
Cholinergic Signaling, Neural Excitability, and Epilepsy


Abstract
:1. Introduction
2. Cholinergic Signaling for Modulating Neural Excitability
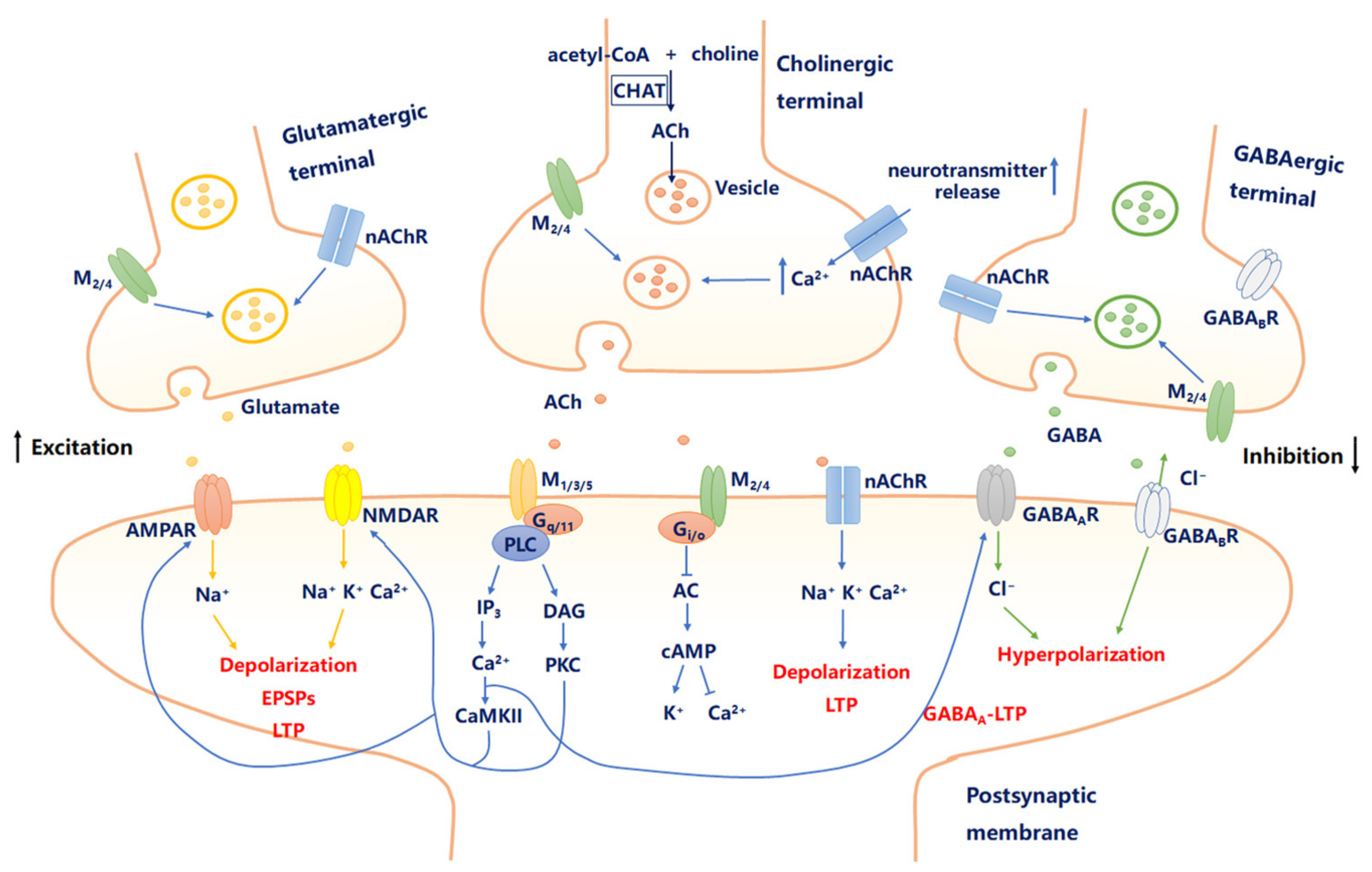
2.1. Cholinergic Signaling in the Brain
2.2. mAChRs and Neural Excitability
2.3. nAChRs and Neural Excitability
3. Cholinergic Signaling in Epilepsy
3.1. mAChRs and Epilepsy
3.2. nAChRs and Epilepsy
3.3. Cholinergic Neurons Circuit in Epilepsy
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thijs, R.D.; Surges, R.; O’Brien, T.J.; Sander, J.W. Epilepsy in adults. Lancet 2019, 393, 689–701. [Google Scholar] [CrossRef]
- Sontheimer, H. Seizure Disorders and Epilepsy. Dis. Nerv. Syst. 2015, 61–95. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Z. An update for epilepsy research and antiepileptic drug development: Toward precise circuit therapy. Pharmacol. Ther. 2019, 201, 77–93. [Google Scholar] [CrossRef]
- Picciotto, M.R.; Higley, M.J.; Mineur, Y.S. Acetylcholine as a neuromodulator: Cholinergic signaling shapes nervous system function and behavior. Neuron 2012, 76, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Langley, J.N. Observations on the physiological action of extracts of the supra-renal bodies. J. Physiol. 1901, 27, 237–256. [Google Scholar] [CrossRef] [Green Version]
- Curia, G.; Longo, D.; Biagini, G.; Jones, R.S.; Avoli, M. The pilocarpine model of temporal lobe epilepsy. J. Neurosci. Methods 2008, 172, 143–157. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Xu, C.; Wang, S.; Tan, N.; Chen, C.; Chen, L.; Wu, X.; Fei, F.; Cheng, H.; et al. Direct Septum-Hippocampus Cholinergic Circuit Attenuates Seizure through Driving Somatostatin Inhibition. Biol. Psychiatry 2020, 87, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Allaway, K.C.; Machold, R. Developmental specification of forebrain cholinergic neurons. Dev. Biol. 2017, 421, 1–7. [Google Scholar] [CrossRef]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef] [Green Version]
- Andrews, J.P.; Yue, Z.; Ryu, J.H.; Neske, G.; McCormick, D.A.; Blumenfeld, H. Mechanisms of decreased cholinergic arousal in focal seizures: In vivo whole-cell recordings from the pedunculopontine tegmental nucleus. Exp. Neurol. 2019, 314, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Motelow, J.E.; Li, W.; Zhan, Q.; Mishra, A.M.; Sachdev, R.N.; Liu, G.; Gummadavelli, A.; Zayyad, Z.; Lee, H.S.; Chu, V.; et al. Decreased subcortical cholinergic arousal in focal seizures. Neuron 2015, 85, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Von Engelhardt, J.; Eliava, M.; Meyer, A.H.; Rozov, A.; Monyer, H. Functional characterization of intrinsic cholinergic interneurons in the cortex. J. Neurosci. 2007, 27, 5633–5642. [Google Scholar] [CrossRef]
- Collier, B. Choline analogues: Their use in studies of acetylcholine synthesis, storage, and release. Can. J. Physiol. Pharmacol. 1986, 64, 341–346. [Google Scholar] [CrossRef]
- Nguyen, M.L.; Cox, G.D.; Parsons, S.M. Kinetic parameters for the vesicular acetylcholine transporter two protons are exchanged for one acetylcholine. Biochemistry 1998, 37, 13400–13410. [Google Scholar] [CrossRef]
- Descarries, L.; Gisiger, V.; Steriade, M. Diffuse transmission by acetylcholine in the CNS. Prog. Neurobiol. 1997, 53, 603–625. [Google Scholar] [CrossRef]
- Fuenzalida, M.; Pérez, M.Á.; Arias, H.R. Role of Nicotinic and Muscarinic Receptors on Synaptic Plasticity and Neurological Diseases. Curr. Pharm. Des. 2016, 22, 2004–2014. [Google Scholar] [CrossRef]
- Dominguez, S.; Fernandez de Sevilla, D.; Buno, W. Postsynaptic activity reverses the sign of the acetylcholine-induced long-term plasticity of GABAA inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, E2741–E2750. [Google Scholar] [CrossRef] [Green Version]
- De Sevilla, D.F.; Buno, W. The Muscarinic Long-Term Enhancement of NMDA and AMPA Receptor-Mediated Transmission at Schaffer Collateral Synapses Develop through Different Intracellular Mechanisms. J. Neurosci. 2010, 30, 11032–11042. [Google Scholar] [CrossRef] [PubMed]
- Morales-Weil, K.; Moreno, M.; Ahumada, J.; Arriagada, J.; Fuentealba, P.; Bonansco, C.; Fuenzalida, M. Priming of GABAergic Long-term Potentiation by Muscarinic Receptors. Neuroscience 2020, 428, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, H.; Young, K.; Hasselmo, M. Modulation of Hippocampal Circuits by Muscarinic and Nicotinic Receptors. Front. Neural Circuits 2017, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandecasteele, M.; Varga, V.; Berenyi, A.; Papp, E.; Bartho, P.; Venance, L.; Freund, T.F.; Buzsaki, G. Optogenetic activation of septal cholinergic neurons suppresses sharp wave ripples and enhances theta oscillations in the hippocampus. Proc. Natl. Acad. Sci. USA 2014, 111, 13535–13540. [Google Scholar] [CrossRef] [Green Version]
- Samengo, I.A.; Curro, D.; Martire, M. Nicotinic receptors modulate the function of presynaptic AMPA receptors on glutamatergic nerve terminals in the trigeminal caudal nucleus. Neurochem. Int. 2015, 90, 166–172. [Google Scholar] [CrossRef] [PubMed]
- McQuiston, A.R. Acetylcholine release and inhibitory interneuron activity in hippocampal CA1. Front. Synaptic. Neurosci. 2014, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, N.; Nichols, R.A. Beyond the Channel: Metabotropic Signaling by Nicotinic Receptors. Trends Pharmacol. Sci. 2018, 39, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Dani, J.A.; Bertrand, D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 699–729. [Google Scholar] [CrossRef]
- Cheng, S.B.; Amici, S.A.; Ren, X.Q.; McKay, S.B.; Treuil, M.W.; Lindstrom, J.M.; Rao, J.; Anand, R. Presynaptic targeting of alpha4beta 2 nicotinic acetylcholine receptors is regulated by neurexin-1beta. J. Biol. Chem. 2009, 284, 23251–23259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, A.; Yamagata, K.; Nakagomi, S.; Uejima, H.; Wiriyasermkul, P.; Ohgaki, R.; Nagamori, S.; Kanai, Y.; Tanaka, H. Nicotine induces dendritic spine remodeling in cultured hippocampal neurons. J. Neurochem. 2014, 128, 246–255. [Google Scholar] [CrossRef] [Green Version]
- Tang, A.H.; Karson, M.A.; Nagode, D.A.; McIntosh, J.M.; Uebele, V.N.; Renger, J.J.; Klugmann, M.; Milner, T.A.; Alger, B.E. Nerve terminal nicotinic acetylcholine receptors initiate quantal GABA release from perisomatic interneurons by activating axonal T-type (Cav3) Ca(2)(+) channels and Ca(2)(+) release from stores. J. Neurosci. 2011, 31, 13546–13561. [Google Scholar] [CrossRef] [Green Version]
- Udakis, M.; Wright, V.L.; Wonnacott, S.; Bailey, C.P. Integration of inhibitory and excitatory effects of alpha7 nicotinic acetylcholine receptor activation in the prelimbic cortex regulates network activity and plasticity. Neuropharmacology 2016, 105, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Schliebs, R.; Zivin, M.; Steinbach, J.; Rothe, T. Changes in cholinergic but not in GABAergic markers in amygdala, piriform cortex, and nucleus basalis of the rat brain following systemic administration of kainic acid. J. Neurochem. 1989, 53, 212–218. [Google Scholar] [CrossRef]
- Müller-Gärtner, H.W.; Mayberg, H.S.; Fisher, R.S.; Lesser, R.P.; Wilson, A.A.; Ravert, H.T.; Dannals, R.F.; Wagner, H.N., Jr.; Uematsu, S.; Frost, J.J. Decreased hippocampal muscarinic cholinergic receptor binding measured by 123I-iododexetimide and single-photon emission computed tomography in epilepsy. Ann. Neurol. 1993, 34, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Yi, F.; DeCan, E.; Stoll, K.; Marceau, E.; Deisseroth, K.; Lawrence, J.J. Muscarinic excitation of parvalbumin-positive interneurons contributes to the severity of pilocarpine-induced seizures. Epilepsia 2015, 56, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Bymaster, F.P.; Carter, P.A.; Yamada, M.; Gomeza, J.; Wess, J.; Hamilton, S.E.; Nathanson, N.M.; McKinzie, D.L.; Felder, C.C. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur. J. Neurosci. 2003, 17, 1403–1410. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Loose, M.D.; Qi, M.; Levey, A.I.; Hille, B.; McKnight, G.S.; Idzerda, R.L.; Nathanson, N.M. Disruption of the m1 receptor gene ablates muscarinic receptor-dependent M current regulation and seizure activity in mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13311–13316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisahn, A.; Yamada, M.; Duttaroy, A.; Gan, J.W.; Deng, C.X.; McBain, C.J.; Wess, J. Muscarinic induction of hippocampal gamma oscillations requires coupling of the M1 receptor to two mixed cation currents. Neuron 2002, 33, 615–624. [Google Scholar] [CrossRef] [Green Version]
- Sheffler, D.J.; Williams, R.; Bridges, T.M.; Xiang, Z.; Kane, A.S.; Byun, N.E.; Jadhav, S.; Mock, M.M.; Zheng, F.; Lewis, L.M.; et al. A novel selective muscarinic acetylcholine receptor subtype 1 antagonist reduces seizures without impairing hippocampus-dependent learning. Mol. Pharmacol. 2009, 76, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.L.; Aroniadou-Anderjaska, V.; Pidoplichko, V.I.; Figueiredo, T.H.; Apland, J.P.; Krishnan, J.K.; Braga, M.F. The M1 Muscarinic Receptor Antagonist VU0255035 Delays the Development of Status Epilepticus after Organophosphate Exposure and Prevents Hyperexcitability in the Basolateral Amygdala. J. Pharmacol. Exp. Ther. 2017, 360, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Palomero-Gallagher, N.; Schleicher, A.; Bidmon, H.-J.; Pannek, H.-W.; Hans, V.; Gorji, A.; Speckmann, E.-J.; Zilles, K. Multireceptor analysis in human neocortex reveals complex alterations of receptor ligand binding in focal epilepsies. Epilepsia 2012, 53, 1987–1997. [Google Scholar] [CrossRef]
- Akyuz, E.; Doganyigit, Z.; Paudel, Y.N.; Kaymak, E.; Yilmaz, S.; Uner, A.; Shaikh, M.F. Increased ACh-Associated Immunoreactivity in Autonomic Centers in PTZ Kindling Model of Epilepsy. Biomedicines 2020, 8, 113. [Google Scholar] [CrossRef]
- Graebenitz, S.; Kedo, O.; Speckmann, E.J.; Gorji, A.; Panneck, H.; Hans, V.; Palomero-Gallagher, N.; Schleicher, A.; Zilles, K.; Pape, H.C. Interictal-like network activity and receptor expression in the epileptic human lateral amygdala. Brain 2011, 134, 2929–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalpando-Vargas, F.; Medina-Ceja, L.; Santerre, A.; Enciso-Madero, E.A. The anticonvulsant effect of sparteine on pentylenetetrazole-induced seizures in rats: A behavioral, electroencephalographic, morphological and molecular study. J. Mol. Histol. 2020, 51, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Smolders, I.; Khan, G.M.; Manil, J.; Ebinger, G.; Michotte, Y. NMDA receptor-mediated pilocarpine-induced seizures characterization in freely moving rats by microdialysis. Br. J. Pharmacol. 1997, 121, 1171–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turski, W.A.; Cavalheiro, E.A.; Turski, L.; Kleinrok, Z. Intrahippocampal bethanechol in rats behavioural, electroencephalographic and neuropathological correlates. Behav. Brain Res. 1983, 7, 361–370. [Google Scholar] [CrossRef]
- Ben-Ari, Y.; Tremblay, E.; Ottersen, O.P. Injections of kainic acid into the amygdaloid complex of the rat an electrographic, clinical and histological study in relation to the pathology of epilepsy. Neuroscience 1980, 5, 515–528. [Google Scholar] [CrossRef]
- Krnjević, K.; Pumain, R.; Renaud, L. The mechanism of excitation by acetylcholine in the cerebral cortex. J. Physiol. 1971, 215, 247–268. [Google Scholar] [CrossRef]
- Peterson, S.L.; Armstrong, J.J. Muscarinic receptors mediate carbachol-induced inhibition of maximal electroshock seizures in the nucleus reticularis pontis oralis. Epilepsia 1999, 40, 20–25. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.L.; Armstrong, J.J.; Walker, M.K. Focal microinjection of carbachol into the periaqueductal gray induces seizures in the forebrain of the rat. Epilepsy Res. 2000, 42, 169–181. [Google Scholar] [CrossRef]
- Damaj, M.I.; Glassco, W.; Dukat, M.; Martin, B.R. Pharmacological characterization of nicotine-induced seizures in mice. J. Pharmacol. Exp. Ther. 1999, 291, 1284–1291. [Google Scholar]
- Iha, H.A.; Kunisawa, N.; Shimizu, S.; Tokudome, K.; Mukai, T.; Kinboshi, M.; Ikeda, A.; Ito, H.; Serikawa, T.; Ohno, Y. Nicotine Elicits Convulsive Seizures by Activating Amygdalar Neurons. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.K.; Kaur, S.; Goel, R.K. Exploring the ameliorative role of α7 neuronal nicotinic acetylcholine receptor modulation in epilepsy and associated comorbidities in post-PTZ-kindled mice. Epilepsy Behav. 2020, 103. [Google Scholar] [CrossRef] [PubMed]
- Löscher, W.; Potschka, H.; Wlaź, P.; Danysz, W.; Parsons, C.G. Are neuronal nicotinic receptors a target for antiepileptic drug development? Studies in different seizure models in mice and rats. Eur. J. Pharmacol. 2003, 466, 99–111. [Google Scholar] [CrossRef]
- Banerjee, J.; Dey, S.; Dixit, A.B.; Tripathi, M.; Doddamani, R.; Sharma, M.C.; Chandra, P.S. α7 nicotinic receptors contributes to glutamatergic activity in the hippocampus of patients with mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE-HS). J. Neural Transm. 2020, 127, 1441–1446. [Google Scholar] [CrossRef]
- De Fusco, M.; Becchetti, A.; Patrignani, A.; Annesi, G.; Gambardella, A.; Quattrone, A.; Ballabio, A.; Wanke, E.; Casari, G. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat. Genet. 2000, 26, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A.; Aracri, P.; Meneghini, S.; Brusco, S.; Amadeo, A. The role of nicotinic acetylcholine receptors in autosomal dominant nocturnal frontal lobe epilepsy. Front. Physiol. 2015, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Bertrand, S.; Steinlein, O.K.; Bertrand, D. Mutated nicotinic receptors responsible for autosomal dominant nocturnal frontal lobe epilepsy are more sensitive to carbamazepine. Epilepsia 1999, 40, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Picard, F.; Bruel, D.; Servent, D.; Saba, W.; Fruchart-Gaillard, C.; Schollhorn-Peyronneau, M.A.; Roumenov, D.; Brodtkorb, E.; Zuberi, S.; Gambardella, A.; et al. Alteration of the in vivo nicotinic receptor density in ADNFLE patients: A PET study. Brain 2006, 129, 2047–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadieux-Dion, M.; Meneghini, S.; Villa, C.; Toffa, D.H.; Wickstrom, R.; Bouthillier, A.; Sandvik, U.; Gustavsson, B.; Mohamed, I.; Cossette, P.; et al. Variants in CHRNB2 and CHRNA4 Identified in Patients with Insular Epilepsy. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2020, 47, 800–809. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Yuan, F.; Yang, Y.; Sun, X.L.; Song, L.; Jiang, W. CHRNA4 variant causes paroxysmal kinesigenic dyskinesia and genetic epilepsy with febrile seizures plus? Seizure 2018, 56, 88–91. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Huang, H.L.; Zhou, H. Study of candidate gene cHRNA4 for familial epilepsy syndrome. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1765–1769. [Google Scholar] [CrossRef]
- Jian, L.; Nagarajan, L.; de Klerk, N.; Ravine, D.; Bower, C.; Anderson, A.; Williamson, S.; Christodoulou, J.; Leonard, H. Predictors of seizure onset in Rett syndrome. J. Pediatr. 2006, 149, 542–547.e543. [Google Scholar] [CrossRef]
- Zhang, Y.; Cao, S.X.; Sun, P.; He, H.Y.; Yang, C.H.; Chen, X.J.; Shen, C.J.; Wang, X.D.; Chen, Z.; Berg, D.K.; et al. Loss of MeCP2 in cholinergic neurons causes part of RTT-like phenotypes via alpha7 receptor in hippocampus. Cell Res. 2016, 26, 728–742. [Google Scholar] [CrossRef] [Green Version]
- McCown, T.J.; Breese, G.R. Effects of apamin and nicotinic acetylcholine receptor antagonists on inferior collicular seizures. Eur. J. Pharmacol. 1990, 187, 49–58. [Google Scholar] [CrossRef]
- Jones, B.E. Activity, modulation and role of basal forebrain cholinergic neurons innervating the cerebral cortex. Prog. Brain Res. 2004, 145, 157–169. [Google Scholar] [CrossRef]
- Rosal Lustosa, I.; Soares, J.I.; Biagini, G.; Lukoyanov, N.V. Neuroplasticity in Cholinergic Projections from the Basal Forebrain to the Basolateral Nucleus of the Amygdala in the Kainic Acid Model of Temporal Lobe Epilepsy. Int. J. Mol. Sci. 2019, 20, 5688. [Google Scholar] [CrossRef] [Green Version]
- Unal, C.T.; Pare, D.; Zaborszky, L. Impact of basal forebrain cholinergic inputs on basolateral amygdala neurons. J. Neurosci. 2015, 35, 853–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girgis, M. Participation of muscarinic cholinergic receptors may be an important requirement of the kindling process. Exp. Neurol. 1980, 70, 458–461. [Google Scholar] [CrossRef]
- Ferencz, I.; Leanza, G.; Nanobashvili, A.; Kokaia, M.; Lindvall, O. Basal forebrain neurons suppress amygdala kindling via cortical but not hippocampal cholinergic projections in rats. Eur. J. Neurosci. 2000, 12, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.; Behrens, C.J.; Heinemann, U. Cholinergic dysfunction in temporal lobe epilepsy. Epilepsia 2007, 48 (Suppl. 5), 126–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, C.; Xu, Z.; Ji, C.; Liang, J.; Wang, Y.; Chen, B.; Wu, X.; Gao, F.; Wang, S.; et al. Depolarized GABAergic Signaling in Subicular Microcircuits Mediates Generalized Seizure in Temporal Lobe Epilepsy. Neuron 2017, 95, 92–105.e105. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Wang, Y.; Zhang, S.; Nao, J.; Liu, Y.; Wang, Y.; Ding, F.; Zhong, K.; Chen, L.; Ying, X.; et al. Subicular pyramidal neurons gate drug resistance in temporal lobe epilepsy. Ann. Neurol. 2019, 86, 626–640. [Google Scholar] [CrossRef]
- Ruan, Y.; Xu, C.; Lan, J.; Nao, J.; Zhang, S.; Fan, F.; Wang, Y.; Chen, Z. Low-frequency Stimulation at the Subiculum is Anti-convulsant and Anti-drug-resistant in a Mouse Model of Lamotrigine-resistant Temporal Lobe Epilepsy. Neurosci. Bull. 2020, 36, 654–658. [Google Scholar] [CrossRef]
- Fei, F.; Wang, X.; Wang, Y.; Chen, Z. Dissecting the role of subiculum in epilepsy: Research update and translational potential. Prog. Neurobiol. 2021, 102029. [Google Scholar] [CrossRef]
- Dautan, D.; Huerta-Ocampo, I.; Witten, I.B.; Deisseroth, K.; Bolam, J.P.; Gerdjikov, T.; Mena-Segovia, J. A major external source of cholinergic innervation of the striatum and nucleus accumbens originates in the brainstem. J. Neurosci. 2014, 34, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Furman, M.; Zhan, Q.; McCafferty, C.; Lerner, B.A.; Motelow, J.E.; Meng, J.; Ma, C.; Buchanan, G.F.; Witten, I.B.; Deisseroth, K.; et al. Optogenetic stimulation of cholinergic brainstem neurons during focal limbic seizures: Effects on cortical physiology. Epilepsia 2015, 56, e198–e202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawley, J.N.; Stivers, J.A.; Martin, J.V.; Mendelson, W.B. Cholinergic induction of seizures in the rat prefrontal cortex. Life Sci. 1986, 38, 2347–2354. [Google Scholar] [CrossRef]
- Stivers, J.A.; Skirboll, L.R.; Long, R.; Crawley, J.N. Anatomical analysis of frontal cortex sites at which carbachol induces motor seizures in the rat. Pharmacol. Biochem. Behav. 1988, 30, 129–136. [Google Scholar] [CrossRef]
- Janiesch, P.C.; Kruger, H.S.; Poschel, B.; Hanganu-Opatz, I.L. Cholinergic control in developing prefrontal-hippocampal networks. J. Neurosci. 2011, 31, 17955–17970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.M.; Bertram, E.H., 3rd. Changes in midline thalamic recruiting responses in the prefrontal cortex of the rat during the development of chronic limbic seizures. Epilepsia 2009, 50, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Sloan, D.M.; Zhang, D.; Bertram, E.H., 3rd. Increased GABAergic inhibition in the midline thalamus affects signaling and seizure spread in the hippocampus-prefrontal cortex pathway. Epilepsia 2011, 52, 523–530. [Google Scholar] [CrossRef]
- Bagri, A.; Di Scala, G.; Sandner, G. Myoclonic and tonic seizures elicited by microinjection of cholinergic drugs into the inferior colliculus. Therapie 1999, 54, 589–594. [Google Scholar]
- Lim, S.A.; Kang, U.J.; McGehee, D.S. Striatal cholinergic interneuron regulation and circuit effects. Front. Synaptic. Neurosci. 2014, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Epilepsy Model | Time Point | Observations | References |
|---|---|---|---|
| Temporal lobes with complex partial seizures | Interictal period | The binding of mAChRs antagonist I-iododexetimide was decreased in the anterior hippocampus. | [31] |
| Patients with drug-resistant focal temporal lobe epilepsy | Interictal period | M2 receptors always increased in various seizures including febrile seizure, hippocampal sclerosis, and other neocortical pathologies. | [38] |
| Patients with intractable temporal lobe epilepsy | Interictal period | An enhancement of M2 receptors binding in the lateral amygdala nuclei of TLE patients, while binding to M3 receptors was reduced. | [40] |
| Kainic acid | 3 days after injection of kainic acid | 1. The reduction of ChAT activity in the piriform cortex, amygdala, and nucleus basalis. 2. The reduction of AChE activity in the piriform cortex. 3. The decrease of mAChRs binding in the piriform cortex, amygdala, and nucleus basalis. 4. The decrease of Na+-dependent high-affinity choline uptake in the piriform cortex and amygdala. | [30] |
| Pilocarpine | 30 min after administration of pilocarpine | 1. M1 KO mice did not display seizures and survived after pilocarpine administration. 2. M2-M5 KO mice all had a seizure (clonic seizures) and died within 1 h after pilocarpine administration. | [33] |
| Pilocarpine | 45 min after administration of pilocarpine | The inability of pilocarpine to evoke seizures in both homozygous and heterozygous M1 mutant mice. | [34] |
| Pilocarpine | 45 min after administration of pilocarpine | 1. VU0255035 suppresses the potentiation of NMDAR currents induced by carbachol in hippocampal pyramidal cells. 2. VU0255035 inhibits pilocarpine-induced seizures. | [36] |
| OPs | 60 min after administration of OPs | VU0255035 retarded the process of status epilepticus after OPs exposure. | [37] |
| PTZ kindling model | 30 min after administration of PTZ | The increase of M2 receptors was observed in PTZ-kindled in the brainstem. | [39] |
| PTZ kindling model | 180 and 240 min after administration of PTZ | Sparteine increases the hippocampal M4 receptor expression. | [41] |
| Epilepsy Model | Time Point | Observations | References |
|---|---|---|---|
| Patients with mesial temporal lobe epilepsy with hippocampal sclerosis | Interictal period | α7 nAChRs were found to regulate hyperfunction of glutamatergic synaptic transmission in the hippocampus. | [52] |
| HEK293 cells co-expressing the human α4 nAChRs and the wild-type and the V287L mutant patient | - | 1. The mutant in β2V287L presynaptic nAChRs triggering neuronal firing, serving as an enhancement of neurotransmitter release. 2. The abnormal mutant in postsynaptic nAChRs may cause hyperexcitability. | [53] |
| Reconstituted in Xenopus oocytes | - | 100 μM CBZ inhibits ACh-evoked currents at the human α4β2 nicotinic receptors, and the ADSHE α4S248F or α4L-776ins3 mutant receptors, with a roughly 3 fold increase in sensitivity to CBZ. | [55] |
| ADSHE patients | Interictal period | An increase of midbrain nAChRs density in the ADSHE. | [56] |
| Patients with insular epilepsy | Interictal period | Mutant nACh receptors increased nicotinic currents in whole-cell recording. | [57] |
| Genetic or focal epilepsy with febrile seizures (GEFS+) patients | Interictal period | CHRNA4 was the pathogenic gene of GEFS+. | [58] |
| Familial partial epilepsy with variable foci (FPEVF) patients | Interictal period | cHRNA4 was the pathogenic gene of FPEVF. | [59] |
| Nicotine | Intraperitoneally injected 15 min before the nicotine treatment. | Nicotine elicits convulsive seizures by activating amygdalar neurons mainly via α7 nACh receptors. | [48] |
| PTZ kindling | Exposed to PTZ injections on day 3, 6, and 9 of treatment to assess seizure severity score. | The amelioration of epilepsy by α7 nAChRs agonist choline chloride in PTZ-kindled mice model. | [50] |
| MES and nicotine-induced seizure test in mice;Amygdala-kindling in rats. | 1.Nicotine-induced seizure starting immediately after nicotine injection and up to 5 min afterwards. 2. MES and kindling assesed interictal period. | 1. Various novel amino-alkyl-cyclohexane derivatives, among which nAChRs antagonists have shown an overlap potency between channel blocking at nAChRs and NMDARs. 2. nAChRs preferring antagonists were strongly relived MES and nicotine-induced seizure in mice. 3. The effect of anticonvulsant in the MES was all reduced by an additional injection of a subconvulsant dose of nicotine. 4. Such efficacious anticonvulsants were not affected in kindled rats | [51] |
| Pilocarpine | EEG activities recorded 7 days post-surgical recovery | 1. Chat-Mecp2−/y mice displayed frequent hyperexcitability discharges. 2. Administration of pilocarpine produces status epilepticus in Chat-Mecp2−/y mice. 3. Administration of α7 nAChRs agonist PNU282987 in the CA1 of the hippocampus increased the seizures onset time. | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Tan, B.; Wang, Y.; Chen, Z. Cholinergic Signaling, Neural Excitability, and Epilepsy. Molecules 2021, 26, 2258. https://doi.org/10.3390/molecules26082258
Wang Y, Tan B, Wang Y, Chen Z. Cholinergic Signaling, Neural Excitability, and Epilepsy. Molecules. 2021; 26(8):2258. https://doi.org/10.3390/molecules26082258
Chicago/Turabian StyleWang, Yu, Bei Tan, Yi Wang, and Zhong Chen. 2021. "Cholinergic Signaling, Neural Excitability, and Epilepsy" Molecules 26, no. 8: 2258. https://doi.org/10.3390/molecules26082258