.; Zhao, R.; Zhe, H. Nrf2-Mediated Metabolic Reprogramming in Cancer. Oxidative Med. Cell. Longev. 2018, 2018, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, Z.; Du, G.; Sun, H.; Liu, H.; Zhou, Z.; Gou, X.; Wu, X.; Yu, X.; Huang, Y. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1α/Notch1 axis. J. Cell. Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-S.; Du, G.-Y.; Zhang, Z.-G.; Zhou, Z.; Sun, H.-L.; Yu, X.-Y.; Shi, Y.-T.; Xiong, D.-N.; Li, H.; Huang, Y.-H. NRF2 facilitates breast cancer cell growth via HIF1ɑ-mediated metabolic reprogramming. Int. J. Biochem. Cell Biol. 2018, 95, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hallis, S.P.; Jung, K.-A.; Ryu, D.; Kwak, M.-K. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019, 24, 101210. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Singh, A.; Tully, E.; Woo, J.; Le, A.; Nguyen, T.; Biswal, S.; Sharma, D.; Gabrielson, E. Nrf2 signaling and autophagy are complementary in protecting breast cancer cells during glucose deprivation. Free Radic. Biol. Med. 2018, 120, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.A.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Kao, K.-J.; Chang, K.-M.; Hsu, H.-C.; Huang, A.T. Correlation of microarray-based breast cancer molecular subtypes and clinical outcomes: Implications for treatment optimization. BMC Cancer 2011, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Hart, P.C.; Ratti, B.A.; Mao, M.; Ansenberger-Fricano, K.; Shajahan-Haq, A.N.; Tyner, A.L.; Minshall, R.D.; Bonini, M.G. Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget 2015, 7, 308–322. [Google Scholar] [CrossRef] [Green Version]
- Loignon, M.; Miao, W.; Hu, L.; Bier, A.; Bismar, T.A.; Scrivens, P.J.; Mann, K.; Basik, M.; Bouchard, A.; Fiset, P.O.; et al. Cul3 overexpression depletes Nrf2 in breast cancer and is associated with sensitivity to carcinogens, to oxidative stress, and to chemotherapy. Mol. Cancer Ther. 2009, 8, 2432–2440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodera, Y.; Motohashi, H.; Takagi, K.; Miki, Y.; Shibahara, Y.; Watanabe, M.; Ishida, T.; Hirakawa, H.; Sasano, H.; Yamamoto, M.; et al. NRF2 immunolocalization in human breast cancer patients as a prognostic factor. Endocr. Relat. Cancer 2013, 21, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartikainen, J.M.; Tengström, M.; Kosma, V.-M.; Kinnula, V.L.; Mannermaa, A.; Soini, Y. Genetic Polymorphisms and Protein Expression of NRF2 and Sulfiredoxin Predict Survival Outcomes in Breast Cancer. Cancer Res. 2012, 72, 5537–5546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartikainen, J.M.; Tengström, M.; Winqvist, R.; Jukkola-Vuorinen, A.; Pylkäs, K.; Kosma, V.-M.; Soini, Y.; Mannermaa, A. KEAP1 Genetic Polymorphisms Associate with Breast Cancer Risk and Survival Outcomes. Clin. Cancer Res. 2015, 21, 1591–1601. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Li, X.; Yu, Y.; Ma, L.; Liu, S.; Zong, X.; Zheng, Q. SETD7 is a prognosis predicting factor of breast cancer and regulates redox homeostasis. Oncotarget 2017, 8, 94080–94090. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Alcivar, A.L.; Ma, J.; Foo, T.K.; Zywea, S.; Mahdi, A.; Huo, Y.; Kensler, T.W.; Gatza, M.L.; Xia, B. NRF2 Induction Supporting Breast Cancer Cell Survival Is Enabled by Oxidative Stress–Induced DPP3–KEAP1 Interaction. Cancer Res. 2017, 77, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.; Goebel, G.; Hackl, H.; Fiegl, H. Reduced mRNA expression levels of NFE2L2 are associated with poor outcome in breast cancer patients. BMC Cancer 2016, 16, 821. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, E.; Calcabrini, C.; Turrini, E.; Sestili, P.; Fimognari, C. Nrf2: A potential therapeutic target for naturally occurring anticancer drugs? Expert Opin. Ther. Targets 2017, 21, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Vinayak, M. Long Term Effect of Curcumin in Restoration of Tumour Suppressor p53 and Phase-II Antioxidant Enzymes via Activation of Nrf2 Signalling and Modulation of Inflammation in Prevention of Cancer. PLoS ONE 2015, 10, e0124000. [Google Scholar] [CrossRef]
- Shin, J.W.; Chun, K.-S.; Kim, D.-H.; Kim, S.-J.; Kim, S.H.; Cho, N.-C.; Na, H.-K.; Surh, Y.-J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820. [Google Scholar] [CrossRef]
- Rushworth, S.A.; Ogborne, R.M.; Charalambos, C.A.; O’Connell, M.A. Role of protein kinase C δ in curcumin-induced antioxidant response element-mediated gene expression in human monocytes. Biochem. Biophys. Res. Commun. 2006, 341, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Andreadi, C.K.; Howells, L.M.; Atherfold, P.A.; Manson, M.M. Involvement of Nrf2, p38, B-Raf, and Nuclear Factor-κB, but Not Phosphatidylinositol 3-Kinase, in Induction of Hemeoxygenase-1 by Dietary Polyphenols. Mol. Pharmacol. 2005, 69, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Gupta, S.; Maru, G.B. Dietary curcumin modulates transcriptional regulators of phase I and phase II enzymes in benzo[a]pyrene-treated mice: Mechanism of its anti-initiating action. Carcinogenesis 2008, 29, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Samykutty, A.; Jackson, C.; Browning, D.D.; Bollag, W.B.; Thangaraju, M.; Takahashi, S.; Singh, S.R. Curcumin inhibits PhIP induced cytotoxicity in breast epithelial cells through multiple molecular targets. Cancer Lett. 2015, 365, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Zhang, Y.; Wang, Y.; Rao, J.; Jiang, X.; Xu, Z. Curcumin inhibits proliferation of breast cancer cells through Nrf2-mediated down-regulation of Fen1 expression. J. Steroid Biochem. Mol. Biol. 2014, 143, 11–18. [Google Scholar] [CrossRef]
- Li, X.; Xie, W.; Xie, C.; Huang, C.; Zhu, J.; Liang, Z.; Deng, F.; Zhu, M.; Zhu, W.; Wu, R.; et al. Curcumin Modulates miR-19/PTEN/AKT/p53 Axis to Suppress Bisphenol A-induced MCF-7 Breast Cancer Cell Proliferation. Phytother. Res. 2014, 28, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Foygel, K.; Sekar, T.V.; Paulmurugan, R. Monitoring the Antioxidant Mediated Chemosensitization and ARE-Signaling in Triple Negative Breast Cancer Therapy. PLoS ONE 2015, 10, e0141913. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Miao, W.; Loignon, M.; Kandouz, M.; Batist, G. Putative chemopreventive molecules can increase Nrf2-regulated cell defense in some human cancer cell lines, resulting in resistance to common cytotoxic therapies. Cancer Chemother. Pharmacol. 2009, 66, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive oxygen species and cancer paradox: To promote or to suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol commonly displays hormesis: Occurrence and biomedical significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef] [PubMed]
- Rai, G.; Mishra, S.; Suman, S.; Shukla, Y. Resveratrol improves the anticancer effects of doxorubicin in vitro and in vivo models: A mechanistic insight. Phytomedicine 2016, 23, 233–242. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, Y.; Wang, J.; Wang, X.; Chen, C.; Yin, D.; Zhao, F.; Yin, J.; Guo, M.; Zhang, L.; et al. Resveratrol represses estrogen-induced mammary carcinogenesis through NRF2-UGT1A8-estrogen metabolic axis activation. Biochem. Pharmacol. 2018, 155, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Shoulson, R.; Chatterjee, A.; Ronghe, A.; Bhat, N.K.; Dim, D.C.; Bhat, H.K. Resveratrol inhibits estrogen-induced breast carcinogenesis through induction of NRF2-mediated protective pathways. Carcinogenesis 2014, 35, 1872–1880. [Google Scholar] [CrossRef] [Green Version]
- Sabzichi, M.; Hamishehkar, H.; Ramezani, F.; Sharifi, S.; Tabasinezhad, M.; Pirouzpanah, M.; Ghanbari, P.; Samadi, N. Luteolin-loaded phytosomes sensitize human breast carcinoma MDA-MB 231 cells to doxorubicin by suppressing Nrf2 mediated signalling. Asian Pac. J. Cancer Prev. 2014, 15, 5311–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Zhang, F.; Sun, Z.; Zhou, W.; Li, Z.-Y.; You, Q.-D.; Guo, Q.-L.; Hu, R. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol. Carcinog. 2013, 52, 824–834. [Google Scholar] [CrossRef]
- Marvin 20.20.0. ChemAxon. 2020. Available online: https://www.chemaxon.com (accessed on 26 February 2021).
- Jasser, S.A.; Blask, D.E.; Brainard, G.C. Light During Darkness and Cancer: Relationships in Circadian Photoreception and Tumor Biology. Cancer Causes Control 2006, 17, 515–523. [Google Scholar] [CrossRef]
- Hill, S.M.; Belancio, V.P.; Dauchy, R.T.; Xiang, S.; Brimer, S.; Mao, L.; Hauch, A.; Lundberg, P.W.; Summers, W.; Yuan, L.; et al. Melatonin: An inhibitor of breast cancer. Endocr. Relat. Cancer 2015, 22, R183–R204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuffa, L.G.D.A.; Carvalho, R.F.; Justulin, L.A.; Cury, S.S.; Seiva, F.R.F.; Jardim-Perassi, B.V.; Zuccari, D.A.P.D.C.; Reiter, R.J. A meta-analysis of microRNA networks regulated by melatonin in cancer: Portrait of potential candidates for breast cancer treatment. J. Pineal Res. 2020, 69, 12693. [Google Scholar] [CrossRef]
- Chuffa, L.G.D.A.; Seiva, F.R.F.; Cucielo, M.S.; Silveira, H.S.; Reiter, R.J.; Lupi, L.A. Mitochondrial functions and melatonin: A tour of the reproductive cancers. Cell. Mol. Life Sci. 2019, 76, 837–863. [Google Scholar] [CrossRef]
- Li, B.; Feng, X.J.; Hu, X.Y.; Chen, Y.P.; Sha, J.C.; Zhang, H.Y.; Fan, H.-G. Effect of melatonin on attenuating the isoflurane-induced oxidative damage is related to PKCα/Nrf2 signaling pathway in developing rats. Brain Res. Bull. 2018, 143, 9–18. [Google Scholar] [CrossRef]
- Shi, S.; Lei, S.; Tang, C.; Wang, K.; Xia, Z. Melatonin attenuates acute kidney ischemia/reperfusion injury in diabetic rats by activation of the SIRT1/Nrf2/HO-1 signaling pathway. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wei, Z.; Liu, W.; Wang, J.; He, X.; Huang, H.; Zhang, J.; Yang, Z. Melatonin protects against arsenic trioxide-induced liver injury by the upregulation of Nrf2 expression through the activation of PI3K/AKT pathway. Oncotarget 2016, 8, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Janjetovic, Z.; Jarrett, S.G.; Lee, E.F.; Duprey, C.; Reiter, R.J.; Slominski, A.T. Melatonin and its metabolites protect human melanocytes against UVB-induced damage: Involvement of NRF2-mediated pathways. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Jena, G.; Tikoo, K.; Kumar, V. Melatonin modulated autophagy and Nrf2 signaling pathways in mice with colitis-associated colon carcinogenesis. Mol. Carcinog. 2016, 55, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin prevents senescence of canine adipose-derived mesenchymal stem cells through activating NRF2 and inhibiting ER stress. Aging 2018, 10, 2954–2972. [Google Scholar] [CrossRef]
- Wang, M.; Xue, Y.; Shen, L.; Qin, P.; Sang, X.; Tao, Z.; Yi, J.; Wang, J.; Liu, P.; Cheng, H. Inhibition of SGK1 confers vulnerability to redox dysregulation in cervical cancer. Redox Biol. 2019, 24, 101225. [Google Scholar] [CrossRef]
- Paroni, R.; Terraneo, L.; Bonomini, F.; Finati, E.; Virgili, E.; Bianciardi, P.; Favero, G.; Fraschini, F.; Reiter, R.J.; Rezzani, R.; et al. Antitumour activity of melatonin in a mouse model of human prostate cancer: Relationship with hypoxia signalling. J. Pineal Res. 2014, 57, 43–52. [Google Scholar] [CrossRef]
- Reiter, R.J.; Rosales-Corral, S.A.; Tan, D.-X.; Acuna-Castroviejo, D.; Qin, L.; Yang, S.-F.; Xu, K. Melatonin, a Full Service Anti-Cancer Agent: Inhibition of Initiation, Progression and Metastasis. Int. J. Mol. Sci. 2017, 18, 843. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tascioglu Aliyev, A.; Panieri, E.; Stepanić, V.; Gurer-Orhan, H.; Saso, L. Involvement of NRF2 in Breast Cancer and Possible Therapeutical Role of Polyphenols and Melatonin. Molecules 2021, 26, 1853. https://doi.org/10.3390/molecules26071853
Tascioglu Aliyev A, Panieri E, Stepanić V, Gurer-Orhan H, Saso L. Involvement of NRF2 in Breast Cancer and Possible Therapeutical Role of Polyphenols and Melatonin. Molecules. 2021; 26(7):1853. https://doi.org/10.3390/molecules26071853
Chicago/Turabian StyleTascioglu Aliyev, Alev, Emiliano Panieri, Višnja Stepanić, Hande Gurer-Orhan, and Luciano Saso. 2021. "Involvement of NRF2 in Breast Cancer and Possible Therapeutical Role of Polyphenols and Melatonin" Molecules 26, no. 7: 1853. https://doi.org/10.3390/molecules26071853