
Simple and Equipment-Free Paper-Based Device for Determination of Mercury in Contaminated Soil


Abstract
:
1. Introduction
2. Results and Discussion
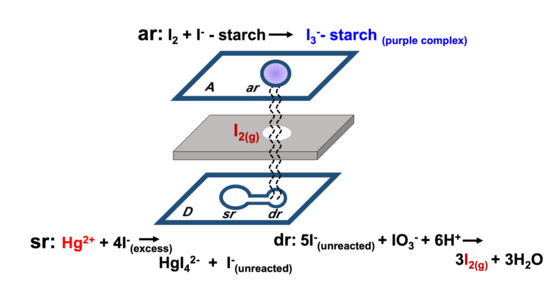
2.1. Principle of Indirect Colorimetric Detection of Mercury
| At the Donor layer | ||
| ‘sr’ reservoir: | Hg2+(aq) + I−(aq) | → HgI42−(aq) + I−(aq) |
| excess | unreacted | |
| ‘dr’ reservoir: | 5I−(aq) + IO3−(aq) + 6H+(aq) | → 3I2(g) + 3H2O(l) |
| unreacted | ||
| At the Acceptor layer | I2(g) + I−-starch | → I3− -starch(aq) |
| purple complex |
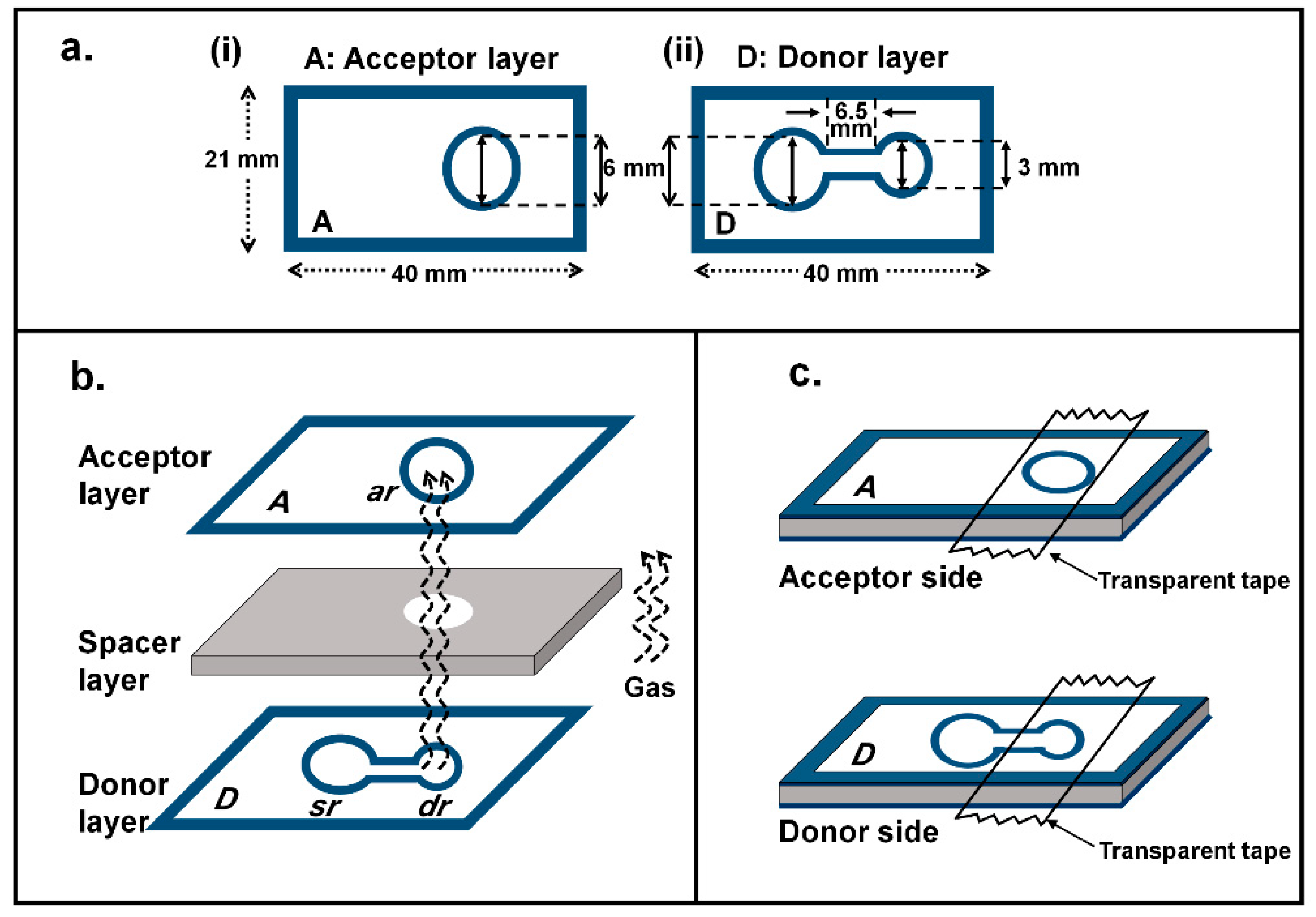
2.2. Optimization of Physical Parameters of the µPAD
2.2.1. Diameter of Donor Reservoir
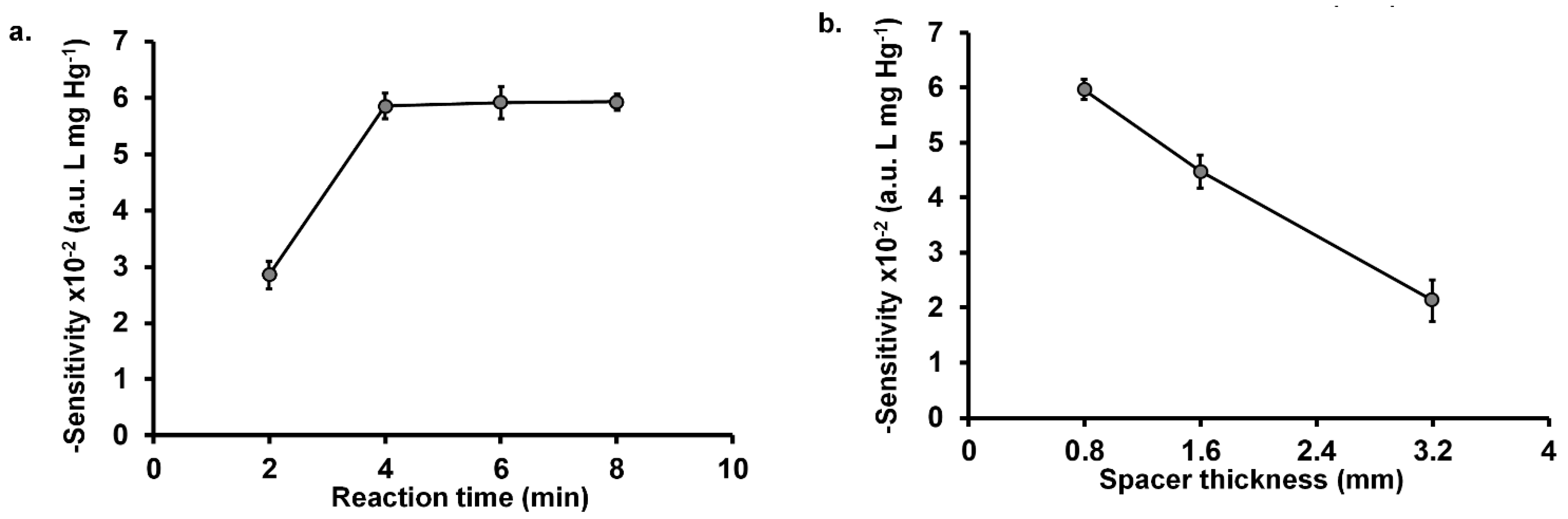
2.2.2. Effect of Reaction Time
2.2.3. Effect of Spacer Thickness
2.3. Optimization of Chemical Parameters of µPAD
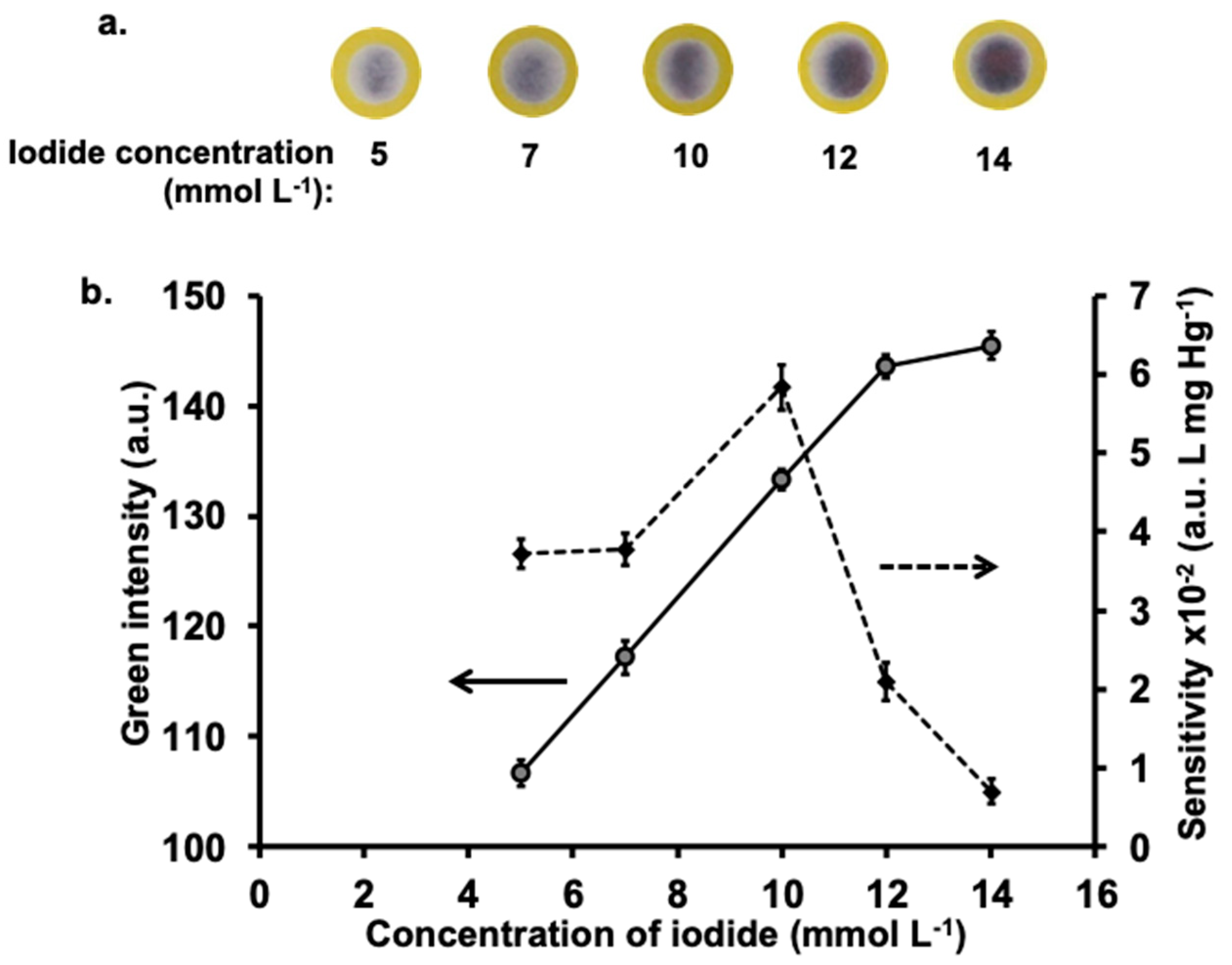
2.3.1. Iodide Concentration
2.3.2. The pH of Mercury Solution
2.4. Analytical Performance and Interference Study
2.4.1. Analytical Features
2.4.2. Interference Study
2.5. Applications and Validation
2.6. Comparison of the Proposed Method with Existing Methods Including Other PADs/µPADs Methods for Determination of Mercury
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of Samples
3.3. Fabrication of µPAD
3.4. Operating Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- United States Environmental Protection Agency. EPA-452/R-97-004, Mercury Study Report to Congress, Volume II: An Inventory of Anthropogenic Mercury Emissions in the United States; U.S. Environmental Protection Agency: Washington, DC, USA, 1997.
- United Nations Environment Programme. A Practical Guide: Reducing Mercury Use in Artisanal and Small-Scale Gold Mining; UNEP: Geneva, Switzerland, 2013. [Google Scholar]
- Esdaile, L.J.; Chalker, J.M. The mercury problem in artisanal and small-scale gold mining. Chem. Eur. J. 2018, 24, 6905–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.D.; Zheng, W. Human exposure and health effects of inorganic and elemental mercury. J. Prev. Med. Public Health 2012, 45, 344–352. [Google Scholar] [CrossRef] [PubMed]
- United States Environmental Protection Agency. Method 245.1, Revision 3.0: Determination of Mercury in Water by Cold Vapor Atomic Absorption Spectrometry; U.S. Environmental Protection Agency: Washington, DC, USA, 1994.
- United States Environmental Protection Agency. Method 7471B, Mercury in Solid or Semisolid Waste (Manual Cold-Vapor Technique); U.S. Environmental Protection Agency: Washington, DC, USA, 1998.
- Almeida, I.L.S.; Oliveira, M.D.R.; Silva, J.B.B.; Coelho, N.M.M. Suitable extraction of soils and sediments for mercury species and determination combined with the cold vapor generation atomic absorption spectrometry technique. Microchem. J. 2016, 124, 326–330. [Google Scholar] [CrossRef]
- Kodamatani, H.; Tomiyasu, T. Selective determination method for measurement of methylmercury and ethylmercury in soil/sediment samples using high-performance liquid chromatography–chemiluminescence detection coupled with simple extraction technique. J. Chromatogr. A 2013, 1288, 155–159. [Google Scholar] [CrossRef]
- Krata, A.; Bulska, E. Critical evaluation of analytical performance of atomic absorption spectrometry and inductively coupled plasma mass spectrometry for mercury determination. Spectrochim. Acta B 2005, 60, 345–350. [Google Scholar] [CrossRef]
- Jiang, Z.; Wen, G.; Fan, Y.; Jiang, C.; Liu, Q.; Huang, Z.; Liang, A. A highly selective nanogold-aptamer catalytic resonance scattering spectral assay for trace Hg2+ using HAuCl4-ascorbic acid as indicator reaction. Talanta 2010, 80, 1287–1291. [Google Scholar] [CrossRef]
- Ackermann, G.; Roder, H. Photometrische Bestimmung von Quecksilber(II) im Nanogrammbereich mit Thio-Michlers Keton. Talanta 1977, 24, 99–103. [Google Scholar] [CrossRef]
- Tupys, A.; Kalembkiewicz, J.; Bazel, Y.; Zapała, L.; Dranka, M.; Ostapiuk, Y.; Tymoshuk, O.; Woźnicka, E. 1-[(5-Benzyl-1,3-thiazol-2-yl)diazenyl]naphthalene-2-ol: X-ray structure, spectroscopic characterization, dissociation studies and application in mercury(II) detection. J. Mol. Struct. 2017, 1127, 722–733. [Google Scholar] [CrossRef]
- Prasertboonyai, K.; Liawraungrath, B.; Pojanakaroon, T.; Liawraungrath, S. Mercury(II) determination in commercial cosmetics and local Thai traditional medicines by flow injection spectrophotometry. Int. J. Cosmet. Sci. 2016, 38, 68–76. [Google Scholar] [CrossRef]
- Tarighat, M.A.; Mohammadi, K. Simultaneous spectrophotometric determination of Cu2+, Hg2+, and Cd2+ ions using 2-(3-hydroxy-1-methylbut-2-enylideneamino) pyridine-3-ol. Environ. Monit. Assess. 2015, 187, 197. [Google Scholar] [CrossRef]
- Meng, S.; Wang, J.; Fan, Y.; Zhao, Q.; Guo, Y. Spectrophotometric determination of trace mercury(II) in cereals with 2,4-bis(4-phenylazophenylaminodiazo) benzenesulfonic acid. J. Anal. Chem. 2013, 68, 488–494. [Google Scholar] [CrossRef]
- Huang, D.; Liu, X.; Lai, C.; Qin, L.; Zhang, C.; Yi, H.; Zeng, G.; Li, B.; Deng, R.; Liu, S.; et al. Colorimetric determination of mercury(II) using gold nanoparticles and double ligand exchange. Microchim. Acta 2019, 186, 31. [Google Scholar] [CrossRef] [PubMed]
- Kataria, R.; Sethuraman, K.; Vashisht, D.; Vashisht, A.; Mehta, S.K.; Gupta, A. Colorimetric detection of mercury ions based on anti-aggregation of gold nanoparticles using 3, 5-dimethyl-1-thiocarboxamidepyrazole. Microchem. J. 2019, 148, 299–305. [Google Scholar] [CrossRef]
- Kong, Y.; Shen, J.; Fan, A. Colorimetric method for the detection of mercury ions based on gold nanoparticles and mercaptophenyl boronic acid. Anal. Sci. 2017, 33, 925–930. [Google Scholar] [CrossRef] [Green Version]
- Kaneta, T.; Alahmad, W.; Varanusupakul, P. Microfluidic paper-based analytical devices with instrument- free detection and miniaturized portable detectors. Appl. Spectrosc. Rev. 2019, 54, 117–141. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.I.G.S.; Jayawardane, B.M.; Kolev, S.D.; McKelvie, I.D. Developments of microfluidic paper-based analytical devices (μPADs) for water analysis: A review. Talanta 2018, 177, 176–190. [Google Scholar] [CrossRef]
- Yang, Y.; Noviana, E.; Nguyen, M.P.; Geiss, B.J.; Dandy, D.S.; Henry, C.S. Paper-based microfluidic devices: Emerging themes and applications. Anal. Chem. 2017, 89, 71–91. [Google Scholar] [CrossRef]
- Ozer, T.; McMahon, C.; Henry, C.S. Advances in paper-based analytical devices. Annu. Rev. Anal. Chem. 2020, 13, 85–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boobphahom, S.; Ly, M.N.; Soum, V.; Pyun, N.; Kwon, O.; Rodthongkum, N.; Shin, K. Recent advances in microfluidic paper-based analytical devices toward high-throughput screening. Molecules 2020, 25, 2970. [Google Scholar] [CrossRef] [PubMed]
- Patidar, R.; Rebary, B.; Paul, P. Colorimetric and fluorogenic recognition of Hg2+ and Cr3+ in acetonitrile and their test paper recognition in aqueous media with the aid of rhodamine based sensors. J. Fluoresc. 2015, 25, 387–395. [Google Scholar] [CrossRef]
- Choi, M.G.; Park, S.Y.; Park, K.Y.; Chang, S.K. Novel Hg2+-selective signaling probe based on resorufin thionocarbonate and its μPAD application. Sci. Rep. 2019, 9, 3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meelapsom, R.; Jarujamrus, P.; Amatatongchai, M.; Chairam, S.; Kulsing, C.; Shen, W. Chromatic analysis by monitoring unmodified silver nanoparticles reduction on double layer microfluidic paper-based analytical devices for selective and sensitive determination of mercury(II). Talanta 2016, 155, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Apilux, A.; Siangproh, W.; Praphairaksit, N.; Chailapakul, O. Simple and rapid colorimetric detection of Hg(II) by a paper-based device using silver nanoplates. Talanta 2012, 97, 388–394. [Google Scholar] [CrossRef]
- Pourreza, N.; Golmohammadi, H.; Rastegarzadeh, S. Highly selective and portable chemosensor for mercury determination in water samples using curcumin nanoparticles in a paper based analytical device. RSC Adv. 2016, 6, 69060–69066. [Google Scholar] [CrossRef]
- Han, K.N.; Choi, J.S.; Kwon, J. Gold nanozyme-based paper chip for colorimetric detection of mercury ions. Sci. Rep. 2017, 7, 2806. [Google Scholar] [CrossRef] [Green Version]
- Jarujamrus, P.; Meelapsom, R.; Pencharee, S.; Obma, A.; Amatatongchai, M.; Ditcharoen, N.; Chairam, S.; Tamuang, S. Use of a smartphone as a colorimetric analyzer in paper-based devices for sensitive and selective determination of mercury in water samples. Anal. Sci. 2018, 34, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Fang, Y.; Mo, Y.; Huang, Y.; Xu, C.; Zhang, Z.; Wang, M. Visual quantification of Hg on a microfluidic paper-based analytical device using distance-based detection technique. AIP Adv. 2017, 7, 085214. [Google Scholar] [CrossRef] [Green Version]
- Phansi, P.; Sumantakul, S.; Wongpakdee, T.; Fukana, N.; Ratanawimarnwong, N.; Sitanurak, J.; Nacapricha, D. Membraneless gas-separation microfluidic paper-based analytical devices for direct quantitation of volatile and nonvolatile compounds. Anal. Chem. 2016, 88, 8749–8756. [Google Scholar] [CrossRef] [PubMed]
- Sitanurak, J.; Fukana, N.; Wongpakdee, T.; Thepchuay, Y.; Ratanawimarnwong, N.; Amornsakchai, T.; Nacapricha, D. T-shirt ink for one-step screen-printing of hydrophobic barriers for 2D- and 3D-microfluidic paper-based analytical devices. Talanta 2019, 205, 120113. [Google Scholar] [CrossRef]
- Skoog, D.A.; West, D.M.; Holler, F.J. Fundamentals of Analytical Chemistry, 7th ed.; Saunders College Publishing: New York, NY, USA, 1997. [Google Scholar]
- Sitanurak, J.; Wangdi, N.; Sonsa-ard, T.; Teerasong, S.; Amornsakchai, T.; Nacapricha, D. Simple and green method for direct quantification of hypochlorite in household bleach with membraneless gas-separation microfluidic paper based analytical device. Talanta 2018, 187, 91–98. [Google Scholar] [CrossRef]
- Miretzky, P.; Bisinoti, M.C.; Jardim, W.F.; Rocha, J.C. Factors affecting Hg (II) adsorption in soils from the Rio Negro basin (Amazon). Quim. Nova 2005, 28, 438–443. [Google Scholar] [CrossRef]
- Kim, C.S.; Rytuba, J.J.; Brown, G.E. EXAFS study of mercury(II) sorption to Fe- and Al-(hydr) oxides: I. Effects of pH. J. Colloid Interface Sci. 2004, 271, 1–15. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, Section 6.2.4, 2nd ed; Available online: http://www.eurachem.org (accessed on 8 March 2021).
- Malehase, T.; Daso, A.P.; Okonkwo, J.O. Determination of mercury and its fractionation products in samples from legacy use of mercury amalgam in gold processing in Randfontein, South Africa. Emerg. Contam. 2016, 2, 157–165. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Guidelines for Drinking-Water Quality, 4th ed.; IWA Publishing: London, UK, 2011. [Google Scholar]
- Fashola, M.O.; Ngole-Jeme, V.M.; Babalola, O.O. Heavy metal pollution from gold mines: Environmental effects and bacterial strategies for resistance. Int. J. Environ. Res. Public Health 2016, 13, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- United State Environmental Protection Agency. USEPA 3050B, Method 3050B. Acid Digestion of Sediments, Sludges and Soils; U.S. Environmental Protection Agency: Washington, DC, USA, 1996.
- World Bank. Project Guidelines: Industry sector guidelines. Pollution Prevention and abatement Handbook; The International Bank for Reconstruction and Development/The World Bank: Washinton, DC, USA, 1998. [Google Scholar]
- AOAC Official Methods of Analysis. Appendix F: Guidelines for Standard Method Performance Requirements; AOAC International: Rockville, MD, USA, 2016; p. 9. [Google Scholar]
- Pourreza, N.; Ghanemi, K. Determination of mercury in water and fish samples by cold vapor atomic absorption spectrometry after solid phase extraction on agar modified with 2-mercaptobenzimidazole. J. Hazard. Mater. 2009, 161, 982–987. [Google Scholar] [CrossRef]
- Elsaesser, A.; Howard, C.V. Toxicology of nanoparticles. Adv. Drug Deliv. Rev. 2012, 64, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, G.; Nurchi, V.M.; Lachowicz, J.I.; Peana, M.; Medici, S.; Zoroddu, M.A. Toxicity of Nanoparticles: Etiology and Mechanisms (Book Chapter 18), Antimicrobial Nanoarchitectonics; Elsevier: Amsterdam, The Neterlands, 2017; pp. 511–546. [Google Scholar]
- Agemian, H.; Chau, A.S.Y. A method for the determination of mercury in sediments by the automated cold vapor atomic absorption technique after digestion. Anal. Chim. Acta 1975, 75, 297–303. [Google Scholar] [CrossRef]
- Bezerra da Silva, Y.J.A.; Araújo do Nascimento, C.W.; Biondi, C.M. Comparison of USEPA digestion methods to heavy metals in soil samples. Environ. Monit. Assess. 2014, 186, 47–53. [Google Scholar] [CrossRef]
- Dushman, S. The rate of the reaction between iodic and hydriodic acids. J. Phys. Chem. 1904, 8, 453–482. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter of Donor Reservoir | Working Range (mg L−1 Hg) | Linear Equation | Schematic Diagram of the Experimental Study | Image of the Acceptor Reservoir (6 mm) for 150 mg L−1 Hg |
|---|---|---|---|---|
| 6 mm | 50–350 | Intensity- = (4.0 ± 0.5) × 10−2 CHg(II) + (123.7 ± 1.1) |  |  |
| 3 mm | 50–350 | Intensity- = (5.5 ± 0.5) × 10−2 CHg(II) + (127.9 ± 1.1) |  |  |
| Foreign Species | Reported Level | Tolerance Limit (mg L−1) | |
|---|---|---|---|
| River Water (mg L−1) a | Soil (mg kg−1) b | ||
| Copper (II) | 0.006–10 | 5–70 | 1000 c |
| Lead (II) | 0.003–0.3 | 10–67 | 1000 c |
| Cadmium (II) | 0.01–0.04 | 6.4–11.7 | 1000 c |
| Iron (II) | 0.03–0.05 | 0.5–10 | 1000 c |
| Nitrate (NO3−) | 5–50 | 8–119 | 1000 c |
| Sulfate (SO42−) | – | 29–130 | 1000 c |
| Cyanide (CN−) | ≤0.2 | 11–44 | 750 |
| Chromium (III) | 0.05–0.2 | 2–60 | 500 |
| Zinc (II) | 0.05–0.1 | 8.9–65.7 | 500 |
| Nickel (II) | 0.03–10 | 3–100 | 500 |
| Iron (III) | ≤7 | 20–30 | 250 |
| Silver (I) | 0.3–1 | 0.2–0.3 | 250 |
| Sulfide (S2−) | Up to 0.05 | Up to 11.7 | 25 |
| Sample | Mercury Concentration (mg L−1 Hg) | % Recovery | ||
|---|---|---|---|---|
| Present a | Added | Found b | ||
| Soil Sample | ||||
| S1 | 121.7 ± 5.3 | 50 | 174.4 ± 4.5 | 105.5 |
| S2 | 126.7 ± 4.3 | 50 | 175.2 ± 3.4 | 97.0 |
| S3 | 126.1 ± 3.7 | 50 | 180.2 ± 4.2 | 108.2 |
| S4 | n.d. | 100 | 105.4 ± 3.6 | 105.4 |
| S5 | n.d. | 100 | 96.9 ± 3.6 | 96.9 |
| S6 | n.d. | 100 | 96.7 ± 4.9 | 96.7 |
| S7 | n.d. | 100 | 104.1 ± 4.3 | 104.1 |
| S8 | n.d. | 100 | 100.3 ± 3.8 | 100.3 |
| S9 | n.d. | 100 | 103.5 ± 4.6 | 103.5 |
| S10 | n.d. | 100 | 105.7 ± 3.7 | 105.7 |
| Water Sample | ||||
| W1 | n.d. | 100 | 90.7 ± 3.9 | 90.7 |
| W2 | n.d. | 100 | 101.2 ± 3.5 | 101.2 |
| W3 | n.d. | 100 | 97.2 ± 2.9 | 97.2 |
| W4 | n.d. | 100 | 102.9 ± 3.2 | 102.9 |
| Technique Class/Reagent | Test Samples | Working Range/LOD | Analysis Time (Excluding Sample Preparation) | Classified as “Equipment-Free” Method | Remark | |
|---|---|---|---|---|---|---|
| 1. Cold vapor-AAS/ SnCl2, NaBH4, KMnO4 (for hydride generation) |
| 0.04–2.4 μg L−1 Hg/0.02 μg L−1 Hg | NR a |  | No | All reagents are commercially available. Hg vapor is generated. |
| 5–40 μg L−1 Hg/0.01 μg L−1 Hg | NR a | ||||
| 1–30 μg L−1 Hg/0.08 mg kg−1 | NR a | ||||
| 2. ColorimetricμPADs or PADs with NPs/ | ||||||
| 2.1 Functionalized AuNPs, TMB, H2O2 |
| 0.2–2000 ng Hg/30 μg L−1 Hg | 10 min |  | Yes | The factionalized AuNPs are not yet commercialized. |
| 2.2 Synthesized-AgNPs |
| 0.01–10 mg L−1 Hg/0.003 mg L−1 Hg | 12 min | AgNPs may be synthesized or purchased. | ||
| 0.05–7 mg L−1 Hg/0.001 mg L−1 Hg | NR | ||||
| 5–75 mg L−1 Hg/0.12 mg L−1 Hg | 45 min | ||||
| 2.3 Synthesized-CcNPs |
| 0.5–20 mg L−1 Hg/0.17 mg L−1 Hg | 15 min | CcNPs are not yet commercialized. | ||
| 3. ColorimetricμPADs or PADs without NPs/ | ||||||
| 3.1 Dithizone in CCl4 |
| 1–30 mg L−1 Hg/0.93 mg L−1 Hg | ≥10 min |  | Yes | Not eco-friendly reagent/solvent. |
| 3.2 Resorufin thiono-carbonate in CH2Cl2 |
| 2–10 mg L−1 Hg/1.18 mg L−1 Hg | NR | Not eco-friendly reagent/solvent. | ||
| 3.3 Rhodamine derivatives in CH2Cl2 |
| 20, 50, 100, 200, 300 mg L−1 Hg (no calibration plot)/NR | ≥15 min | Need synthesized chemicals. Not eco-friendly solvent. | ||
| 3.4 This work/ KI, KIO3, H2SO4, starch |
| 50–300 mg L−1 Hg/20 mg L−1 Hg | 10 min | All reagents are common and are all commercially available. No serious toxicity from skin exposure (KI, KIO3, starch) except 0.2 mol L−1 H2SO4. | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nashukha, H.L.; Sitanurak, J.; Sulistyarti, H.; Nacapricha, D.; Uraisin, K. Simple and Equipment-Free Paper-Based Device for Determination of Mercury in Contaminated Soil. Molecules 2021, 26, 2004. https://doi.org/10.3390/molecules26072004
Nashukha HL, Sitanurak J, Sulistyarti H, Nacapricha D, Uraisin K. Simple and Equipment-Free Paper-Based Device for Determination of Mercury in Contaminated Soil. Molecules. 2021; 26(7):2004. https://doi.org/10.3390/molecules26072004
Chicago/Turabian StyleNashukha, Hikmanita Lisan, Jirayu Sitanurak, Hermin Sulistyarti, Duangjai Nacapricha, and Kanchana Uraisin. 2021. "Simple and Equipment-Free Paper-Based Device for Determination of Mercury in Contaminated Soil" Molecules 26, no. 7: 2004. https://doi.org/10.3390/molecules26072004