
Modular Synthetic Approach to Carboranyl‒Biomolecules Conjugates

 , ,
, , 
Abstract
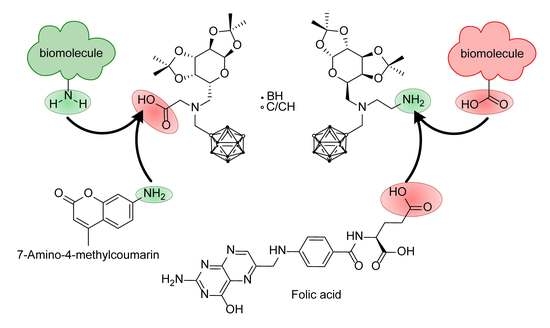
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Locher, G.L. Biological Effects and Therapeutic Possibilities of Neutrons. Am. J. Roentgenol. Radi. 1936, 36, 1–18. [Google Scholar]
- Hatanaka, H. A Revised Boron-Neutron Capture Therapy for Malignant Brain Tumors. J. Neurol. 1975, 209, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H.J.; Goldhaber, M. Detection of Nuclear Disintegration in a Photographic Emulsion. Nature 1935, 135, 341. [Google Scholar] [CrossRef]
- Chadwick, J.; Goldhaber, M. Disintegration by Slow Neutrons. Nature 1935, 135, 65. [Google Scholar] [CrossRef]
- Barth, R.F.; Soloway, A.H.; Fairchild, R.G. Boron Neutron Capture Therapy of Cancer. Cancer Res. 1990, 50, 1061–1070. [Google Scholar] [CrossRef]
- Soloway, A.H.; Tjarks, W.; Barnum, B.A.; Rong, F.-G.; Barth, R.F.; Codogni, I.M.; Wilson, J.G. The Chemistry of Neutron Capture Therapy. Chem. Rev. 1998, 98, 1515–1562. [Google Scholar] [CrossRef]
- Hartman, T.; Carlsson, J. Radiation Dose Heterogeneity in Receptor and Antigen Mediated Boron Neutron Capture Therapy. Radiother. Oncol. 1994, 31, 61–75. [Google Scholar] [CrossRef]
- Coderre, J.A.; Turcotte, J.C.; Riley, K.J.; Binns, P.J.; Harling, O.K.; Kiger, W.S. Boron Neutron Capture Therapy: Cellular Targeting of High Linear Energy Transfer Radiation. Technol. Cancer Res. Treat. 2003, 2, 355–375. [Google Scholar] [CrossRef]
- Sears, V.F. Neutron Scattering Lengths and Cross Sections. Neutron News 1992, 3, 26–37. [Google Scholar] [CrossRef]
- Hawthorne, M.F. The Role of Chemistry in the Development of Boron Neutron Capture Therapy of Cancer. Angew. Chem. Int. Ed. 1993, 32, 950–984. [Google Scholar] [CrossRef]
- Adams, L.; Tomlinson, S.; Wang, J.; Hosmane, S.N.; Maguire, J.A.; Hosmane, N.S. Novel Approach to Boron-10 Enriched Decaborane(14): An Important Advance in Synthetic Boron Hydride Chemistry. Inorg. Chem. Commun. 2002, 5, 765–767. [Google Scholar] [CrossRef]
- Yinghuai, Z.; Widjaja, E.; Lo Sia, S.P.; Zhan, W.; Carpenter, K.; Maguire, J.A.; Hosmane, N.S.; Hawthorne, M.F. Ruthenium(0) Nanoparticle-Catalyzed Isotope Exchange between 10B and 11B Nuclei in Decaborane(14). J. Am. Chem. Soc. 2007, 129, 6507–6512. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, J. Possible Existence of a Neutron. Nature 1932, 129, 312. [Google Scholar] [CrossRef]
- Petry, W.; Neuhaus, J. Neutronen nach Maß. Phys. J. 2007, 6, 31–37. [Google Scholar]
- Kettenbach, K.; Schieferstein, H.; Grunewald, C.; Iffland, D.; Reffert, L.M.; Hampel, G.; Schütz, C.L.; Bings, N.H.; Ross, T.L. Synthesis and Evaluation of Boron Folates for Boron-Neutron-Capture-Therapy (BNCT). Radiochim. Acta 2015, 103, 799–809. [Google Scholar] [CrossRef]
- Rong, F.-G.; Soloway, A.H.; Ikeda, S.; Ives, D.H. Synthesis and Biochemical Activity of Hydrophilic Carborane-Containing Pyrimidine Nucleosides as Potential Agents for DNA Incorporation and BNCT. Nucleos. Nucleot. 1997, 16, 379–401. [Google Scholar] [CrossRef]
- Kueffer, P.J.; Maitz, C.A.; Khan, A.A.; Schuster, S.A.; Shlyakhtina, N.I.; Jalisatgi, S.S.; Brockman, J.D.; Nigg, D.W.; Hawthorne, M.F. Boron Neutron Capture Therapy Demonstrated in Mice Bearing EMT6 Tumors Following Selective Delivery of Boron by Rationally Designed Liposomes. Proc. Natl. Acad. Sci. USA 2013, 110, 6512–6517. [Google Scholar] [CrossRef]
- Ahrens, V.M.; Frank, R.; Boehnke, S.; Schütz, C.L.; Hampel, G.; Iffland, D.S.; Bings, N.H.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Receptor-mediated Uptake of Boron-rich Neuropeptide Y Analogues for Boron Neutron Capture Therapy. ChemMedChem 2015, 10, 164–172. [Google Scholar] [CrossRef]
- Calabrese, G.; Daou, A.; Barbu, E.; Tsibouklis, J. Towards Carborane-functionalised Structures for the Treatment of Brain Cancer. Drug Discov. Today 2018, 23, 63–75. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron Delivery Agents for Neutron Capture Therapy of Cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed]
- Bleuel, D.L.; Donahue, R.J.; Ludewigt, B.A.; Vujic, J. Designing Accelerator-based Epithermal Neutron Beams for Boron Neutron Capture Therapy. Med. Phys. 1998, 25, 1725–1734. [Google Scholar] [CrossRef] [PubMed]
- Onishi, T.; Kumada, H.; Takada, K.; Naito, F.; Kurihara, T.; Sakae, T. Investigation of the Neutron Spectrum Measurement Method for Dose Evaluation in Boron Neutron Capture Therapy. Appl. Radiat. Isot. 2018, 140, 5–11. [Google Scholar] [CrossRef]
- Durisi, E.; Alikaniotis, K.; Borla, O.; Bragato, F.; Costa, M.; Giannini, G.; Monti, V.; Visca, L.; Vivaldo, G.; Zanini, A. Design and Simulation of an Optimized E-linac Based Neutron Source for BNCT Research. Appl. Radiat. Isot. 2015, 106, 63–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kato, T.; Hirose, K.; Tanaka, H.; Mitsumoto, T.; Motoyanagi, T.; Arai, K.; Harada, T.; Takeuchi, A.; Kato, R.; Yajima, S.; et al. Design and Construction of an Accelerator-based Boron Neutron Capture Therapy (AB-BNCT) Facility with Multiple Treatment Rooms at the Southern Tohoku BNCT Research Center. Appl. Radiat. Isot. 2019, 156, 108961. [Google Scholar] [CrossRef]
- Mori, Y.; Suzuki, A.; Yoshino, K.; Kakihana, H. Complex Formation of p-Boronophenylalanine with some Monosaccharides. Pigment Cell Res. 1989, 2, 273–277. [Google Scholar] [CrossRef]
- Stella Pharma Corporation. Available online: https://stella-pharma.co.jp/en (accessed on 6 March 2021).
- Stella Pharma Corporation. News Release: STELLA PHARMA will Launch Steboronine®, the World’s First BNCT Drug, on May 20, 2020. Available online: https://stella-pharma.co.jp/cp-bin/wordpress5/wp-content/uploads/2020/05/Steboronine-launched_ENG.pdf (accessed on 6 March 2021).
- Frank, R.; Ahrens, V.M.; Boehnke, S.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Charge-Compensated Metallacarborane Building Blocks for Conjugation with Peptides. ChemBioChem 2016, 17, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Köbberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. Selective Neuropeptide Y Conjugates with Maximized Carborane Loading as Promising Boron Delivery Agents for Boron Neutron Capture Therapy. J. Med. Chem. 2020, 63, 2358–2371. [Google Scholar] [CrossRef]
- Hoppenz, P.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Lerchen, H.-G.; Köbberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. A Selective Carborane-Functionalized Gastrin-Releasing Peptide Receptor Agonist as Boron Delivery Agent for Boron Neutron Capture Therapy. J. Org. Chem. 2020, 85, 1446–1457. [Google Scholar] [CrossRef]
- Hattori, Y.; Kusaka, S.; Mukumoto, M.; Uehara, K.; Asano, T.; Suzuki, M.; Masunaga, S.; Ono, K.; Tanimori, S.; Kirihata, M. Biological Evaluation of Dodecaborate-Containing L-Amino Acids for Boron Neutron Capture Therapy. J. Med. Chem. 2012, 55, 6980–6984. [Google Scholar] [CrossRef]
- Pan, X.Q.; Wang, H.; Shukla, S.; Sekido, M.; Adams, D.M.; Tjarks, W.; Barth, R.F.; Lee, R.J. Boron-Containing Folate Receptor-Targeted Liposomes as Potential Delivery Agents for Neutron Capture Therapy. Bioconjug. Chem. 2002, 13, 435–442. [Google Scholar] [CrossRef]
- Dubey, R.; Kushal, S.; Mollard, A.; Vojtovich, L.; Oh, P.; Levin, M.D.; Schnitzer, J.E.; Zharov, I.; Olenyuk, B.Z. Tumor Targeting, Trifunctional Dendritic Wedge. Bioconjug. Chem. 2015, 26, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Doi, A.; Kawabata, S.; Iida, K.; Yokoyama, K.; Kajimoto, Y.; Kuroiwa, T.; Shirakawa, T.; Kirihata, M.; Kasaoka, S.; Maruyama, K.; et al. Tumor-specific Targeting of Sodium Borocaptate (BSH) to Malignant Glioma by Transferrin-PEG Liposomes: A Modality for Boron Neutron Capture Therapy. J. Neurooncol. 2008, 87, 287–294. [Google Scholar] [CrossRef]
- Mier, W.; Gabel, D.; Haberkorn, U.; Eisenhut, M. Conjugation of the closo-Borane Mercaptoundecahydrododecaborate (BSH) to a Tumour Selective Peptide. Z. Anorg. Allg. Chem. 2004, 630, 1258–1262. [Google Scholar] [CrossRef]
- Liu, L.; Barth, R.F.; Adams, D.M.; Soloway, A.H.; Reisfeld, R.A. Bispecific Antibodies as Targeting Agents for Boron Neutron Capture Therapy of Brain Tumors. J. Hematother. 1995, 4, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.V.; Zinin, A.I.; Malysheva, N.N.; Kononov, L.O.; Sivaev, I.B.; Bregadze, V.I. Conjugates of Polyhedral Boron Compounds with Carbohydrates. 1. New Approach to the Design of Selective Agents for Boron Neutron Capture Therapy of Cancer. Russ. Chem. Bull. Int. Ed. 2003, 52, 2766–2769. [Google Scholar] [CrossRef]
- Marepally, S.R.; Yao, M.-L.; Kabalka, G.W. Boronated Carbohydrate Derivatives as Potential Boron Neutron Capture Therapy Reagents. Future Med. Chem. 2013, 5, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Varadarajan, A.; Hawthorne, M.F. Novel Carboranyl Amino Acids and Peptides: Reagents for Antibody Modification and Subsequent Neutron-Capture Studies. Bioconjug. Chem. 1991, 2, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Timofeev, S.V.; Bregadze, V.I.; Osipov, S.N.; Titanyuk, I.D.; Petrovskii, P.V.; Starikova, Z.A.; Glukhov, I.V.; Beletskaya, I.P. New Carborane-containing Amino Acids and Their Derivatives. Crystal Structures of N-protected Carboranylalaninates. Russ. Chem. Bull. Int. Ed. 2007, 56, 791–797. [Google Scholar] [CrossRef]
- Stogniy, M.Y.; Zakharova, M.V.; Sivaev, I.B.; Godovikov, I.A.; Chizov, A.O.; Bregadze, V.I. Synthesis of New Carborane-based Amino Acids. Polyhedron 2013, 55, 117–120. [Google Scholar] [CrossRef]
- Michiue, H.; Sakurai, Y.; Kondo, N.; Kitamatsu, M.; Bin, F.; Nakajima, K.; Hirota, Y.; Kawabata, S.; Nishiki, T.; Ohmori, I.; et al. The Acceleration of Boron Neutron Capture Therapy Using Multi-linked Mercaptoundecahydrododecaborate (BSH) Fused Cell-penetrating Peptide. Biomaterials 2014, 35, 3396–3405. [Google Scholar] [CrossRef] [PubMed]
- Worm, D.J.; Els-Heindl, S.; Kellert, M.; Kuhnert, R.; Saretz, S.; Koebberling, J.; Riedl, B.; Hey-Hawkins, E.; Beck-Sickinger, A.G. A Stable meta-Carborane Enables the Generation of Boron-rich Peptide Agonists Targeting the Ghrelin Receptor. J. Pept. Sci. 2018, 32, e3119. [Google Scholar] [CrossRef]
- Lerouge, F.; Viñas, C.; Teixidor, F.; Núñez, R.; Abreu, A.; Xochitiotzi, E.; Santillan, R.; Farfán, N. High Boron Content Carboranyl-functionalized Aryl Ether Derivatives Displaying Photoluminescent Properties. Dalton Trans. 2007, 92, 1898–1903. [Google Scholar] [CrossRef]
- Feng, B.; Tomizawa, K.; Michiue, H.; Miyatake, S.-I.; Han, X.-J.; Fujimura, A.; Seno, M.; Kirihata, M.; Matsui, H. Delivery of Sodium Borocaptate to Glioma Cells Using Immunoliposome Conjugated with Anti-EGFR Antibodies by ZZ-His. Biomaterials 2009, 30, 1746–1755. [Google Scholar] [CrossRef]
- Ciofani, G.; Raffa, V.; Menciassi, A.; Cuschieri, A. Folate Functionalized Boron Nitride Nanotubes and their Selective Uptake by Glioblastoma Multiforme Cells: Implications for their Use as Boron Carriers in Clinical Boron Neutron Capture Therapy. Nanoscale Res. Lett. 2008, 4, 113–121. [Google Scholar] [CrossRef]
- Kellert, M.; Worm, D.J.; Hoppenz, P.; Sárosi, M.B.; Lönnecke, P.; Riedl, B.; Koebberling, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Modular Triazine-based Carborane-containing Carboxylic Acids—Synthesis and Characterisation of Potential Boron Neutron Capture Therapy Agents Made of Readily Accessible Building Blocks. Dalton Trans. 2019, 48, 10834–10844. [Google Scholar] [CrossRef]
- Kellert, M.; Hoppenz, P.; Lönnecke, P.; Worm, D.J.; Riedl, B.; Koebberling, J.; Beck-Sickinger, A.G.; Hey-Hawkins, E. Tuning a Modular System-Synthesis and Characterisation of a Boron-rich s-Triazine-based Carboxylic Acid and Amine Bearing a Galactopyranosyl Moiety. Dalton Trans. 2020, 49, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kellert, M.; Lönnecke, P.; Riedl, B.; Koebberling, J.; Hey-Hawkins, E. Enlargement of a Modular System-Synthesis and Characterization of an s-Triazine-based Carboxylic Acid Ester Bearing a Galactopyranosyl Moiety and an Enormous Boron Load. Molecules 2019, 24, 3288. [Google Scholar] [CrossRef]
- He, H.; Li, D.-W.; Yang, L.-Y.; Fu, L.; Zhu, X.-J.; Wong, W.-K.; Jiang, F.-L.; Liu, Y. A Novel Bifunctional Mitochondria-targeted Anticancer Agent with High Selectivity for Cancer Cells. Sci. Rep. 2015, 5, 13543. [Google Scholar] [CrossRef] [PubMed]
- Bryden, F.; Savoie, H.; Rosca, E.V.; Boyle, R.W. PET/PDT Theranostics: Synthesis and Biological Evaluation of a Peptide-targeted Gallium Porphyrin. Dalton Trans. 2015, 44, 4925–4932. [Google Scholar] [CrossRef]
- Griffith, D.; Morgan, M.P.; Marmion, C.J. A Novel Anti-cancer Bifunctional Platinum Drug Candidate with Dual DNA Binding and Histone Deacetylase Inhibitory Activity. Chem. Comm. 2009, 6735–6737. [Google Scholar] [CrossRef]
- Chen, L.; Wang, X.; Ji, F.; Bao, Y.; Wang, J.; Guo, L.; Li, Y. New Bifunctional-pullulan-based Micelles with Good Biocompatibility for Efficient Co-delivery of Cancer-suppressing p53 Gene and Doxorubicin to Cancer Cells. RSC Adv. 2015, 5, 94719–94731. [Google Scholar] [CrossRef]
- Soni, R.; Soman, S.S. Design and Synthesis of Aminocoumarin Derivatives as DPP-IV Inhibitors and Anticancer Agents. Bioorg. Chem. 2018, 79, 277–284. [Google Scholar] [CrossRef]
- Achilli, C.; Jadhav, S.A.; Guidetti, G.F.; Ciana, A.; Abbonante, V.; Malara, A.; Fagnoni, M.; Torti, M.; Balduini, A.; Balduini, C.; et al. Folic Acid-Conjugated 4-Amino-Phenylboronate, a Boron-Containing Compound Designed for Boron Neutron Capture Therapy, is an Unexpected Agonist for Human Neutrophils and Platelets. Chem. Biol. Drug Des. 2014, 83, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Leamon, C.P.; Reddy, J.A. The Folate Receptor as a Rational Therapeutic Target for Personalized Cancer Treatment. Drug Resist. Updat. 2014, 17, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Ohta, K.; Suzuki, T.; Ohta, S.; Endo, Y. Design and Synthesis of Novel Androgen Receptor Antagonists with Sterically Bulky Icosahedral Carboranes. Bioorgan. Med. Chem. 2005, 13, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, V.N.; Rys, E.G.; Tyutyunov, A.A.; Starikova, Z.A.; Korlyukov, A.A.; Ol’shevskaya, V.A.; Sung, D.D.; Ponomaryov, A.B.; Petrovskii, P.V.; Hey-Hawkins, E. The First Carborane Triflates: Synthesis and Reactivity of 1-Trifluoromethanesulfonylmethyl- and 1,2-Bis(trifluoromethanesulfonylmethyl)-o-carborane. Dalton Trans. 2005, 903–908. [Google Scholar] [CrossRef]
- Saltan, F.; Akat, H. Synthesis and Thermal Degradation Kinetics of D-(+)-Galactose Containing Polymers. Polímeros 2013, 23, 697–704. [Google Scholar] [CrossRef]
- Brackhagen, M.; Boye, H.; Vogel, C. Synthesis of (6-2H)- and 6-Deoxy-6-fluoro-L-galactose Derivatives. J. Carbohyd. Chem. 2001, 20, 31–43. [Google Scholar] [CrossRef]
- Wuts, P.G.M.; Greene, T.W. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley-Interscience: Hoboken, NJ, USA, 2007; ISBN 9780471697541. [Google Scholar]
- Kokotos, G.; Theodorou, V.; Tzougraki, C. Synthesis and In Vitro Cytotoxicity of Aminocoumarin Platinum(II) Complexes. Bioorg. Med. Chem. Lett. 1997, 7, 2165–2168. [Google Scholar] [CrossRef]
- Kaur, M.; Kohli, S.; Sandhu, S.; Bansal, Y.; Bansal, G. Coumarin: A Promising Scaffold for Anticancer Agents. Anticancer Agents Med. Chem. 2015, 15, 1032–1048. [Google Scholar] [CrossRef]
- Klenkar, J.; Molnar, M. Natural and Synthetic Coumarins as Potential Anticancer Agents. J. Chem. Pharm. Res. 2015, 7, 1223–1238. [Google Scholar]
- Majnooni, M.B.; Fakhri, S.; Smeriglio, A.; Trombetta, D.; Croley, C.R.; Bhattacharyya, P.; Sobarzo-Sánchez, E.; Farzaei, M.H.; Bishayee, A. Antiangiogenic Effects of Coumarins against Cancer: From Chemistry to Medicine. Molecules 2019, 24, 4278. [Google Scholar] [CrossRef]
- Shukla, S.; Wu, G.; Chatterjee, M.; Yang, W.; Sekido, M.; Diop, L.A.; Müller, R.; Sudimack, J.J.; Lee, R.J.; Barth, R.F.; et al. Synthesis and Biological Evaluation of Folate Receptor-targeted Boronated PAMAM Dendrimers as Potential Agents for Neutron Capture Therapy. Bioconjug. Chem. 2003, 14, 158–167. [Google Scholar] [CrossRef]
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting Folate Receptor Alpha for Cancer Treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef]
- Quéléver, G.; Burlet, S.; Garino, C.; Pietrancosta, N.; Laras, Y.; Kraus, J.-L. Simple Coupling Reaction between Amino Acids and Weakly Nucleophilic Heteroaromatic Amines. J. Comb. Chem. 2004, 6, 695–698. [Google Scholar] [CrossRef]
- El-Faham, A.; Albericio, F. Peptide Coupling Reagents, More than a Letter Soup. Chem. Rev. 2011, 111, 6557–6602. [Google Scholar] [CrossRef] [PubMed]
- Trindade, A.F.; Frade, R.F.M.; Maçôas, E.M.S.; Graça, C.; Rodrigues, C.A.B.; Martinho, J.M.G.; Afonso, C.A.M. “Click and Go”: Simple and fast Folic Acid Conjugation. Org. Biomol. Chem. 2014, 12, 3181–3190. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Aoyagi, K.; Yamamoto, Y. Tetrabutylammonium Fluoride Promoted Novel Reactions of o-Carborane: Inter- and Intramolecular Additions to Aldehydes and Ketones and Annulation via Enals and Enones. J. Am. Chem. Soc. 1998, 120, 1167–1171. [Google Scholar] [CrossRef]
- Tiwari, N.; Sharma, R.K.; Gupta, P.; Misra, S.; Misra-Bhattacharya, S.; Butcher, R.J.; Singh, K.; Katiyar, D. Synthesis, Structure Elucidation, Homology Modeling and Antifilarial Activity of 7-Benzamidocoumarin Derivatives. ChemistrySelect 2019, 4, 3300–3307. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; de Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts. IUPAC Recommendations 2001. Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar] [CrossRef]
- MestReNova, v12.00-20080; Mestrelab Research S.L.: Santiago de Compostela, Spain, 2017.




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kellert, M.; Friedrichs, J.-S.J.; Ullrich, N.A.; Feinhals, A.; Tepper, J.; Lönnecke, P.; Hey-Hawkins, E. Modular Synthetic Approach to Carboranyl‒Biomolecules Conjugates. Molecules 2021, 26, 2057. https://doi.org/10.3390/molecules26072057
Kellert M, Friedrichs J-SJ, Ullrich NA, Feinhals A, Tepper J, Lönnecke P, Hey-Hawkins E. Modular Synthetic Approach to Carboranyl‒Biomolecules Conjugates. Molecules. 2021; 26(7):2057. https://doi.org/10.3390/molecules26072057
Chicago/Turabian StyleKellert, Martin, Jan-Simon Jeshua Friedrichs, Nadine Anke Ullrich, Alexander Feinhals, Jonas Tepper, Peter Lönnecke, and Evamarie Hey-Hawkins. 2021. "Modular Synthetic Approach to Carboranyl‒Biomolecules Conjugates" Molecules 26, no. 7: 2057. https://doi.org/10.3390/molecules26072057
APA StyleKellert, M., Friedrichs, J.-S. J., Ullrich, N. A., Feinhals, A., Tepper, J., Lönnecke, P., & Hey-Hawkins, E. (2021). Modular Synthetic Approach to Carboranyl‒Biomolecules Conjugates. Molecules, 26(7), 2057. https://doi.org/10.3390/molecules26072057