
Validation by Molecular Dynamics of the Major Components of Sugarcane Vinasse, On a Surface of Calcium Carbonate (Calcite)

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Models and Computational Methods
2.1. Three-Dimensional Structures
2.2. Molecular Modeling
2.2.1. Molecular Optimization Using DFT
2.2.2. Molecular Optimization Using CHARMM22
2.3. Molecular Orientation
2.4. Solvation Process
2.5. Simulation Settings
3. Results and Discussion
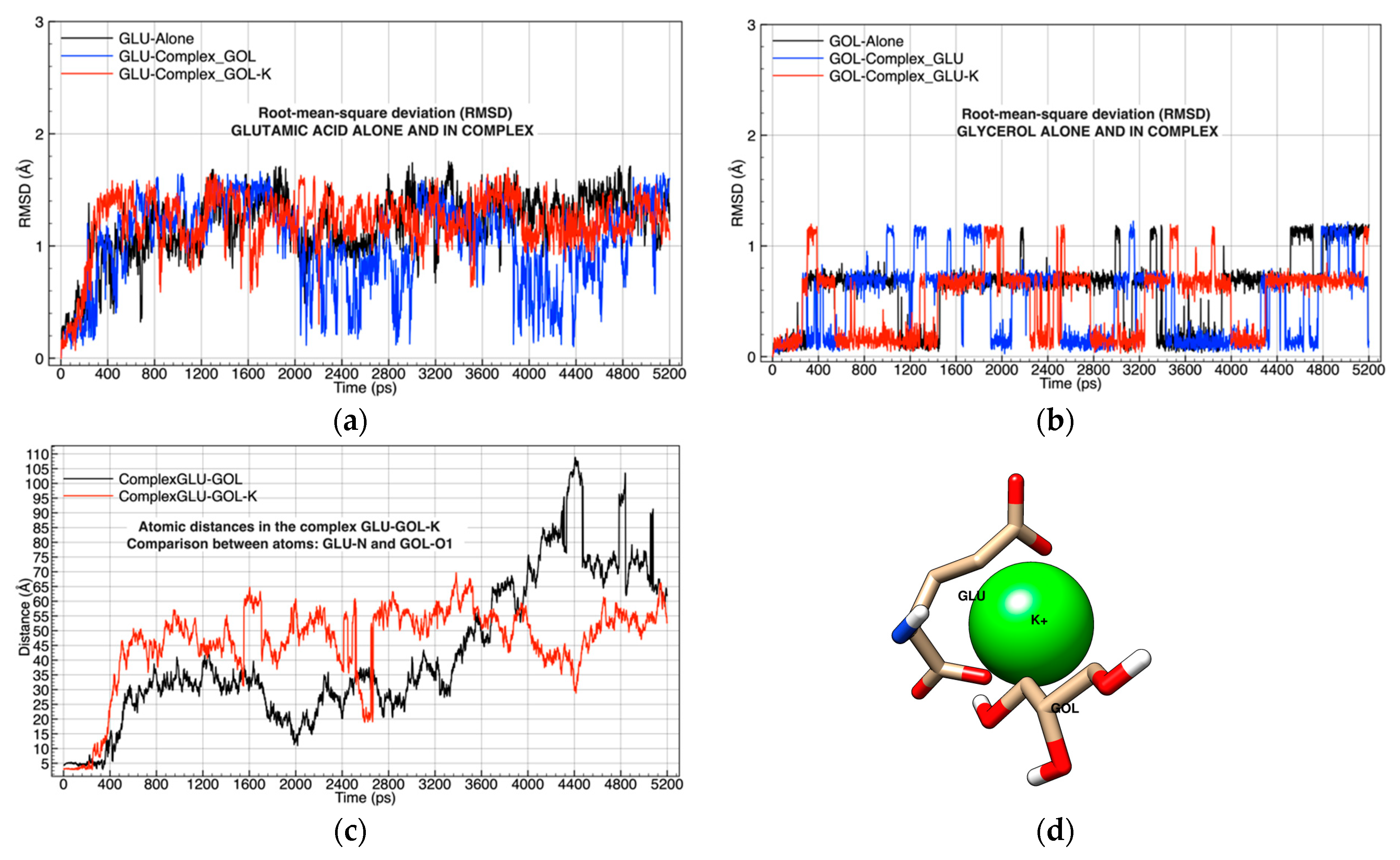
3.1. Chemical Complexes of the Major Sugarcane Vinasse Components
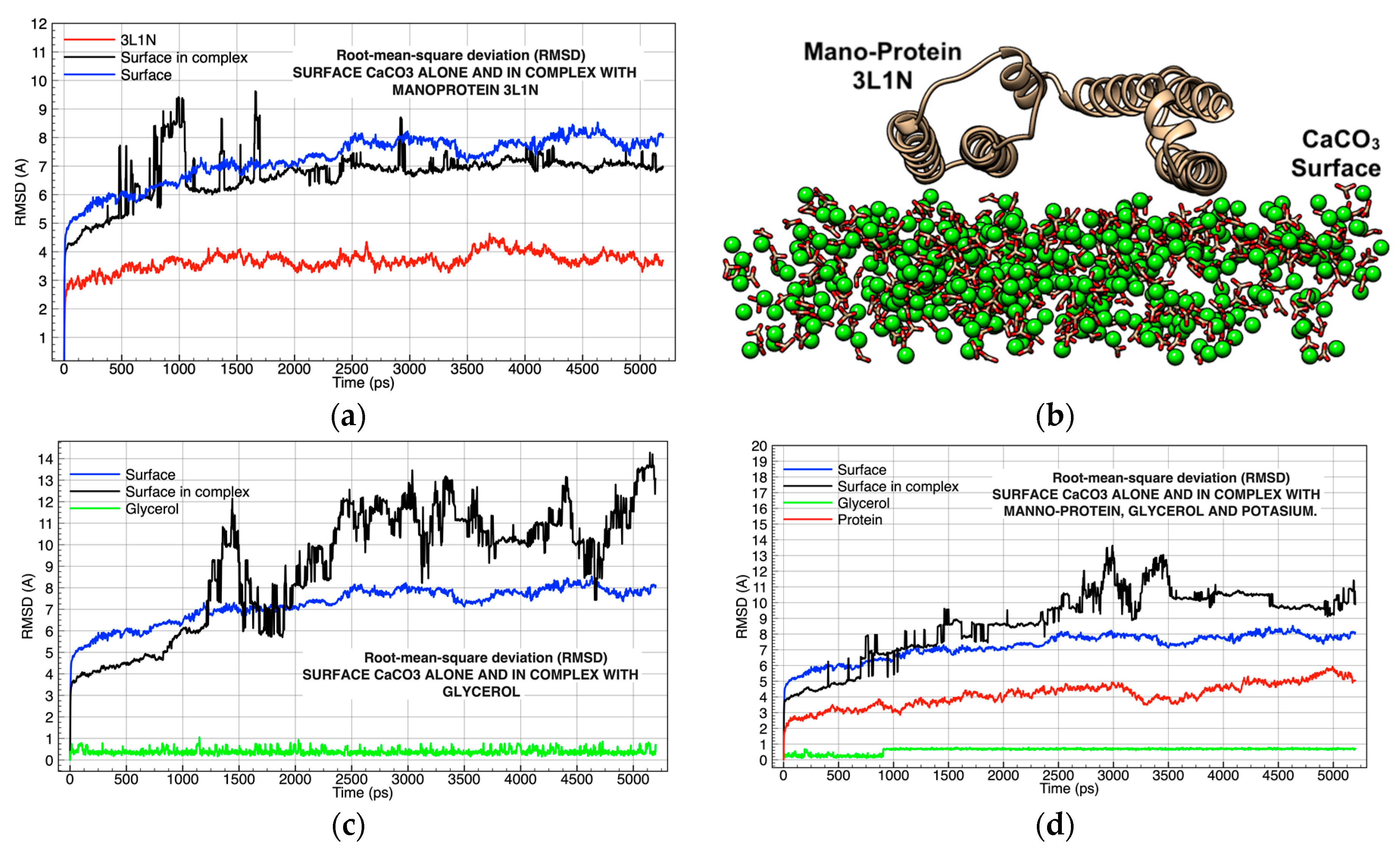
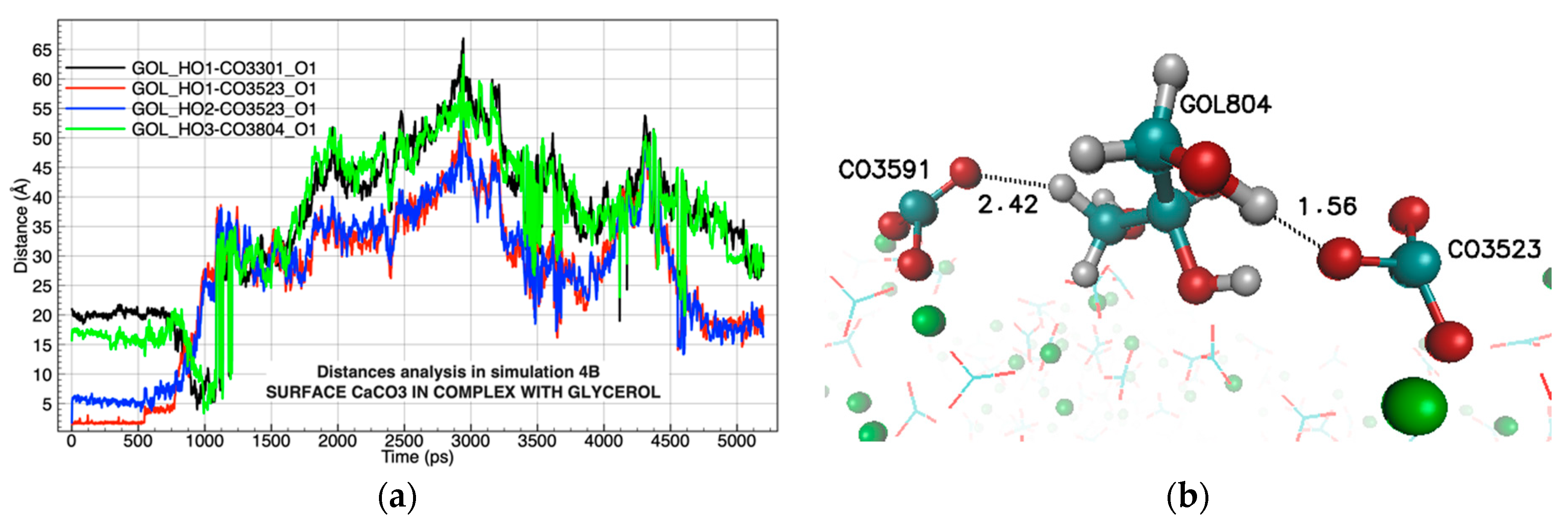
3.2. The Major Components of Sugarcane Vinasse on a Calcium Carbonate Surface
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Rodrigues Reis, C.E.; Hu, B. Vinasse from Sugarcane Ethanol Production: Better Treatment or Better Utilization? Front. Energy Res. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Ortegón, G.P.; Arboleda, F.M.; Candela, L.; Tamoh, K.; Valdes-Abellan, J. Vinasse Application to Sugar Cane Fields. Effect on the Unsaturated Zone and Groundwater at Valle Del Cauca (Colombia). Sci. Total Environ. 2016, 539, 410–419. [Google Scholar] [CrossRef] [Green Version]
- Lewicki, W. Introduction to Vinasses (Cms) from Sugarbeet and Sugar Cane Molasses Fermentation. Int. Sugar J. 2001, 103, 123–128. [Google Scholar]
- Christofoletti, C.A.; Escher, J.P.; Correia, J.E.; Marinho, J.F.U.; Fontanetti, C.S. Sugarcane Vinasse: Environmental Implications of Its Use. Waste Manag. 2013, 33, 2752–2761. [Google Scholar] [CrossRef] [PubMed]
- Scull, I.; Savón, L.; Gutiérrez, O.; Valiño, E.; Orta, I.; Mora, P.O.; Orta, H.; Ramos, Y.; Molineda, A.; Coto, G.; et al. Physic-chemical composition of concentrated vinasse for their assessment in animal diets. Cuba. J. Agric. Sci. 2012, 46, 385–389. [Google Scholar]
- Hidalgo, K.; Bocourt, R.; Mora, L.; Albelo, A. Physical-Chemical and Microbiological Characterization of the Concentrated Vinasse from Alcohol Distillery Caracterización Físico-Química y Microbiológica de La Vinaza Concentrada de Destilería de Alcohol. Cuba. J. Agric. Sci. 2017, 51, 4. [Google Scholar]
- Silva, R.O.; Torres, C.M.; Bonfim-Rocha, L.; Lima, O.C.M.; Coutu, A.; Jiménez, L.; Jorge, L.M.M. Multi-Objective Optimization of an Industrial Ethanol Distillation System for Vinasse Reduction—A Case Study. J. Clean. Prod. 2018, 183, 956–963. [Google Scholar] [CrossRef]
- Martinez-Echevarria Romero, M.J.; García-Travé, G.; Rubio-Gámez, M.C.; Moreno-Navarro, F.; Pérez-Mira, D. Valorization of Vinasse as Binder Modifier in Asphalt Mixtures. DYNA 2015, 82, 52–56. [Google Scholar] [CrossRef]
- Ahmed, A.M.; Mohamed, M.A.; Omar, A.F.; Shawky, M.H. Improvement of the concrete characteristics by using sugar industry wastes (vinasse). Jes. J. Eng. Sci. 2018, 46, 142–159. [Google Scholar] [CrossRef]
- Davies, M.; Nowotka, M.; Papadatos, G.; Dedman, N.; Gaulton, A.; Atkinson, F.; Bellis, L.; Overington, J.P. ChEMBL Web Services: Streamlining Access to Drug Discovery Data and Utilities. Nucleic Acids Res. 2015, 43, W612–W620. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, D.R.; Berrisford, J.M.; Conroy, M.J.; Clark, A.R.; Gupta, D.; Mukhopadhyay, A. Protein Structure Databases. In Encyclopedia of Bioinformatics and Computational Biology; Elsevier: Amsterdam, Netherlands, 2019; pp. 460–471. ISBN 978-0-12-811432-2. [Google Scholar]
- wwPDB consortium; Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Christie, C.; Duarte, J.M.; Dutta, S.; et al. Protein Data Bank: The Single Global Archive for 3D Macromolecular Structure Data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [Green Version]
- Guvench, O.; Mallajosyula, S.S.; Raman, E.P.; Hatcher, E.; Vanommeslaeghe, K.; Foster, T.J.; Jamison, F.W.; MacKerell, A.D. CHARMM Additive All-Atom Force Field for Carbohydrate Derivatives and Its Utility in Polysaccharide and Carbohydrate–Protein Modeling. J. Chem. Theory Comput. 2011, 7, 3162–3180. [Google Scholar] [CrossRef] [Green Version]
- Strub, M.-P.; Hoh, F.; Sanchez, J.-F.; Strub, J.M.; Böck, A.; Aumelas, A.; Dumas, C. Selenomethionine and Selenocysteine Double Labeling Strategy for Crystallographic Phasing. Structure 2003, 11, 1359–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calais, J.-L. Density-Functional Theory of Atoms and Molecules. R.G. Parr and W. Yang, Oxford University Press, New York, Oxford, 1989. IX + 333 Pp. Price £45.00: Book Review. Int. J. Quantum Chem. 1993, 47, 101. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density Functional Theory of Electronic Structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Hratchian, H.P.; Nielsen, A.B. Gaussian 09: Programmer’s Reference; Gaussian: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP Charges to Calculate Conformational Energies, Hydrogen Bond Energies, and Free Energies of Solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanommeslaeghe, K.; MacKerell, A.D. CHARMM Additive and Polarizable Force Fields for Biophysics and Computer-Aided Drug Design. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2015, 1850, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Hartley, H. Origin of the Word ‘Protein’. Nature 1951, 168, 244. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zoete, V.; Cuendet, M.A.; Grosdidier, A.; Michielin, O. SwissParam: A Fast Force Field Generation Tool for Small Organic Molecules. J. Comput. Chem. 2011, 32, 2359–2368. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Root Mean Square Deviation (RMSD). In Wiley StatsRef: Statistics Reference Online; Balakrishnan, N.; Colton, T.; Everitt, B.; Piegorsch, W.; Ruggeri, F.; Teugels, J.L. (Eds.) John Wiley & Sons, Ltd.: Chichester, UK, 2014; p. stat00728. ISBN 978-1-118-44511-2. [Google Scholar]
- Liao, S.; Tung, E.T.K.; Zheng, W.; Chong, K.; Xu, Y.; Dai, P.; Guo, Y.; Bartlam, M.; Yuen, K.-Y.; Rao, Z. Crystal Structure of the Mp1p Ligand Binding Domain 2 Reveals Its Function as a Fatty Acid-Binding Protein. J. Biol. Chem. 2010, 285, 9211–9220. [Google Scholar] [CrossRef] [Green Version]
- Numata, K. How to Define and Study Structural Proteins as Biopolymer Materials. Polym. J. 2020, 52, 1043–1056. [Google Scholar] [CrossRef]
- Kadota, K.; Furukawa, R.; Shirakawa, Y.; Shimosaka, A.; Hidaka, J. Effect of Surface Properties of Calcium Carbonate on Aggregation Process Investigated by Molecular Dynamics Simulation. J. Mater. Sci. 2014, 49, 1724–1733. [Google Scholar] [CrossRef]
- Bruneval, F.; Donadio, D.; Parrinello, M. Molecular Dynamics Study of the Solvation of Calcium Carbonate in Water. J. Phys. Chem. B 2007, 111, 12219–12227. [Google Scholar] [CrossRef] [PubMed]
- Konopacka-Łyskawa, D.; Kościelska, B.; Karczewski, J. Effect of Some Organic Solvent–Water Mixtures Composition on Precipitated Calcium Carbonate in Carbonation Process. J. Cryst. Growth 2015, 418, 25–31. [Google Scholar] [CrossRef]
- Kandeel, A.M.; Sanad, S.A.; Mohammed, S. Physico-Chemical, Mechanical and Mycological Properties of Cement Pastes Incorporating Vinasse. Chem. Res. J. 2019, 4, 75–85. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Complexes Modeled | Simulation | Components | Force Fields | Number of Water Molecules |
|---|---|---|---|---|
| GOL-GLU-K | 1A | GOL | AMBER: General AMBER force field (GAFF) [14] Primary protein model. ff14SB [15] | 530 |
| 1B | GLU | 605 | ||
| 1C | GLU-GOL | 671 | ||
| 1D | GOL-GLU-K | 670 | ||
| GOL-3L1N-K | 2A | 3L1N | 12681 | |
| 2B | GOL-3L1N | 12911 | ||
| 2C | GOL-3L1N-K | 12910 | ||
| Calcite | 3A | Calcite | CHARMM: General FF (CGenFF) [16] Empirical force field parameterization for proteins [17], lipids [18] and carbohydrates [19]. | 9240 |
| 3B | CaCO3 | 510 | ||
| SufaceCaCO3- GOL-3L1N-K | 4A | SufaceCaCO3 | 8447 | |
| 4B | SufaceCaCO3-GOL | 8446 | ||
| 4C | SufaceCaCO3-3L1N | 8305 | ||
| 4D | SufaceCaCO3-GOL-3L1N-K | 8303 |
| Simulation 4D: SufaceCaCO3-GOL-3L1N-K | Simulation 4D: SufaceCaCO3-GOL-3L1N-K | ||||
|---|---|---|---|---|---|
| Donor | Acceptor | Occupancy | Donor | Acceptor | Occupancy |
| ARG833 | CO3445 | 100.00% | GOL957 | CO3513 | 100.00% |
| LYS921 | CO3435 | 76.59% | Simulation 4B: SufaceCaCO3-GOL | ||
| LYS929 | CO3441 | 72.02% | GOL804 | CO3523 | 8.27% |
| LYS921 | CO3149 | 60.72% | GOL804 | CO3591 | 8.22% |
| LYS907 | CO3656 | 46.88% | GOL804 | CO3205 | 2.07% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas Álvarez, O.E.; Nicolás Vázquez, M.I.; Oñate-Garzón, J.; Arango, C.A. Validation by Molecular Dynamics of the Major Components of Sugarcane Vinasse, On a Surface of Calcium Carbonate (Calcite). Molecules 2021, 26, 2353. https://doi.org/10.3390/molecules26082353
Rojas Álvarez OE, Nicolás Vázquez MI, Oñate-Garzón J, Arango CA. Validation by Molecular Dynamics of the Major Components of Sugarcane Vinasse, On a Surface of Calcium Carbonate (Calcite). Molecules. 2021; 26(8):2353. https://doi.org/10.3390/molecules26082353
Chicago/Turabian StyleRojas Álvarez, Oscar Eduardo, María Inés Nicolás Vázquez, Jose Oñate-Garzón, and Carlos A. Arango. 2021. "Validation by Molecular Dynamics of the Major Components of Sugarcane Vinasse, On a Surface of Calcium Carbonate (Calcite)" Molecules 26, no. 8: 2353. https://doi.org/10.3390/molecules26082353
APA StyleRojas Álvarez, O. E., Nicolás Vázquez, M. I., Oñate-Garzón, J., & Arango, C. A. (2021). Validation by Molecular Dynamics of the Major Components of Sugarcane Vinasse, On a Surface of Calcium Carbonate (Calcite). Molecules, 26(8), 2353. https://doi.org/10.3390/molecules26082353