
Synthesis and Identification of Epoxy Derivatives of 5-Methylhexahydroisoindole-1,3-dione and Biological Evaluation



Abstract
1. Introduction
2. Results and Discussions
2.1. Synthesis
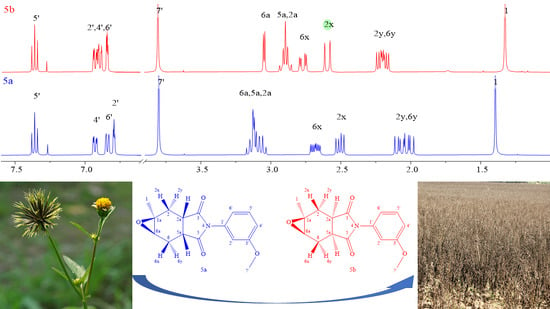
2.1.1. Structure Identification of Compound (5b)
2.1.2. Structure Identification of Compound (5a)
2.2. Computational Analysis
2.3. Biological Assay
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. Synthesis of Amide (2)
3.2.2. Synthesis of 2-(3-methoxyphenyl)-5-methyl-3a,4,7,7a-tetrahydro-1H-isoindole-1,3(2H)-dione (4)
3.2.3. Synthesis of Epoxides 5a and 5b
(1aS*,2aS*,5aR*,6aR*)-4-(3-methoxyphenyl)-1a-methylhexahydro-3H-oxireno[2,3-f]isoindole-3,5(4H)-dione (5a)
(1aS*,2aR*,5aS*,6aR*)-4-(3-methoxyphenyl)-1a-methylhexahydro-3H-oxireno[2,3-f]isoindole-3,5(4H)-dione (5b)
3.3. Computation
3.4. Biological Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- FAO. High-Level Expert Forum-How to Feed World 2050; FAO: Rome, Italy, 2009; pp. 1–4. [Google Scholar]
- Chauhan, B.S. Grand Challenges in Weed Management. Front. Agron. 2020, 1, 1–4. [Google Scholar] [CrossRef]
- Hargreaves, M.K.; Pritchard, J.G.; Dave, H.R. Cyclic carboxylic monoimides. Chem. Rev. 1970, 70, 439–469. [Google Scholar] [CrossRef]
- Kim, J.; Hong, S.H. Synthesis of cyclic imides from nitriles and diols using hydrogen transfer as a substrate-activating strategy. Org. Lett. 2014, 16, 4404–4407. [Google Scholar] [CrossRef]
- da Silva, G.F.; dos Anjos, M.F.; Rocha, L.W.; Ferreira, L.F.G.R.; Stiz, D.S.; Corrêa, R.; Santin, J.R.; Cechinel Filho, V.; Hernandes, M.Z.; Quintão, N.L.M. Anti-hypersensitivity effects of the phthalimide derivative N-(4methyl-phenyl)-4-methylphthalimide in different pain models in mice. Biomed. Pharmacother. 2017, 96, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Meazza, G.; Bettarini, F.; La Porta, P.; Piccardi, P.; Signorini, E.; Portoso, D.; Fornara, L. Synthesis and herbicidal activity of novel heterocyclic protoporphyrinogen oxidase inhibitors. Pest Manag. Sci. 2004, 60, 1178–1188. [Google Scholar] [CrossRef] [PubMed]
- Alvarenga, E.S.; Santos, J.O.; Moraes, F.C.; Carneiro, V.M.T. Quantum mechanical approach for structure elucidation of novel halogenated sesquiterpene lactones. J. Mol. Struct. 2019, 1180, 41–47. [Google Scholar] [CrossRef]
- Krivdin, L.B. Computational 1H NMR: Part 1. Theoretical background. Magn. Reson. Chem. 2019, 57, 897–914. [Google Scholar] [CrossRef]
- Krivdin, L.B. Computational 1H NMR: Part 2. Chemical applications. Magn. Reson. Chem. 2020, 58, 5–14. [Google Scholar] [CrossRef]
- Pinto, B.N.S.; Teixeira, M.G.; Alvarenga, E.S. Synthesis and structural elucidation of a phthalide analog using NMR analysis and DFT calculations. Magn. Reson. Chem. 2019, 1–7. [Google Scholar] [CrossRef]
- Channar, P.A.; Arshad, N.; Larik, F.A.; Farooqi, S.I.; Saeed, A.; Hökelek, T.; Batool, B.; Ujan, R.; Ali, H.S.; Flörke, U. 4-(4-Bromophenyl)thiazol-2-amine: Crystal structure determination, DFT calculations, visualizing intermolecular interactions using Hirshfeld surface analysis, and DNA binding studies. J. Phys. Org. Chem. 2019, 32, 1–15. [Google Scholar] [CrossRef]
- Moraes, F.C.; Alvarenga, E.S.; Demuner, A.J.; Viana, V.M. Assignment of the relative and absolute stereochemistry of two novel epoxides using NMR and DFT-GIAO calculations. J. Mol. Struct. 2018, 1164, 109–115. [Google Scholar] [CrossRef]
- Teixeira, M.G.; Alvarenga, E.S. Characterization of novel isobenzofuranones by DFT calculations and 2D NMR analysis. Magn. Reson. Chem. 2016, 623–631. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning the stereochemistry of pairs of diastereoisomers using GIAO NMR shift calculation. J. Org. Chem. 2009, 74, 4597–4607. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Martínez-Casares, R.M.; Méndez, H.I.P.; Alvarez, N.M.; Mendoza, E.S.; Oba, A.S.; Vázquez, L.H. Structural study of 1-(2′, 3′-O-isopropylidene-(α-d-allo and -β-l-talofuranosyluron)-5′-cyanohydrin)uracil stereoisomers by NMR spectroscopy and theoretical methods. Magn. Reson. Chem. 2017, 55, 766–772. [Google Scholar] [CrossRef]
- Minch, M.J. Orientational dependence of vicinal proton-proton NMR coupling constants: The Karplus relationship. Concepts Magn. Reson. 1994, 6, 41–56. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Willoughby, P.H.; Jansma, M.J.; Hoye, T.R. Addendum: A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2020, 03216, 41596. [Google Scholar] [CrossRef] [PubMed]
- Lopes, D.T.; Hoye, T.R.; Alvarenga, E.S. Characterization of stereoisomeric 5-(2-nitro-1-phenylethyl)furan-2(5H)-ones by computation of 1H and 13C NMR chemical shifts and electronic circular dichroism spectra. Magn. Reson. Chem. 2020, 59, 1–9. [Google Scholar] [CrossRef]
- de Alvarenga, E.S.; Saliba, W.A.; Milagres, B.G. Montagem de câmara com lâmpada de ultravioleta de baixo custo. Quim. Nova 2005, 28, 927–928. [Google Scholar] [CrossRef]
- Maestro; Schrödinger Release 2019. Available online: https://www.schrodinger.com/products/maestro (accessed on 11 February 2019).
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Channon, J.A.; Paterson, I.; Goodman, J.M. The stereochemical assignment of acyclic polyols: A computational study of the NMR data of a library of stereopentad sequences from polyketide natural products. Tetrahedron 2010, 66, 6437–6444. [Google Scholar] [CrossRef]
- Resende, G.C.; Alvarenga, E.S.; Galindo, J.C.G.; Macias, F.A. Synthesis and phytotoxicity of 4,5 functionalized tetrahydrofuran-2-ones. J. Braz. Chem. Soc. 2012, 23, 2266–2270. [Google Scholar] [CrossRef][Green Version]
- Teixeira, M.G.; Alvarenga, E.S.; Lopes, D.T.; Oliveira, D.F. Herbicidal activity of isobenzofuranones and in silico identification of their enzyme target. Pest Manag. Sci. 2019, 75, 3331–3339. [Google Scholar] [CrossRef]
- Sartori, S.K.; Alvarenga, E.S.; Franco, C.A.; Ramos, D.S.; Oliveira, D.F. One-pot synthesis of anilides, herbicidal activity and molecular docking study. Pest Manag. Sci. 2018, 74, 1637–1645. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assign. | δC (ppm) | Assign. | δH (ppm) m, J (Hz) | COSY | NOESY |
|---|---|---|---|---|---|
| C1 | 21.9 | H1 | 1.32, s | - | - |
| C1A | 56.7 | - | - | - | - |
| C2 | 28.0 | H2x | 2.59, d, J = 15.0 Hz | H2y | H2y |
| - | - | H2y | 2.18, dd, J = 15.0 and 7.0 Hz | H2a, H2x | H2x, H2a |
| C2a | 35.3 | H2a | 2.89, nfom | H2y | H2y |
| C5a | 36.7 | H5a | 2.89, nfom | H6y | H6y |
| C6 | 23.6 | H6x | 2.77, dd, J = 15.0 and 4.0 Hz, | H6y, H6a | H6y |
| - | - | H6y | 2.22, dd, J = 15.0 and 7.0 Hz | H6x, H5a | H6x, H5a |
| C6a | 57.6 | H6a | 3.04, d, J = 4.0 Hz | H6x | H6x |
| C2′ | 112.4 | H2′ | 6.84, t, J = 2.0 Hz | H2′ | - |
| C4′ | 114.6 | H4′ | 6.92, ddd, J = 8.0, 2.0, and 1.0 Hz | H2′, H5′, H6′ | - |
| C5′ | 129.8 | H5′ | 7.36, t, J = 8.0 Hz | H4′, H6′ | - |
| C6′ | 119.0 | H6′ | 6.89, ddd, J = 8.0, 2.0, and 1.0 Hz | H2′, H4′, H5′ | - |
| C7′ | 55.4 | H7′ | 3.80, s | - | - |
| C1′ | 133.8 | - | - | - | - |
| C3′ | 160.1 | - | - | - | - |
| C3 | 179.6 | - | - | - | - |
| C5 | 179.7 | - | - | - | - |
| Nuclei | 5a | 5b | ||
|---|---|---|---|---|
| Dihedral Angle (ϕ) | J (Hz) * | Dihedral Angle (ϕ) | J (Hz) * | |
| 2y-2a | 160 | 13 | 44 | 6.5 |
| 2x-2a | 42 | 7 | 70 | 1 |
| 6y-5a | 160 | 13 | 44 | 6 |
| 6x-5a | 41 | 7 | 73 | 0.9 |
| 6y-6a | 75 | 0.7 | 75 | 0.7 |
| 6x-6a | 42 | 7 | 44 | 6 |
| Assign. | δC (ppm) | Assign. | δH (ppm) m, J (Hz) | COSY | NOESY |
|---|---|---|---|---|---|
| C1 | 21.8 | H1 | 1.40, s | - | H2y, H2x, H6a |
| C1A | 55.0 | - | - | - | - |
| C2 | 28.8 | H2x | 2.50, dd, J = 15.0 and 8.0 Hz | H2y, H2a | H2y, H6x |
| - | - | H2y | 2.01, dd, J = 15.0 and 10.0 Hz | H2x, H2a | H2x |
| C2a | 35.3 | H2a | 2.98–3.20, m | H2x, H2y | H2x |
| C5a | 36.9 | H5a | 2.98–3.20, m | H6x, H6y | H6x |
| C6 | 23.8 | H6x | 2.68, ddd, J = 15.0, 8.0, and 4.0 Hz | H6y, H5a, H6a | H5a, H2x, H6y |
| - | - | H6y | 2.08, dd, J = 15.0 and 10.0 Hz | H6x, H5a | H6x, H6a |
| C6a | 56.1 | H6a | 2.98–3.20, m | H6x, H6y | H1, H6y |
| C2′ | 112.2 | H2′ | 6.79, t, J = 2.0 Hz | H4′, H6′ | H5′, H7′ |
| C4′ | 114.5 | H4′ | 6.93, ddd, J = 8.0, 2.0, and 1.0 Hz | H2′, H5′ | H5′, H7′ |
| C5′ | 129.9 | H5′ | 7.36, t, J = 8.0 Hz | H4′, H6′ | H4′, H6′ |
| C6′ | 118.6 | H6′ | 6.84, ddd, J = 8.0, 2.0, and 1.0 Hz | H2′, H5′ | H5′ |
| C7′ | 55.4 | H7′ | 3.80, s | - | H2′, H4′ |
| C1′ | 132.7 | - | - | - | - |
| C3′ | 160.1 | - | - | - | - |
| C3 | 178.5 | - | - | - | - |
| C5 | 178.6 | - | - | - | - |
| Pairwise Comparisons | CMAEproton d | CMAEcarbon e | CP3proton | CP3carbon | CP3 all Data c | DP4 all Data f |
|---|---|---|---|---|---|---|
| 5aexpt vs. 5acalc | 0.04 ppm | 1.66 ppm | 88.1% | 100% | 100% | 100% |
| 5aexpt vs. 5bcalc | 0.10 ppm | 1.91 ppm | 11.9% | 0% | 0 | 0% |
| 5bexpt vs. 5bcalc | 0.04 ppm | 1.66 ppm | 88.1% | 100% | 100% | 93.9% |
| 5bexpt vs. 5acalc | 0.10 ppm | 1.91 ppm | 11.9% | 0% | 0 | 6.1% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrent, K.B.A.; Alvarenga, E.S. Synthesis and Identification of Epoxy Derivatives of 5-Methylhexahydroisoindole-1,3-dione and Biological Evaluation. Molecules 2021, 26, 1923. https://doi.org/10.3390/molecules26071923
Torrent KBA, Alvarenga ES. Synthesis and Identification of Epoxy Derivatives of 5-Methylhexahydroisoindole-1,3-dione and Biological Evaluation. Molecules. 2021; 26(7):1923. https://doi.org/10.3390/molecules26071923
Chicago/Turabian StyleTorrent, Kariny B. A., and Elson S. Alvarenga. 2021. "Synthesis and Identification of Epoxy Derivatives of 5-Methylhexahydroisoindole-1,3-dione and Biological Evaluation" Molecules 26, no. 7: 1923. https://doi.org/10.3390/molecules26071923
APA StyleTorrent, K. B. A., & Alvarenga, E. S. (2021). Synthesis and Identification of Epoxy Derivatives of 5-Methylhexahydroisoindole-1,3-dione and Biological Evaluation. Molecules, 26(7), 1923. https://doi.org/10.3390/molecules26071923