
Nature of the Hydrogen Bond Enhanced Halogen Bond



Abstract
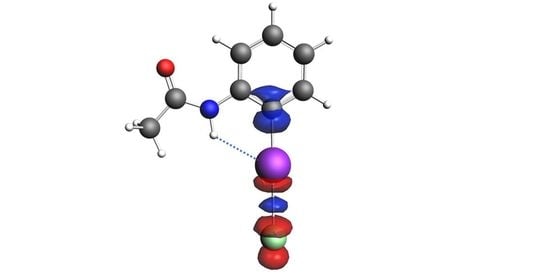
:
1. Introduction
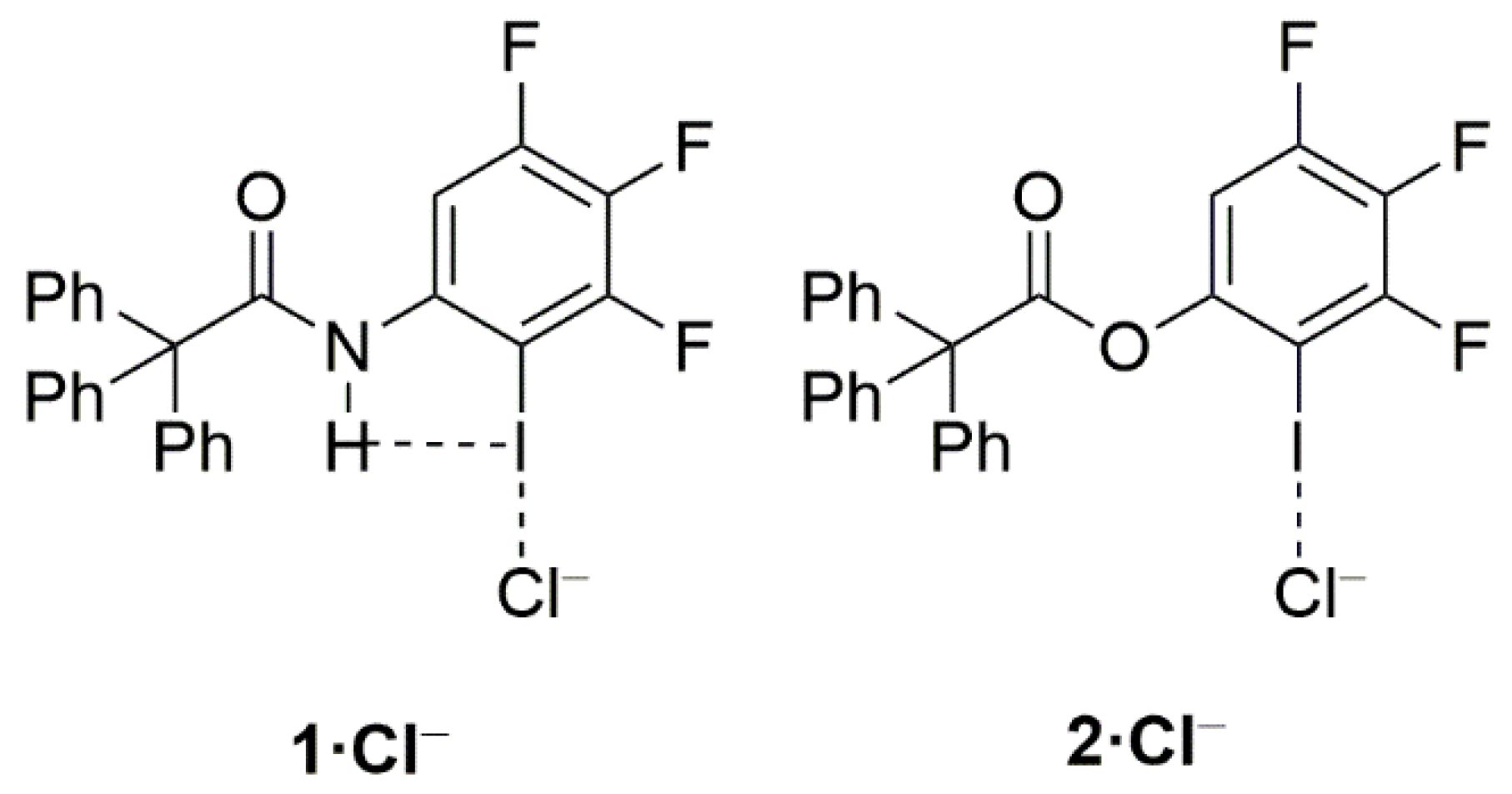
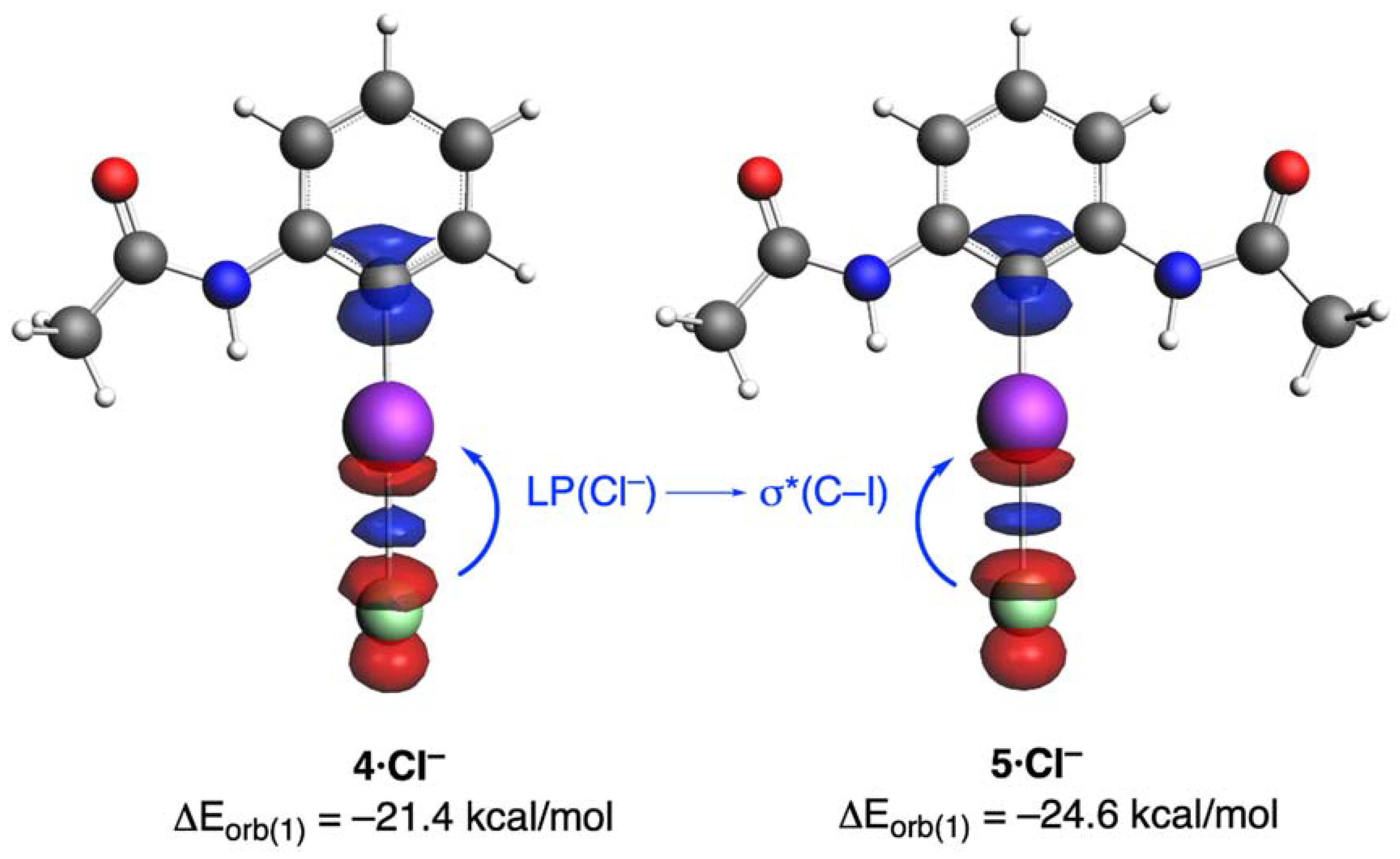
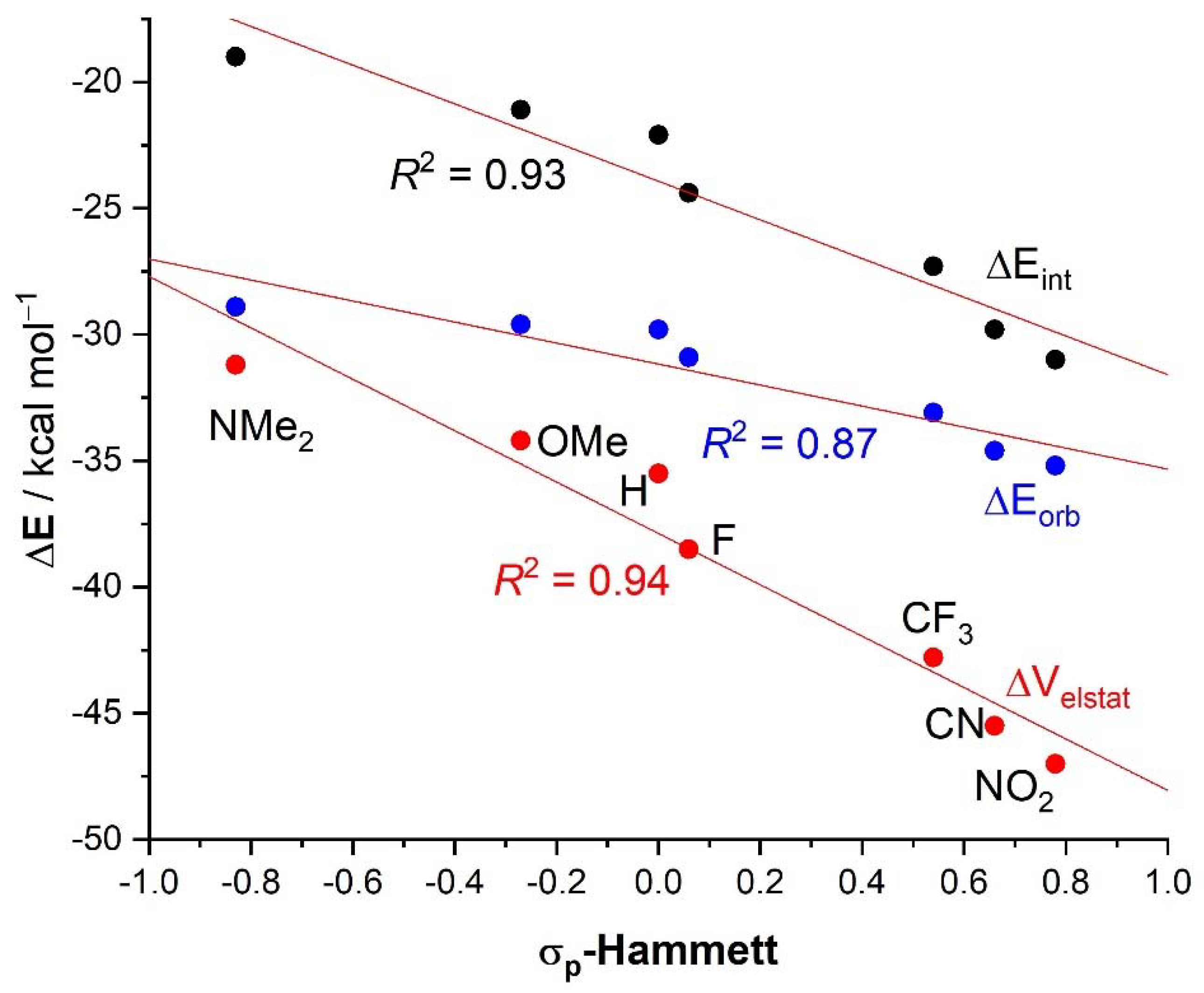
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hobza, P.; Rezác, J. (Eds.) Noncovalent Interactions. Chem. Rev. 2016, 116, 4911–4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halogen Bonding I; Metrangol, P.; Resnati, G. (Eds.) Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Halogen Bonding II; Metrangol, P.; Resnati, G. (Eds.) Springer: Berlin/Heidelberg, Germany, 2015. [Google Scholar]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R.; Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef] [PubMed]
- Pancholi, J.; Beer, P.D. Halogen bonding motifs for anion recognition. Coord. Chem. Rev. 2020, 416, 213281. [Google Scholar] [CrossRef]
- Danelius, E.; Andersson, H.; Jarvoll, P.; Lood, K.; Gräfenstein, J.; Erdélyi, M. Halogen Bonding: A Powerful Tool for Modulation of Peptide Conformation. Biochemistry 2017, 56, 3265–3272. [Google Scholar] [CrossRef]
- Sutar, R.L.; Huber, S.M. Catalysis of Organic Reactions through Halogen Bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Breugst, M.; Koenig, J.J. σ-Hole Interactions in Catalysis. Eur. J. Org. Chem. 2020, 5473–5487. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Decato, D.A.; Riel, A.M.S.; May, J.H.; Bryantsev, V.S.; Berryman, O.B. Theoretical, Solid-State and Solution Quantification of the Hydrogen Bond Enhanced Halogen Bond. Angew. Chem. Int. Ed. 2021, 60, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Landis, C.R.; Weinhold, C.F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Outeiral, C.; Vincent, M.A.; Martín Pendás, A.; Popelier, P.L.A. Revitalizing the concept of bond order through delocalization measures in real space. Chem. Sci. 2018, 9, 5517–5529. [Google Scholar] [CrossRef] [Green Version]
- von Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Zhao, L.; von Hopffgarten, M.; Andrada, D.M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2018, 8, e1345. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. Chem. Open 2012, 1, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Kurczab, R.; Mitoraj, M.P.; Michalak, A.; Ziegler, T. Theoretical analysis of the resonance assisted hydrogen bond based on the combined extended transition state method and natural orbitals for chemical valence scheme. J. Phys. Chem. A 2010, 114, 8581–8590. [Google Scholar] [CrossRef]
- Stasyuk, O.A.; Sedlak, R.; Fonseca Guerra, C.; Hobza, P. Comparison of the DFT-SAPT and Canonical EDA Schemes for the Energy Decomposition of Various Types of Noncovalent Interactions. J. Chem. Theory Comput. 2018, 14, 3440–3450. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Fernández, I.; Bickelhaupt, F.M. How Dihalogens Catalyze Michael Addition Reactions. Angew. Chem. Int. Ed. 2019, 58, 8922–8926. [Google Scholar] [CrossRef] [Green Version]
- Kellett, C.W.; Kennepohl, P.; Berlinguette, C.P. π-Covalency in the halogen bond. Nat. Commun. 2020, 11, 3310. [Google Scholar]
- Politzer, P.; Murray, J.S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Lv, H.; Zhuo, H.-Y.; Li, Q.-Z.; Yang, X.; Li, W.-Z.; Cheng, J.-B. Halogen bonds with N-heterocyclic carbenes as halogen acceptors: A partially covalent character. Mol. Phys. 2014, 112, 3024–3032. [Google Scholar] [CrossRef]
- Sanyal, S.; Esterhuysen, C. Nature of Halogen Bond Adducts of Carbones with XCF3 (X = Cl, Br, I) species. Polyhedron 2021. [Google Scholar] [CrossRef]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A Combined Charge and Energy Decomposition Scheme for Bond Analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef]
- Mayer, R.J.; Ofial, A.R.; Mayr, H.; Legault, C.Y. Lewis Acidity Scale of Diaryliodonium Ions toward Oxygen, Nitrogen, and Halogen Lewis Bases. J. Am. Chem. Soc. 2020, 142, 5221–5233. [Google Scholar] [CrossRef]
- Reinhard, D.L.; Heinen, F.; Stoesser, J.; Engelage, E.; Huber, S.M. Tuning the Halogen Bonding Strength of Cyclic Diaryliodonium Salts. Helv. Chem. Acta 2021, 104, e2000221. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Wittkopp, A.; Schreiner, P.R. Metal-Free, Noncovalent Catalysis of Diels–Alder Reactions by Neutral Hydrogen Bond Donors in Organic Solvents and in Water. Chem. Eur. J. 2003, 9, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M.; Fernández, I. Bifunctional Hydrogen Bond Donor-Catalyzed Diels–Alder Reactions: Origin of Stereoselectivity and Rate Enhancement. Chem. Eur. J. 2021, 27, 5180–5190. [Google Scholar] [CrossRef]
- Jakab, G.; Tancon, C.; Zhang, Z.; Lippert, K.M.; Schreiner, P.R. (Thio)urea Organocatalyst Equilibrium Acidities in DMSO. Org. Lett. 2012, 14, 1724–1727. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. ; et. al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yan, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huzinaga, S.; Klobukowski, M. Well-tempered Gaussian basis sets for the calculation of matrix Hartree-Fock wavefunctions. Chem. Phys. Lett. 1993, 212, 260–264. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll. 2010. Available online: http://tkgristmill.com (accessed on 9 March 2021).
- Shaik, S.S.; Hiberty, P.C. A Chemist’s Guide to Valence Bond. Theory; Wiley: New York, NY, USA, 2007. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Politzer, P. A perspective on quantum mechanics and chemical concepts in describing noncovalent interactions. Phys. Chem. Chem. Phys. 2018, 20, 30076–30082. [Google Scholar] [CrossRef] [PubMed]
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. 2020. Available online: http://www.scm.com (accessed on 9 March 2021).
- Snijders, J.G.; Vernooijs, P.; Baerends, E.J. Roothaan-Hartree-Fock-Slater atomic wave functions: Single-zeta, double-zeta, and extended Slater-type basis sets for 87Fr-103Lr. At. Data Nucl. Data Tables 1981, 26, 483–509. [Google Scholar] [CrossRef]
- Krijn, J.; Baerends, E.J. Fit Functions in the HFS-Method, Internal Report; Vrije Universiteit: Amsterdam, The Netherlands, 1984. (In Dutch) [Google Scholar]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; Ehlers, A.; Baerends, E.J. Geometry optimizations in the zero order regular approximation for relativistic effects. J. Chem. Phys. 1999, 110, 8943–8953. [Google Scholar] [CrossRef] [Green Version]

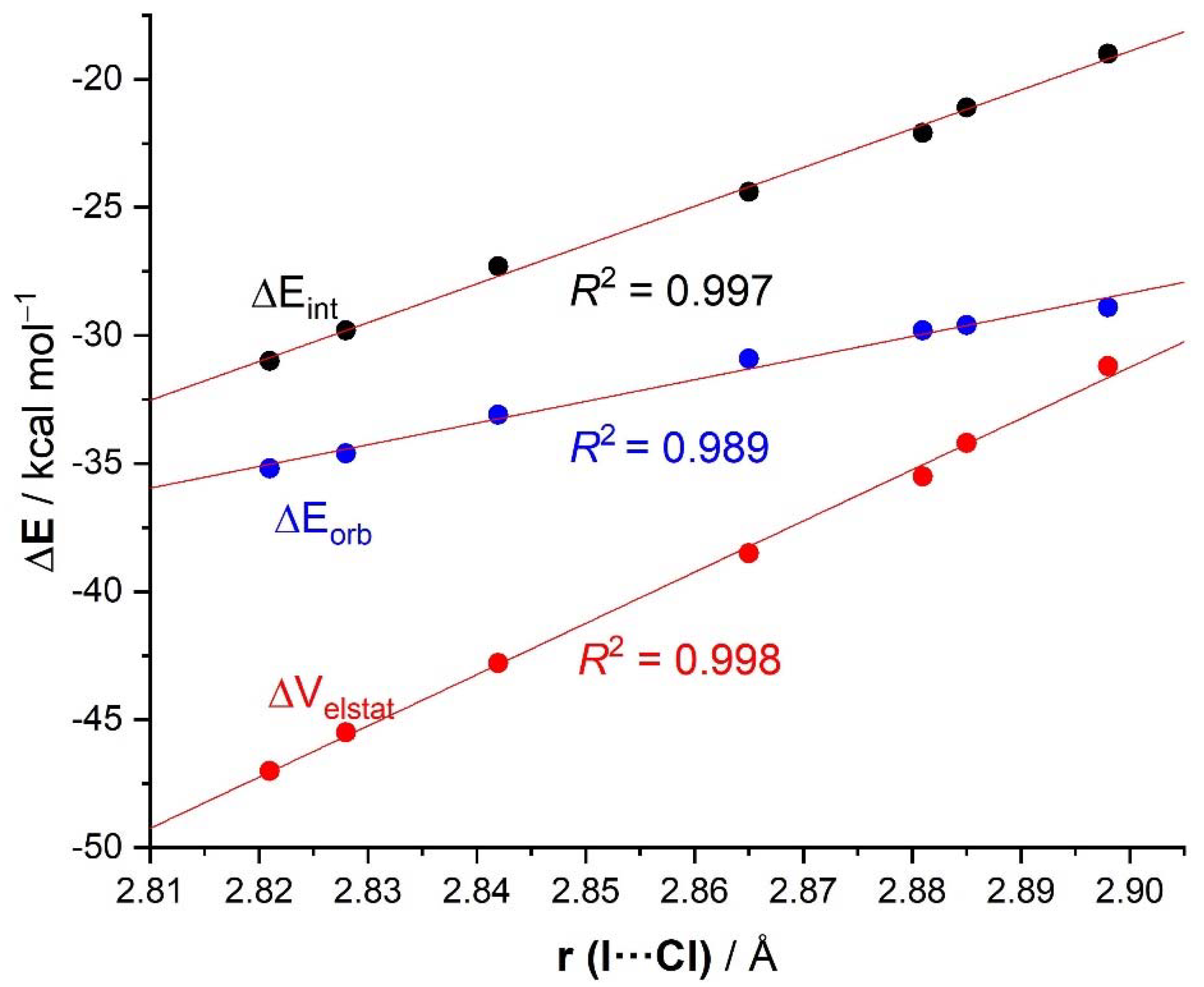

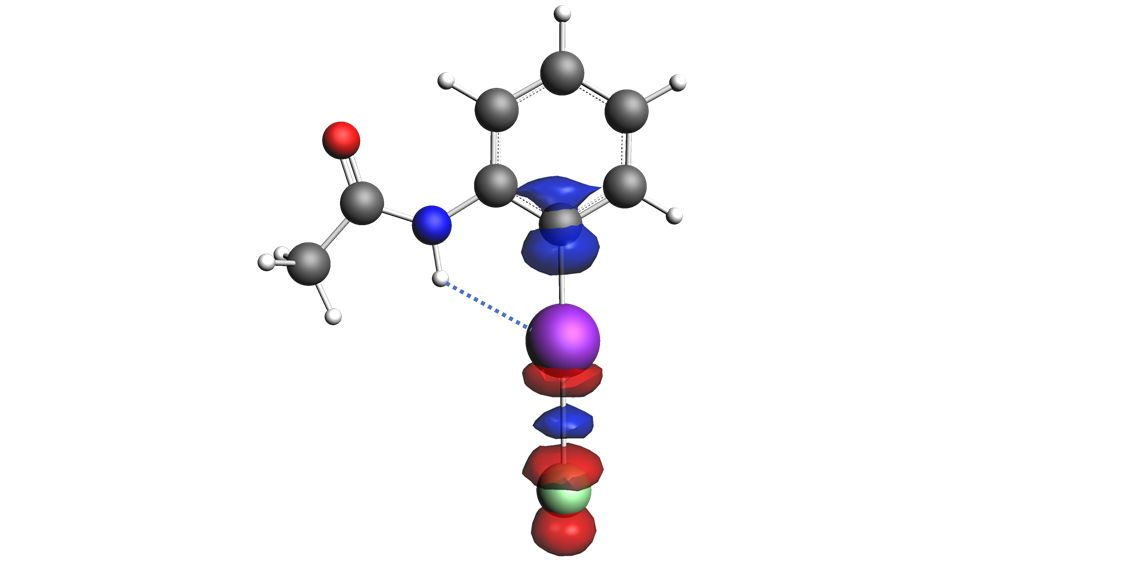{kind=link}
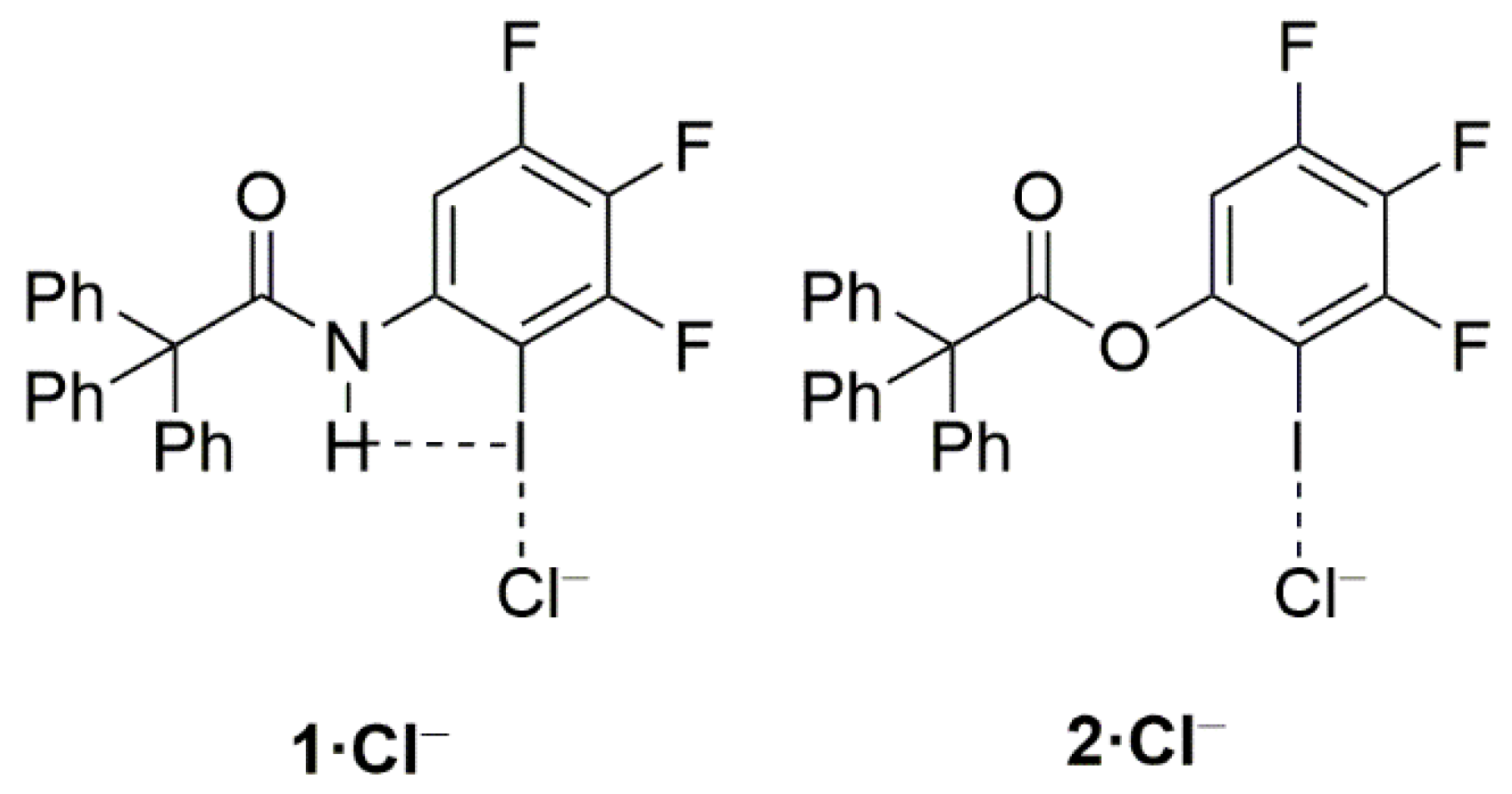{kind=link}
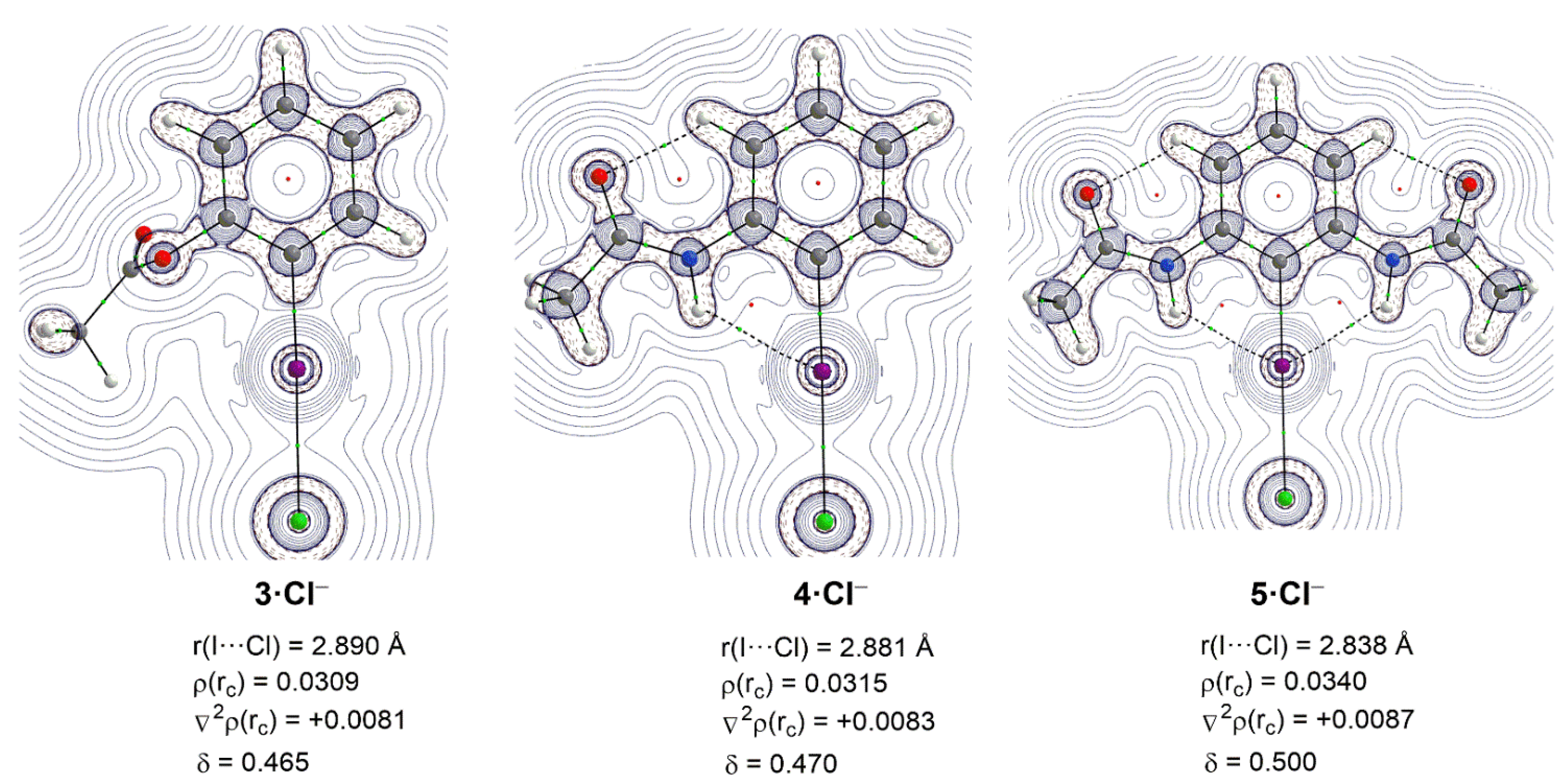{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | 3·Cl− | 4·Cl− | 5·Cl− |
|---|---|---|---|
| ΔEint | −19.0 | −22.1 | −28.8 |
| ΔEPauli | 42.0 | 43.6 | 49.7 |
| ΔVelstat a | −31.8 (52.2%) | −35.5 (54.0%) | −45.1 (57.4%) |
| ΔEorb a | −28.7 (47.0%) | −29.8 (45.4%) | −33.0 (42.0%) |
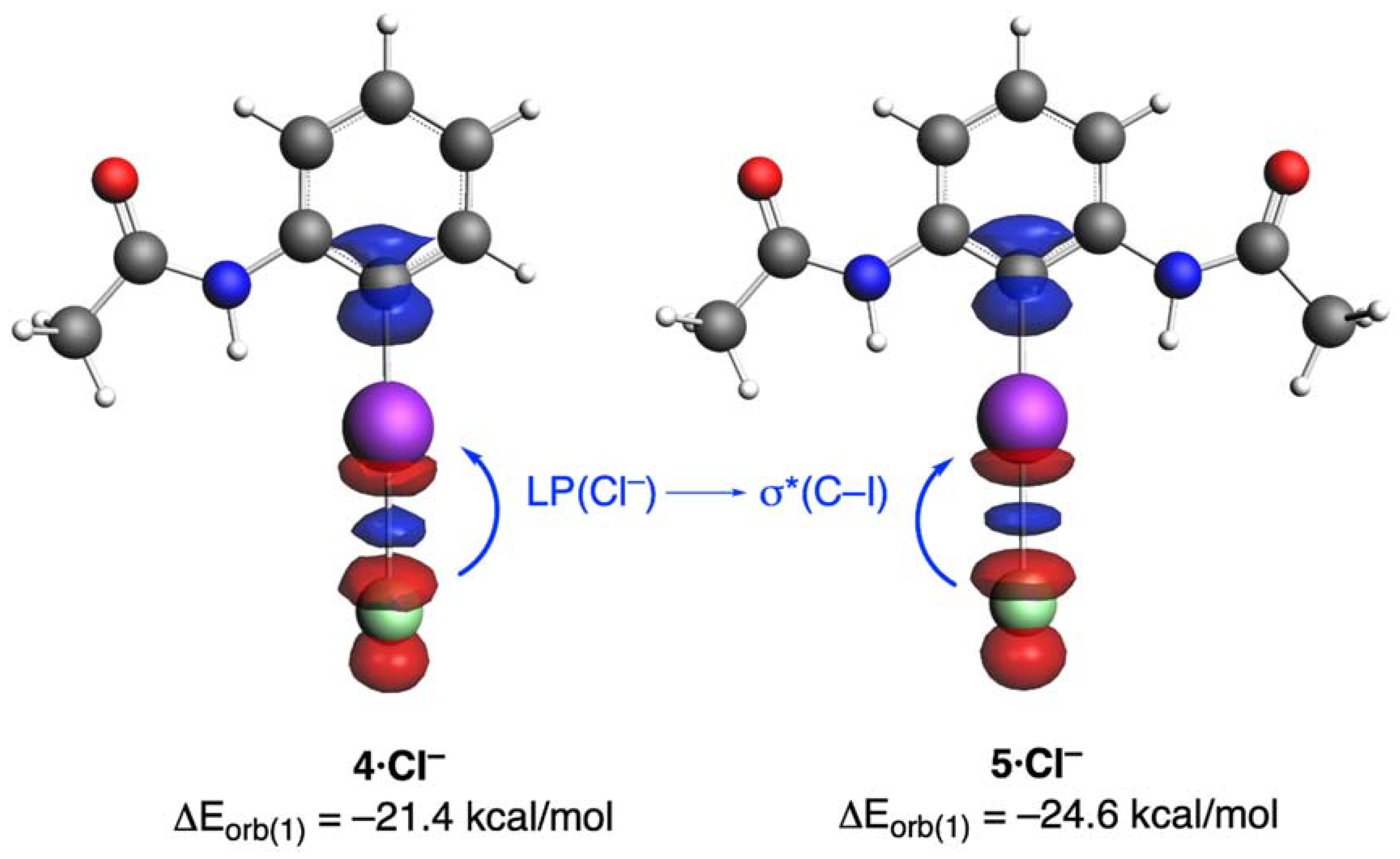
| ΔEorb(1) b | −20.6 (71.8%) | −21.4 (71.8%) | −24.6 (74.5%) |
| ΔEorb(2) b | −2.4 (8.4%) | −2.3 (7.7%) | −2.3 (7.0%) |
| ΔEorb(rest) b | −5.8 (20.2%) | −6.1 (20.5%) | −8.4 (25.5%) |
| ΔEdisp a | −0.5 (0.8%) | −0.4 (0.6%) | −0.5 (0.6%) |
| Complex | 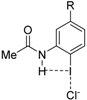 | 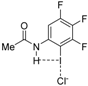 | 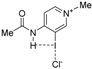 | ||||||
|---|---|---|---|---|---|---|---|---|---|
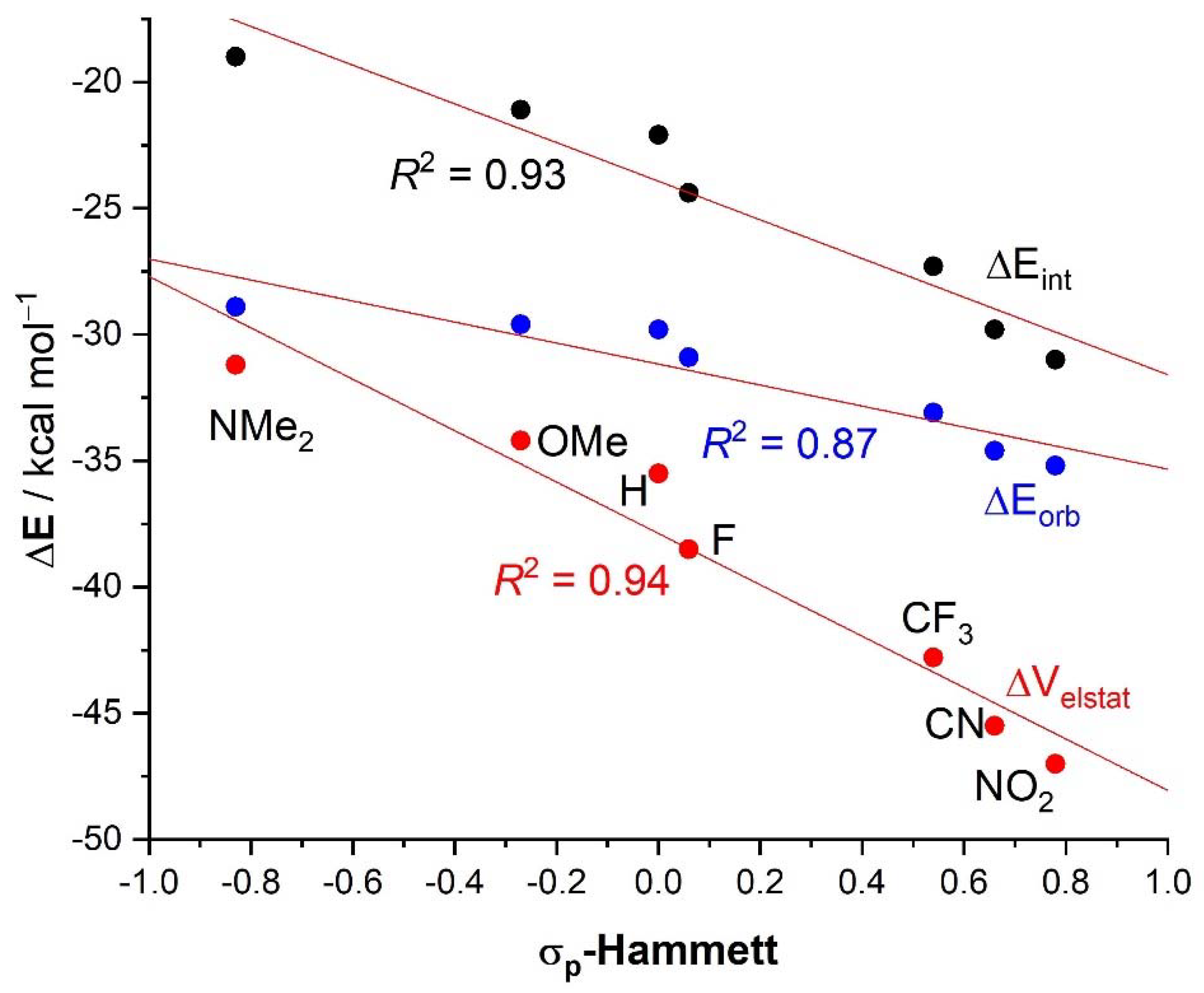
| 4·Cl− | 4−NMe2·Cl− | 4−OMe·Cl− | 4−F·Cl− | 4−CF3·Cl− | 4−CN·Cl− | 4−NO2·Cl− | 1·Cl− | 6·Cl− | |
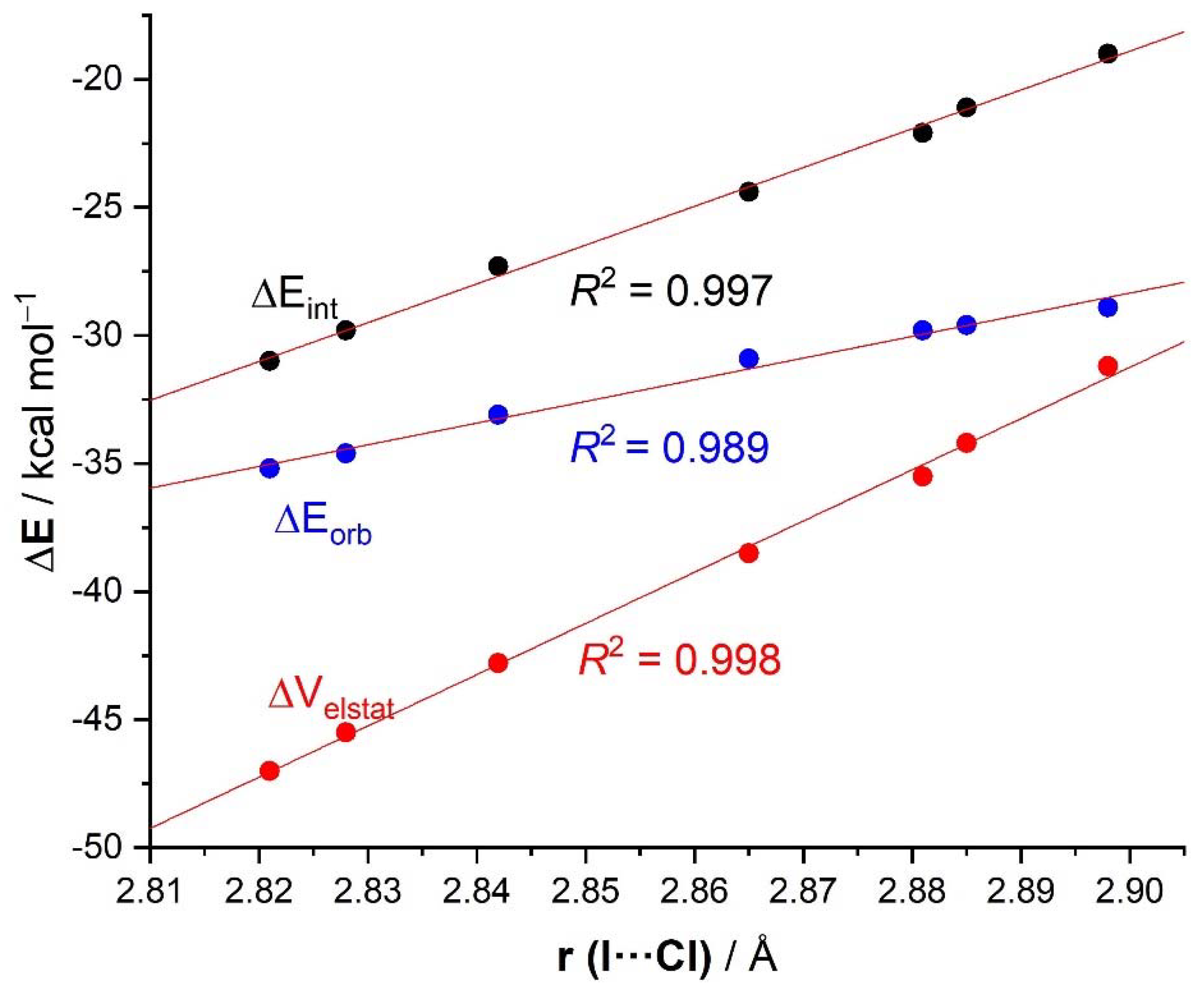
| ΔEint | −22.1 | −19.0 | −21.1 | −24.4 | −27.3 | −29.8 | −31.0 | −31.1 | −96.2 |
| ΔEPauli | 43.6 | 41.5 | 43.1 | 45.4 | 48.6 | 50.6 | 51.6 | 54.5 | 86.0 |
| ΔVelstat a | −35.5 (54.0%) | −31.2 (51.6%) | −34.2 (53.3%) | −38.5 (55.2%) | −42.8 (56.1%) | −45.5 (56.5%) | −47.0 (56.9%) | −48.1 (56.2%) | −121.0 (66.4%) |
| ΔEorb a | −29.8 (45.4%) | −28.9 (47.8%) | −29.6 (46.1%) | −30.9 (44.3%) | −33.1 (43.4%) | −34.6 (43.0%) | −35.2 (42.6%) | −37.1 (43.3%) | −60.8 (33.4%) |
| ΔEorb(1) b | −21.4 (71.8%) | −20.2 (69.9%) | −21.1 (71.3%) | −22.3 (72.2%) | −23.8 (71.9%) | −24.7 (71.4%) | −25.0 (71.0%) | −27.5 (74.1%) | −46.2 (76.0%) |
| ΔEorb(2) b | −2.3 (7.7%) | −2.5 (8.7%) | −2.3 (7.8%) | −2.3 (7.4%) | −2.6 (7.9%) | −3.0 (8.7%) | −3.4 (9.7%) | −2.4 (6.5%) | −3.6 (5.9%) |
| ΔEorb(rest) b | −6.1 (20.5%) | −6.1 (21.1%) | −6.2 (20.9%) | −6.3 (20.4%) | −6.6 (19.9%) | −6.8 (19.7%) | −6.8 (19.3%) | −7.2 (19.4%) | −11.0 (18.1%) |
| ΔEdisp | −0.4 (0.6%) | −0.4 (0.7%) | −0.4 (0.6%) | −0.4 (0.6%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.5%) | −0.4 (0.2%) |
| r(I···Cl)/Å | 2.881 | 2.898 | 2.885 | 2.865 | 2.842 | 2.828 | 2.821 | 2.796 | 2.632 |
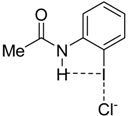 | 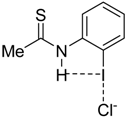 | 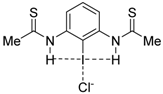 | 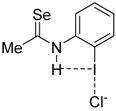 | |
|---|---|---|---|---|
| 4·Cl− | 7·Cl− | 8·Cl− | 9·Cl− | |
| ΔEint | −22.1 | −24.9 | −35.5 | −25.8 |
| ΔEPauli | 43.6 | 47.0 | 56.2 | 48.2 |
| ΔVelstat a | −35.5 (54.0%) | −39.2 (54.5%) | −52.0 (56.8%) | −40.4 (54.7%) |
| ΔEorb a | −29.8 (45.4%) | −32.3 (44.9%) | −39.0 (42.6%) | −33.1 (44.8%) |
| ΔEorb(1) b | −21.4 (71.8%) | −23.1 (71.5%) | −27.8 (71.3%) | −23.6 (71.2%) |
| ΔEorb(2) b | −2.3 (7.7%) | −2.4 (7.4%) | −2.5 (6.4%) | −2.4 (7.3%) |
| ΔEorb(rest) b | −6.1 (20.5%) | −6.8 (21.1%) | −8.7 (22.3%) | −7.1 (21.5%) |
| ΔEdisp | −0.4 (0.6%) | −0.4 (0.6%) | −0.5 (0.5%) | −0.4 (0.5%) |
| r(I···Cl)/Å | 2.881 | 2.855 | 2.796 | 2.847 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Portela, S.; Fernández, I. Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules 2021, 26, 1885. https://doi.org/10.3390/molecules26071885
Portela S, Fernández I. Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules. 2021; 26(7):1885. https://doi.org/10.3390/molecules26071885
Chicago/Turabian StylePortela, Susana, and Israel Fernández. 2021. "Nature of the Hydrogen Bond Enhanced Halogen Bond" Molecules 26, no. 7: 1885. https://doi.org/10.3390/molecules26071885
APA StylePortela, S., & Fernández, I. (2021). Nature of the Hydrogen Bond Enhanced Halogen Bond. Molecules, 26(7), 1885. https://doi.org/10.3390/molecules26071885