
Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques



Abstract
:1. Introduction
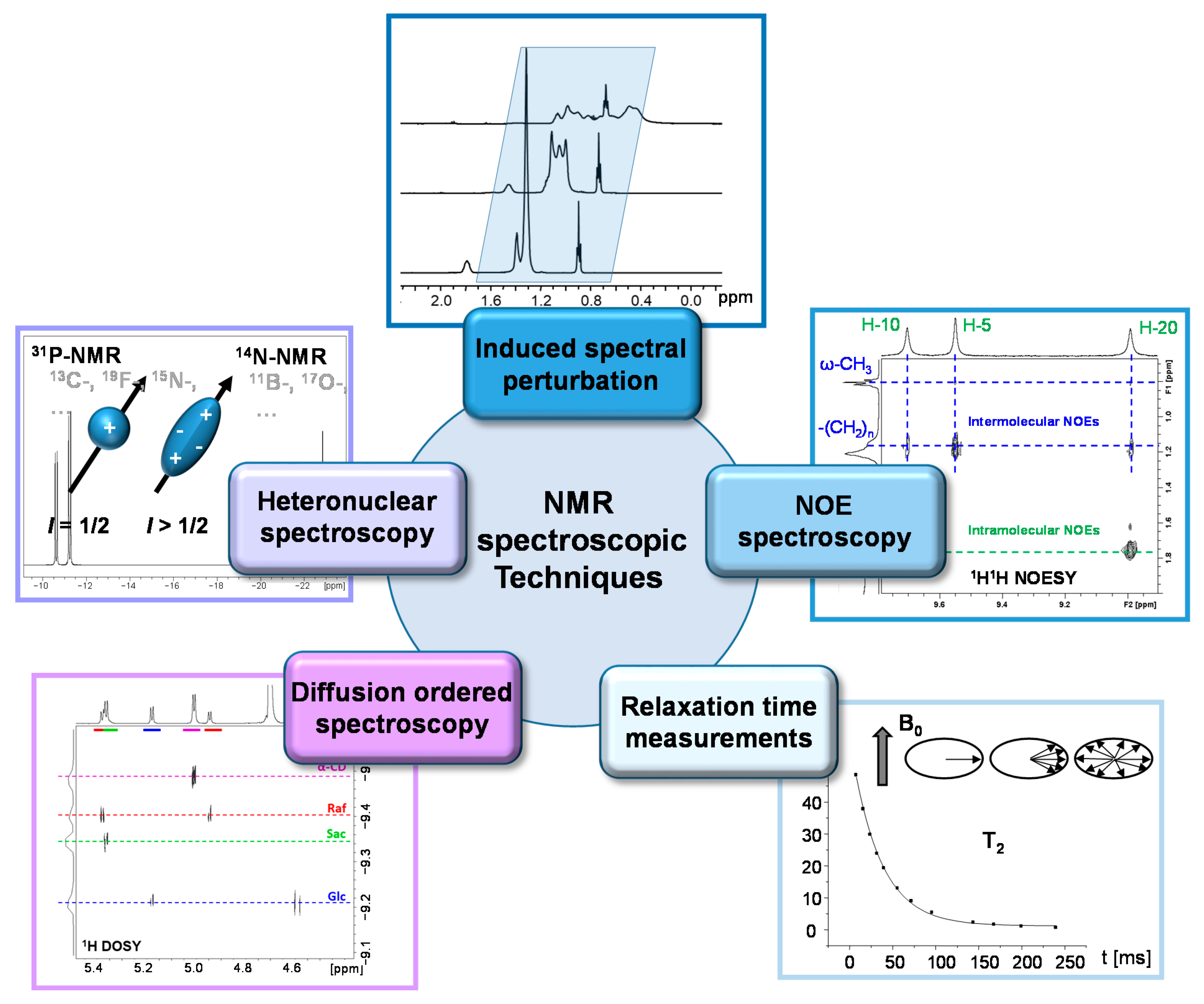
2. NMR Parameters and Methods for Studying Porphyrinic Compounds and Their Surroundings
2.1. NMR Basics
2.2. Induced Spectral Perturbation
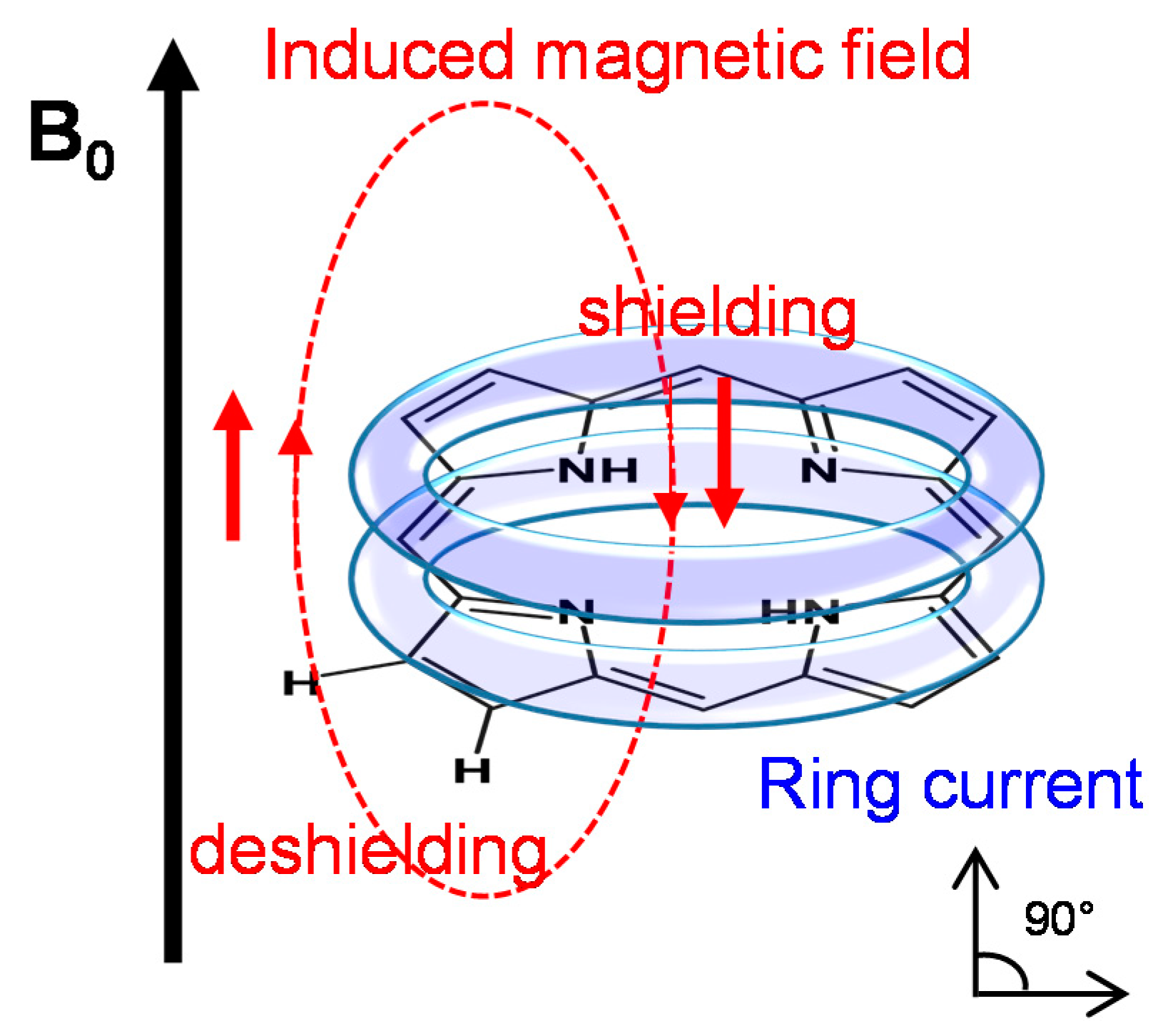
2.2.1. The Porphyrin Ring Current Effect
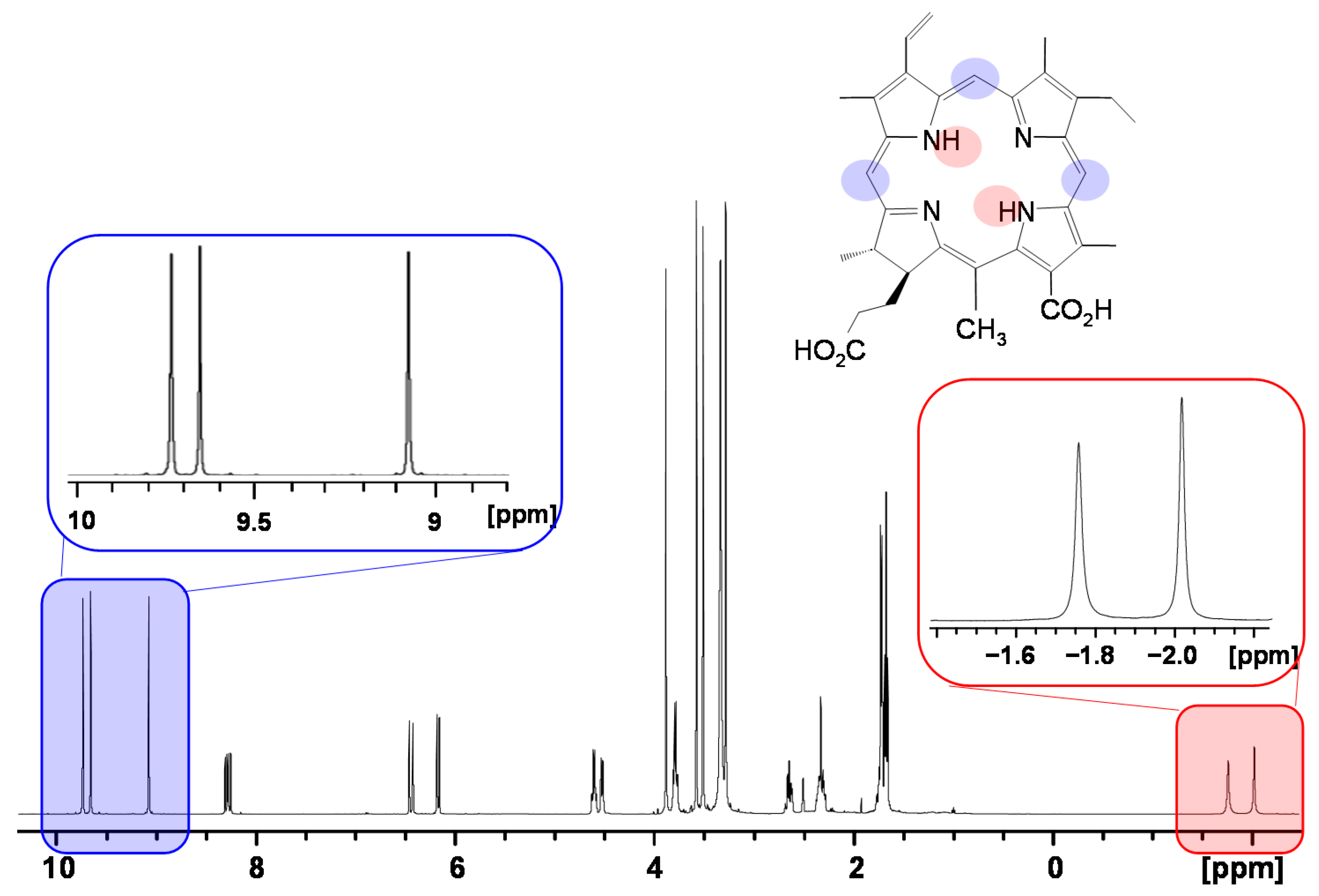
2.2.2. Induced Changes onto the NMR Spectrum of the Porphyrin
2.2.3. Induced Changes onto the NMR Spectrum of the Macromolecule
2.3. Nuclear Overhauser Enhancement Spectroscopy
2.4. NMR Relaxation Times (T1 and T2)
2.5. Diffusion-Ordered Spectroscopy
2.6. Heteronuclear NMR Spectroscopy
3. Applications to Study Porphyrin–Macromolecule Interactions
3.1. Biomolecules
3.1.1. Phospholipids (Membrane Models and Liposomal Drug Delivery Vehicles)
3.1.2. Proteins
3.1.3. Nucleic Acids (DNA, RNA)
3.2. Carrier Polymers
3.2.1. Polyvinylpyrrolidone (PVP)
3.2.2. Cyclodextrins (CDs)
3.2.3. Surfactant Micelles
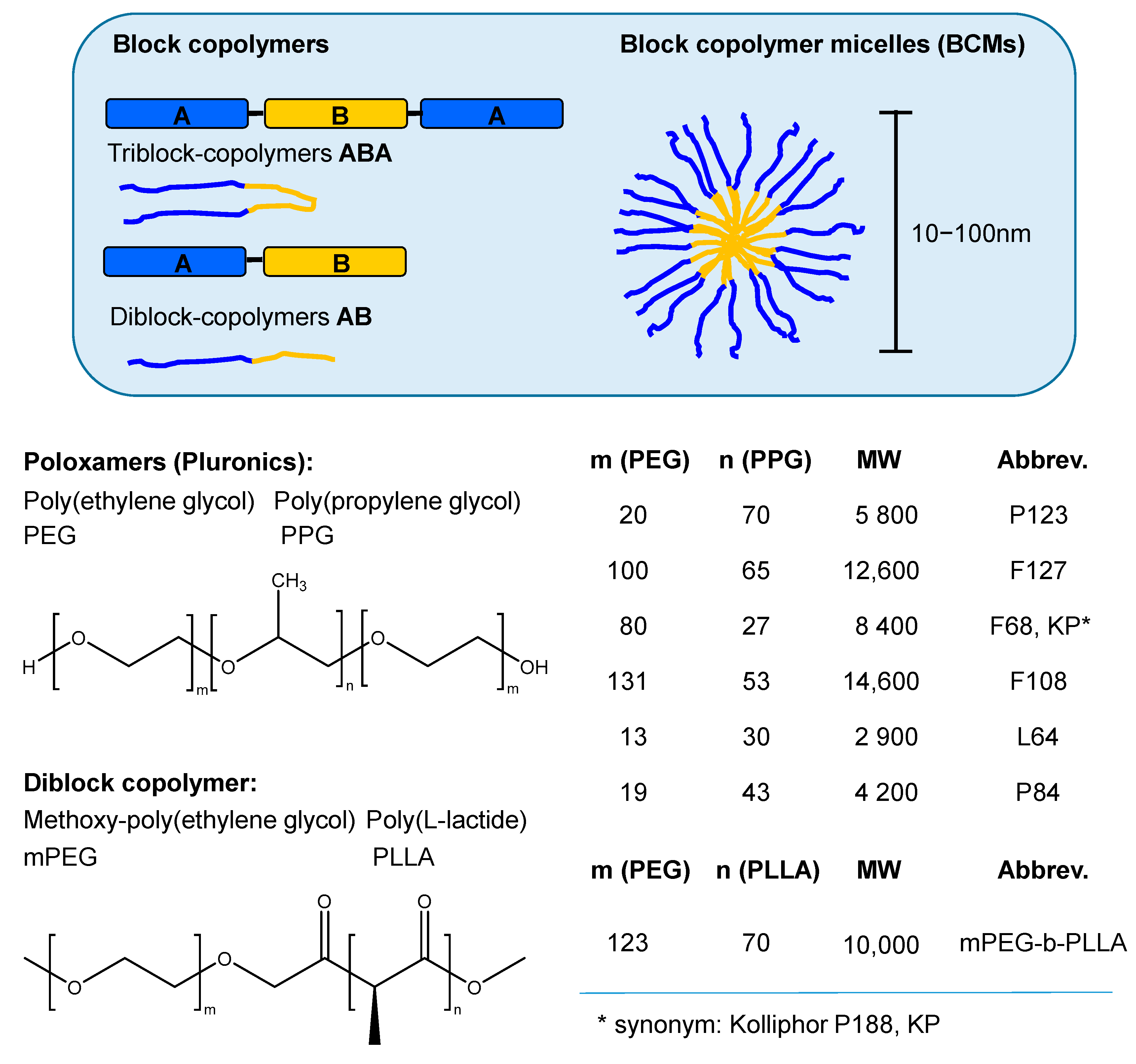
3.2.4. Block Copolymer Micelles (BCMs)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. The Following Abbreviations were used in the Text
References
- Lemberg, R. Porphyrins in Nature. In Fortschritte der Chemie Organischer Naturstoffe/Progress in the Chemistry of Organic Natural Products/Progrés dans la Chimie des Substances Organiques Naturelles; Ƶechmeister, L., Ed.; Springer Vienna: Vienna, Austria, 1954; pp. 299–349. [Google Scholar]
- Williams, R.J.P. The Properties Of Metalloporphyrins. Chem. Rev. 1956, 56, 299–328. [Google Scholar] [CrossRef]
- Boucher, L.J. Coordination Chemistry of Porphyrins. In Coordination Chemistry of Macrocyclic Compounds; Melson, G.A., Ed.; Springer US: Boston, MA, USA, 1979; pp. 517–536. [Google Scholar]
- Battersby, A.R. Tetrapyrroles: The pigments of life. Nat. Prod. Rep. 2000, 17, 507–526. [Google Scholar] [CrossRef]
- Lesage, S.; Xu, H.A.O.; Durham, L. The occurrence and roles of porphyrins in the environment: Possible implications for bioremediation. Hydrol. Sci. J. 1993, 38, 343–354. [Google Scholar] [CrossRef]
- Bryant, D.A.; Hunter, C.N.; Warren, M.J. Biosynthesis of the modified tetrapyrroles-the pigments of life. J. Biol. Chem. 2020, 295, 6888–6925. [Google Scholar] [CrossRef] [Green Version]
- Poulos, T.L. Heme enzyme structure and function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latunde-Dada, G.O. Iron: Biosynthesis and Significance of Heme. In Encyclopedia of Food and Health; Caballero, B., Finglas, P.M., Toldrá, F., Eds.; Academic Press: Oxford, UK, 2016; pp. 452–460. [Google Scholar]
- Scheer, H. An Overview of Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications. In Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions and Applications; Grimm, B., Porra, R.J., Rüdiger, W., Scheer, H., Eds.; Springer: Dordrecht, The Netherlands, 2006; pp. 1–26. [Google Scholar]
- Tanaka, A.; Tanaka, R. Chapter Six—The biochemistry, physiology, and evolution of the chlorophyll cycle. In Advances in Botanical Research; Grimm, B., Ed.; Academic Press: London, UK, 2019; Volume 90, pp. 183–212. [Google Scholar]
- Hunter, C.N.; Thurnauer, F.D.M.C.; Beatty, J.T. The Purple Phototrophic Bacteria, 1st ed.; Springer: Dordrecht, The Netherlands, 2009; p. 1013. [Google Scholar]
- Graham, R.M.; Deery, E.; Warren, M.J. Vitamin B12: Biosynthesis of the Corrin Ring. In Tetrapyrroles: Birth, Life and Death; Springer: New York, NY, USA, 2009; pp. 286–299. [Google Scholar]
- Kadish, K.M.; Smith, K.M.; Guilard, R. The Porphyrin Handbook. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Amsterdam, The Netherlands, 2003. [Google Scholar]
- Min Park, J.; Lee, J.H.; Jang, W.-D. Applications of porphyrins in emerging energy conversion technologies. Coord. Chem. Rev. 2020, 407, 213157. [Google Scholar] [CrossRef]
- Costa, E.; Silva, R.; Oliveira da Silva, L.; de Andrade Bartolomeu, A.; Brocksom, T.J.; de Oliveira, K.T. Recent applications of porphyrins as photocatalysts in organic synthesis: Batch and continuous flow approaches. Beilstein J. Org. Chem. 2020, 16, 917–955. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Díaz, M.V.; de la Torre, G.; Torres, T. Lighting porphyrins and phthalocyanines for molecular photovoltaics. Chem. Commun. 2010, 46, 7090–7108. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Song, W.; Rieffel, J.; Lovell, J.F. Emerging applications of porphyrins in photomedicine. Front. Phys. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imran, M.; Ramzan, M.; Qureshi, A.K.; Khan, M.A.; Tariq, M. Emerging Applications of Porphyrins and Metalloporphyrins in Biomedicine and Diagnostic Magnetic Resonance Imaging. Biosensors 2018, 8, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsolekile, N.; Nelana, S.; Oluwafemi, O.S. Porphyrin as Diagnostic and Therapeutic Agent. Molecules 2019, 24, 2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part one-photosensitizers, photochemistry and cellular localization. Photodiagnosis Photodyn. Ther. 2004, 1, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Castano, A.P.; Demidova, T.N.; Hamblin, M.R. Mechanisms in photodynamic therapy: Part three-Photosensitizer pharmacokinetics, biodistribution, tumor localization and modes of tumor destruction. Photodiagnosis Photodyn. Ther. 2005, 2, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhou, T.; Bai, R.; Xie, Y. Chemical approaches for the enhancement of porphyrin skeleton-based photodynamic therapy. J. Enzym. Inhib. Med. Chem. 2020, 35, 1080–1099. [Google Scholar] [CrossRef]
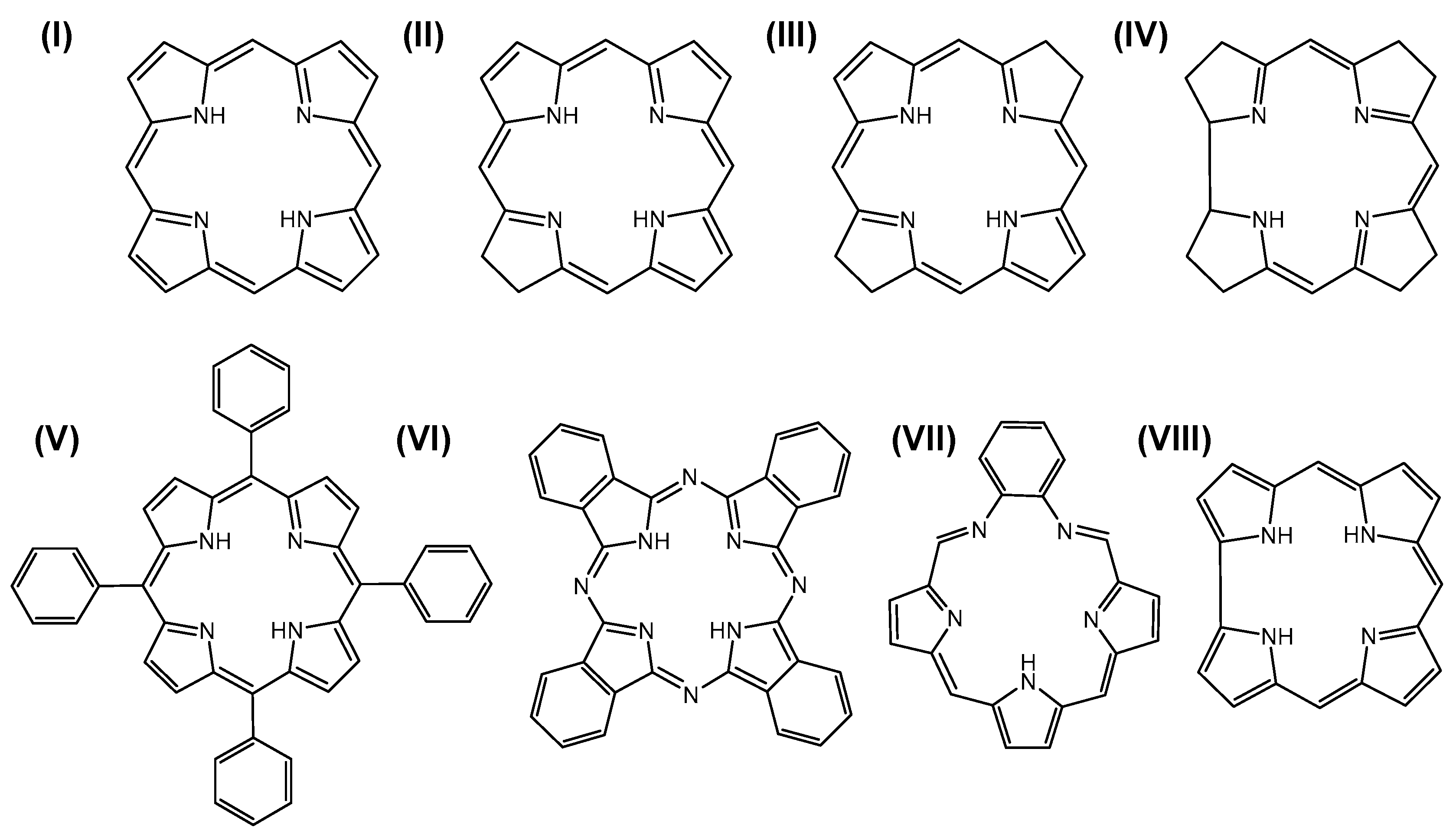
- Costa, L.D.; Costa, J.I.T.; Tomé, A.C. Porphyrin Macrocycle Modification: Pyrrole Ring-Contracted or -Expanded Porphyrinoids. Molecules 2016, 21, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, J.S.; Hsu, H.C.; Schreiman, I.C. Synthesis of tetraphenylporphyrins under very mild conditions. Tetrahedron Lett. 1986, 27, 4969–4970. [Google Scholar] [CrossRef]
- Lo, P.-C.; Rodríguez-Morgade, M.S.; Pandey, R.K.; Ng, D.K.P.; Torres, T.; Dumoulin, F. The unique features and promises of phthalocyanines as advanced photosensitisers for photodynamic therapy of cancer. Chem. Soc. Rev. 2020, 49, 1041–1056. [Google Scholar] [CrossRef] [PubMed]
- Mody, T.D.; Sessler, J.L. Texaphyrins: A new approach to drug development. J. Porphyr. Phthalocyanines 2001, 05, 134–142. [Google Scholar] [CrossRef]
- Teo, R.D.; Hwang, J.Y.; Termini, J.; Gross, Z.; Gray, H.B. Fighting Cancer with Corroles. Chem. Rev. 2017, 117, 2711–2729. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J. An update on photodynamic therapy applications. J. Clin. Laser Med. Surg. 2002, 20, 3–7. [Google Scholar] [CrossRef]
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107. [Google Scholar] [CrossRef]
- Cieplik, F.; Deng, D.; Crielaard, W.; Buchalla, W.; Hellwig, E.; Al-Ahmad, A.; Maisch, T. Antimicrobial photodynamic therapy—What we know and what we don’t. Crit. Rev. Microbiol. 2018, 44, 571–589. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.M.B.; Tomé, J.P.C.; Faustino, M.A.F.; Neves, M.G.P.M.S.; Tomé, A.C.; Cavaleiro, J.A.S.; Costa, L.; Alves, E.; Oliveira, A.; Cunha, Â.; et al. Antimicrobial photodynamic activity of porphyrin derivatives: Potential application on medical and water disinfection. J. Porphyr. Phthalocyanines 2009, 13, 574–577. [Google Scholar] [CrossRef]
- Jocham, D.; Stepp, H.; Waidelich, R. Photodynamic Diagnosis in Urology: State-of-the-Art. Eur. Urol. 2008, 53, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Zumbraegel, A.; Bichler, K.-H.; Krause, F.S.; Feil, G.; Nelde, H.J. The Photodynamic Diagnosis (PDD) for Early Detection of Carcinoma and Dysplasia of the Bladder. In Bladder Disease, Part A: Research Concepts and Clinical Applications; Atala, A., Slade, D., Eds.; Springer: Boston, MA, USA, 2003; pp. 61–66. [Google Scholar]
- Fritsch, C.; Lang, K.; Neuse, W.; Ruzicka, T.; Lehmann, P. Photodynamic diagnosis and therapy in dermatology. Skin Pharmacol. Appl. Skin Physiol. 1998, 11, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.L.; Haedicke, I.E.; Cheng, W.; Tchouala Nofiele, J.; Zhang, X.A. Gadolinium-free T1 contrast agents for MRI: Tunable pharmacokinetics of a new class of manganese porphyrins. J. Magn. Reson. Imaging 2014, 40, 1474–1480. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y. Metalloporphyrins and Functional Analogues as MRI Contrast Agents. Curr. Med. Imaging Rev. 2008, 4, 96–112. [Google Scholar] [CrossRef] [Green Version]
- Nasim, V.; Amir, R.J. An Overview of Labeled Porphyrin Molecules in Medical Imaging. Recent Pat. Top. Imaging 2015, 5, 3–12. [Google Scholar] [CrossRef]
- Yap, S.Y.; Price, T.W.; Savoie, H.; Boyle, R.W.; Stasiuk, G.J. Selective radiolabelling with 68Ga under mild conditions: A route towards a porphyrin PET/PDT theranostic agent. Chem. Commun. 2018, 54, 7952–7954. [Google Scholar] [CrossRef] [PubMed]
- Merkes, J.M.; Zhu, L.; Bahukhandi, S.B.; Rueping, M.; Kiessling, F.; Banala, S. Photoacoustic Imaging Probes Based on Tetrapyrroles and Related Compounds. Int. J. Mol. Sci. 2020, 21, 3082. [Google Scholar] [CrossRef] [PubMed]
- Abuteen, A.; Zanganeh, S.; Akhigbe, J.; Samankumara, L.P.; Aguirre, A.; Biswal, N.; Braune, M.; Vollertsen, A.; Röder, B.; Brückner, C.; et al. The evaluation of NIR-absorbing porphyrin derivatives as contrast agents in photoacoustic imaging. Phys. Chem. Chem. Phys. 2013, 15, 18502–18509. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lovell, J.F. Porphyrins as theranostic agents from prehistoric to modern times. Theranostics 2012, 2, 905–915. [Google Scholar] [CrossRef] [PubMed]
- Jenni, S.; Sour, A. Molecular Theranostic Agents for Photodynamic Therapy (PDT) and Magnetic Resonance Imaging (MRI). Inorganics 2019, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Jerjes, W.; Theodossiou, T.A.; Hirschberg, H.; Høgset, A.; Weyergang, A.; Selbo, P.K.; Hamdoon, Z.; Hopper, C.; Berg, K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. J. Clin. Med. 2020, 9, 528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norum, O.-J.; Selbo, P.K.; Weyergang, A.; Giercksky, K.-E.; Berg, K. Photochemical internalization (PCI) in cancer therapy: From bench towards bedside medicine. J. Photochem. Photobiol. B: Biol. 2009, 96, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, Y.; Kaskel, S. Porphyrin-Based Metal-Organic Frameworks for Biomedical Applications. Angew. Chem. Int. Ed. Engl. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Jo, Y.-U.; Na, K. Photodynamic therapy with smart nanomedicine. Arch. Pharmacal Res. 2020, 43, 22–31. [Google Scholar] [CrossRef]
- Moret, F.; Reddi, E. Strategies for optimizing the delivery to tumors of macrocyclic photosensitizers used in photodynamic therapy (PDT). J. Porphyr. Phthalocyanines 2017, 21, 239–256. [Google Scholar] [CrossRef] [Green Version]
- Sztandera, K.; Gorzkiewicz, M.; Klajnert-Maculewicz, B. Nanocarriers in photodynamic therapy-in vitro and in vivo studies. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1509. [Google Scholar] [CrossRef]
- Calixto, G.M.; Bernegossi, J.; de Freitas, L.M.; Fontana, C.R.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for Photodynamic Therapy of Cancer: A Review. Molecules 2016, 21, 342. [Google Scholar] [CrossRef]
- Montaseri, H.; Kruger, C.A.; Abrahamse, H. Recent Advances in Porphyrin-Based Inorganic Nanoparticles for Cancer Treatment. Int. J. Mol. Sci. 2020, 21, 3358. [Google Scholar] [CrossRef]
- Düzgüneş, N.; Piskorz, J.; Skupin-Mrugalska, P.; Goslinski, T.; Mielcarek, J.; Konopka, K. Photodynamic therapy of cancer with liposomal photosensitizers. Ther. Deliv. 2018, 9, 823–832. [Google Scholar] [CrossRef]
- Derycke, A.S.L.; de Witte, P.A.M. Liposomes for photodynamic therapy. Adv. Drug Del. Rev. 2004, 56, 17–30. [Google Scholar] [CrossRef]
- Huang, H.C.; Mallidi, S.; Obaid, G.; Sears, B.; Tangutoori, S.; Hasan, T. 23—Advancing photodynamic therapy with biochemically tuned liposomal nanotechnologies. In Applications of Nanoscience in Photomedicine; Hamblin, M.R., Avci, P., Eds.; Chandos Publishing: Oxford, UK, 2015; pp. 487–510. [Google Scholar]
- Demazeau, M.; Gibot, L.; Mingotaud, A.-F.; Vicendo, P.; Roux, C.; Lonetti, B. Rational design of block copolymer self-assemblies in photodynamic therapy. Beilstein J. Nanotechnol. 2020, 11, 180–212. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, B.F.O.; Pereira, N.A.M.; Valente, A.J.M.; Pinho, E.; Melo, T.M.V.D.; Pineiro, M. A Review on (Hydro)Porphyrin-Loaded Polymer Micelles: Interesting and Valuable Platforms for Enhanced Cancer Nanotheranostics. Pharmaceutics 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pehlivan, E.G.; Ek, Y.; Topkaya, D.; Tazebay, U.H.; Dumoulin, F. Effect of PVP formulation on the in vitro photodynamic efficiency of a photosensitizing phthalocyanine. J. Porphyr. Phthalocyanines 2019, 23, 1587–1591. [Google Scholar] [CrossRef]
- Chin, W.W.L.; Heng, P.W.S.; Thong, P.S.P.; Bhuvaneswari, R.; Hirt, W.; Kuenzel, S.; Soo, K.C.; Olivo, M. Improved formulation of photosensitizer chlorin e6 polyvinylpyrrolidone for fluorescence diagnostic imaging and photodynamic therapy of human cancer. Eur. J. Pharm. Biopharm. 2008, 69, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Mazzaglia, A.; Micali, N.; Scolaro, L.M.; Sciortino, M.T.; Sortino, S.; Villari, V. Design of photosensitizer/cyclodextrin nanoassemblies: Spectroscopy, intracellular delivery and photodamage. J. Porphyr. Phthalocyanines 2010, 14, 661–677. [Google Scholar] [CrossRef]
- Ben Mihoub, A.; Larue, L.; Moussaron, A.; Youssef, Z.; Colombeau, L.; Baros, F.; Frochot, C.; Vanderesse, R.; Acherar, S. Use of Cyclodextrins in Anticancer Photodynamic Therapy Treatment. Molecules 2018, 23, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouterman, M. 1—Optical Spectra and Electronic Structure of Porphyrins and Related Rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; pp. 1–165. [Google Scholar]
- Ernst, R.R.; Bodenhausen, G.; Wokaun, A. Principles of Nuclear Magnetic Resonance in One and Two Dimensions; Clarendon Press: Oxford, UK, 1987. [Google Scholar]
- Becker, E.D.; Bradley, R.B. Effects of ‘‘Ring Currents’’ on the NMR Spectra of Porphyrins. J. Chem. Phys. 1959, 31, 1413–1414. [Google Scholar] [CrossRef]
- Scheer, H.; Katz, J.J. Nuclear magnetic resonance spectroscopy of porphyrins and metalloporphyrins. In Porphyrins and Metalloporphyrins; Smith, K.M., Ed.; Elsevier: New York, NY, USA, 1975; pp. 399–524. [Google Scholar]
- Keeler, J. Understanding NMR Spectroscopy, 2nd ed.; John Wiley and Sons: Chichester, UK, 2010. [Google Scholar]
- Günther, H. NMR Spectroscopy: Basic Principles, Concepts, and Applications in Chemistry, 3rd ed.; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Friebolin, H. Basic One- and Two-Dimensional NMR Spectroscopy. 5th edition ed.; Wiley-VCH: Weinheim, Germany, 2010; p. 418. [Google Scholar]
- Karplus, M. Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys. 1959, 30, 11–15. [Google Scholar] [CrossRef]
- Karplus, M. Vicinal Proton Coupling in Nuclear Magnetic Resonance. J. Am. Chem. Soc. 1963, 85, 2870–2871. [Google Scholar] [CrossRef]
- Minch, M.J. Orientational dependence of vicinal proton-proton NMR coupling constants: The Karplus relationship. Concepts Magn. Reson. 1994, 6, 41–56. [Google Scholar] [CrossRef]
- Pretsch, E.; Clerc, T.; Seibl, J.; Simon, W. Protonenresonanzspektroskopie. In Tabellen zur Strukturaufklärung Organischer Verbindungen Mit Spektroskopischen Methoden; Springer: Berlin/Heidelberg, Germany, 1981; pp. 108–181. [Google Scholar]
- Pauling, L. The Diamagnetic Anisotropy of Aromatic Molecules. J. Chem. Phys. 1936, 4, 673–677. [Google Scholar] [CrossRef] [Green Version]
- Pople, J.A. Proton Magnetic Resonance of Hydrocarbons. J. Chem. Phys. 1956, 24, 1111. [Google Scholar] [CrossRef]
- Waugh, J.S.; Fessenden, R.W. Nuclear Resonance Spectra of Hydrocarbons: The Free Electron Model. J. Am. Chem. Soc. 1957, 79, 846–849. [Google Scholar] [CrossRef]
- Waugh, J.; Fessendn, R. Additions and Corrections: Nuclear Resonance Spectra of Hydrocarbons: The Free Electron Model. J. Am. Chem. Soc. 1958, 80, 6697–6699. [Google Scholar] [CrossRef]
- Johnson, C.E.; Bovey, F.A. Calculation of Nuclear Magnetic Resonance Spectra of Aromatic Hydrocarbons. J. Chem. Phys. 1958, 29, 1012–1014. [Google Scholar] [CrossRef]
- Abraham, R.J. A ring current model for the heme ring. J. Magn. Reson. 1981, 43, 491–494. [Google Scholar] [CrossRef]
- Abraham, R.J.; Bedford, G.R.; Wright, B. The NMR spectra of the porphyrins. 17—Metalloporphyrins as diamagnetic shift reagents, structural and specificity studies. Org. Magn. Reson 1982, 18, 45–52. [Google Scholar] [CrossRef]
- Abraham, R.J.; Bedford, G.R.; McNeillie, D.; Wright, B. The NMR spectra of the porphyrins 16—zinc(II) meso-tetraphenylporphyrin (Zn TPP) as a diamagnetic shift reagent. A quantitative ring current model. Org. Magn. Reson 1980, 14, 418–425. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. NMR and EPR, 1st ed.; Elsevier Science Publishing Co Inc.: San Diego, CA, USA, 1999; Volume 5, p. 360. [Google Scholar]
- Hunter, C.A.; Sanders, J.K.M. The nature of.pi.-.pi. interactions. J. Am. Chem. Soc. 1990, 112, 5525–5534. [Google Scholar] [CrossRef]
- Abraham, R.J.; Smith, K.M. NMR spectra of porphyrins. 21. Applications of the ring-current model to porphyrin and chlorophyll aggregation. J. Am. Chem. Soc. 1983, 105, 5734–5741. [Google Scholar] [CrossRef]
- Abraham, R.J.; Rowan, A.E.; Smith, N.W.; Smith, K.M. NMR spectra of the porphyrins. Part 42. The synthesis and aggregation behaviour of some chlorophyll analogues. J. Chem. Soc. Perkin Trans. 2 1993, 1047–1059. [Google Scholar] [CrossRef]
- Hynninen, P.H.; Lötjönen, S. Effects of π−π interactions on the 1H-NMR spectra and solution structures of pheophytin a and a′ dimers. Biochim. Biophys. Acta 1993, 1183, 374–380. [Google Scholar] [CrossRef]
- Vermathen, M.; Marzorati, M.; Bigler, P. Self-assembling properties of porphyrinic photosensitizers and their effect on membrane interactions probed by NMR spectroscopy. J. Phys. Chem. B 2013, 117, 6990–7001. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Yu, Z.; Li, P.; Merz, K.M. Using Ligand-Induced Protein Chemical Shift Perturbations To Determine Protein–Ligand Structures. Biochemistry 2017, 56, 2349–2362. [Google Scholar] [CrossRef]
- Hunashal, Y.; Cantarutti, C.; Giorgetti, S.; Marchese, L.; Molinari, H.; Niccolai, N.; Fogolari, F.; Esposito, G. Exploring exchange processes in proteins by paramagnetic perturbation of NMR spectra. Phys. Chem. Chem. Phys. 2020, 22, 6247–6259. [Google Scholar] [CrossRef]
- Mayo, B.C. Lanthanide shift reagents in nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 1973, 2, 49–74. [Google Scholar] [CrossRef]
- von Ammon, R.; Fischer, R.D. Shift Reagents in NMR Spectroscopy. Angew. Chem. Int. Ed. 1972, 11, 675–692. [Google Scholar] [CrossRef]
- Neuhaus, D.; Williamson, M.P. The Nuclear Overhauser Effect in Structural and Conformational Analysis; Wiley: New York, NY, USA, 2000; p. 656. [Google Scholar]
- Macura, S.; Huang, Y.; Suter, D.; Ernst, R.R. Two-dimensional chemical exchange and cross-relaxation spectroscopy of coupled nuclear spins. J. Magn. Reson. 1981, 43, 259–281. [Google Scholar] [CrossRef]
- Macura, S.; Ernst, R.R. Elucidation of cross relaxation in liquids by two-dimensional N.M.R. spectroscopy. Mol. Phys. 1980, 41, 95–117. [Google Scholar] [CrossRef]
- Kumar, A.; Ernst, R.R.; Wüthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Butts, C.P.; Jones, C.R.; Towers, E.C.; Flynn, J.L.; Appleby, L.; Barron, N.J. Interproton distance determinations by NOE—Surprising accuracy and precision in a rigid organic molecule. Org. Biomol. Chem. 2011, 9, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.R.; Butts, C.P.; Harvey, J.N. Accuracy in determining interproton distances using Nuclear Overhauser Effect data from a flexible molecule. Beilstein J. Org. Chem. 2011, 7, 145–150. [Google Scholar] [CrossRef]
- Vögeli, B.; Segawa, T.F.; Leitz, D.; Sobol, A.; Choutko, A.; Trzesniak, D.; van Gunsteren, W.; Riek, R. Exact Distances and Internal Dynamics of Perdeuterated Ubiquitin from NOE Buildups. J. Am. Chem. Soc. 2009, 131, 17215–17225. [Google Scholar] [CrossRef] [PubMed]
- Bothner-By, A.A.; Stephens, R.L.; Lee, J.; Warren, C.D.; Jeanloz, R.W. Structure determination of a tetrasaccharide: Transient nuclear Overhauser effects in the rotating frame. J. Am. Chem. Soc. 1984, 106, 811–813. [Google Scholar] [CrossRef]
- Bauer, C.J.; Frenkiel, T.A.; Lane, A.N. A comparison of the ROESY and NOESY experiments for large molecules, with application to nucleic acids. J. Magn. Reson. 1990, 87, 144–152. [Google Scholar] [CrossRef]
- Williamson, M.P. Nuclear Magnetic Resonance Spectroscopy | Nuclear Overhauser Effect. In Encyclopedia of Analytical Science, 3rd ed.; Worsfold, P., Poole, C., Townshend, A., Miró, M., Eds.; Academic Press: Oxford, UK, 2019; pp. 264–271. [Google Scholar]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echoes in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef] [Green Version]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef]
- Johnson, C.S. Diffusion ordered nuclear magnetic resonance spectroscopy: Principles and applications. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 34, 203–256. [Google Scholar] [CrossRef]
- Groves, P. Diffusion ordered spectroscopy (DOSY) as applied to polymers. Polym. Chem. 2017, 8, 6700–6708. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Balayssac, S.; Delsuc, M.-A.; Gilard, V.; Prigent, Y.; Malet-Martino, M. Two-dimensional DOSY experiment with Excitation Sculpting water suppression for the analysis of natural and biological media. J. Magn. Reson. 2009, 196, 78–83. [Google Scholar] [CrossRef]
- Simpson, A.J. Determining the molecular weight, aggregation, structures and interactions of natural organic matter using diffusion ordered spectroscopy. Magn. Reson. Chem. 2002, 40, S72–S82. [Google Scholar] [CrossRef]
- Colbourne, A.A.; Morris, G.A.; Nilsson, M. Local Covariance Order Diffusion-Ordered Spectroscopy: A Powerful Tool for Mixture Analysis. J. Am. Chem. Soc. 2011, 133, 7640–7643. [Google Scholar] [CrossRef] [PubMed]
- Pagès, G.; Gilard, V.; Martino, R.; Malet-Martino, M. Pulsed-field gradient nuclear magnetic resonance measurements (PFG NMR) for diffusion ordered spectroscopy (DOSY) mapping. Analyst 2017, 142, 3771–3796. [Google Scholar] [CrossRef] [PubMed]
- Cohen, Y.; Avram, L.; Evan-Salem, T.; Frish, L. Diffusion NMR in Supramolecular Chemistry. In Analytical Methods in Supramolecular Chemistry; Schalley, C., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 163–219. [Google Scholar]
- Kharlamov, S.V.; Latypov, S.K. Modern diffusion-ordered NMR spectroscopy in chemistry of supramolecular systems: The scope and limitations. Russ. Chem. Rev 2010, 79, 635–653. [Google Scholar] [CrossRef]
- Bakkour, Y.; Darcos, V.; Li, S.; Coudane, J. Diffusion ordered spectroscopy (DOSY) as a powerful tool for amphiphilic block copolymer characterization and for critical micelle concentration (CMC) determination. Polym. Chem. 2012, 3, 2006–2010. [Google Scholar] [CrossRef]
- Khodov, I.A.; Alper, G.A.; Mamardashvili, G.M.; Mamardashvili, N.Z. Hybrid multi-porphyrin supramolecular assemblies: Synthesis and structure elucidation by 2D DOSY NMR studies. J. Mol. Struct. 2015, 1099, 174–180. [Google Scholar] [CrossRef]
- Johnston, M.R.; Latter, M.J. Characterization of porphyrin supramolecular complexes using NMR diffusion spectroscopy. J. Porphyr. Phthalocyanines 2002, 6, 757–762. [Google Scholar] [CrossRef]
- Chatzigiannis, C.M.; Kiriakidi, S.; Tzakos, A.G.; Mavromoustakos, T. 2D DOSYTwo-dimensional diffusion-ordered NMR spectroscopy (2D DOSY) NMR: A Valuable Tool to Confirm the Complexation in Drug Delivery Systems. In Supramolecules in Drug Discovery and Drug Delivery: Methods and Protocols; Mavromoustakos, T., Tzakos, A.G., Durdagi, S., Eds.; Springer: New York, NY, USA, 2021; pp. 235–246. [Google Scholar]
- Momot, K.I.; Kuchel, P.W. Pulsed field gradient nuclear magnetic resonance as a tool for studying drug delivery systems. Concepts Magn. Reson. Part A 2003, 19, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Bruker Corporation. Almanac—Bruker Corporation [Brochure]: Analytical Tables and Product Overview; Innovation with Integrity; Analytical Solutions; T8-T13; Bruker Corporation: Billerica, MA, USA, 2012. [Google Scholar]
- Harris, R.K. Nmr and the periodic table. Chem. Soc. Rev. 1976, 5, 1–22. [Google Scholar] [CrossRef]
- Patching, S.G. NMR active nuclei for biological and biomedical applications. J. Diagn. Imaging Ther. 2016, 3, 7–48. [Google Scholar] [CrossRef] [Green Version]
- Cavanagh, J.; Fairbrother, W.J.; Palmer, A.G.; Rance, M.; Skelton, N.J. Chapter 7—Heteronuclear nmr experiments. In Protein NMR Spectroscopy, 2nd ed.; Cavanagh, J., Fairbrother, W.J., Palmer, A.G., Rance, M., Skelton, N.J., Eds.; Academic Press: Burlington, NJ, USA, 2007; pp. 533–678. [Google Scholar]
- Watts, A. NMR of Lipids. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1727–1738. [Google Scholar]
- Nieto, L.; Jiménez-Barbero, J. Carbohydrate NMR Spectroscopy. In Encyclopedia of Biophysics; Roberts, G.C.K., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 228–232. [Google Scholar]
- Marion, D. An introduction to biological NMR spectroscopy. Mol. Cell. Proteom. MCP 2013, 12, 3006–3025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindon, J.C. Nuclear Magnetic Resonance Spectroscopy | Fluorine-19. In Encyclopedia of Analytical Science, 3rd ed.; Worsfold, P., Poole, C., Townshend, A., Miró, M., Eds.; Academic Press: Oxford, UK, 2016; pp. 141–151. [Google Scholar]
- Goslinski, T.; Piskorz, J. Fluorinated porphyrinoids and their biomedical applications. J. Photochem. Photobiol. C 2011, 12, 304–321. [Google Scholar] [CrossRef]
- Oz, G.; Pountney, D.L.; Armitage, I.M. NMR spectroscopic studies of I = 1/2 metal ions in biological systems. Biochem Cell Biol 1998, 76, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Moan, J.; Berg, K.; Kvam, E.; Western, A.; Malik, Z.; Rück, A.; Schneckenburger, H. Intracellular Localization of Photosensitizers. In Ciba Foundation Symposium 146—Photosensitizing Compounds: Their Chemistry, Biology and Clinical Use; Bock, G., Harnett, S., Eds.; Wiley Online Library: Chichester, UK, 2007; pp. 95–111. [Google Scholar]
- Lavi, A.; Weitman, H.; Holmes, R.T.; Smith, K.M.; Ehrenberg, B. The Depth of Porphyrin in a Membrane and the Membrane’s Physical Properties Affect the Photosensitizing Efficiency. Biophys. J. 2002, 82, 2101–2110. [Google Scholar] [CrossRef] [Green Version]
- Warschawski, D.E.; Arnold, A.A.; Beaugrand, M.; Gravel, A.; Chartrand, É.; Marcotte, I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim. Biophys. Acta 2011, 1808, 1957–1974. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, G.; Mouret, L.; Chevance, S.; Le Rumeur, E.; Bondon, A. NMR of molecules interacting with lipids in small unilamellar vesicles. Eur. Biophys. J. 2007, 36, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, G.; Chevance, S.; Le Rumeur, E.; Bondon, A. Proton NMR detection of porphyrins and cytochrome C in small unilamellar vesicles: Role of the dissociation kinetic constant. Biophys. J. 2006, 90, L55–L57. [Google Scholar] [CrossRef] [PubMed]
- Vermathen, M.; Vermathen, P.; Simonis, U.; Bigler, P. Time-dependent interactions of the two porphyrinic compounds chlorin e6 and mono-L-aspartyl-chlorin e6 with phospholipid vesicles probed by NMR spectroscopy. Langmuir 2008, 24, 12521–12533. [Google Scholar] [CrossRef] [PubMed]
- Barsukov, L.I.; Victorov, A.V.; Vasilenko, I.A.; Evstigneeva, R.P.; Bergelson, L.D. Investigation of the inside-outside distribution, intermembrane exchange and transbilayer movement of phospholipids in sonicated vesicles by shift reagent NMR. Biochim. Biophys. Acta 1980, 598, 153–168. [Google Scholar] [CrossRef]
- Vermathen, M.; Marzorati, M.; Vermathen, P.; Bigler, P. pH-dependent distribution of chlorin e6 derivatives across phospholipid bilayers probed by NMR spectroscopy. Langmuir 2010, 26, 11085–11094. [Google Scholar] [CrossRef]
- Marzorati, M.; Bigler, P.; Vermathen, M. Interactions between selected photosensitizers and model membranes: An NMR classification. Biochim. Biophys. Acta 2011, 1808, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Temizel, E.; Sagir, T.; Ayan, E.; Isik, S.; Ozturk, R. Delivery of lipophilic porphyrin by liposome vehicles: Preparation and photodynamic therapy activity against cancer cell lines. Photodiagnosis Photodyn. Ther. 2014, 11, 537–545. [Google Scholar] [CrossRef]
- Sadasivam, M.; Avci, P.; Gupta, G.K.; Lakshmanan, S.; Chandran, R.; Huang, Y.Y.; Kumar, R.; Hamblin, M.R. Self-assembled liposomal nanoparticles in photodynamic therapy. Eur. J. Nanomed. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, A.; Hino, S.; Mae, T.; Tsuchiya, Y.; Sugikawa, K.; Tsukamoto, M.; Yasuhara, K.; Shigeto, H.; Funabashi, H.; Kuroda, A.; et al. Porphyrin-uptake in liposomes and living cells using an exchange method with cyclodextrin. RSC Adv. 2015, 5, 105279–105287. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, T.; Tsuchiya, Y.; Horiguchi, B.; Sugikawa, K.; Komaguchi, K.; Ikeda, A. 1H NMR Determination of Incorporated Porphyrin Location in Lipid Membranes of Liposomes. Bull. Chem. Soc. Jpn. 2018, 91, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Bertini, I.; Turano, P.; Vila, A.J. Nuclear magnetic resonance of paramagnetic metalloproteins. Chem. Rev. 1993, 93, 2833–2932. [Google Scholar] [CrossRef]
- Stojanović, S.; Isenović, E.R.; Zarić, B.L. Non-canonical interactions of porphyrins in porphyrin-containing proteins. Amino Acids 2012, 43, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.; Yanamala, N.; Tan, Y.L.; Gardner, E.E.; Tirupula, K.C.; Balem, F.; Sheves, M.; Nietlispach, D.; Klein-Seetharaman, J. Structural and Functional Consequences of the Weak Binding of Chlorin e6 to Bovine Rhodopsin. Photochem. Photobiol. 2019, 95, 787–802. [Google Scholar] [CrossRef]
- Gjuroski, I.; Girousi, E.; Meyer, C.; Hertig, D.; Stojkov, D.; Fux, M.; Schnidrig, N.; Bucher, J.; Pfister, S.; Sauser, L.; et al. Evaluation of polyvinylpyrrolidone and block copolymer micelle encapsulation of serine chlorin e6 and chlorin e4 on their reactivity towards albumin and transferrin and their cell uptake. J. Control. Release 2019, 316, 150–167. [Google Scholar] [CrossRef] [PubMed]
- Fiel, R.J.; Howard, J.C.; Mark, E.H.; Gupta, N.D. Interaction of DNA with a porphyrin ligand: Evidence for intercalation. Nucleic Acids Res. 1979, 6, 3093–3118. [Google Scholar] [CrossRef] [Green Version]
- Fiel, R.J.; Munson, B.R. Binding of meso-tetra (4-N-methylpyridyl) porphine to DNA. Nucleic Acids Res. 1980, 8, 2835–2842. [Google Scholar] [CrossRef] [Green Version]
- Carvlin, M.J.; Fiel, R.J. Intercalative and nonintercalative binding of large cationic porphyrin ligands to calf thymus DNA. Nucleic Acids Res. 1983, 11, 6121–6139. [Google Scholar] [CrossRef] [Green Version]
- Banville, D.L.; Marzilli, L.G.; Wilson, W.D. 31P NMR and viscometric studies of the interaction of meso-tetra(4-N-methyl-pyridyl)porphine and its Ni(II) and Zn(II) derivatives with DNA. Biochem. Biophys. Res. Commun. 1983, 113, 148–154. [Google Scholar] [CrossRef]
- Fiel, R.J. Porphyrin—Nucleic Acid Interactions: A Review. J. Biomol. Struct. Dyn. 1989, 6, 1259–1274. [Google Scholar] [CrossRef] [PubMed]
- Pratviel, G. Porphyrins in complex with DNA: Modes of interaction and oxidation reactions. Coord. Chem. Rev. 2016, 308, 460–477. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, Z.; Hu, H.; Yang, Q.; Hou, X.; Wu, P. DNA-modulated photosensitization: Current status and future aspects in biosensing and environmental monitoring. Anal. Bioanal. Chem. 2019, 411, 4415–4423. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-M.; Lu, Q.-Q.; Yao, S.; Su, H.-F.; Liu, H.-J.; Wang, Z.-J.; Wu, F.-S.; Wang, K. N-Methylpyridylporphyrin tailed with folate conjugate as a potential lysosomal-targeted photosensitizer: Synthesis, DNA interaction, singlet oxygen and subcellular localization. J. Porphyr. Phthalocyanines 2019, 23, 679–684. [Google Scholar] [CrossRef]
- Hirakawa, K.; Hirano, T.; Nishimura, Y.; Arai, T.; Nosaka, Y. Control of Singlet Oxygen Generation Photosensitized by meso-Anthrylporphyrin through Interaction with DNA. Photochem. Photobiol. 2011, 87, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Harada, M.; Okazaki, S.; Nosaka, Y. Controlled generation of singlet oxygen by a water-soluble meso-pyrenylporphyrin photosensitizer through interaction with DNA. Chem. Commun. 2012, 48, 4770–4772. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Nishimura, Y.; Arai, T.; Okazaki, S. Singlet Oxygen Generating Activity of an Electron Donor Connecting Porphyrin Photosensitizer Can Be Controlled by DNA. J. Phys. Chem. B 2013, 117, 13490–13496. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, K.; Taguchi, M.; Okazaki, S. Relaxation Process of Photoexcited meso-Naphthylporphyrins while Interacting with DNA and Singlet Oxygen Generation. J. Phys. Chem. B 2015, 119, 13071–13078. [Google Scholar] [CrossRef]
- López-Cebral, R.; Martín-Pastor, M.; Seijo, B.; Sanchez, A. Progress in the characterization of bio-functionalized nanoparticles using NMR methods and their applications as MRI contrast agents. Prog. Nucl. Magn. Reson. Spectrosc. 2014, 79, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Haaf, F.; Sanner, A.; Straub, F. Polymers of N-Vinylpyrrolidone: Synthesis, Characterization and Uses. Polym. J. 1985, 17, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Hong, Y.; Shen, L.; Wu, F.; Lin, X. Multifunctional Role of Polyvinylpyrrolidone in Pharmaceutical Formulations. AAPS PharmSciTech 2021, 22, 34. [Google Scholar] [CrossRef]
- Franco, P.; De Marco, I. The Use of Poly(N-vinyl pyrrolidone) in the Delivery of Drugs: A Review. Polymers 2020, 12, 1114. [Google Scholar] [CrossRef] [PubMed]
- Copley, L.; van der Watt, P.; Wirtz, K.W.; Parker, M.I.; Leaner, V.D. Photolon™, a chlorin e6 derivative, triggers ROS production and light-dependent cell death via necrosis. Int. J. Biochem. Cell Biol. 2008, 40, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Zhiyentayev, T.M.; Boltaev, U.T.; Solov’eva, A.B.; Aksenova, N.A.; Glagolev, N.N.; Chernjak, A.V.; Melik-Nubarov, N.S. Complexes of Chlorin e6 with Pluronics and Polyvinylpyrrolidone: Structure and Photodynamic Activity in Cell Culture. Photochem. Photobiol. 2014, 90, 171–182. [Google Scholar] [CrossRef]
- Tsvetkov, V.B.; Solov’eva, A.B.; Melik-Nubarov, N.S. Computer modeling of the complexes of Chlorin e6 with amphiphilic polymers. Phys. Chem. Chem. Phys. 2014, 16, 10903–10913. [Google Scholar] [CrossRef] [PubMed]
- Hädener, M.; Gjuroski, I.; Furrer, J.; Vermathen, M. Interactions of Polyvinylpyrrolidone with Chlorin e6-Based Photosensitizers Studied by NMR and Electronic Absorption Spectroscopy. J. Phys. Chem. B 2015, 119, 12117–12128. [Google Scholar] [CrossRef]
- Solov’eva, A.B.; Khasanova, O.V.; Aksenova, N.A.; Chernyak, A.V.; Volkov, V.I.; Timofeeva, V.A.; Timashev, P.S. The Influence of Effect of Polysaccharides and Polyvinylpyrrolidone on the Photocatalytic Activity of Chlorin e6 in Tryptophan Oxidation. Russ. J. Phys. Chem. A 2019, 93, 2507–2514. [Google Scholar] [CrossRef]
- Gjuroski, I.; Furrer, J.; Vermathen, M. How Does the Encapsulation of Porphyrinic Photosensitizers into Polymer Matrices Affect Their Self-Association and Dynamic Properties? Chemphyschem 2018, 19, 1089–1102. [Google Scholar] [CrossRef]
- Aksenova, N.A.; Oles, T.; Sarna, T.; Glagolev, N.N.; Chernjak, A.V.; Volkov, V.I.; Kotova, S.L.; Melik-Nubarov, N.S.; Solovieva, A.B. Development of novel formulations for photodynamic therapy on the basis of amphiphilic polymers and porphyrin photosensitizers. Porphyrin-polymer complexes in model photosensitized processes. Laser Phys. 2012, 22, 1642–1649. [Google Scholar] [CrossRef]
- Solovieva, A.B.; Aksenova, N.A. Porphyrin-Polymer Complexes in Model Photosensitized Processes and in Photodynamic Therapy. In Additives in Polymers; Berlin, A.A., Rogovina, S.Z., Zaikov, G.E., Eds.; Apple Academic Press: New York, NY, USA, 2015; pp. 77–123. [Google Scholar]
- Küçük, T.; Alpugan, S.; Davarcı, D.; Pehlivan, E.G.; Bayır, S.; Tazebay, U.H.; Dumoulin, F. Photoproperties, PVP formulation and 19F NMR of a Zn phthalocyanine with 24 magnetically pseudo-equivalent fluorine atoms. J. Porphyr. Phthalocyanines 2019, 23, 611–618. [Google Scholar] [CrossRef]
- Schneider, H.-J.; Hacket, F.; Rüdiger, V.; Ikeda, H. NMR Studies of Cyclodextrins and Cyclodextrin Complexes. Chem. Rev. 1998, 98, 1755–1786. [Google Scholar] [CrossRef]
- Gonzalez, M.C.; Weedon, A.C. Preparation and properties of a linked porphyrin–cyclodextrin. Can. J. Chem. 1985, 63, 602–608. [Google Scholar] [CrossRef]
- Kuroda, Y.; Hiroshige, T.; Sera, T.; Shiroiwa, Y.; Tanaka, H.; Ogoshi, H. Cyclodextrin-sandwiched porphyrin. J. Am. Chem. Soc. 1989, 111, 1912–1913. [Google Scholar] [CrossRef]
- Dick, D.L.; Rao, T.V.S.; Sukumaran, D.; Lawrence, D.S. Molecular encapsulation: Cyclodextrin-based analogs of heme-containing proteins. J. Am. Chem. Soc. 1992, 114, 2664–2669. [Google Scholar] [CrossRef]
- Kano, K.; Kitagishi, H.; Dagallier, C.; Kodera, M.; Matsuo, T.; Hayashi, T.; Hisaeda, Y.; Hirota, S. Iron Porphyrin−Cyclodextrin Supramolecular Complex as a Functional Model of Myoglobin in Aqueous Solution. Inorg. Chem. 2006, 45, 4448–4460. [Google Scholar] [CrossRef] [PubMed]
- Carofiglio, T.; Fornasier, R.; Lucchini, V.; Rosso, C.; Tonellato, U. Very strong binding and mode of complexation of water-soluble porphyrins with a permethylated β-cyclodextrin. Tetrahedron Lett. 1996, 37, 8019–8022. [Google Scholar] [CrossRef]
- Kadish, K.M.; Maiya, G.B.; Araullo, C.; Guilard, R. Micellar effects on the aggregation of tetraanionic porphyrins. Spectroscopic characterization of free-base meso-tetrakis(4-sulfonatophenyl)porphyrin, (TPPS)H2, and (TPPS)M (M = zinc(II), copper(II), and vanadyl) in aqueous micellar media. Inorg. Chem. 1989, 28, 2725–2731. [Google Scholar] [CrossRef]
- Ribó, J.M.; Farrera, J.-A.; Valero, M.L.; Virgili, A. Self-assembly of cyclodextrins with meso-tetrakis(4-sulfonatophenyl)porphyrin in aqueous solution. Tetrahedron 1995, 51, 3705–3712. [Google Scholar] [CrossRef]
- Sur, S.K.; Bryant, R.G. Spin-Lattice Relaxation Enhancement of Water Protons by Manganese Porphyrins Complexed with Cyclodextrins. J. Phys. Chem. 1995, 99, 4900–4905. [Google Scholar] [CrossRef]
- Mosseri, S.; Mialocq, J.C.; Perly, B.; Hambright, P. Porhyrins-cyclodextrin 1 Photooxidation of zinc tetrakis(4-sulfonatophenyl)porphyrin in cyclodextrin cavities: The characterization of ZnTSPP dication Photolysis, radiolysis, and NMR studies. J. Phys. Chem. 1991, 95, 2196–2203. [Google Scholar] [CrossRef]
- Mosinger, J.; Kliment, V., Jr.; Sejbal, J.; Kubát, P.; Lang, K. Host-guest complexes of anionic porphyrin sensitizers with cyclodextrins. J. Porphyr. Phthalocyanines 2002, 6, 514–526. [Google Scholar] [CrossRef]
- Kano, K.; Nishiyabu, R.; Asada, T.; Kuroda, Y. Static and Dynamic Behavior of 2:1 Inclusion Complexes of Cyclodextrins and Charged Porphyrins in Aqueous Organic Media. J. Am. Chem. Soc. 2002, 124, 9937–9944. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.L.; Wu, J.J.; Liang, W.J.; Chao, J.B. Study on the Association Phenomenon of Cyclodextrin to Porphyrin J-aggregates by NMR Spectroscopy. J. Incl. Phenom. Macrocycl. Chem. 2007, 58, 221–226. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Farrera, J.-A.; Garcia-Ortega, H.; Ramirez-Gutierrez, O.; Ribo, J.M. Heteroassociation of meso-sulfonatophenylporphyrins with β- and γ-cyclodextrin. J. Porphyr. Phthalocyanines 2001, 05, 465–473. [Google Scholar] [CrossRef]
- Venema, F.; Rowan, A.E.; Nolte, R.J.M. Binding of Porphyrins in Cyclodextrin Dimers. J. Am. Chem. Soc. 1996, 118, 257–258. [Google Scholar] [CrossRef] [Green Version]
- Mosinger, J.; Slavětínská, L.; Lang, K.; Coufal, P.; Kubát, P. Cyclodextrin carriers of positively charged porphyrin sensitizers. Org. Biomol. Chem. 2009, 7, 3797–3804. [Google Scholar] [CrossRef]
- Xiliang, G.; Shaomin, S.; Chuan, D.; Feng, F.; Wong, M.S. Comparative study on the inclusion behavior between meso-tetrakis(4-N-ethylpyridiniurmyl)porphyrin and β-cyclodextrin derivatives. Spectrochim. Acta A 2005, 61, 413–418. [Google Scholar] [CrossRef]
- Khurana, R.; Kakatkar, A.S.; Chatterjee, S.; Barooah, N.; Kunwar, A.; Bhasikuttan, A.C.; Mohanty, J. Supramolecular Nanorods of (N-Methylpyridyl) Porphyrin With Captisol: Effective Photosensitizer for Anti-bacterial and Anti-tumor Activities. Front. Chem. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.-J.; Chao, J.-B.; Pan, J.-H. Study on the interaction of 5-pyridine-10,15,20-tris-(p-chlorophenyl)porphyrin with cyclodextrins and DNA by spectroscopy. Spectrochim. Acta A 2007, 68, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Cosma, P.; Catucci, L.; Fini, P.; Dentuto, P.L.; Agostiano, A.; Angelini, N.; Scolaro, L.M. Tetrakis(4-pyridyl)porphyrin supramolecular complexes with cyclodextrins in aqueous solution. Photochem. Photobiol. 2006, 82, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Notsu, S.; Sugikawa, K.; Ikeda, A. Reversible Supramolecular System of Porphyrin Exchange between Inclusion in Cyclodextrin and Intercalation in DNA by Change in pH. ChemistrySelect 2018, 3, 5900–5904. [Google Scholar] [CrossRef]
- Ikeda, A.; Satake, S.; Mae, T.; Ueda, M.; Sugikawa, K.; Shigeto, H.; Funabashi, H.; Kuroda, A. Photodynamic Activities of Porphyrin Derivative–Cyclodextrin Complexes by Photoirradiation. ACS Med. Chem. Lett. 2017, 8, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Kitagishi, H.; Hatada, S.; Itakura, T.; Maki, Y.; Maeda, Y.; Kano, K. Cellular uptake of octaarginine-conjugated tetraarylporphyrin included by per-O-methylated β-cyclodextrin. Org. Biomol. Chem. 2013, 11, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Králová, J.; Kejík, Z.; Bříza, T.; Poučková, P.; Král, A.; Martásek, P.; Král, V. Porphyrin−Cyclodextrin Conjugates as a Nanosystem for Versatile Drug Delivery and Multimodal Cancer Therapy. J. Med. Chem. 2010, 53, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, A.; Purrello, R.; Rizzarelli, E.; Sortino, S.; Vecchio, G. Spectroscopic and self-association behavior of a porphyrin-β-cyclodextrin conjugate. New J. Chem. 2007, 31, 1499–1506. [Google Scholar] [CrossRef]
- Mineo, P. A porphyrin/β-cyclodextrin conjugated nano-system having a pan–lid molecular structure for smart drug carrier applications. Org. Biomol. Chem. 2014, 12, 3663–3670. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, H.Y.; Sun, H.L.; Liu, Y. Supramolecular nanoassemblies of an amphiphilic porphyrin-cyclodextrin conjugate and their morphological transition from vesicle to network. Chemistry 2015, 21, 4457–4464. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Zhang, H.-Y.; Liu, B.-W.; Liu, Y. Construction of a Supramolecular Polymer by Bridged Bis(permethyl-β-cyclodextrin)s with Porphyrins and Its Highly Efficient Magnetic Resonance Imaging. Macromolecules 2013, 46, 4268–4275. [Google Scholar] [CrossRef]
- Liu, Y.; Ke, C.-F.; Zhang, H.-Y.; Cui, J.; Ding, F. Complexation-Induced Transition of Nanorod to Network Aggregates: Alternate Porphyrin and Cyclodextrin Arrays. J. Am. Chem. Soc. 2008, 130, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, B.; Zhu, M.; Grayson, S.M.; Schmehl, R.; Jayawickramarajah, J. Water-soluble porphyrin nanospheres: Enhanced photo-physical properties achieved via cyclodextrin driven double self-inclusion. Chem. Commun. 2014, 50, 4853–4855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerichelli, G.; Mancinit, G. NMR techniques applied to micellar systems. Curr. Opin. Colloid Interface Sci. 1997, 2, 641–648. [Google Scholar] [CrossRef]
- Söderman, O.; Stilbs, P.; Price, W.S. NMR studies of surfactants. Concepts in Magnetic Resonance Part A 2004, 23A, 121–135. [Google Scholar] [CrossRef]
- Cui, X.; Mao, S.; Liu, M.; Yuan, H.; Du, Y. Mechanism of Surfactant Micelle Formation. Langmuir 2008, 24, 10771–10775. [Google Scholar] [CrossRef] [PubMed]
- Medhi, O.K.; Mazumdar, S.; Mitra, S. Proton NMR and optical spectra and magnetic properties of four-coordinated intermediate-spin, five-coordinated high-spin, and six-coordinated low-spin iron(II) hemes encapsulated in aqueous detergent micelles: Model for hemoproteins. Inorg. Chem. 1989, 28, 3243–3248. [Google Scholar] [CrossRef]
- Mazumdar, S. Proton and carbon-13 NMR studies on the structure of micelles encapsulating hemes in aqueous sodium dodecyl sulfate solutions. J. Phys. Chem. 1990, 94, 5947–5953. [Google Scholar] [CrossRef]
- Mazumdar, S.; Medhi, O.K.; Mitra, S. Stability and characterization of iron(III) and iron(II) heme peptides encapsulated in aqueous detergent micelles: Proton NMR and UV-visible spectroscopic studies. Inorg. Chem. 1991, 30, 700–705. [Google Scholar] [CrossRef]
- Mazumdar, S.; Mitra, S. Biomimetic chemistry of hemes inside aqueous micelles. In Structures and Biological Effects; Springer: Berlin/Heidelberg, Germany, 1993; pp. 115–145. [Google Scholar]
- Maiti, N.C.; Mazumdar, S.; Periasamy, N. Dynamics of Porphyrin Molecules in Micelles. Picosecond Time-Resolved Fluorescence Anisotropy Studies. J. Phys. Chem. 1995, 99, 10708–10715. [Google Scholar] [CrossRef]
- Gandini, S.C.M.; Yushmanov, V.E.; Borissevitch, I.E.; Tabak, M. Interaction of the Tetra(4-sulfonatophenyl)porphyrin with Ionic Surfactants: Aggregation and Location in Micelles. Langmuir 1999, 15, 6233–6243. [Google Scholar] [CrossRef]
- Yushmanov, V.E. Aggregation of Fe(III)TPPS4 on Biological Structures Is pH-Dependent, Suggesting Oxo-Bridging in the Aggregates. Inorg. Chem. 1999, 38, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Maiya, B.G.; Araullo-McAdams, C. Spectroscopic characterization of meso-tetrakis(1-methylpyridinium-4-yl)porphyrins, [(TMpyP)H2]4+ and [(TMpyP)M]4+, in aqueous micellar media, where M = VO2+, Cu(II), and Zn(II). J. Phys. Chem. 1991, 95, 427–431. [Google Scholar] [CrossRef]
- Monsú Scolaro, L.; Donato, C.; Castriciano, M.; Romeo, A.; Romeo, R. Micellar aggregates of platinum(II) complexes containing porphyrins. Inorg. Chim. Acta 2000, 300–302, 978–986. [Google Scholar] [CrossRef]
- Vermathen, M.; Louie, E.A.; Chodosh, A.B.; Ried, S.; Simonis, U. Interactions of Water-Insoluble Tetraphenylporphyrins with Micelles Probed by UV−Visible and NMR Spectroscopy. Langmuir 2000, 16, 210–221. [Google Scholar] [CrossRef]
- Maiti, N.C.; Mazumdar, S.; Periasamy, N. J- and H-Aggregates of Porphyrin−Surfactant Complexes: Time-Resolved Fluorescence and Other Spectroscopic Studies. J. Phys. Chem. B 1998, 102, 1528–1538. [Google Scholar] [CrossRef]
- Qiu, W.-G.; Li, Z.-F.; Bai, G.-M.; Meng, S.-N.; Dai, H.-X.; He, H. Interaction of water-soluble cationic porphyrin with anionic surfactant. Spectrochim. Acta A 2007, 68, 1164–1169. [Google Scholar] [CrossRef]
- Gandini, S.C.M.; Yushmanov, V.E.; Tabak, M. Interaction of Fe(III)- and Zn(II)-tetra(4-sulfonatophenyl) porphyrins with ionic and nonionic surfactants: Aggregation and binding. J. Inorg. Biochem. 2001, 85, 263–277. [Google Scholar] [CrossRef]
- Mamardashvili, G.M.; Kaigorodova, E.Y.; Khodov, I.y.A.; Scheblykin, I.; Mamardashvili, N.Z.; Koifman, O.I. Micelles encapsulated Co(III)-tetra(4-sulfophenyl)porphyrin in aqueous CTAB solutions: Micelle formation, imidazole binding and redox Co(III)/Co(II) processes. J. Mol. Liq. 2019, 293, 111471. [Google Scholar] [CrossRef]
- Mamardashvili, G.M.; Kaigorodova, E.Y.; Simonova, O.R.; Lazovskiy, D.A.; Mamardashvili, N.Z. Interaction of the Sn(IV)-tetra(4-sulfonatophenyl)porphyrin axial complexes with cetyltrimethylammonium bromide: Aggregation and location in micelles, fluorescence properties and photochemical stability. J. Mol. Liq. 2020, 318, 113988. [Google Scholar] [CrossRef]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Kumari, P.; Lakhani, P.M.; Ghosh, B. Recent advances in polymeric micelles for anti-cancer drug delivery. Eur. J. Pharm. Sci. 2016, 83, 184–202. [Google Scholar] [CrossRef] [PubMed]
- Sakai-Kato, K.; Nishiyama, N.; Kozaki, M.; Nakanishi, T.; Matsuda, Y.; Hirano, M.; Hanada, H.; Hisada, S.; Onodera, H.; Harashima, H.; et al. General considerations regarding the in vitro and in vivo properties of block copolymer micelle products and their evaluation. J. Control. Release 2015, 210, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Houdaihed, L.; Evans, J.C.; Allen, C. Overcoming the Road Blocks: Advancement of Block Copolymer Micelles for Cancer Therapy in the Clinic. Mol. Pharm. 2017, 14, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Fraenza, C.C.; Mattea, C.; Farrher, G.D.; Ordikhani-Seyedlar, A.; Stapf, S.; Anoardo, E. Rouse dynamics in PEO-PPO-PEO block-copolymers in aqueous solution as observed through fast field-cycling NMR relaxometry. Polymer 2018, 150, 244–253. [Google Scholar] [CrossRef]
- Walderhaug, H.; Söderman, O. NMR studies of block copolymer micelles. Curr. Opin. Colloid Interface Sci. 2009, 14, 171–177. [Google Scholar] [CrossRef]
- Ma, J.-H.; Guo, C.; Tang, Y.-L.; Liu, H.-Z. 1H NMR Spectroscopic Investigations on the Micellization and Gelation of PEO−PPO−PEO Block Copolymers in Aqueous Solutions. Langmuir 2007, 23, 9596–9605. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-H.; Guo, C.; Tang, Y.-L.; Wang, J.; Zheng, L.; Liang, X.-F.; Chen, S.; Liu, H.-Z. Salt-Induced Micellization of a Triblock Copolymer in Aqueous Solution: A 1H Nuclear Magnetic Resonance Spectroscopy Study. Langmuir 2007, 23, 3075–3083. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Håkansson, B.; Söderman, O.; Topgaard, D. Influence of Polydispersity on the Micellization of Triblock Copolymers Investigated by Pulsed Field Gradient Nuclear Magnetic Resonance. Macromolecules 2007, 40, 8250–8258. [Google Scholar] [CrossRef]
- Pfister, S.; Sauser, L.; Gjuroski, I.; Furrer, J.; Vermathen, M. Monitoring the encapsulation of chlorin e6 derivatives into polymer carriers by NMR spectroscopy. J. Porphyr. Phthalocyanines 2019, 23, 1576–1586. [Google Scholar] [CrossRef]
- Izunobi, J.U.; Higginbotham, C.L. Polymer Molecular Weight Analysis by 1H NMR Spectroscopy. J. Chem. Educ. 2011, 88, 1098–1104. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kabanov, A.V. Pluronic block copolymers: Evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Cacaccio, J.; Durrani, F.; Cheruku, R.R.; Borah, B.; Ethirajan, M.; Tabaczynski, W.; Pera, P.; Missert, J.R.; Pandey, R.K. Pluronic F-127: An Efficient Delivery Vehicle for 3-(1’-hexyloxy)ethyl-3-devinylpyropheophorbide-a (HPPH or Photochlor). Photochem. Photobiol. 2020, 96, 625–635. [Google Scholar] [CrossRef]
- Mike Motloung, B.; Edward Sekhosana, K.; Managa, M.; Prinsloo, E.; Nyokong, T. The photophysicochemical properties and photodynamic therapy activity of phenyldiazenyl phenoxy substituted phthalocyanines when incorporated into Pluronic® F127 micelles. Polyhedron 2019, 174, 114157. [Google Scholar] [CrossRef]
- Wenceslau, A.C.; Ferreira, G.L.Q.C.; Hioka, N.; Caetano, W. Spectroscopic studies of pyridil and methoxyphenyl porphyrins in homogeneous and Pluronic®-based nanostructured systems. J. Porphyr. Phthalocyanines 2015, 19, 1168–1176. [Google Scholar] [CrossRef]
- D’Souza, A.A.; Shegokar, R. Polyethylene glycol (PEG): A versatile polymer for pharmaceutical applications. Expert Opin. Drug Deliv. 2016, 13, 1257–1275. [Google Scholar] [CrossRef] [PubMed]
- Zalipsky, S. Chemistry of polyethylene glycol conjugates with biologically active molecules. Adv. Drug Del. Rev. 1995, 16, 157–182. [Google Scholar] [CrossRef]
- Harris, J.M.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Swierczewska, M.; Lee, K.C.; Lee, S. What is the future of PEGylated therapies? Expert. Opin. Emerg. Drugs 2015, 20, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Steinbeck, C.A.; Hedin, N.; Chmelka, B.F. Interactions of Charged Porphyrins with Nonionic Triblock Copolymer Hosts in Aqueous Solutions. Langmuir 2004, 20, 10399–10412. [Google Scholar] [CrossRef] [PubMed]
- Lamch, Ł.; Tylus, W.; Jewgiński, M.; Latajka, R.; Wilk, K.A. Location of Varying Hydrophobicity Zinc(II) Phthalocyanine-Type Photosensitizers in Methoxy Poly(ethylene oxide) and Poly(l-lactide) Block Copolymer Micelles Using 1H NMR and XPS Techniques. J. Phys. Chem. B 2016, 120, 12768–12780. [Google Scholar] [CrossRef] [PubMed]
- Lepre, C.A.; Moore, J.M.; Peng, J.W. Theory and applications of NMR-based screening in pharmaceutical research. Chem. Rev. 2004, 104, 3641–3676. [Google Scholar] [CrossRef]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrin (Guest) | Macromolecule (Host) | NMR Technique | Result | Ref |
|---|---|---|---|---|
| Phospholipids | ||||
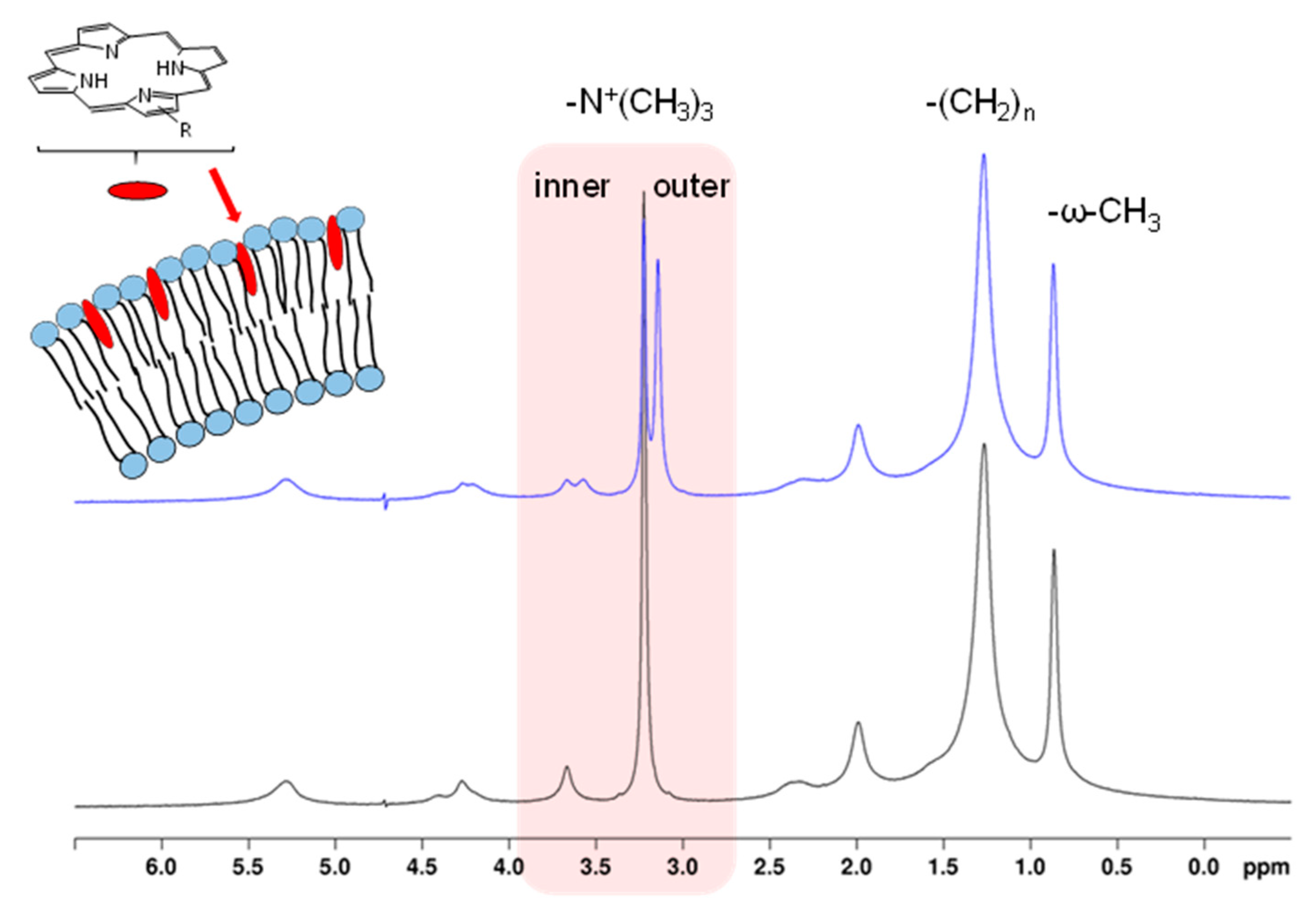
| Ce6 Ce6 derivatives | DOPC-SUVs | 1H NMR chem. shift perturbation of host Time-dependent 1H NMR chem. shift perturbation of host | Ce6 attached to PL-bilayer head group Transmembrane kinetics of Ce6 (flip-flop) pH dependence of kinetics | [131,133] |
| Ce6, Ce6 derivatives PPIX, DPIX, HPIX and derivatives | DOPC-SUVs | 1H NMR chem. shift perturbation of host | Porphyrin aggregate structure determines membrane interaction | [84] |
| Ce6 derivatives PPIX, DPIX, HPIX and derivatives TPP derivatives | DOPC-SUVs | 1H NMR chem. shift perturbation of host Time-dependent 1H NMR chem. shift perturbation of host | Different patterns of bilayer localization and transmembrane kinetics depending on porphyrin structure and substitution Patterns used for classification of membrane interactions | [134] |
| TPP Zn-TPP | DMPC liposomes | 1H NMR spectral appearance of guest | Transfer from CD complex to liposome | [138] |
| TPP | Egg-PC liposomes | 1H NMR chem. shift perturbation of host | Liposomal localization (hydrophobic core) | [139] |
| Proteins | ||||
| Ce6 | Bovine rhodopsin 19F-/15N-Trp-labeled rhodopsin | 1H-, 19F- and 15N-NMR chem. shift perturbation of host 1H-15N NMR HSQC 1H NMR spectral appearance of guest | Weak binding of Ce6 to rhodopsin localized at cytoplasmic domain | [142] |
| Ce6 SerCe | HSA Tf | 1H NMR spectral appearance of guest | Binding to both HSA and Tf PVP encapsulation prevents binding BCM encapsulation prevents only Tf binding | [143] |
| Nucleic acids | ||||
| TMPyP, Ni(II)TMPyP, Zn(II)TMPyP | DNA | 31P NMR chem. shift perturbation of host | TMPyP, Ni(II)TMPyP intercalate, Zn(II)TMPyP binds to the outside of DNA | [147] |
| Cationic TMPyP derivatives | DNA | 31P-, 1H NMR chem. shift perturbation of host | Review: Three binding modes (intercalation, outside binding, outside binding with self-stacking) | [148] |
| Porphyrin (Guest) | Macromolecule (Host) | NMR Technique | Result | Ref |
|---|---|---|---|---|
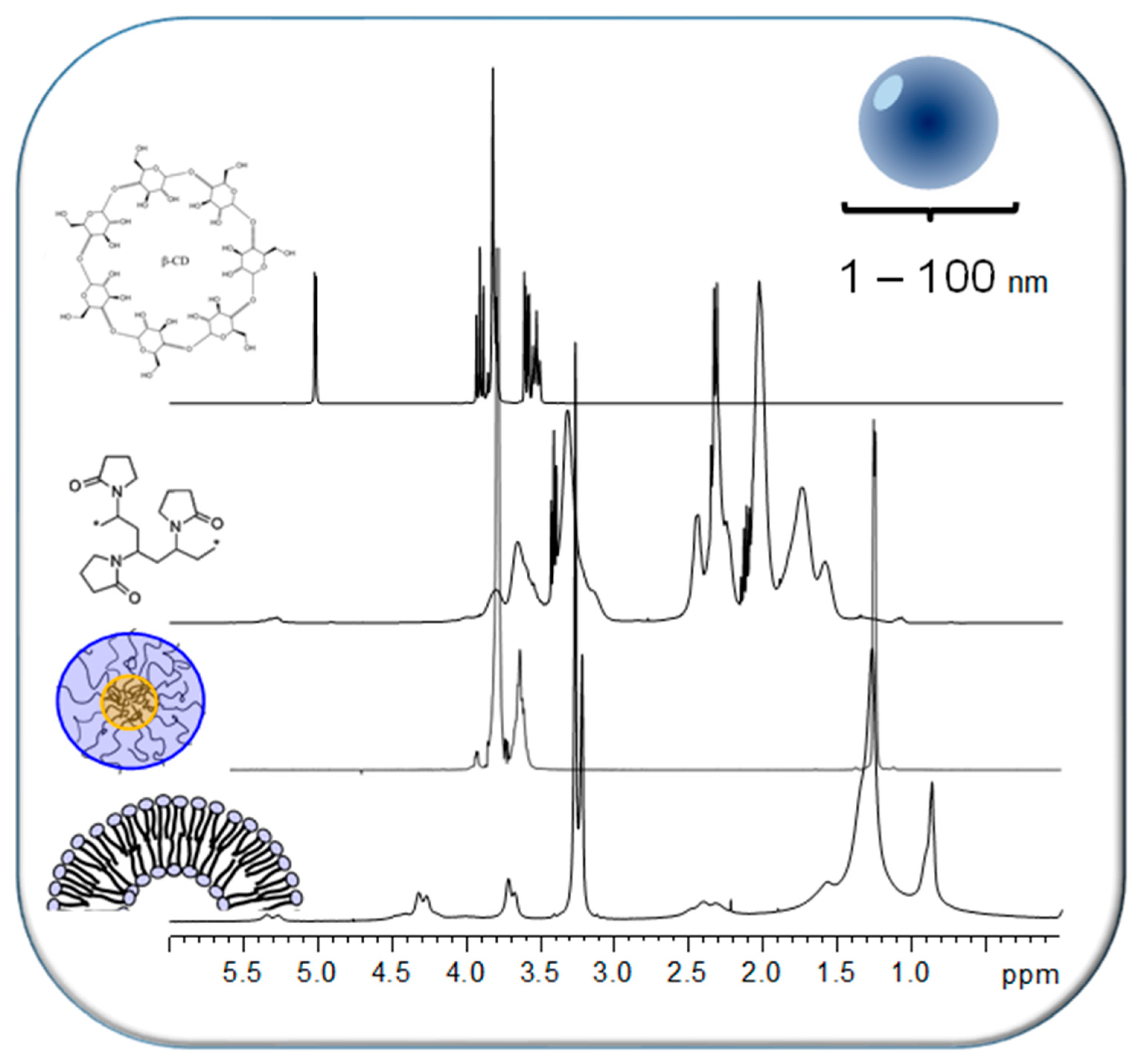
| Ce6 | PVP (MW 25 kDa) | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Ce6 mainly interacts with the hydrophobic vinyl-backbone of PVP Disaggregation upon interaction with PVP | [161] |
| Ce6 | PVP (MW 40 kDa) | 1H NMR spectral appearance of guest | Disaggregation upon interaction with PVP | [164] |
| Ce6 SerCe LysCe TyrCe ArgCe Ce6-amino-hexanoic amide | PVP (MW 10 kDa) | 1H NMR spectral appearance of guest 1H NMR chem. shift titration with host 1H DOSY of host–guest mixture 2D 1H1H NOESY of host–guest mixture | Disaggregation upon interaction with PVP Determination of binding constant Host and guest have same diffusion properties Identification of host and guest protons in close proximity | [163] |
| SerCe | PVP (MW 10 kDa) | T2 relaxation time measurements of host and guest | Change and assimilation of dynamic properties of host and guest Motional restriction of guest | [163,165] |
| Ce4 | PVP (MW 10 kDa) | 1H NMR spectral appearance of guest 1H NMR chem. shift titration with host 1H DOSY of host–guest mixture | Disaggregation upon interaction with PVP Determination of binding constant Host and guest have same diffusion properties | [143] |
| Ce4, SerCe | PVP (MW 10 kDa), HSA, Tf | 1H NMR spectral appearance of guest | PVP-encapsulated guest is protected from protein binding | [143] |
| DMG | PVP (MW 40 kDa) | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host | Disaggregation upon interaction with PVP Hydrophobic and hydrophilic interactions between host and guest | [166,167] |
| PPIX, DPIX, HPIX and derivatives | PVP (MW 10 kDa) | 1H NMR spectral appearance of guest | Different extent of disaggregation upon PVP interaction | [165] |
| HPIX, DPIXDS, DPIXDSME | PVP (MW 10 kDa) | 1H NMR chem. shift titration with host 1H DOSY of host–guest mixture T2 relaxation time measurements of host and guest | Determination of binding curves Host and guest have same diffusion properties Restricted mobility of encapsulated guest | [165] |
| Fluorinated ZnPc (ZnPcF24) | PVP | 19F NMR spectral appearance of guest | Guest exists as aggregate in PVP | [168] |
| Porphyrin (Guest) | Macromolecule (Host) | NMR Technique | Result | Ref |
|---|---|---|---|---|
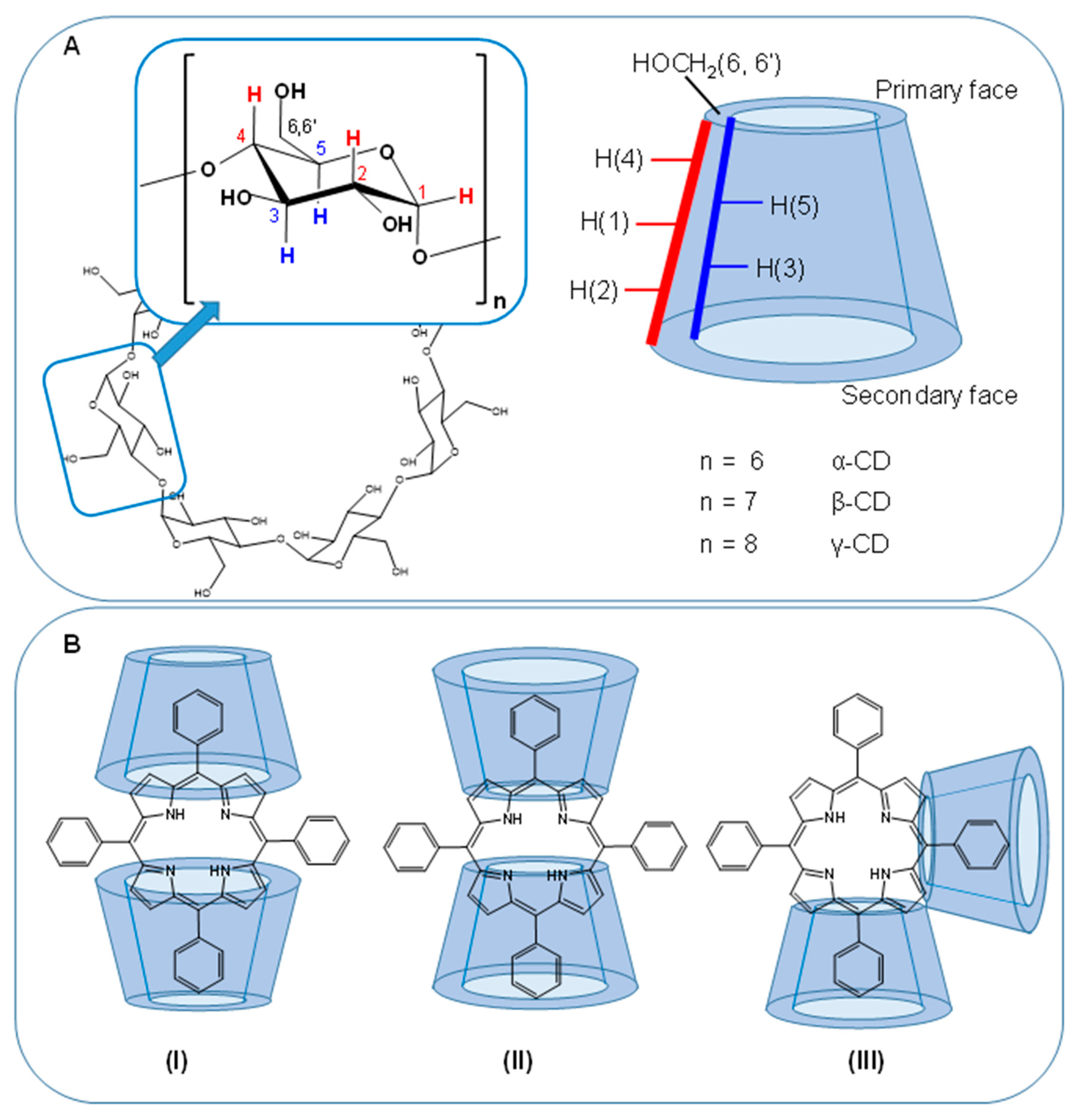
| TPPS4 | α-, β-, γ-CD | 1H NMR chem. shift perturbation of host 2D 1H1H ROESY of host–guest mixture | 2:1 (CD:TPPS4) inclusion complexes with β- and γ-CD, no complex with α-CD β-CD: through secondary face γ -CD: through primary face | [176] |
| TPPS4, Mn(III)TPPS4 | α-, β-, γ-CD | 1H and 13C NMR chem. shift perturbation of host 2D 1H1H ROESY of host–guest mixture | Strongest binding for β-CD | [177] |
| Zn(II)TPPS4 | β-CD | 1H NMR chem. shift perturbation of host | Formation of inclusion complex | [178] |
| Zn(II)TPPS4 Pd(II)TPPS4 TPPC4 | β-, γ-CD HP-β-CD HP-γ-CD | 1H and 13C NMR chem. shift perturbation of host 2D 1H1H ROESY of host–guest mixture | Formation of inclusion complexes: (HP)β-CD: through secondary face (HP)γ -CD: through primary face Weak binding to CD exterior | [179] |
| TPPS4 TPPOC3PSa TPPC4 TPPOC3Py | β-Cd TMe-β-CD | 1H and 13C NMR chem. shift perturbation of host 2D 1H1H ROESY 1H NMR spectral appearance of guest 13C-T1 relaxation time of host and guest | β-CD, TMe-β-CD: anionic porphyrin guests binding more favorable than cationic Disaggregation; formation of trans-type 2:1 complexes with TMe-β-CD TPPS4: stronger binding to TMe-β-CD than to β-CD Motional restriction of CD and TPP-phenyl rings (less pronounced inside cavity) | [180] |
| TPPS4 Acidic conditions | β-CD Me-β-CD HP-β-CD | 1H and 13C NMR chem. shift perturbation of host 2D 1H1H ROESY 1H NMR spectral appearance of guest | β-CD, HP-β-CD: inclusion via secondary face Me-β-CD: inclusion via primary face Disaggregation of J-aggregates upon complexation | [181] |
| TPPS4 TPPS3 TPPS2o TPPS2a TMPyP | β-, γ-CD | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host 2D 1H1H ROESY | Inclusion complexes formed with all but TPPS2a and TMPyP TPPS4, TPPS3, TPPS2o: Partial disaggregation Sulfonatophenyl- but not phenyl-group included in the case of mixed substituents Inclusion via secondary face for β-CD and primary for γ-CD | [182] |
| TPPS4 TPPC4 | CD dimers with flexible spacers | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host | Adjacently (“syn”) and oppositely (“anti”) capped TPPS4 | [183] |
| TMPyP TPPS4H22+ | α-, β-, γ-Cd TMe-β-CD SO3-β-CD DiMeSO3-β-CD | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest 1H NMR chem. shift titration with host 2D 1H1H ROESY | TMPyP: External binding to native CDs and TMe-β-CD TPPS4H22+: Inclusion with β-CD via secondary and with γ-CD via primary face Fast exchange between free/complex form | [184] |
| TEPyP | β-CD HP-β-CD SBE-CD | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest 2D 1H1H ROESY | Inclusion with β-CD and HP-β-CD from the primary face | [185] |
| TMPyP | SBE-CD | 1H NMR spectral appearance of guest | Complex formation with SBE-CD Fast exchange between free/complex form | [186] |
| PyTPP | β-CD Tme-β-CD | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Inclusion with TMe-β-CD (from the primary face) but hardly with β-CD | [187] |
| TPyP | HP-β-CD Di-Me-β-Cd TMe-β-CD | 1H NMR spectral appearance of guest 2D 1H1H NOESY | 1:1 complex with TMe-β-CD from the secondary face | [188] |
| TPyP | TMe-β-CD | 1H NMR spectral appearance of guest at different pH | 1:2 (TPyP:CD) complex pH-dependent release (acidic condition) | [189] |
| TPP, TPPC4 THPPb, TAPPc TMeOPPd | TMe-β-CD | 1H NMR spectral appearance of host–guest mixture | All form 1:2 (TPP:CD) inclusion complexes with TMe-β-CD | [190] |
| octa-arginine-TPP (R8-TPP) | TMe-β-CD | 1H NMR chem. shift titration with host 1H NMR spectral appearance of guest 2D 1H1H ROESY | Trans-type 1:2 (TPP:CD) inclusion complex with the non-substituted phenyl groups via secondary face Slow free/complex exchange rate | [191] |
| TPPS4 | ZnTPP- DAPM-β-CDe | 1H NMR spectral appearance of host–guest mixture 2D 1H1H NOESY 1H-DOSY | ZnTPP- β-CD forms self-inclusion and inclusion complexes with TPPS4 yielding vesicles and networks | [195] |
| Mn(III)TPP PEGylated | bridged bis(TMe-β-CD) | 2D 1H1H NOESY | Inclusion complexes, formation of supramolecular polymers | [196] |
| TPPS4 | ZnTPP-, DAPM-β-CDe DAPM-TMe-β-CDe | 1H NMR spectral appearance of host–guest mixture 2D 1H1H NOESY | ZnTPP- β-CD and -TMe-β-CD form self-inclusion and inclusion complexes; the latter are dissolved in favor of TPPS4 inclusion Formation of networks and nanorods | [197] |
| Porphyrin (Guest) | Macromolecule (Host) | NMR Technique | Result | Ref |
|---|---|---|---|---|
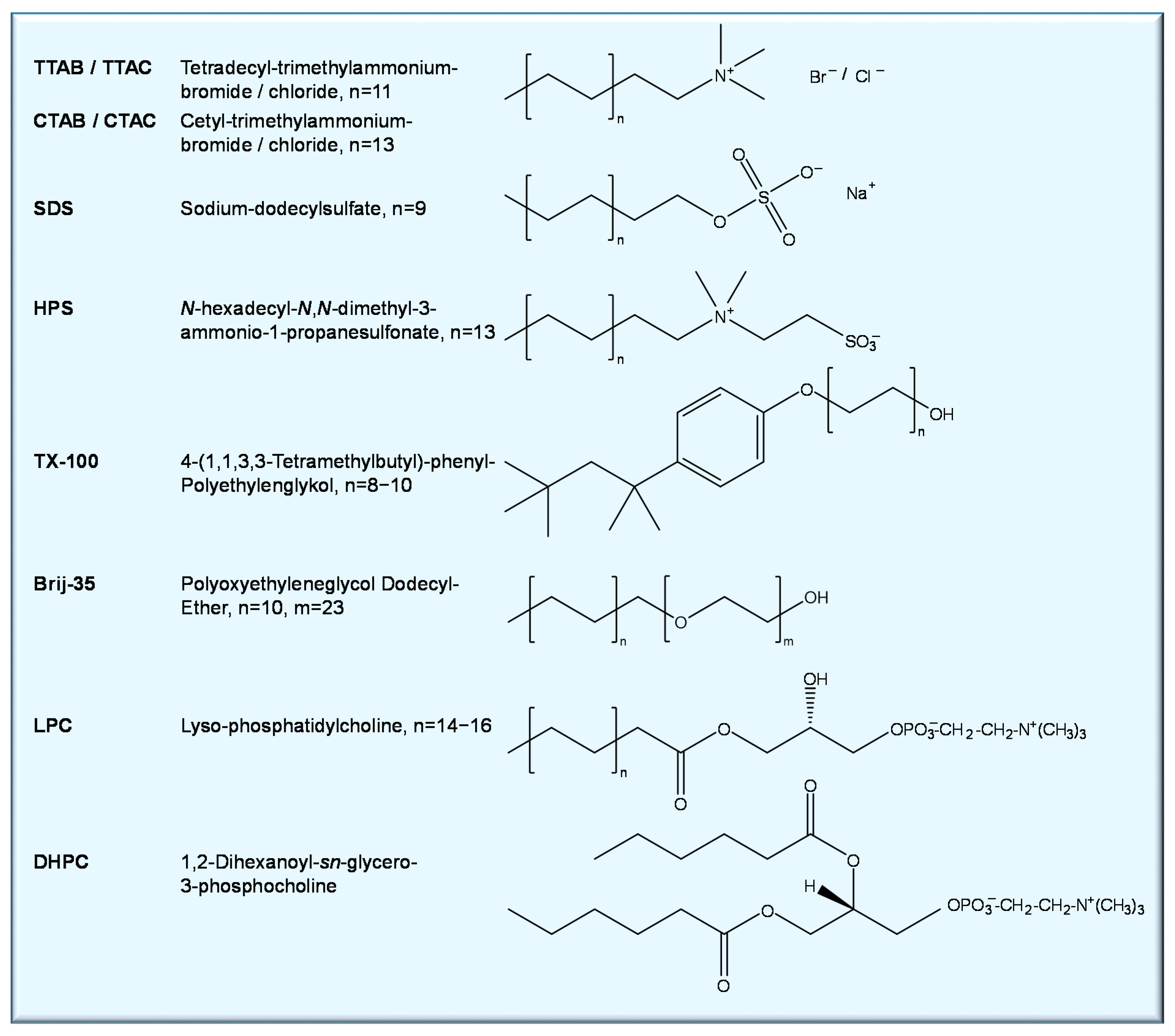
| TPPS4 Zn(II)TPPS4 Cu(II)TPPS4 VO2+TPPS4 | CTAB SDS TX-100 | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Encapsulation in hydrophobic core of CTAB and TX-100 micelles Disaggregation in CTAB, TX-100 SDS promotes aggregation, no insertion | [175] |
| TPPS4 | CTAC HPS LPC | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest 1D NOE spectroscopy 1H T1 relaxation times | CTAC and HPS micelles: TPPS4 localizes in the hydrophobic core Reduced molecular mobility of TPPS4 LPC micelles: TPPS4 intercalates involving the polar head region Fast exchange between free and bound states | [207] |
| TPPS4 Fe(III)TPPS4 Zn(II)TPPS4 | CTAC LPC | 1H NMR chem. shift perturbation of host; pH dependence 1H NMR spectral appearance of guest 1H T1 relaxation time | TPPS4, Zn(II)-, Fe(III)TPPS4: Incorporate into CTAC and LPC micelles in the core region pH-dependent aggregation of Fe(III)TPPS4 in micelles | [208] |
| TMPyP Zn(II)TMPyP Cu(II)TMPyP VO2+TMPyP | CTAB SDS TX-100 | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | All solubilized by SDS micelles but not by CTAB and TX-100 Monomerization in SDS | [209] |
| Pt(Cy2dim)Me]4(TpyP) a | SDS TX-100 | 1H NMR chem. shift perturbation of host | Location in hydrophobic region of SDS micelles; Low solubility in TX-100 | [210] |
| p-(OH)-phenyl-TPP p-(COOH)-phenyl-TPP p-(NH2)-phenyl-TPP p-(NO2)-phenyl-TPP | TTAB SDS TX-100 | 1H NMR chem. shift perturbation of host 1H NMR chem. shift titration with guest 1H NMR spectral appearance of guest | Insertion inhibited by electrostatic repulsion p-(NO2)-TPP not inserted in any micelles p-(OH)-phenyl-TPP inserted in all micelles most efficiently | [211] |
| TPPS4 | CTAB | 1H NMR spectral appearance of guest 1H NMR chem. shift titration with host | Below cmc: Premicellar aggregates Above cmc: Micellar insertion and monomerization | [212] |
| TPPOC3Py | SDS | 1H NMR chem. shift perturbation of host | Below cmc: Premicellar aggregates Above cmc: Intercalation among SDS chains, monomerization | [213] |
| Fe(III)TPPS4 Zn(II)TPPS4, | CTAC HPS Brij-35 TX-100 | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Fe(III)TPPS4, Zn(II)TPPS4, embedded in hydrophobic core of the micelles | [214] |
| Co(III)TPPS4(imidazole)2 | CTAB | 1D selective NOE spectroscopy | Closer location near the CTAB micellar core | [215] |
| Sn(IV)TPPS4(OH)2 Sn(IV)TPPS4(Met)2 Sn(IV)TPPS4(Tyr)2 | CTAB | 1D selective NOE spectroscopy | Closer location near the CTAB micellar core | [216] |
| Ce6, Ce4 Ce6 derivatives DPIX, PPIX, HPIX and derivatives | DHPC | 1H NMR spectral appearance of guest | Chlorin derivatives: Monomerized in DHPC micelles Porphyrin derivatives: Only weak interactions, no disaggregation | [84] |
| Porphyrin (Guest) | Macromolecule (Host) | NMR Technique | Result | Ref |
|---|---|---|---|---|
| TPPS4 | P123 | 1H NMR chem. shift perturbation of host 1H13C HSQC 1H NMR spectral appearance of guest 1D ROE spectroscopy DOSY (HSQC-res.) T-dependent NMR | <20 °C: TPPS4 strong interactions with PPG units Small TPPS4-P123 aggregates >35 °C: stronger interactions with PEG units TPPS4-P123 micelles in fast exchange with smaller TPPS4-P123 aggregates | [236] |
| ZnPc ZnPcS4 ZnPcF16 | mPEG-b-PLLA | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest 1D NOE spectroscopy | ZnPc localized in micellar core ZnPcS4 and ZnPcF16 localize in micellar corona | [237] |
| DMG | F127 pure PEG | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Disaggregation DMG localizes in hydrophobic core of F127 micelles DMG forms complex with PEG | [166] |
| Ce6 | F127 pure PEG | 1H NMR chem. shift perturbation of host 1H NMR spectral appearance of guest | Disaggregation Ce6 localizes in the PEG-PPG-interface region of F127 micelles | [161] |
| Ce6 SerCe, LysCe, TyrCe, ArgCe DPIX DPIXDS, DPIXDSME PPIX HPIX, iHPIX | KP | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host 1H NMR chem. shift titration with host 1H DOSY 1D NOE spectroscopy | Chlorins: disaggregation Porphyrins: only HPIX, DPIXDS, DPIXDSME disaggregate Determination of binding curves Dynamic exchange between free and micellar state Preferential localization in micellar core | [165] |
| Ce4 | KP | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host 1H NMR chem. shift titration with host 1H DOSY 2D NOESY | Disaggregation Localization in hydrophobic core of KP micelles Lack of dynamic exchange | [143] |
| SerCe Ce4 CeMED CeDM CeTM | KP F108 F127 L64 P84 | 1H NMR spectral appearance of guest 1H NMR chem. shift perturbation of host 1H DOSY 1H T1 / T2 relaxation | Loading efficiency inversely correlated with chlorin hydrophobicity Interaction mainly with hydrophobic core Differently pronounced dynamic exchange Disaggregation accompanied by changes in the dynamics | [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gjuroski, I.; Furrer, J.; Vermathen, M. Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques. Molecules 2021, 26, 1942. https://doi.org/10.3390/molecules26071942
Gjuroski I, Furrer J, Vermathen M. Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques. Molecules. 2021; 26(7):1942. https://doi.org/10.3390/molecules26071942
Chicago/Turabian StyleGjuroski, Ilche, Julien Furrer, and Martina Vermathen. 2021. "Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques" Molecules 26, no. 7: 1942. https://doi.org/10.3390/molecules26071942
APA StyleGjuroski, I., Furrer, J., & Vermathen, M. (2021). Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques. Molecules, 26(7), 1942. https://doi.org/10.3390/molecules26071942