
Development of a Multimatrix UHPLC-MS/MS Method for the Determination of Paracetamol and Its Metabolites in Animal Tissues

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of LC-MS/MS Conditions
2.2. Optimization of Sample Preparation
2.3. Method Validation
2.4. Application to Real Samples
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Preparation of the Standard Stock Solution and Working Solutions
3.3. Sample Preparation
3.4. LC-MS-MS Analysis
3.5. Method Validation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malaise, O.; Bruyere, O.; Reginster, J.Y. Intravenous paracetamol: A review of efficacy and safety in therapeutic use. Future Neurol. 2007, 2, 673–688. [Google Scholar] [CrossRef]
- Jozwiak-Bebenista, M.; Nowak, J.Z. Paracetamol: Mechanism of action, applications and safety concern. Acta Pol. Pharm. Drug Res. 2014, 71, 11–23. [Google Scholar]
- Josephy, P.D. The molecular toxicology of acetaminophen. Drug Metab. Rev. 2005, 37, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Scott, K.F.; Day, R.O. Tolerability of paracetamol. Drug Saf. 2005, 28, 227–240. [Google Scholar] [CrossRef]
- Graham, G.G.; Davies, M.J.; Day, R.O.; Mohamudally, A.; Scott, K.F. The modern pharmacology of paracetamol: Therapeutic actions, mechanism of action, metabolism, toxicity and recent pharmacological findings. Inflammopharmacology 2013, 21, 201–232. [Google Scholar] [CrossRef] [PubMed]
- Cuzzolin, L.; Antonucci, R.; Fanos, V. Paracetamol (Acetaminophen) Efficacy and Safety in the Newborn. Curr. Drug Metab. 2013, 14, 178–185. [Google Scholar]
- Swales, J.G.; Temesi, D.G.; Denn, M.; Murphy, K. Determination of paracetamol in mouse, rat and dog plasma samples by laser diode thermal desorption-APCI-MS/MS. Bioanalysis 2012, 4, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Mian, P.; Allegaert, K.; Spriet, I.; Tibboel, D.; Petrovic, M. Paracetamol in Older People: Towards Evidence-Based Dosing? Drugs Aging 2018, 35, 603–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.C.; Wang, H.; Shi, K.; Li, J.M.; Zong, Y.; Du, R. Hepatoprotective effect of baicalein against acetaminophen-induced acute liver injury in mice. Molecules 2019, 24, 131. [Google Scholar] [CrossRef] [Green Version]
- Cranswick, N.; Coghlan, D. Paracetamol Efficacy and Safety in Children. Am. J. Ther. 2000, 7, 135–141. [Google Scholar] [CrossRef]
- Veterinary Medicines Evaluation Unit Paracetamol; Summary Report; European Agency for the Evaluation of Medicinal Products: Amsterdam, The Netherlands, 1999; pp. 1–6.
- DG Health and Food Safety. Overview Report Welfare of Cattle on Dairy Farms; European Commission: Belgium, Brussels, 2017; pp. 1–14. [Google Scholar]
- European Parliament and the Council of the European Union COMMISSION REGULATION (EU) No 37/2010. Off. J. Eur. Union. 2010, pp. 1–72. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0201&from=NL (accessed on 30 March 2021).
- Bosch, M.E.; Ruiz, A.J.S.; Ojeda, C.B. Determination of paracetamol : Historical evolution. J. Pharm. Biomed. Anal. 2006, 42, 291–321. [Google Scholar] [CrossRef] [PubMed]
- Critchley, J.; Nimmo, G.; Gregson, C.; Woolhouse, N.; Prescott, L. Inter-subject and ethnic differences in paracetamol metabolism. Br. J. Clin. Pharmacol. 1986, 22, 649–657. [Google Scholar] [CrossRef] [Green Version]
- Hao, W.; Zhang, Y.; Fan, J.; Liu, H.; Shi, Q.; Liu, W.; Peng, Q.; Zang, G. Copper nanowires modified with graphene oxide nanosheets for simultaneous voltammetric determination of ascorbic acid, dopamine and acetaminophen. Molecules 2019, 24, 2320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Guan, H.; Li, R.; Liao, X.; Zhao, F.; Wang, M.; Li, J.; Xu, G.; He, X.; Zhang, J.; et al. Auriculatone sulfate effectively protects mice against acetaminophen-induced liver injury. Molecules 2019, 24, 3642. [Google Scholar] [CrossRef] [Green Version]
- Ilavenil, S.; Al-Dhabi, N.A.; Srigopalram, S.; Kim, Y.O.; Agastian, P.; Baru, R.; Choi, K.C.; Arasu, M.V. Acetaminophen induced hepatotoxicity in Wistar rats - A proteomic approach. Molecules 2016, 21, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, H.; Yuan, H.; Ruan, Z.; Jiang, B. Simultaneous determination of paracetamol, pseudoephedrine, dextrophan and chlorpheniramine in human plasma by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2010, 878, 682–688. [Google Scholar] [CrossRef]
- Lu, W.; Zhao, S.; Gong, M.; Sun, L.; Ding, L. Simultaneous determination of acetaminophen and oxycodone in human plasma by LC–MS/MS and its application to a pharmacokinetic study. J. Pharm. Anal. 2018, 8, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Xie, Z.; Pan, B.; Zhu, C.; Yao, M.; Xu, X.; Wan, J. LC-MS-MS simultaneous determination of paracetamol, pseudoephedrine and chlorpheniramine in human plasma: Application to a pharmacokinetic study. Chromatographia 2008, 67, 687–694. [Google Scholar] [CrossRef]
- Tan, Q.; Zhu, R.; Li, H.; Wang, F.; Yan, M.; Dai, L. Simultaneous quantitative determination of paracetamol and its glucuronide conjugate in human plasma and urine by liquid chromatography coupled to electrospray tandem mass spectrometry: Application to a clinical pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 893–894, 162–167. [Google Scholar] [CrossRef]
- Hewavitharana, A.K.; Lee, S.; Dawson, P.A.; Markovich, D.; Shaw, P.N. Development of an HPLC-MS/MS method for the selective determination of paracetamol metabolites in mouse urine. Anal. Biochem. 2008, 374, 106–111. [Google Scholar] [CrossRef]
- An, J.H.; Lee, H.J.; Jung, B.H. Quantitative analysis of acetaminophen and its six metabolites in rat plasma using liquid chromatography/tandem mass spectrometry. Biomed. Chromatogr. 2012, 26, 1596–1604. [Google Scholar] [CrossRef]
- Neirinckx, E.; Vervaet, C.; De Boever, S.; Remon, J.P.; Gommeren, K.; Daminet, S.; De Backer, P.; Croubels, S. Species comparison of oral bioavailability, first-pass metabolism and pharmacokinetics of acetaminophen. Res. Vet. Sci. 2010, 89, 113–119. [Google Scholar] [CrossRef]
- Janus, K.; Grochowina, B.; Antoszek, J.; Suszycki, S.; Muszczynski, Z. The effect of food or water deprivation on paracetamol pharmacokinetics in calves. J. Vet. Pharmacol. Ther. 2003, 26, 291–296. [Google Scholar] [CrossRef]
- Sartini, I.; Łebkowska-Wieruszewska, B.; Lisowski, A.; Poapolathep, A.; Cuniberti, B.; Giorgi, M. Pharmacokinetics of acetaminophen after intravenous and oral administration in fasted and fed Labrador Retriever dogs. J. Vet. Pharmacol. Ther. 2021, 44, 28–35. [Google Scholar] [CrossRef]
- Zhang, X.; Li, R.; Hu, W.; Zeng, J.; Jiang, X.; Wang, L. A reliable LC-MS/MS method for the quantification of N-acetyl-p-benzoquinoneimine, acetaminophen glutathione and acetaminophen glucuronide in mouse plasma, liver and kidney: Method validation and application to a pharmacokinetic study. Biomed. Chromatogr. 2018, 32, e4331. [Google Scholar] [CrossRef]
- Li, W.; Doherty, J.P.; Kulmatycki, K.; Smith, H.T.; Tse, F.L. Simultaneous LC-MS/MS quantitation of acetaminophen and its glucuronide and sulfate metabolites in human dried blood spot samples collected by subjects in a pilot clinical study. Bioanalysis 2012, 4, 1429–1443. [Google Scholar] [CrossRef]
- Jian, N.; Li, R.; Li, J.; Liang, S.; Xu, Q.; Wang, C. Simple, efficient, and eco-friendly sample preparation for simultaneous determination of paracetamol and chloramphenicol in meat. J. Sep. Sci. 2019, 42, 2696–2705. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Peng, T.; Li, X.J.; Chen, D.D.; Dai, H.H.; Deng, X.J.; Yue, Z.F.; Wang, G.M.; Shen, J.Z.; Xia, X.; et al. Simultaneous determination of thirty non-steroidal anti-inflammatory drug residues in swine muscle by ultra-high-performance liquid chromatography with tandem mass spectrometry. J. Chromatogr. A 2012, 1219, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Wageningen University & Research. Available online: https://www.wur.nl/en/show/SANTE-2018-11188-TEXT-Rev0.htm (accessed on 1 April 2021).
- Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results (2002/657/EC). Off. J. Eur. Union L 2002, 221, 8–36.
- Shao, B.; Jia, X.; Zhang, J.; Meng, J.; Wu, Y.; Duan, H.; Tu, X. Multi-residual analysis of 16 β-agonists in pig liver, kidney and muscle by ultra performance liquid chromatography tandem mass spectrometry. Food Chem. 2009, 114, 1115–1121. [Google Scholar] [CrossRef]
- Jedziniak, P.; Pietruk, K.; Śledzińska, E.; Olejnik, M.; Szprengier-Juszkiewicz, T.; Żmudzki, J. Rapid method for the determination of metamizole residues in bovine muscle by LC-MS/MS. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2013, 30, 977–982. [Google Scholar] [CrossRef]
- Zeleny, R.; Harbeck, S.; Schimmel, H. Validation of a liquid chromatography-tandem mass spectrometry method for the identification and quantification of 5-nitroimidazole drugs and their corresponding hydroxy metabolites in lyophilised pork meat. J. Chromatogr. A 2009, 1216, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.L.; Kang, Y. Matrix effect in high-performance liquid chromatography-tandem mass spectrometry analysis of antibiotics in environmental water samples. J. Sep. Sci. 2013, 36, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food. EUR 28099 EN; Institute for Reference Materials and Measurements: Haier, Belgium, 2016. [Google Scholar]
- Taylor, P.J. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography–electrospray–tandem mass spectrometry. Clin. Biochem. 2005, 38, 328–334. [Google Scholar] [CrossRef] [PubMed]
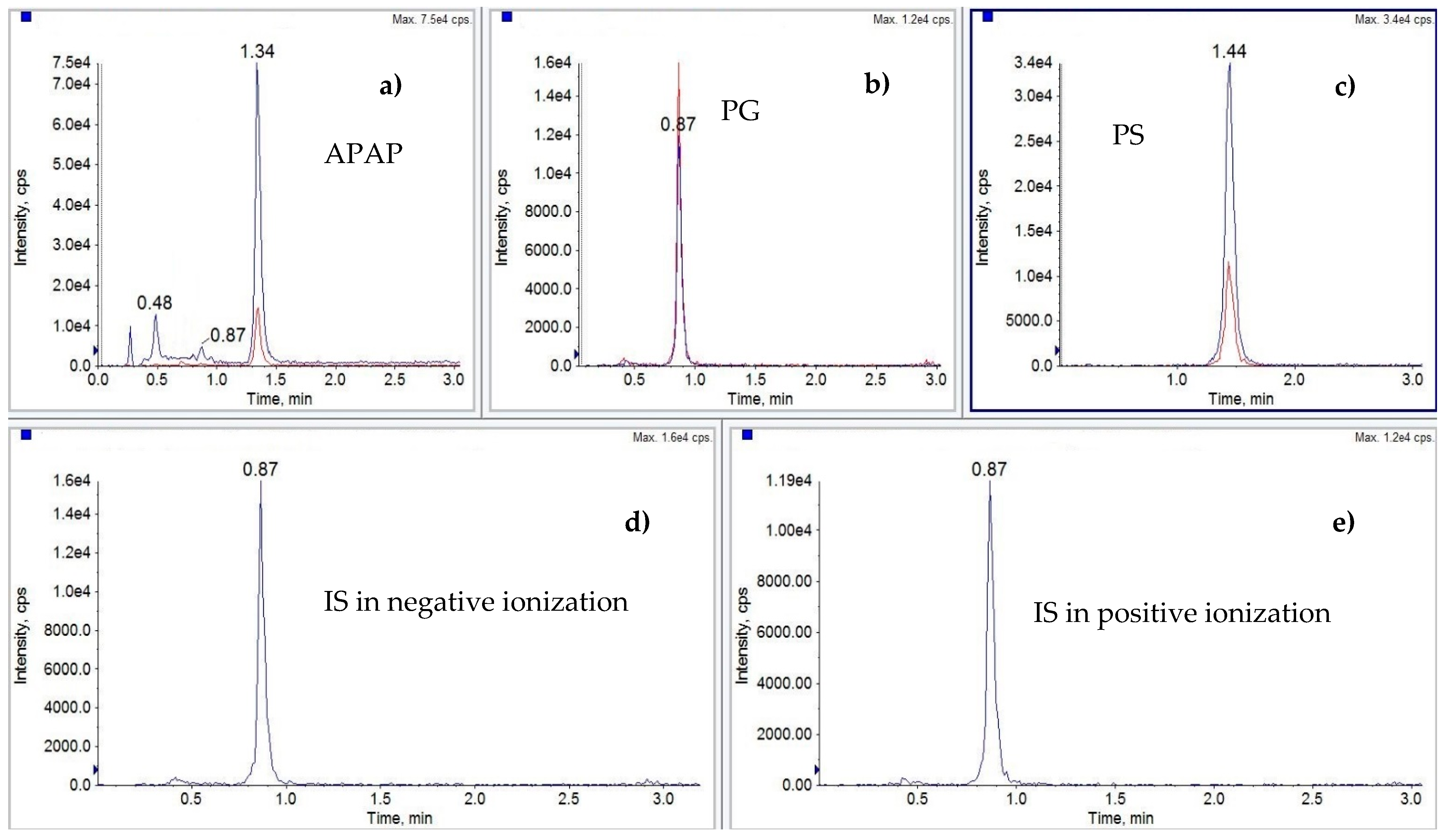
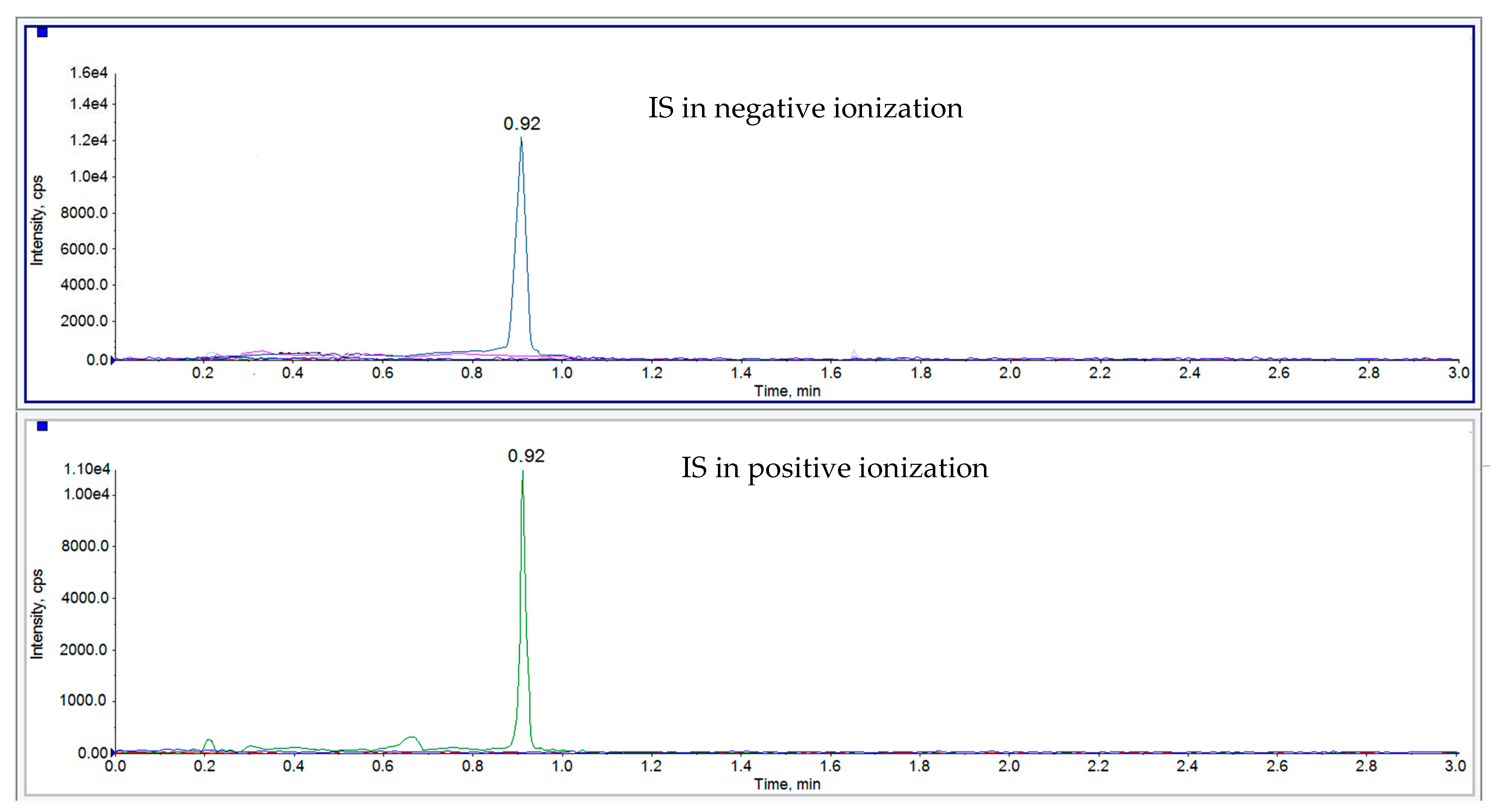
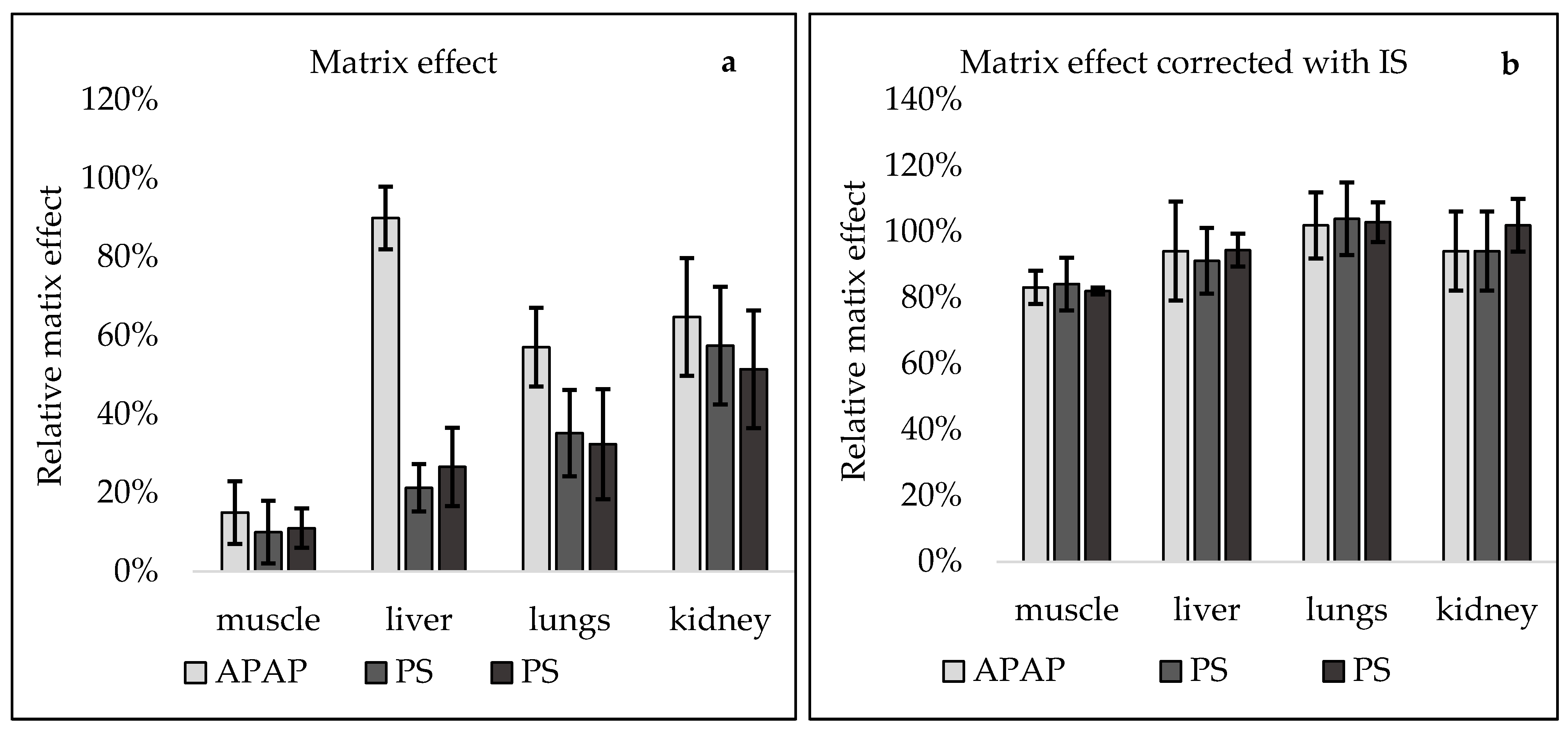

{kind=link}
{kind=link}
{kind=link}
| Analyte | Ionization | Parent ion [m/z] | Daughter ion(s) [m/z] Quantitative/Qualitative | Retention Time (min) | DP [V] | CE [eV] |
|---|---|---|---|---|---|---|
| Paracetamol (APAP) | positive | 152.0 | 110.1/65.1 | 1.34 | 45.0 | 23.0/45.0 |
| Paracetamol sulfate potasium (PS) | negative | 230.0 | 150.0/107.0 | 1.44 | −75.0 | −28.0/−46.0 |
| p-Acetamidophenyl β-D-glucuronide sodium salt (PG) | negative | 326.0 | 150.0/107.0 | 0.87 | −50.0 | −61.0/−38.0 |
| 4-Acetamidophenyl β-D-glucuronide-d3 sodium salt (PG-d3) IS | negative positive | 351.0 353.0 | 150.0 177.0 | 0.87 | -130.0 45.0 | −40.0 23.0 |
| Muscle | ||||||
|---|---|---|---|---|---|---|
| Analyte | Repeatability*, (CV,%) | Within-lab Reproducibility*, (CV,%) | LOQ (µg/kg) | Recovery* (%) | CCα (%) | CCβ (%) |
| APAP | 8.3 ± 2.9 | 10.8 ± 1.5 | 50.0 | 101.4 ± 6.6 | 63.3 | 73.1 |
| PS | 11,0 ± 1.4 | 14.0 ± 5.0 | 50.0 | 98.2 ± 4.4 | 76.9 | 86.7 |
| PG | 17.9 ± 4.3 | 21.0 ± 5.0 | 50.0 | 98.2 ± 4.4 | 76.9 | 86.7 |
| Liver | ||||||
| APAP | 10.8 ± 4.9 | 11.2 ± 4.5 | 50.0 | 110.3 ± 4.6 | 69.3 | 79.5 |
| PS | 9.5 ± 4.2 | 12.3 ± 6.3 | 50.0 | 114.0 ± 3.4 | 66.1 | 76.3 |
| PG | 11.5 ± 5.2 | 15.9 ± 6.3 | 50.0 | 108.0 ± 3.4 | 69.1 | 79.3 |
| Lungs | ||||||
| APAP | 9.8 ± 2.1 | 10.2 ± 2.5 | 50.0 | 110.6 ± 4.6 | 68.2 | 78.4 |
| PS | 5.4 ± 1.2 | 7.3 ± 2.1 | 50.0 | 109.0 ± 3.4 | 64.2 | 74.4 |
| PG | 15.5 ± 3.2 | 18.9 ± 4.1 | 50.0 | 91.0 ± 3.4 | 69.1 | 79.3 |
| Kidneys | ||||||
| APAP | 7.3 ± 2.1 | 9.7 ± 2.5 | 50.0 | 97.6 ± 4.6 | 69.4 | 78.9 |
| PS | 8.2 ± 3.2 | 12.1 ± 3.1 | 50.0 | 114.0 ± 3.9 | 61.2 | 75.1 |
| PG | 12.9 ± 5.2 | 17.5 ± 6.3 | 50.0 | 91.0 ± 3.4 | 67.3 | 77.7 |
| Matrix | APAP µg/kg | PS µg/kg | PG µg/kg |
|---|---|---|---|
| Muscle | 174 ± 24 | 136 ± 30 | 106 ± 21 |
| Liver | 918 ± 73 | < LOQ | 765 ± 30 |
| Lung | 494 ± 71 | 966 ± 86 | 1120 ± 97 |
| Kidneys | < LOQ | < LOQ | 257 ± 86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietruk, K.; Gbylik-Sikorska, M.; Łebkowska-Wieruszewska, B.; Gajda, A.; Giorgi, M.; Sartini, I.; Jedziniak, P. Development of a Multimatrix UHPLC-MS/MS Method for the Determination of Paracetamol and Its Metabolites in Animal Tissues. Molecules 2021, 26, 2046. https://doi.org/10.3390/molecules26072046
Pietruk K, Gbylik-Sikorska M, Łebkowska-Wieruszewska B, Gajda A, Giorgi M, Sartini I, Jedziniak P. Development of a Multimatrix UHPLC-MS/MS Method for the Determination of Paracetamol and Its Metabolites in Animal Tissues. Molecules. 2021; 26(7):2046. https://doi.org/10.3390/molecules26072046
Chicago/Turabian StylePietruk, Konrad, Małgorzata Gbylik-Sikorska, Beata Łebkowska-Wieruszewska, Anna Gajda, Mario Giorgi, Irene Sartini, and Piotr Jedziniak. 2021. "Development of a Multimatrix UHPLC-MS/MS Method for the Determination of Paracetamol and Its Metabolites in Animal Tissues" Molecules 26, no. 7: 2046. https://doi.org/10.3390/molecules26072046