
Characterization of Natural and Alkaline-Oxidized Proanthocyanidins in Plant Extracts by Ultrahigh-Resolution UHPLC-MS/MS





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
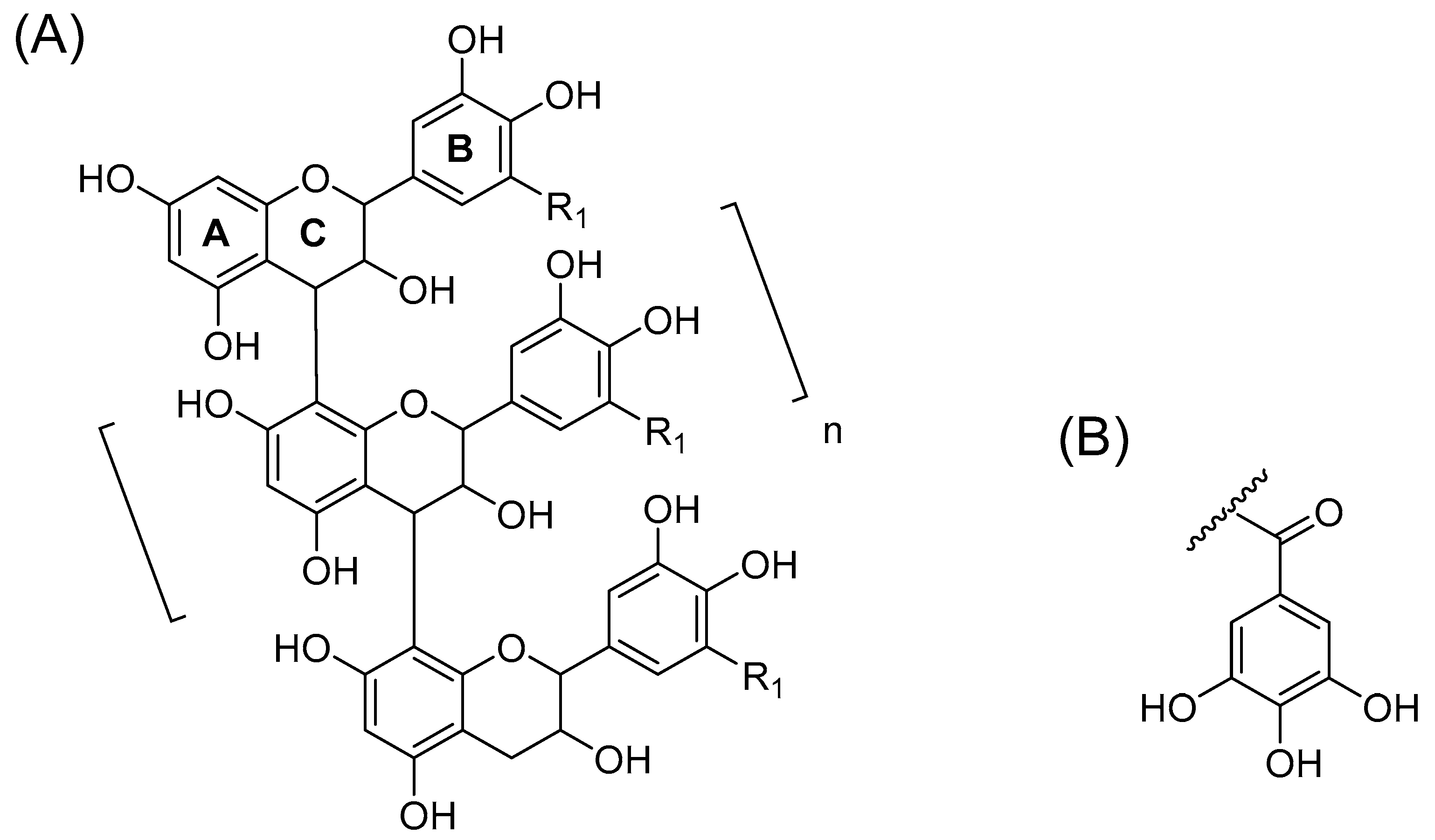
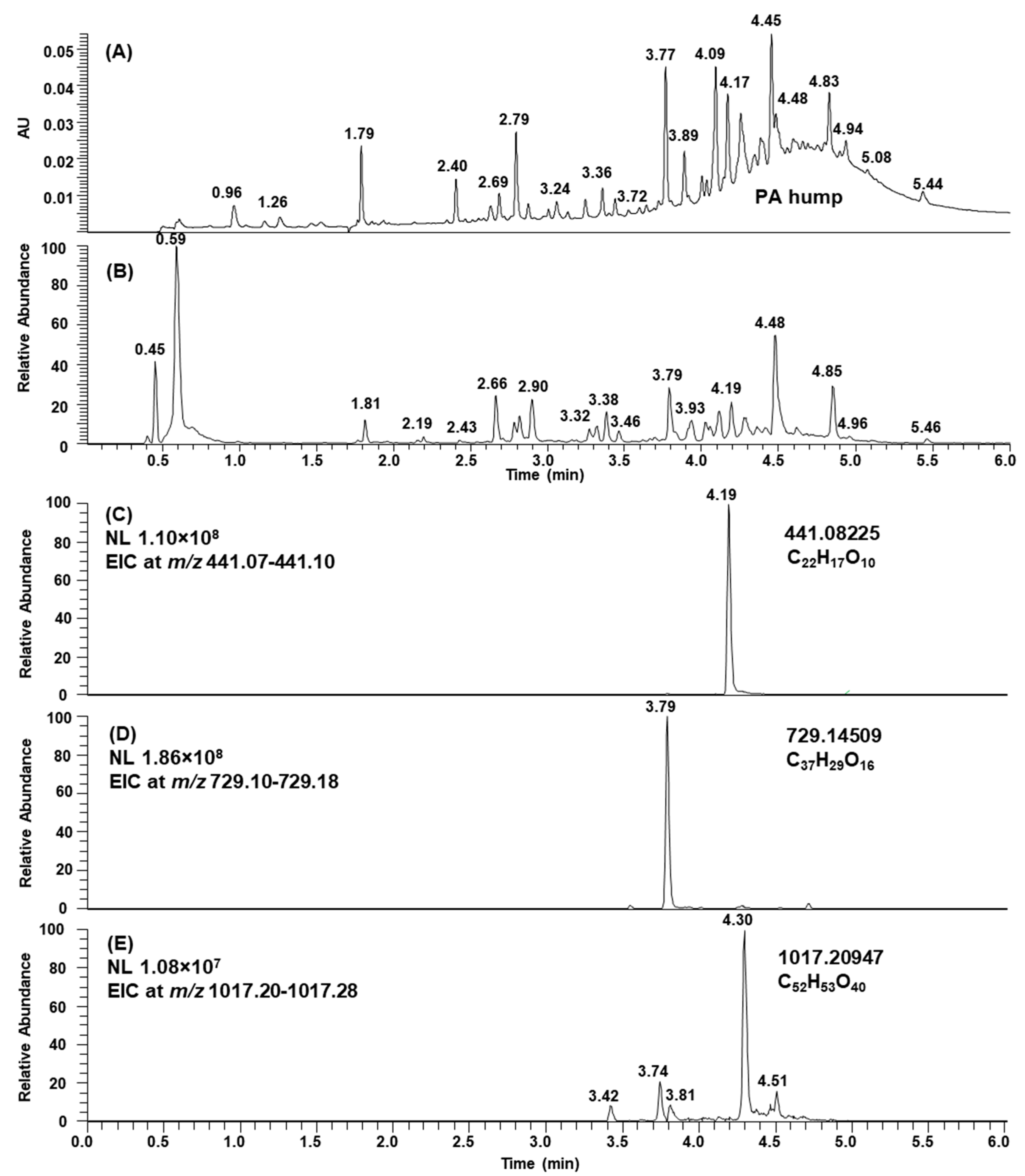
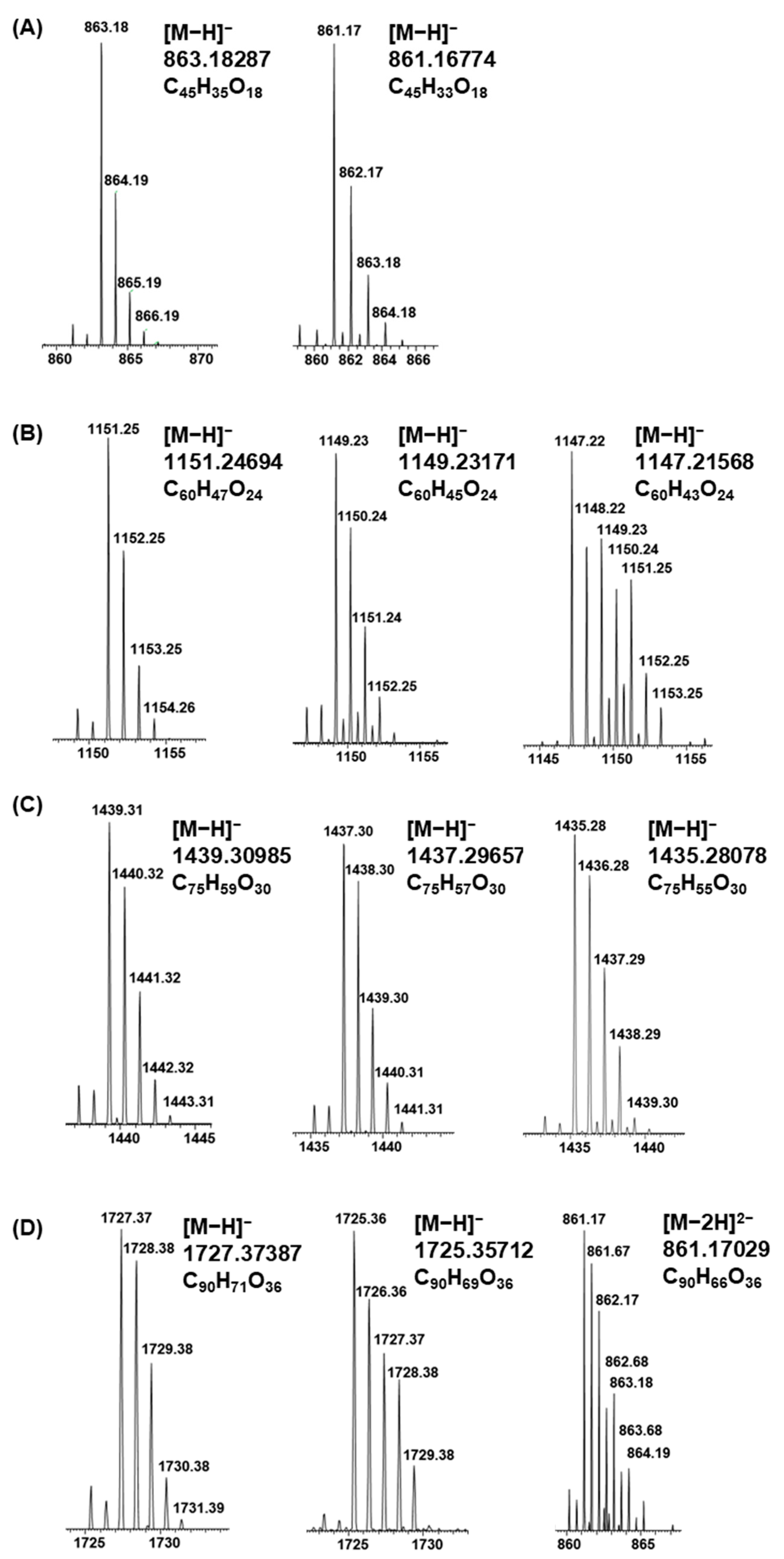
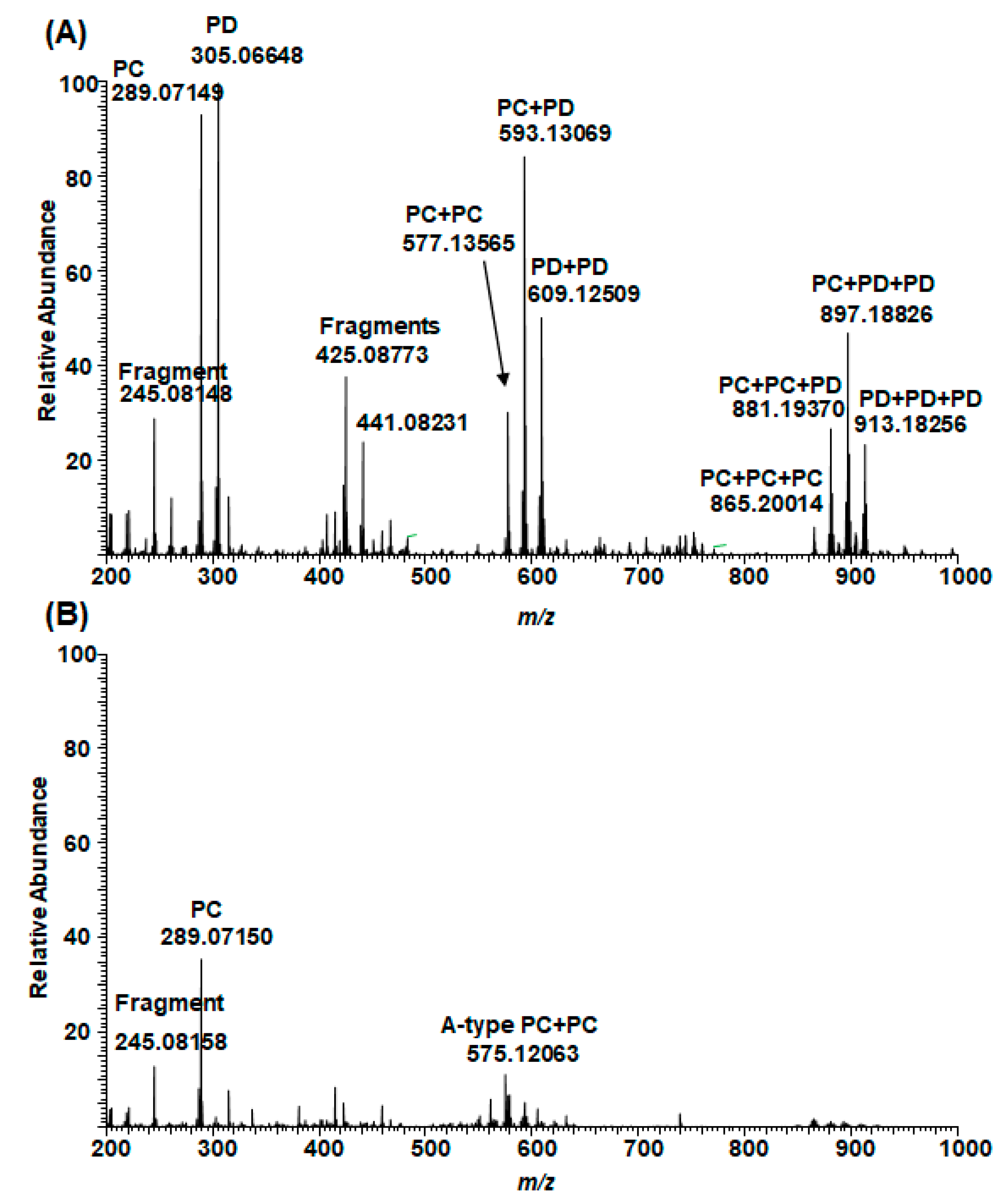
2.1. B-Type PCs in the Initial Non-Oxidized Plant Extracts
2.2. A-Type PCs in the Initial Non-Oxidized Plant Extracts
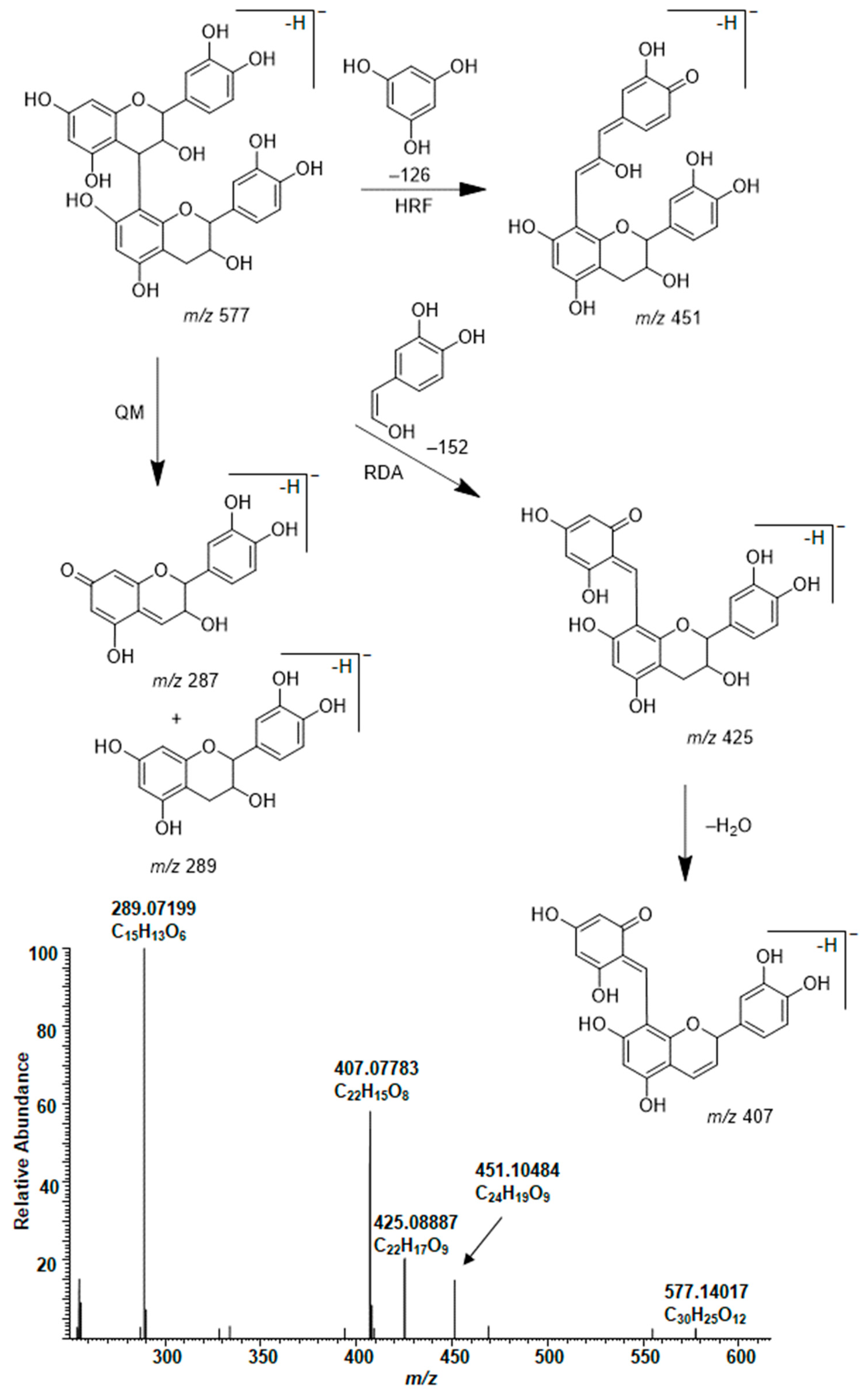
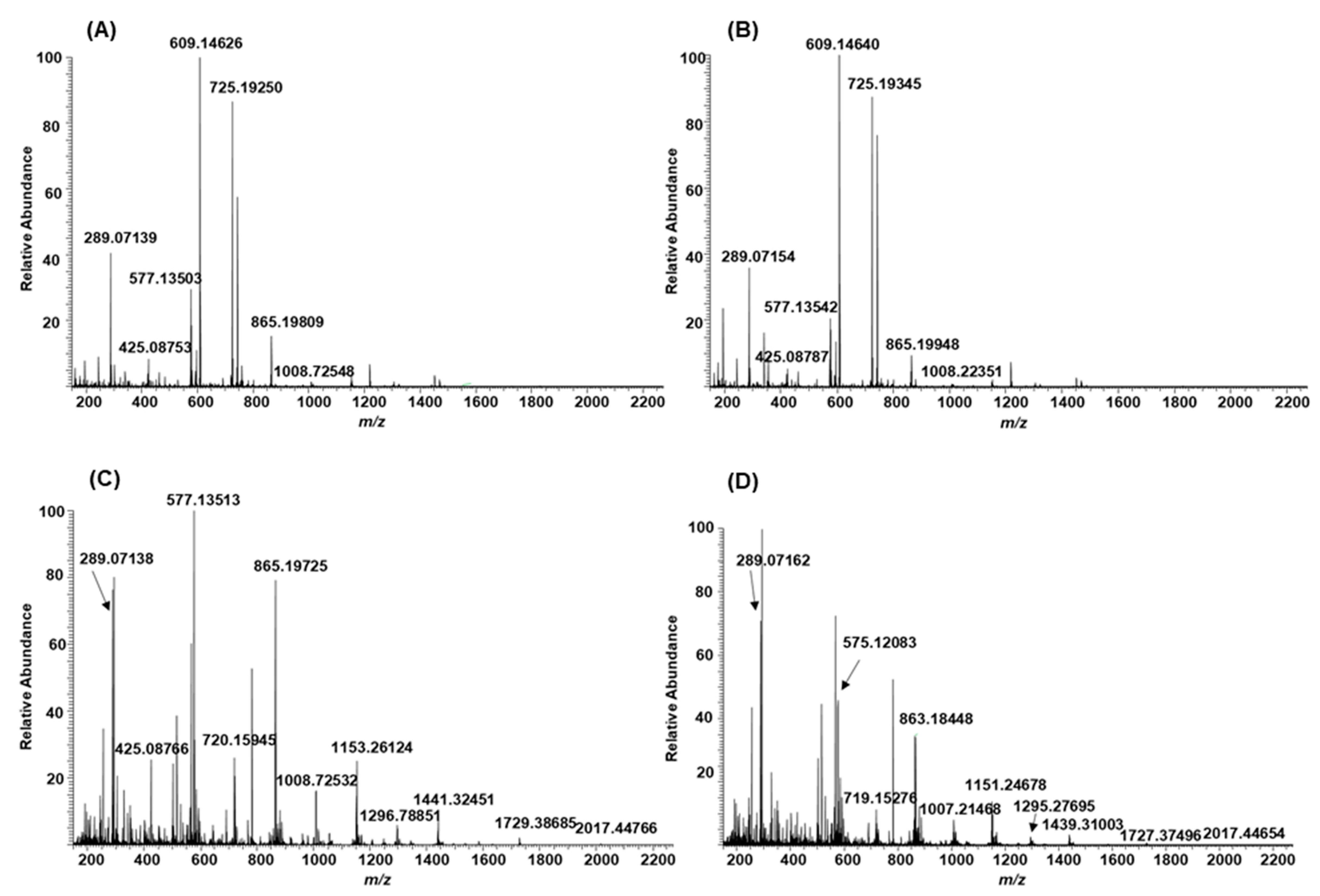
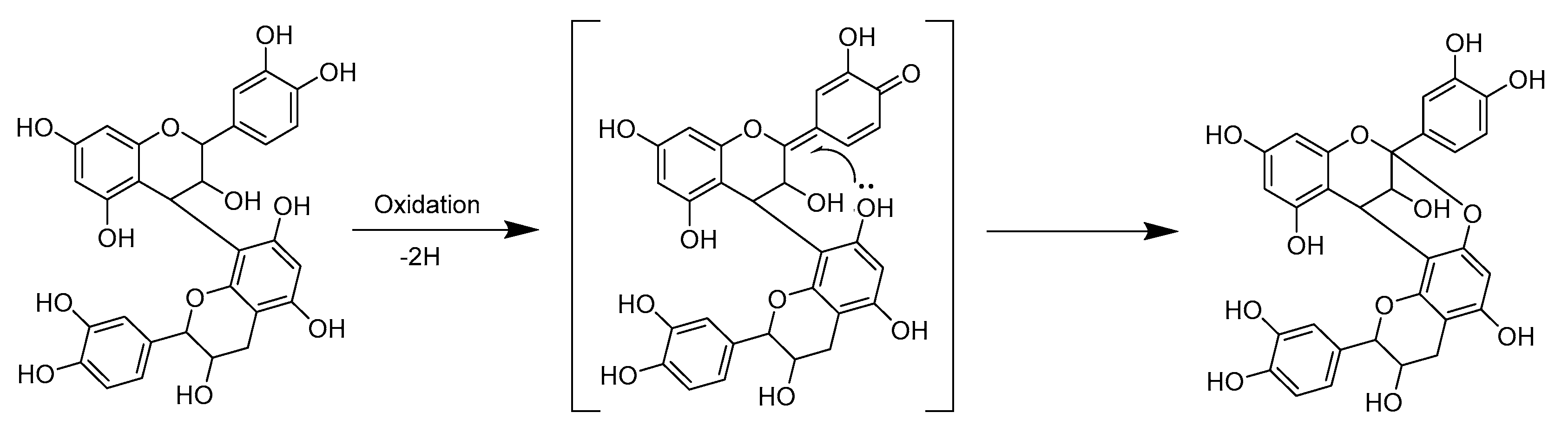
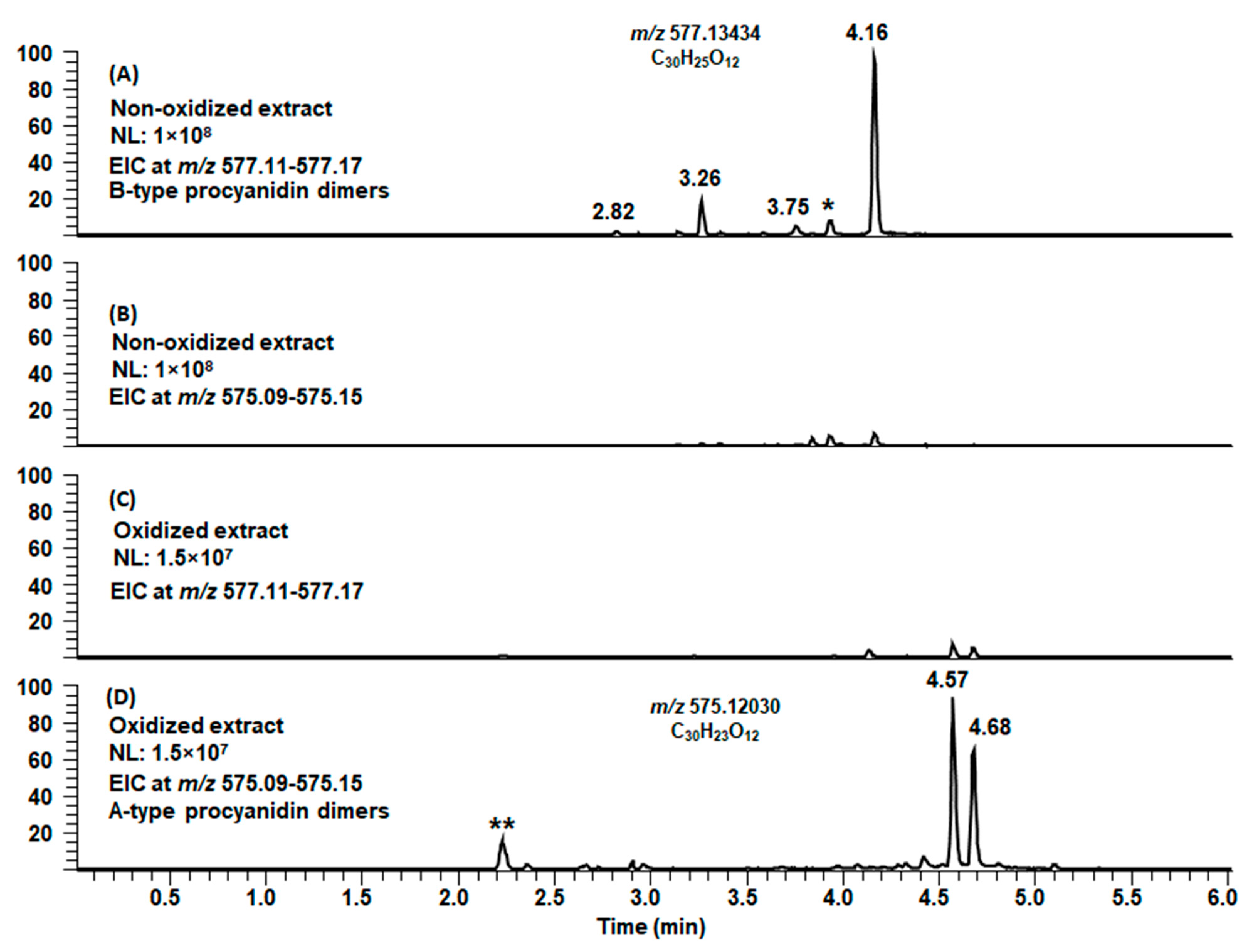
2.3. Modifications of B-Type PCs in Plant Extracts Due to the Alkaline Oxidation
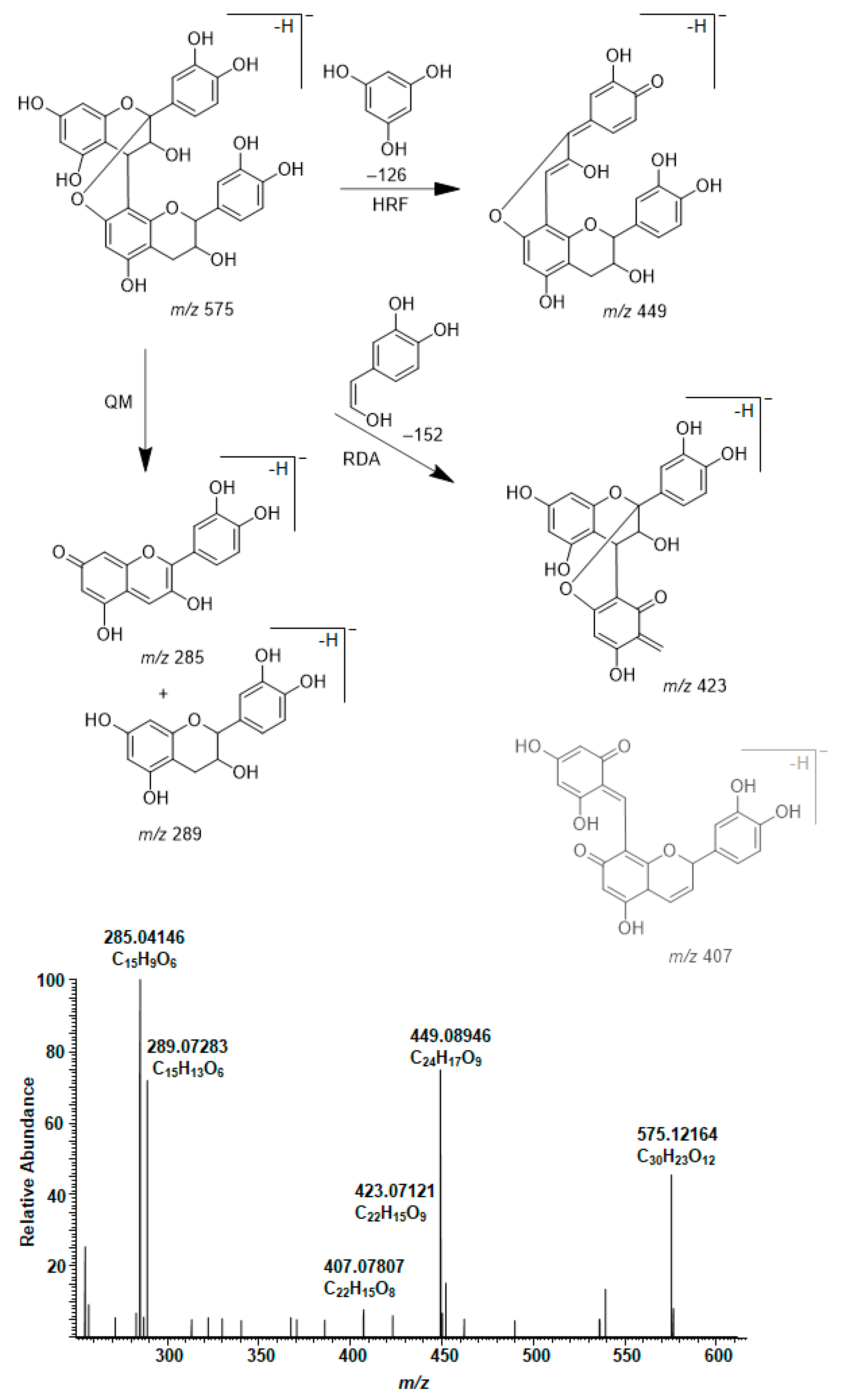
2.4. Modifications of A-Type PCs in Plant Extracts due to the Alkaline Oxidation
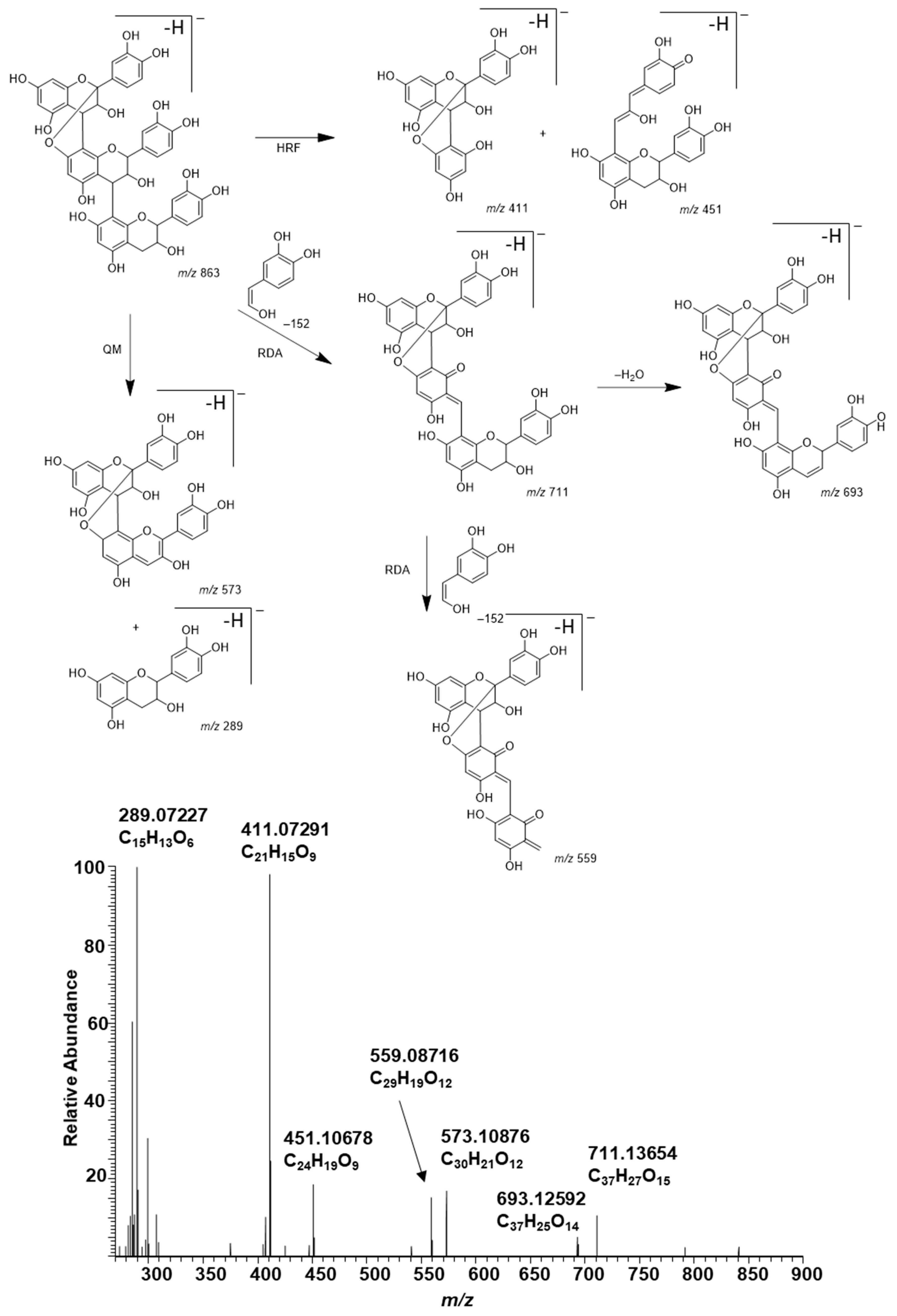
2.5. Plant Extracts with PAs Having Both PC and PD Subunits and Their Modifications Due to the Alkaline Oxidation
2.6. Galloylated PAs in Plant Extracts and Their Modifications Due to the Alkaline Oxidation
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hemingway, R.W. Structural variation in proanthocyanidins and their derivatives. In Chemistry and Significance of Condensed Tannins; Hemingway, R.W., Karchesy, J.J., Eds.; Plenum Press: New York, NY, USA, 1989; pp. 83–107. [Google Scholar]
- Yazaki, Y. Utilization of flavonoid compounds from bark and wood: A review. Nat. Prod. Commun. 2015, 10, 513–520. [Google Scholar] [CrossRef]
- Tannin Market Size, Share & Trends Analysis Report by Sources (Plants, Brown Algae), by Product (Hydrolysable, Non-Hydrolysable, Phlorotannins), by Application (Leather Tanning, Wine Production, Wood Adhesives), & Segment Forecasts, 2014–2015. Available online: https://www.grandviewresearch.com/industry-analysis/tannin-market (accessed on 5 February 2021).
- Prigione, V.; Spina, F.; Tigini, V.; Giovando, S.; Varese, G.C. Biotransformation of industrial tannins by filamentous fungi. Appl. Microbiol. Biotechnol. 2018, 102, 10361–10375. [Google Scholar] [CrossRef]
- Pizzi, A. Tannin-based adhesives. J. Macromol. Sci. Part C 1980, 18, 247–325. [Google Scholar] [CrossRef]
- Pizzi, A. Tannins: Prospectives and actual industrial applications. Biomolecules 2019, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Kumar, S. Applications of tannins in industry. In Tannins–Structural Properties, Biological Properties and Current Knowledge; Aires, A., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Kumar, R.S.; Manimegalai, G. Fruit and vegetable processing industries and environment. In Industrial Pollution & Management; Kumar, A., Ed.; APH Publishing Corporation: New Delhi, India, 2004; pp. 97–117. [Google Scholar]
- Federici, F.; Fava, F.; Kalogerakis, N.; Mantzavinos, D. Valorisation of agro-industrial by-products, effluents and waste: Concept, opportunities and the case of olive mill waste waters. J. Chem. Technol. Biotechnol. 2009, 84, 895–900. [Google Scholar] [CrossRef]
- Aires, A.; Carvalho, R.; Saavedra, M.J. Valorization of solid wastes from chestnut industry processing: Extraction and optimization of polyphenols, tannins and ellagitannins and its potential for adhesives, cosmetic and pharmaceutical industry. Waste Manag. 2016, 48, 457–464. [Google Scholar] [CrossRef] [PubMed]
- de Hoyos-Martínez, P.L.; Merle, J.; Labidi, J.; Charrier-El Bouhtoury, F. Tannins extraction: A key point for their valorization and cleaner production. J. Clean. Prod. 2019, 206, 1138–1155. [Google Scholar] [CrossRef]
- García, D.E.; Glasser, W.G.; Pizzi, A.; Paczkowski, S.P.; Laborie, M.-P. Modification of condensed tannins: From polyphenol chemistry to materials engineering. New J. Chem. 2016, 40, 36–49. [Google Scholar] [CrossRef]
- Imran, I.; Karonen, M.; Salminen, J.-P.; Engström, M.T. Modification of natural proanthocyanidin oligomers and polymers via chemical oxidation under alkaline conditions. ACS Omega 2021, 6, 4726–4739. [Google Scholar] [CrossRef] [PubMed]
- Mouls, L.; Fulcrand, H. UPLC-ESI-MS study of the oxidation markers released from tannin depolymerization: Toward a better characterization of the tannin evolution over food and beverage processing. J. Mass Spectrom. 2012, 47, 1450–1457. [Google Scholar] [CrossRef]
- Ferreira, D.; Steynberg, J.P.; Burger, J.F.W.; Bezuidenhoudt, B.C.B. Oxidation and rearrangement reactions of condensed tannins. In Plant Polyphenols: Synthesis, Properties, Significance; Hemingway, R., Laks, P., Branham, S., Eds.; Plenum: New York, NY, USA, 1992; pp. 349–384. [Google Scholar]
- Laks, P.E.; Hemingway, R.W.; Conner, A.H. Condensed tannins. Base-catalysed reactions of polymeric procyanidins with phloroglucinol: Intramolecular rearrangements. J. Chem. Soc. Perkin Trans. Chem. 1987, 1875–1881. [Google Scholar] [CrossRef]
- Burger, J.F.W.; Kolodziej, H.; Hemingwayc, R.W.; Steynberg, J.P.; Young, D.A.; Ferreira, D. Oligomeric flavanoids. Part 15a. Base-catalyzed pyran rearrangements of procyanidin B-2, and evidence for the oxidative transformation of B- to A-type procyanidins. Tetrahedron 1990, 46, 5733–5740. [Google Scholar] [CrossRef]
- Hibi, Y.; Yanase, E. Oxidation of procyanidins with various degrees of condensation: Influence on the color-deepening phenomenon. J. Agric. Food Chem. 2019, 67, 4940–4946. [Google Scholar] [CrossRef]
- Vázquez, G.; Antorrena, G.; Parajó, J. Studies on the utilization of Pinus pinaster bark. Wood Sci. Technol. 1987, 21, 155–166. [Google Scholar] [CrossRef]
- Fradinho, D.M.; Pascoal Neto, C.; Evtuguin, D.; Jorge, F.C.; Irle, M.A.; Gil, M.H.; Pedrosa de Jesus, J. Chemical characterisation of bark and of alkaline bark extracts from maritime pine grown in Portugal. Ind. Crops Prod. 2002, 16, 23–32. [Google Scholar] [CrossRef]
- Salminen, J.-P.; Karonen, M. Chemical ecology of tannins and other phenolics: We need a change in approach. Funct. Ecol. 2011, 25, 325–338. [Google Scholar] [CrossRef]
- Vihakas, M.; Pälijärvi, M.; Karonen, M.; Roininen, H.; Salminen, J.-P. Rapid estimation of the oxidative activities of individual phenolics in crude plant extracts. Phytochemistry 2014, 103, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Pälijärvi, M.; Karonen, M.; Salminen, J.-P. Oxidatively active plant phenolics detected by UHPLC-DAD-MS after enzymatic and alkaline oxidation. J. Chem. Ecol. 2018, 44, 483–496. [Google Scholar] [CrossRef]
- Poncet-Legrand, C.; Cabane, B.; Bautista-Ortín, A.B.; Carrillo, S.; Fulcrand, H.; Pérez, J.; Vernhet, A. Tannin oxidation: Intra-versus intermolecular reactions. Biomacromolecules 2010, 11, 2376–2386. [Google Scholar] [CrossRef]
- Vernhet, A.; Carrillo, S.; Poncet-Legrand, C. Condensed tannin changes induced by autoxidation: Effect of the initial degree of polymerization and concentration. J. Agric. Food Chem. 2014, 62, 7833–7842. [Google Scholar] [CrossRef]
- Sui, Y.; Zheng, Y.; Li, X.; Li, S.; Xie, B.; Sun, Z. Characterization and preparation of oligomeric procyanidins from Litchi chinensis pericarp. Fitoterapia 2016, 112, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Leppä, M.M.; Karonen, M.; Tähtinen, P.; Engström, M.T.; Salminen, J.-P. Isolation of chemically well-defined semipreparative liquid chromatography fractions from complex mixtures of proanthocyanidin oligomers and polymers. J. Chromatogr. A 2018, 1576, 67–79. [Google Scholar] [CrossRef]
- Lin, L.-Z.; Sun, J.; Chen, P.; Monagas, M.J.; Harnly, J.M. UHPLC-PDA-ESI/HRMSn profiling method to identify and quantify oligomeric proanthocyanidins in plant products. J. Agric. Food Chem. 2014, 62, 9387–9400. [Google Scholar] [CrossRef]
- Navarro-Hoyos, M.; Lebrón-Aguilar, R.; Quintanilla-López, J.E.; Cueva, C.; Hevia, D.; Quesada, S.; Azofeifa, G.; Moreno-Arribas, M.V.; Monagas, M.; Bartolomé, B. Proanthocyanidin characterization and bioactivity of extracts from different parts of Uncaria tomentosa L. (Cat’s Claw). Antioxidants 2017, 6, 12. [Google Scholar] [CrossRef]
- Karonen, M.; Liimatainen, J.; Sinkkonen, J. Birch inner bark procyanidins can be resolved with enhanced sensitivity by hydrophilic interaction HPLC-MS. J. Sep. Sci. 2011, 34, 3158–3165. [Google Scholar] [CrossRef]
- Friedrich, W.; Eberhardt, A.; Galensa, R. Investigation of proanthocyanidins by HPLC with electrospray ionization mass spectrometry. Eur. Food Res. Technol. 2000, 211, 56–64. [Google Scholar] [CrossRef]
- Karonen, M.; Loponen, J.; Ossipov, V.; Pihlaja, K. Analysis of procyanidins in pine bark with reversed-phase and normal-phase high-performance liquid chromatography-electrospray ionization mass spectrometry. Anal. Chim. Acta 2004, 522, 105–112. [Google Scholar] [CrossRef]
- Gu, L.; Kelm, M.A.; Hammerstone, J.F.; Beecher, G.; Holden, J.; Haytowitz, D.; Prior, R.L. Screening of foods containing proanthocyanidins and their structural characterization using LC-MS/MS and thiolytic degradation. J. Agric. Food Chem. 2003, 51, 7513–7521. [Google Scholar] [CrossRef]
- Rue, E.A.; Rush, M.D.; van Breemen, R.B. Procyanidins: A comprehensive review encompassing structure elucidation via mass spectrometry. Phytochem. Rev. 2018, 17, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Hernández, A.M.; Muñiz-Márquez, D.B.; Wong-Paz, J.E.; Aguilar-Zárate, P.; de la Rosa-Hernández, M.; Larios-Cruz, R.; Aguilar, C.N. Characterization by HPLC–ESI–MS2 of native and oxidized procyanidins from litchi (Litchi chinensis) pericarp. Food Chem. 2019, 291, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Gariboldi, E.; Mascetti, D.; Galli, G.; Caballion, P.; Bosisio, E. LC-UV-electrospray-MS-MS mass spectrometry analysis of plant constituents inhibiting xanthine oxidase. Pharm. Res. 1998, 15, 936–943. [Google Scholar] [CrossRef]
- Chen, L.; Yuan, P.; Chen, K.; Jia, Q.; Li, Y. Oxidative conversion of B- to A-type procyanidin trimer: Evidence for quinone methide mechanism. Food Chem. 2014, 154, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Kurihara, M.; Fukuhara, K.; Tanaka, T.; Suzuki, T.; Miyata, N.; Toyoda, M. Conversion of procyanidin B-type (catechin dimer) to A-type: Evidence for abstraction of C-2 hydrogen in catechin during radical oxidation. Tetrahedron Lett. 2000, 41, 485–488. [Google Scholar] [CrossRef]
- Osman, A.M.; Wong, K.K.Y. Laccase (EC 1.10.3.2) catalyses the conversion of procyanidin B-2 (epicatechin dimer) to type A-2. Tetrahedron Lett. 2007, 48, 1163–1167. [Google Scholar] [CrossRef]
- Kothe, L.; Zimmermann, B.F.; Galensa, R. Temperature influences epimerization and composition of flavanol monomers, dimers and trimers during cocoa bean roasting. Food Chem. 2013, 141, 3656–3663. [Google Scholar] [CrossRef]
- Tanaka, T.; Kondou, K.; Kouno, I. Oxidation and epimerization of epigallocatechin in banana fruits. Phytochemistry 2000, 53, 311–316. [Google Scholar] [CrossRef]
- Valcic, S.; Burr, J.A.; Timmermann, B.N.; Liebler, D.C. Antioxidant chemistry of green tea catechins. New oxidation products of (-)-epigallocatechin gallate and (-)-epigallocatechin from their reactions with peroxyl radicals. Chem. Res. Toxicol. 2000, 13, 801–810. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karonen, M.; Imran, I.B.; Engström, M.T.; Salminen, J.-P. Characterization of Natural and Alkaline-Oxidized Proanthocyanidins in Plant Extracts by Ultrahigh-Resolution UHPLC-MS/MS. Molecules 2021, 26, 1873. https://doi.org/10.3390/molecules26071873
Karonen M, Imran IB, Engström MT, Salminen J-P. Characterization of Natural and Alkaline-Oxidized Proanthocyanidins in Plant Extracts by Ultrahigh-Resolution UHPLC-MS/MS. Molecules. 2021; 26(7):1873. https://doi.org/10.3390/molecules26071873
Chicago/Turabian StyleKaronen, Maarit, Iqbal Bin Imran, Marica T. Engström, and Juha-Pekka Salminen. 2021. "Characterization of Natural and Alkaline-Oxidized Proanthocyanidins in Plant Extracts by Ultrahigh-Resolution UHPLC-MS/MS" Molecules 26, no. 7: 1873. https://doi.org/10.3390/molecules26071873
APA StyleKaronen, M., Imran, I. B., Engström, M. T., & Salminen, J.-P. (2021). Characterization of Natural and Alkaline-Oxidized Proanthocyanidins in Plant Extracts by Ultrahigh-Resolution UHPLC-MS/MS. Molecules, 26(7), 1873. https://doi.org/10.3390/molecules26071873