
Simultaneous Determination of As, Bi, Sb, Se, Te, Hg, Pb and Sn by Small-Sized Electrothermal Vaporization Capacitively Coupled Plasma Microtorch Optical Emission Spectrometry Using Direct Liquid Microsampling

 ,
,  ,
,  , ,
, ,  , ,
, ,
Abstract

1. Introduction
2. Results
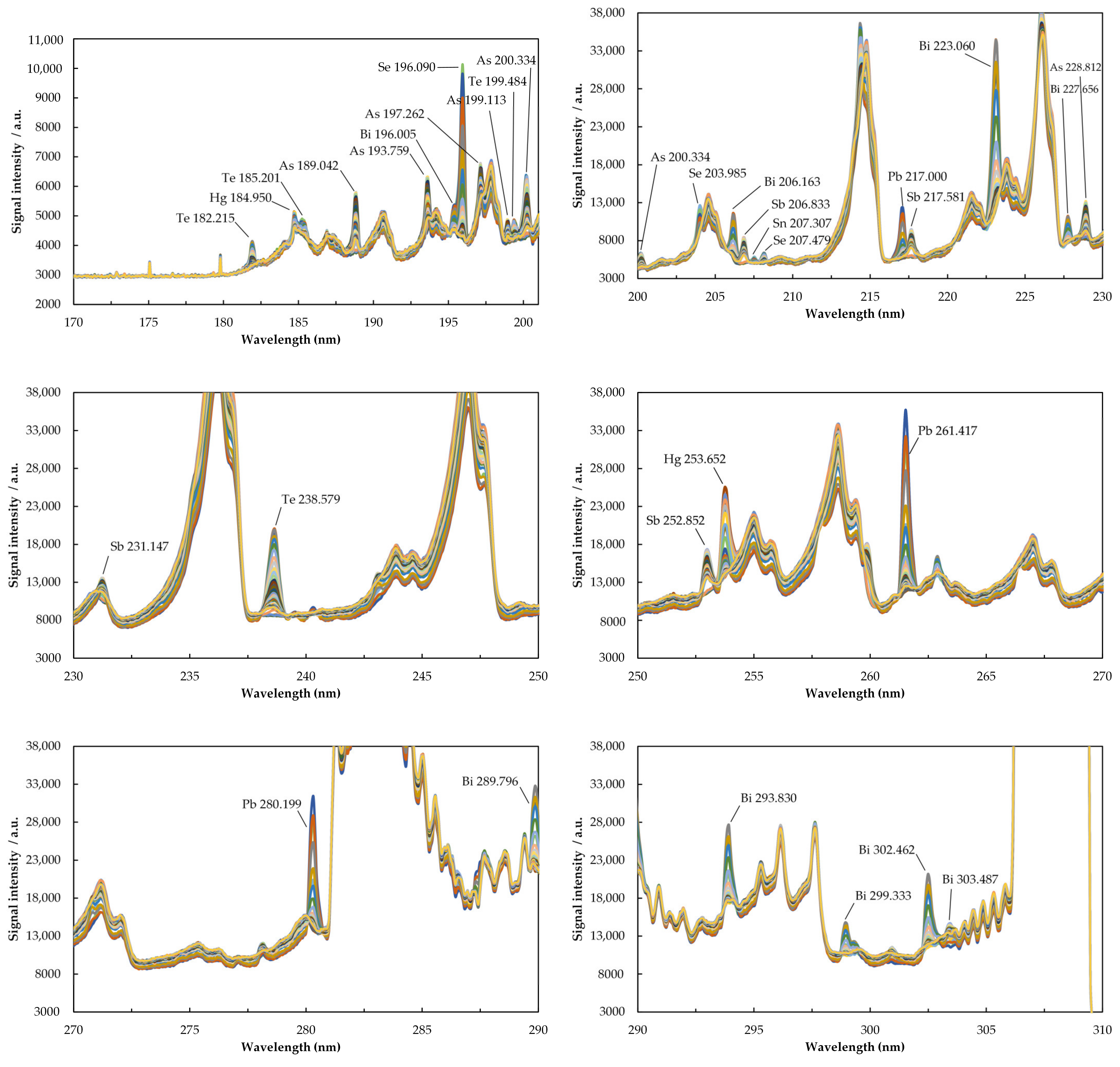
2.1. Characteristics of the Emission Spectrum of Elements
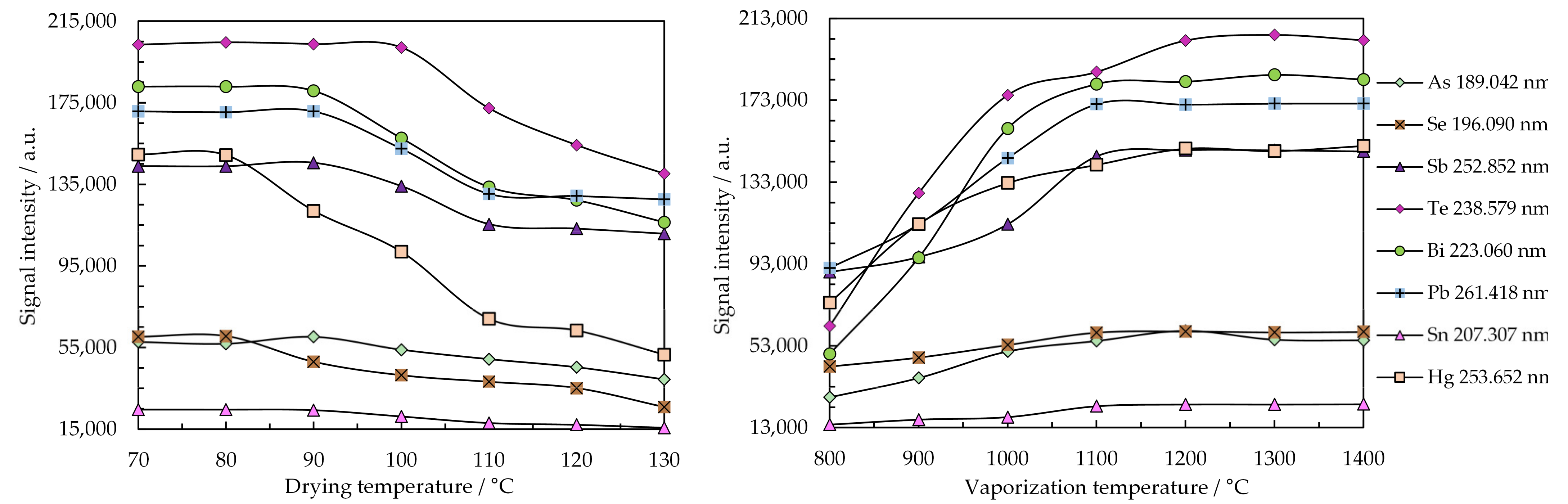
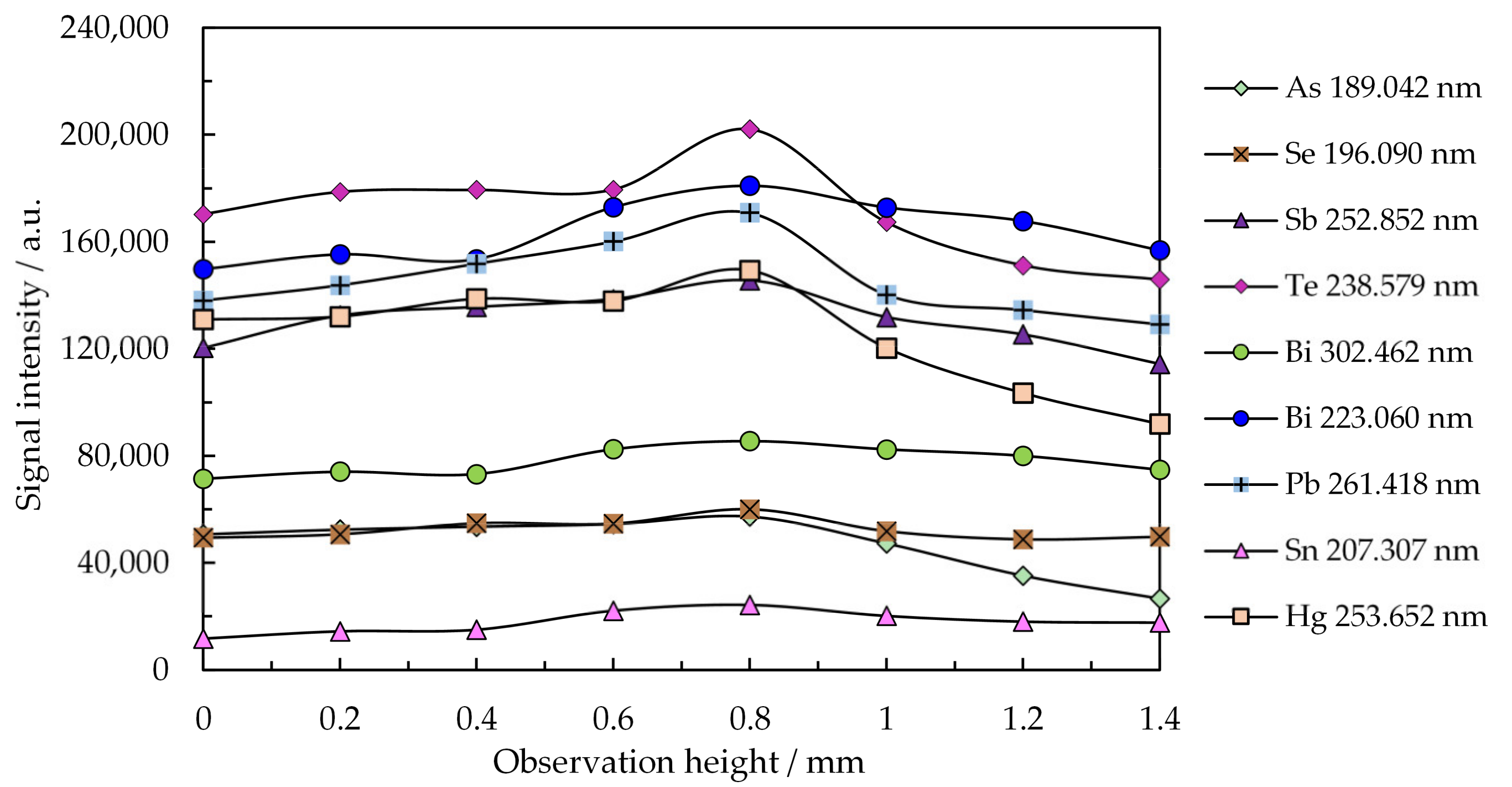
2.2. Optimization of Working Parameters for the SSETV-µCCP-OES Instrument
2.3. Figures of Merit and Validation of the SSETV-µCCP-OES Method
2.4. Application of the SSETV-µCCP-OES Method for the Analysis of Cave and River Sediment
3. Discussions
3.1. Excitation Capability of the Capacitively Coupled Plasma Microtorch
3.2. Capability for Simultaneous Determination by SSETV-µCCP-OES and Influence of Working Conditions
3.3. Limits of Detection by SSETV-µCCP-OES and Comparison with Other Methods
3.4. Accuracy of the SSETV-µCCP-OES Method and Non-Spectral Interferences
3.5. Analysis of Cave and River Sediment Samples by SSETV-µCCP-OES Method
4. Materials and Methods
4.1. Instrumentation
4.2. Reagents and Solutions
4.3. Certified Reference Materials and Test Samples
4.4. Sites Description and Sampling
4.5. Sample Preparation and Analytical Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Liu, X.Y.; Xiao, M.S.; Miao, Y.Q.; Ouyang, R.Z. Characteristics, applications and determination of bismuth. J. Nanosci. Nanotech. 2016, 16, 6679–6689. [Google Scholar] [CrossRef]
- Mahdy, M.A.; Mahdy, I.A.; El Zawawi, I.K. Characterization of Pb24 Te76 quantum dot thin film synthetized by inert gas condensation. Spectrochim. Acta. Part A Mol. Biomol. Spectrosc. 2015, 134, 302–309. [Google Scholar] [CrossRef]
- Akanda, M.R.; Osman, A.M.; Nazal, M.K.; Aziz, M.A. Review—Recent advancements in the utilization of Indium Tin Oxide (ITO) in electroanalysis without surface modification. J. Electrochem. Soc. 2020, 167, 037534. [Google Scholar] [CrossRef]
- Agency for Toxic Substances and Disease Registry (ATSDR). Public Health Statement Tin and Tin Compounds. 2005. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp55-c1-b.pdf (accessed on 29 April 2021).
- Hadrup, N.; Ravn-Haren, G. Acute human toxicity and mortality after selenium ingestion: A review. Trace Elem. Med. Biol. 2020, 58, 126425. [Google Scholar] [CrossRef] [PubMed]
- Gebreeyessus, G.D.; Zewge, F.A. review on environmental selenium issues. SN Appl. Sci. 2019, 1, 55. [Google Scholar] [CrossRef]
- Steeland, K.; Boffetta, P. Lead and cancer in humans: Where are we now? Am. J. Ind. Med. 2000, 38, 295–299. [Google Scholar] [CrossRef]
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, R.B.; Friedli, H.R.; Leaner, J.; Mason, R.; Mukherjee, A.B.; Stracher, G.B.; Streets, D.G.; et al. Global mercury emissions to the atmosphere from anthropogenic and natural sources. Atmos. Chem. Phys. 2010, 10, 5951–5964. [Google Scholar] [CrossRef]
- Abernathy, C.O.; Liu, Y.P.; Longfellow, D.; Aposhian, H.V.; Beck, B.; Fowler, B.; Goyer, R.; Menzer, R.; Rossaman, T.; Thompson, C.; et al. Arsenic: Health effects, mechanisms of actions, and research issues. Environ. Health Perspect. 1999, 107, 593–597. [Google Scholar] [CrossRef] [PubMed]
- International agency for Research on Cancer, Summary and Evaluations. Antimony Trioxide and Antimony Trisulfide. 1989. Available online: http://www.inchem.org/documents/iarc/vol47/47–11.html (accessed on 29 April 2021).
- Welna, M.; Szymczycha-Madeja, A.; Pohl, P. Non-chromatographic speciation of As by HG technique-analysis of samples with different matrices. Molecules 2020, 25, 4944. [Google Scholar] [CrossRef]
- Pohl, P.; Greda, K.; Dzimitrowicz, A.; Welna, M.; Szymczycha-Madeja, A.; Lesniewicz, A.; Jamroz, P. Cold atmospheric plasma-induced chemical vapor generation in trace element analysis by spectrometric methods. TRAC Trend Anal. Chem. 2019, 113, 234–245. [Google Scholar] [CrossRef]
- Slachcinski, M. Modern chemical and photochemical vapor generators for use in optical emission and mass spectrometry. J. Anal. At. Spectrom. 2019, 34, 257–273. [Google Scholar] [CrossRef]
- Li, Z.X.; Guo, Y.A. Simultaneous determination of trace arsenic, antimony, bismuth and selenium in biological samples by hydride generation-four-channel atomic fluorescence spectrometry. Talanta 2005, 65, 1318–1325. [Google Scholar] [CrossRef]
- Cava-Montesinos, P.; Cervera, M.L.; Pastor, A.; de la Guardia, M. Determination of As, Sb, Se, Te and Bi in milk by slurry sampling hydride generation atomic fluorescence spectrometry. Talanta 2004, 62, 173–182. [Google Scholar] [CrossRef]
- Deng, X.; Li, R.H.; Deng, S.P. Determination of the total content of arsenic, antimony, selenium and mercury in Chinese herbal food by chemical vapor generation-four-channel non-dispersive atomic fluorescence spectrometry. J. Fluoresc. 2020, 30, 949–954. [Google Scholar] [CrossRef]
- Mansur, E.T.; Barnes, S.J.; Savard, D.; Webb, P.C. Determination of Te, As, Bi, Sb and Se (TABS) in geological reference materials and geoPT proficiency test materials by hydride generation-atomic fluorescence spectrometry (HG-AFS). Geostand. Geoanal. Res. 2020, 44, 147–167. [Google Scholar] [CrossRef]
- Ruiz-de-Cenzano, M.; Rochina-Marco, A.; Cervera, M.L.; de la Guardia, M. Evaluation of the content of antimony, arsenic, bismuth, selenium, tellurium and their inorganic forms in commercially baby foods. Biol. Trace Elem. Res. 2017, 180, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Frentiu, T.; Butaciu, S.; Darvasi, E.; Ponta, M.; Senilă, M.; Levei, E.; Frentiu, M. Sono-induced cold vapour generation interfaced with capacitively coupled plasma microtorch optical emission spectrometry: Analytical characterization and comparison with atomic fluorescence spectrometry. J. Anal. At. Spectrom. 2015, 30, 1161–1168. [Google Scholar] [CrossRef]
- Valdivia, A.C.; Guererro, M.M.L.; Alonso, E.V.; Pavoin, J.M.C.; de Torres, A.G. Determination of As, Sb and Hg in water samples by flow injection coupled HR CS ETAAS with an in situ hydride generator. Microchem. J. 2018, 138, 109–115. [Google Scholar] [CrossRef]
- Hesse, S.; Ristau, T.; Einax, J.W. Chemical vapor generation by coupling high-pressure liquid flow injection to high-resolution continuum source hydride generation atomic absorption spectrometry for the determination of arsenic. Microchem. J. 2015, 123, 42–50. [Google Scholar] [CrossRef]
- Chirita, L.; Covaci, E.; Mot, A.; Ponta, M.; Gandea, A.; Frentiu, T. Determination of selenium in food and environmental samples by hydride generation high-resolution continuum source quartz furnace atomic absorption spectrometry. J. Anal. At. Spectrom. 2021, 36, 267–272. [Google Scholar] [CrossRef]
- Xi, M.Y.; Liu, R.; Wu, P.; Xu, K.L.; Hou, X.D.; Lv, Y. Atomic absorption spectrometric determination of trace tellurium after hydride trapping on platinum-coated tungsten coil. Microchem. J. 2010, 95, 320–325. [Google Scholar] [CrossRef]
- Welna, M.; Szymczycha-Madeja, A.; Pohl, P. Critical evaluation of strategies for single and simultaneous determinations of As, Bi, Sb and Se by hydride generation inductively coupled plasma optical emission spectrometry. Talanta 2017, 167, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Nunes, M.A.G.; Floes, E.M.M.; Paniz, J.N.G.; Dressler, V.L. Simultaneous determination of As, Bi, Sb, Se, Sn and Te in lead alloy using flow injection-hydride generation-inductively coupled plasma mass spectrometry. Anal. Methods. 2016, 8, 6805–6814. [Google Scholar] [CrossRef]
- Yilmaz, V.; Arslan, Z.; Rose, L. Determination of lead by hydride generation inductively coupled plasma mass spectrometry (HG-ICP-MS): On-line generation of plumbane using potassium hexacyanomanganate (III). Anal. Chim. Acta. 2013, 761, 18–26. [Google Scholar] [CrossRef]
- Yesiller, S.U.; Yalcın, S. Optimization of chemical and instrumental parameters in hydride generation laser-induced breakdown spectrometry for the determination of arsenic, antimony, lead and germanium in aqueous samples. Anal. Chim. Acta. 2013, 770, 7–17. [Google Scholar] [CrossRef]
- Sturgeon, R.E. Photochemical vapor generation: A radical approach to analyte introduction for atomic spectrometry. J. Anal. At. Spectrom. 2017, 32, 2319–2340. [Google Scholar] [CrossRef]
- Lavilla, I.; Romero, V.; Costas, I.; Bendicho, C. Greener derivatization in analytical chemistry. TRAC Trend Anal. Chem. 2014, 61, 1–10. [Google Scholar] [CrossRef]
- Zheng, C.B.; Ma, Q.; Wu, L.; Hou, X.D.; Sturgeon, R.E. UV photochemical vapor generation-atomic fluorescence spectrometric determination of conventional hydride generation elements. Microchem. J. 2010, 95, 32–37. [Google Scholar] [CrossRef]
- Burylin, M.Y.; Romanovskii, K.A.; Kaigorodova, E.A. Determination of selenium in drinking water by electrothermal atomic absorbtion spectrometry after photochemical generation, distillation, and preconcentration of its gaseous compounds in a graphite furnace. J. Anal. Chem. 2020, 75, 1408–1414. [Google Scholar] [CrossRef]
- Potes, M.D.; Nakadi, F.V.; Frois, C.F.G.; Vale, M.G.R.; da Silva, M.M. Investigation of the conditions for selenium determination by photochemical vapor generation coupled to graphite atomic absorption spectrometry. Microchem. J. 2019, 147, 324–332. [Google Scholar] [CrossRef]
- Romanovskiy, K.A.; Bolshov, M.A.; Munz, A.V.; Temerdashev, Z.A.; Burylin, M.Y.; Sirota, K.A. A novel photochemical vapor generation for ICP-MS determination of As, Bi, Hg, Sb, Se and Te. Talanta 2018, 187, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Jia, Y.T.; Shi, Z.M.; Chen, Y.L.; Ni, S.J.; Wang, R.L.; Tang, Y.R.; Gao, Y. Enhanced photochemical vapor generation for determination of bismuth by inductively coupled plasma mass spectrometry. Anal. Chem. 2018, 90, 13557–13563. [Google Scholar] [CrossRef] [PubMed]
- Gorska, M.; Greda, K.; Pohl, P. Determination of bismuth by optical emission spectrometry with liquid anode/cathode atmospheric pressure glow discharge. J. Anal. At. Spectrom. 2021, 36, 165–177. [Google Scholar] [CrossRef]
- Gorska, M.; Greda, K.; Pohl, P. On the coupling of hydride generation (HG) with flowing liquid anode atmospheric pressure glow discharge (FLA-APGD) for determination of traces of As, Bi, Hg, Sb and Se by optical emission spectrometry (OES). Talanta 2021, 222, 121510. [Google Scholar] [CrossRef] [PubMed]
- Annibaldi, A.; Truzzi, C.; Carnevali, O.; Pignalosa, P.; Api, M.; Scarponi, G.; Illuminati, S. Determination of Hg in farmed and wild Atlantic Bluefin Tuna (Thunnus thynnus L.) muscle. Molecules 2019, 24, 1273. [Google Scholar] [CrossRef] [PubMed]
- Senila, M.; Cadar, O.; Senila, L.; Hoaghia, A.; Miu, I. Mercury determination in natural zeolites by thermal decomposition atomic absorption spectrometry: Method validation in compliance with requirements for use as dietary supplements. Molecules 2019, 24, 4023. [Google Scholar] [CrossRef]
- Zacharia, A.N.; Arabadji, M.V.; Chebotarev, A.N. Direct electrothermal atomic absorption determination of trace elements in body fluids (Review). J. Appl. Spectrosc. 2017, 84, 1–7. [Google Scholar] [CrossRef]
- de Oliveira, R.M.; Antunes, A.C.N.; Vieira, M.A.; Medina, A.L.; Ribeiro, A.S. Evaluation of sample preparation methods for determination of As, Cd, Pb, and Se in rice samples by GF-AAS. Microchem. J. 2016, 124, 402–409. [Google Scholar] [CrossRef]
- Senila, M.; Cadar, O.; Miu, I. Development and validation of a spectrometric method for Cd and Pb determination in zeolites and safety evaluation. Molecules 2020, 25, 2591. [Google Scholar] [CrossRef]
- Guerrero, M.M.L.; Cordero, M.T.S.; Alonso, E.V.; Pavon, J.M.C.; de Torres, A.G. High resolution continuum source atomic absorption spectrometry and solid phase extraction for the simultaneous separation/preconcentration and sequential monitoring of Sb, Bi, Sn and Hg in low concentrations. J. Anal. At. Spectrom. 2015, 30, 1169–1178. [Google Scholar] [CrossRef]
- Schneider, M.; Cadorim, H.R.; Welz, B.; Carasek, E.; Feldmann, J. Determination of arsenic in agricultural soil samples using high-resolution continuum source graphite furnace atomic absorption spectrometry and direct solid sample analysis. Talanta 2018, 188, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, S.; Dash, K.; Dhavile, S.M.; Sahayam, A.C. Determination of traces of As, B, Bi, Ga, Ge, P, Pb, Sb, Se, Si and Te in high-purity nickel using inductively coupled plasma-optical emission spectrometry (ICP-OES). Talanta 2015, 131, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Shchukin, V.M.; Zhigilei, E.S.; Erina, A.A.; Shvetsova, Y.N.; Kuz’mina, N.E.; Luttseva, A.I. Validation of an ICP-MS method for the determination of mercury, lead, cadmium, and arsenic in medicinal plants and related drug preparation. Pharm. Chem. J. 2020, 54, 968–976. [Google Scholar] [CrossRef]
- Komorowicz, I.; Sajnog, A.; Baralkiewicz, D. Total arsenic and arsenic species determination in freshwater fish by ICP-DRC-MS and HPLC/ICP-DRC-MS techniques. Molecules 2019, 24, 607. [Google Scholar] [CrossRef] [PubMed]
- Pluhacek, T.; Rucka, M.; Maier, V. A direct LA-ICP-MS screening of elemental impurities in pharmaceutical products in compliance with USP and ICH-Q3 D. Anal. Chim. Acta 2018, 1078, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.Y.; Gao, X.; Chen., A.M.; Lin, J.Q. Effect of spatial confinement on Pb measurements in soil by femtosecond laser-induced breakdown spectroscopy. Appl. Phys. B 2020, 126, 7. [Google Scholar] [CrossRef]
- Wu, P.; Wen, X.D.; He, L.; He, Y.H.; Chen, M.Z.; Hou, X.D. Evaluation of tungsten coil electrothermal vaporization-Ar/H2 flame atomic fluorescence spectrometry for determination of eight traditional hydride-forming elements and cadmium without chemical vapor generation. Talanta 2008, 74, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Beuachemin, D. Electrothermal vaporization. In Sample Introduction Systems in ICPMS and ICPOES, 1st ed.; Beauchemin, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 411–467. [Google Scholar]
- Hanna, S.N.; Jones, B.T. A review of tungsten coil electrothermal vaporization as a sample introduction technique in atomic spectrometry. Appl. Spectrosc. Rev. 2011, 46, 624–635. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Nunes, M.A.G.; Costa, A.B.; Pozebon, D.; Duarte, F.A.; Dressler, V.L. Multielement determination in medicinal plants using electrothermal vaporization coupled to ICP-OES. Anal. Methods 2017, 9, 3497–3504. [Google Scholar] [CrossRef]
- Henn, A.S.; Flores, E.M.M.; Dressler, V.L.; Mesko, M.F.; Feldmann, J.; Mello, P.A. Feasibility of As, Sb, Se and Te determination in coal by solid sampling electrothermal vaporization inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2018, 33, 1384–1393. [Google Scholar] [CrossRef]
- Althobiti, R.A.; Beauchemin, D. Direct multi-element analysis of natural toothbrush by electrothermal vaporization into inductively coupled plasma optical emission spectrometry. J. Anal. At. Spectrom. 2021, 36, 535–539. [Google Scholar] [CrossRef]
- Liu, S.; Yu, Y.L.; Wang, J.H. Advances in discharge-based microplasmas for the analysis of trace species by atomic spectrometry. J. Anal. At. Spectrom. 2017, 32, 2118–2126. [Google Scholar] [CrossRef]
- Swiderski, K.; Pohl, P.; Jamroz, P. A miniaturized atmospheric pressure glow microdischarge system generated in contact with a hanging drop electrode – a new approach to spectrochemical analysis of liquid microsamples. J. Anal. At. Spectrom. 2019, 34, 1287–1293. [Google Scholar] [CrossRef]
- Peng, X.X.; Guo, X.H.; Ge, F.; Wang, Z. Battery-operated portable high-throughput solution cathode glow discharge optical emission spectrometry for environmental metal detection. J. Anal. At. Spectrom. 2019, 34, 394–400. [Google Scholar] [CrossRef]
- Pohl, P.; Jamroz, P.; Swiderski, K.; Dymitrowicz, A.; Lesniewicz, A. Critical evaluation of recent achievements in low power glow discharge generated at atmospheric pressure between a flowing liquid cathode and a metallic anode for element analysis by optical emission spectrometry. TRAC Trend Anal. Chem. 2017, 88, 119–133. [Google Scholar] [CrossRef]
- Weagant, S.; Chen, V.; Karanassios, V. Battery-operated, argon-hydrogen microplasma on hybrid, postage stamp-sized plastic-quartz chips for elemental analysis of liquid microsamples using a portable optical emission spectrometer. Anal. Bioanal. Chem. 2011, 401, 2865–2880. [Google Scholar] [CrossRef] [PubMed]
- Weagant, S.; Dulai, G.; Li, L.; Karanassios, V. Characterization of rapidly-prototyped, battery-operated, argon-hydrogen micro-plasma on a hybrid chip for elemental analysis of microsamples by portable optical emission spectrometry. Spectrochim Acta Part B 2015, 106, 75–80. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, Y.; Zheng, C.; Hou, X.D. Electrothermal vaporization for universal liquid sample introduction to dielectric barrier discharge microplasma for portable atomic emission spectrometry. Anal. Chem. 2014, 86, 5220–5224. [Google Scholar] [CrossRef]
- Li, N.; Wu, Z.C.; Wang, Y.Y.; Zhang, J.; Zhang, X.N.; Zhang, H.N.; Wu, W.H.; Gao, J.; Jiang, J. Portable dielectric barrier discharge-atomic emission spectrometer. Anal. Chem. 2017, 89, 2205–2210. [Google Scholar] [CrossRef] [PubMed]
- Frentiu, T.; Darvasi, E.; Butaciu, S.; Ponta, M.; Petreus, D.; Mihaltan, A.I.; Frentiu, M. A miniaturized capacitively coupled plasma microtorch optical emission spectrometer and a Rh coiled-filament as small-sized electrothermal vaporization device for simultaneous determination of volatile elements from liquid microsamples: Spectral and analytical characterization. Talanta 2014, 129, 72–78. [Google Scholar]
- Frentiu, T.; Darvasi, E.; Butaciu, S.; Ponta, M.; Petreus, D.; Etz, R.; Frentiu, M. Application of low-cost electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometry for simultaneous determination of Cd and Pb in environmental samples. Microchem. J. 2015, 121, 192–198. [Google Scholar] [CrossRef]
- Butaciu, S.; Frentiu, T.; Senila, M.; Darvasi, E.; Cadar, S.; Ponta, M.; Petreus, D.; Etz, R.; Frentiu, M. Determination of Cd in food using an electrothermal vaporization capacitively coupled plasma microtorch optical emission microspectrometer: Compliance with European legislation and comparison with graphite furnace atomic absorption spectrometry. Food Control 2016, 61, 227–234. [Google Scholar] [CrossRef]
- Frentiu, T.; Butaciu, S.; Darvasi, E.; Ponta, M.; Frentiu, M.; Petreus, D. A microanalytical method based on electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometry for multielemental determination: Comparison with inductively coupled plasma optical emission spectrometry. Chem. Pap. 2017, 71, 91–102. [Google Scholar] [CrossRef]
- Angyus, S.B.; Darvasi, E.; Ponta, M.; Petreus, D.; Etz, R.; Senila, M.; Frentiu, M.; Frentiu, T. Interference-free, green microanalytical method for total mercury and methylmercury determination in biological and environmental samples using small-sized electrothermal vaporization capacitively coupled plasma microtorch optical emission spectrometry. Talanta 2020, 217, 121067. [Google Scholar] [CrossRef] [PubMed]
- Frentiu, T.; Petreus, D.; Senila, M.; Mihaltan, A.I.; Darvasi, E.; Ponta, M.; Plaian, E.; Cordos, E.A. Low power capacitively coupled plasma microtorch for simultaneous multielemental determination by atomic emission using microspectrometres. Microchem. J. 2011, 97, 188–195. [Google Scholar] [CrossRef]
- Frentiu, T.; Ponta, M.; Darvasi, E.; Butaciu, S.; Cadar, S.; Senila, M.; Mathe, A.; Frentiu, M.; Petreus, D.; Etz, R.; et al. Miniaturized Analyzer for Simultaneous Determination of Elements from Liquid Micro Samples by Optical Emission Spectrometry. US Patent 131066 B1, 30 July 2020. Available online: http://pub.osim.ro/publication-server/pdf-document?PN=RO131066%20RO%20131066&iDocId=13576&iepatch=.pdf (accessed on 30 April 2021).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element/Line Type | λ (nm) | Excitation Energy (eV) | Relative Intensity (%) | |
|---|---|---|---|---|
| Maya2000 Pro | QE65 Pro 1 | |||
| As(I) R | 189.042 | 6.56 | 38 | - |
| 193.759 | 6.40 | 32 | 4 | |
| 197.262 | 6.28 | 25 | 5 | |
| 199.113 | 7.54 | 13 | 4 | |
| 200.334 | 7.54 | 34 | 3 | |
| 228.812 | 6.77 | 100 | 4 | |
| Bi(I) | 196.005 | 6.33 | 9 | 3 |
| 206.163 | 6.13 | 37 | 1 | |
| 223.060 | 5.56 | 100 | 3 | |
| 227.656 | 5.44 | 12 | 2 | |
| 289.796 | 5.69 | 59 | 3 | |
| 293.830 | 6.13 | 43 | 2 | |
| 299.333 | 5.56 | 21 | 2 | |
| 302.462 | 6.01 | 47 | 2 | |
| 303.487 | - | 19 | 3 | |
| Sb(I) R | 206.833 | 5.99 | 46 | 3 |
| 217.581 | 5.70 | 80 | 2 | |
| 231.147 | 5.36 | 66 | 2 | |
| 252.852 | 6.12 | 100 | 3 | |
| 326.751 | 5.83 | - | 1 | |
| 338.315 | 5.70 | - | 1 | |
| Se(I) | 196.090 | 6.32 | 100 | 3 |
| 203.985 | 6.32 | 34 | 3 | |
| 207.479 | 5.97 | 10 | 9 | |
| Te(I) | 182.215 | 6.81 | 8 | - |
| 185.201 | 7.28 | 5 | - | |
| 199.484 | 6.80 | 6 | 4 | |
| 238.579 | 5.78 | 100 | 2 | |
| Hg(I) R | 184.950 | 6.70 | 10 | - |
| 253.652 | 4.89 | 100 | 5 | |
| Pb(I) R | 217.000 | 5.71 | 33 | 3 |
| 261.418 | 5.71 | 100 | 3 | |
| 280.199 | 5.71 | 78 | 3 | |
| 363.957 | 4.38 | - | 1 | |
| 368.346 | 4.38 | - | 1 | |
| Sn(I) | 207.307 | 5.97 | 100 | 4 |
| 326.233 | 4.87 | - | 8 | |
| Element | λ (nm) | Maya2000 Pro | QE65 Pro | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Slope (mL µg−1) | R2 | sy/x (a.u) | LOD in Liquid (ng mL−1) | LOD in Solid 1 (mg kg−1) | Slope (mL µg−1) | R2 | sy/x (a.u) | LOD in Liquid (ng mL−1) | LOD in Solid 1 (mg kg−1) | ||
| As | 189.042 | 21398 | 0.9999 | 100 | 14 | 0.35 | - | - | - | - | - |
| 193.759 | 18020 | 0.9995 | 150 | 25 | 0.63 | 649 | 0.9994 | 8 | 40 2 | 1.00 2 | |
| 197.262 | 12824 | 0.9992 | 130 | 30 | 0.75 | 641 | 0.9993 | 10 | 49 | 1.22 | |
| 200.334 | 18188 | 0.9996 | 120 | 20 | 0.50 | 543 | 0.9991 | 10 | 60 | 1.50 | |
| Bi | 223.060 | 60608 | 0.9998 | 300 | 15 | 0.37 | 1560 | 0.9987 | 50 | 100 | 2.50 |
| 293.830 | 35759 | 0.9998 | 300 | 25 | 0.63 | - | - | - | - | - | |
| 302.462 | 28486 | 0.9997 | 310 | 33 | 0.82 | - | - | - | - | - | |
| Sb | 206.833 | 103961 | 0.9983 | 620 | 18 | 0.45 | 1789 | 0.9980 | 30 | 50 | 1.25 |
| 217.581 | 116758 | 0.9987 | 683 | 17 | 0.43 | 2816 | 0.9992 | 38 | 40 2 | 1.00 2 | |
| 252.852 | 145462 | 0.9994 | 385 | 8 | 0.20 | 4623 | 0.9993 | 40 | 30 | 0.75 | |
| Se | 196.026 | 22186 | 0.9999 | 100 | 13 | 0.33 | 1024 | 0.9991 | 52 | 152 | 3.75 |
| 203.985 | 6878 | 0.9991 | 100 | 44 | 1.10 | - | - | - | - | - | |
| Te | 182.215 | 4567 | 0.9997 | 385 | 140 | 3.50 | - | - | - | - | - |
| 238.579 | 79690 | 0.9998 | 833 | 30 | 0.75 | 1660 | 0.9970 | 277 | 501 | 12.52 | |
| Hg | 253.652 | 981663 | 0.9999 | 230 | 0.7 | 0.02 | 34235 | 0.9996 | 171 | 15 2 | 0.37 2 |
| Pb | 261.417 | 170830 | 0.9998 | 289 | 5 | 0.13 | 4506 | 0.9994 | 45 | 30 | 0.75 |
| 280.199 | 132913 | 0.9998 | 270 | 6 | 0.15 | 3418 | 0.9994 | 30 | 30 | 0.75 | |
| 217.000 | 57162 | 0.9992 | 520 | 27 | 0.67 | 1607 | 0.9991 | 54 | 101 | 2.53 | |
| 368.346 | - | - | - | - | - | 2431 | 0.9997 | 16 | 20 2 | 0.50 2 | |
| Sn | 207.307 | 8116 | 0.9992 | 8 | 3 | 0.08 | 300 | 0.9990 | 1 | 10 | 0.25 |
| 326.233 | - | - | - | - | - | 2727 | 0.9999 | 4 | 4.4 2 | 0.11 2 | |
| Element | λ (nm) | Maya2000 Pro | QE65 Pro | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Slope (mL µg−1) | R2 | sy/x (a.u) | LOD in Liquid (ng mL−1) | LOD in Solid 1 (mg kg−1) | Slope (mL µg−1) | R2 | sy/x (a.u) | LOD in Liquid (ng mL−1) | LOD in Solid 1 (mg kg−1) | ||
| As | 189.042 | 682 | 0.9993 | 16 | 70 | 1.75 | - | - | - | - | - |
| 193.759 | 589 | 0.9990 | 18 | 90 | 2.25 | - | - | - | - | - | |
| 197.626 | 500 | 0.9984 | 16 | 96 | 2.40 | - | - | - | - | - | |
| 200.334 | 668 | 0.9991 | 18 | 80 | 2.00 | - | - | - | - | - | |
| Bi | 223.060 | 7032 | 0.9994 | 70 | 30 | 0.75 | 153 | 0.9989 | 6 | 124 | 3.10 |
| 293.830 | 2807 | 0.9993 | 47 | 50 | 1.25 | - | - | - | - | - | |
| 302.462 | 1005 | 0.9993 | 25 | 75 | 1.88 | - | - | - | - | - | |
| Sb | 206.833 | 3320 | 0.9979 | 34 | 31 | 0.78 | - | - | - | - | - |
| 217.581 | 3638 | 0.9982 | 35 | 29 | 0.73 | - | - | - | - | - | |
| 252.852 | 4322 | 0.9984 | 25 | 17 | 0.43 | 149 | 0.9954 | 10 | 200 | 5.00 | |
| Se | 196.026 | 1970 | 0.9991 | 46 | 70 | 1.75 | - | - | - | - | - |
| 203.985 | 1069 | 0.9981 | 36 | 100 | 2.50 | - | - | - | - | - | |
| Te | 182.215 | 299 | 0.9869 | 33 | 330 | 8.25 | - | - | - | - | - |
| 238.579 | 3689 | 0.9972 | 69 | 56 | 1.40 | 100 | 0.9888 | 15 | 450 | 11.25 | |
| Hg | 253.652 | 58955 | 0.9987 | 197 | 10 | 0.25 | 2165 | 0.9905 | 50 | 70 | 1.80 |
| Pb | 217.000 | 6701 | 0.9952 | 114 | 51 | 1.28 | 137 | 0.9907 | 10 | 220 | 5.50 |
| 261.417 | 21508 | 0.9957 | 143 | 20 | 0.50 | 502 | 0.9913 | 14 | 80 | 2.00 | |
| 280.199 | 17586 | 0.9961 | 170 | 29 | 0.73 | 384 | 0.9944 | 12 | 90 | 2.25 | |
| 368.346 | - | - | - | - | - | 260 | 0.9975 | 14 | 160 | 4.00 | |
| Sn | 207.307 | 450 | 0.9985 | 6 | 40 | 1.00 | 12 | 0.9932 | 1 | 250 | 6.25 |
| 326.233 | - | - | - | - | - | 323 | 0.9947 | 15 | 140 | 3.50 | |
| CRM | Certified Value ± U (mg kg−1) 1 | Found Value ± CI (mg kg−1) 2 Standard Addition | Found Value ± CI (mg kg−1) 2 External Calibration | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| As | Hg | Pb | Sb | As | Hg | Pb | Sb | As | Hg | Pb | Sb | |
| ERM–CC141 | 7.5 ± 1.4 | 0.08 ± 0.008 | - | - | 8.0 ± 1.5 | 0.08 ± 0.009 | - | - | 7.9 ± 1.5 | 0.07 ± 0.005 | - | - |
| CRM04850 G | 123 ± 3.4 | 28 ± 1.13 | 86.9 ± 2.42 | 139 ± 13.9 | 122 ± 4.3 | 28 ± 1.60 | 86.9 ± 2.45 | 142 ± 14.1 | 124 ± 4.3 | 29 ± 1.62 | 84.9 ± 2.5 | 138.5 ± 14.0 |
| LGC6141 | 13.2 ± 3.5 | - | 75.8 ± 16 | - | 12.1 ± 1.1 | 73.9 ± 2.30 | - | 13.8 ± 0.8 | - | 75.8 ± 3.65 | - | |
| Metranal–32 | 26.1 ± 1.1 | 0.120 | 35.5 ± 0.9 | - | 25.6 ± 1.1 | 0.13 ± 0.005 | 36.2 ± 2.5 | - | 25.7 ± 1.4 | 0.12 ± 0.01 | 33.4 ± 2.4 | - |
| Metranal–34 | 42.4 ± 2.2 | 0.21 | 83.1 ± 2.3 | - | 43.0 ± 2.4 | 0.20 ± 0.012 | 82.6 ± 3.5 | - | 44.5 ± 2.3 | 0.22 ± 0.02 | 82.8 ± 2.7 | - |
| BCR–142 R | - | - | 25.7 ± 1.6 | - | - | - | 25.8 ± 2.16 | - | - | - | 25.8 ± 1.7 | - |
| BCR–287 A | - | - | - | 0.04 ± 0.015 | - | - | - | <0.20 | - | - | - | <0.20 |
| NC SDC 78301 | 56 ± 10 | 0.22 ± 0.04 | 79 ± 12 | - | 55 ± 10 | 0.21 ± 0.05 | 79 ± 13 | - | 55 ± 12 | 0.19 ± 0.04 | 82 ± 13 | - |
| ERM–CC580 | - | 132 ± 3 | - | - | - | 128 ± 8 | - | - | - | 125 ± 4 | - | - |
| CRM025 | 339 ± 20 | 99.8 ± 18 | 1447 ± 88 | <3.2 | 341 ± 20 | 104 ± 19 | 1366 ± 93 | 3.1 ± 0.2 | 357 ± 21 | 94.9 ± 18 | 1370 ± 92 | 3.5 ± 0.4 |
| BCR–280 R | 33.4 ± 2.9 | 1.46 ± 0.2 | - | - | 35.0 ± 3.7 | 1.45 ± 0.3 | - | - | 29.6 ± 3.1 | 1.26 ± 0.2 | - | - |
| RTCSQC001 | 43.1 ± 0.7 | 2.86 ± 0.1 | 144 ± 2 | 42.0 ± 4.1 | 45.2 ± 2.0 | 3.02 ± 0.2 | 134 ± 5 | 43.1 ± 4.7 | 44.2 ± 0.9 | 2.99 ± 0.1 | 139 ± 3 | 48.9 ± 4.2 |
| Rec. range (%) | 92–107 | 95–108 | 93–102 | 97–103 | 89–105 | 86–105 | 94–104 | 100–116 | ||||
| Pooled rec. (%) | 101 ± 10 | 101 ± 13 | 98 ± 8 | 101 ± 9 | 101 ± 10 | 96 ± 12 | 98 ± 8 | 108 ± 10 | ||||
| CRM | Certified Value ± U (mg kg−1) 1 | Found Value ± CI (mg kg−1) 2 Standard Addition | Found Value ± CI (mg kg−1) 2 External Calibration | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Bi | Se | Te | Sn | Bi | Se | Te | Sn | Bi | Se | Te | Sn | |
| CRM048 | - | 178 ± 5.68 | - | 93.5 ± 3.26 | - | 176 ± 5.91 | - | 96.6 ± 3.26 | - | 176 ± 6.10 | - | 95.4 ± 6.07 |
| BCR 142 R | - | - | - | - | - | - | - | - | - | - | - | |
| BCR 287 | 67.3 ± 1.1 | - | - | - | 67.3 ± 4.6 | - | - | - | 68.0 ± 5.3 | - | - | |
| NC SDC 78301 | - | 0.39 ± 0.1 | - | - | - | 0.35 ± 0.1 | - | - | - | 0.40 ± 0.1 | - | - |
| CRM025 | - | 518 ± 31 | - | - | - | 536 ± 38 | - | - | - | 550 ± 32 | - | - |
| SQC001 | - | 154 ± 3 | - | 215 ± 8 | - | 156 ± 7 | - | 212 ± 12 | - | 148 ± 7 | - | 203 ± 9 |
| Rec. range (%) | 94–107 | 90–103 | - | 99–103 | 98–104 | 96–106 | - | 94–102 | ||||
| Pooled rec. (%) | 100 ± 7 | 98 ± 15 | - | 101 ± 5 | 101 ± 3 | 101 ± 13 | - | 98 ± 3 | ||||
| Sample Origin | Sample Code | Concentration ± CI (mg kg−1) 1 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| As | Bi | Sb | Se | Te | Hg | Pb | Sn | ||
| Lesu Cave | 1 | <0.35 | 20.86 ± 0.53 | 7.02 ± 0.28 | <0.33 | 8.38 ± 0.18 | 0.23 ± 0.01 | 588.19 ± 21.52 | 6.77 ± 0.33 |
| 2 | 1.52 ± 0.05 | 36.90 ± 2.30 | 1.20 ± 0.06 | <0.33 | 5.37 ± 0.13 | 0.27 ± 0.01 | 10.29 ± 0.44 | 9.63 ± 0.19 | |
| Movile Cave | 3 | <0.35 | 107.34 ± 2.65 | 6.53 ± 0.26 | 6.07 ± 0.27 | 8.49 ± 0.15 | 0.17 ± 0.01 | 16.13 ± 0.41 | 5.94 ± 0.30 |
| Muierilor Cave | 4 | 1.72 ± 0.04 | 57.79 ± 1.91 | 2.61 ± 0.04 | 3.39 ± 0.13 | 5.93 ± 0.30 | 1.49 ± 0.04 | 83.25 ± 4.26 | 4.83 ± 0.17 |
| 5 | 2.20 ± 0.06 | 130.87 ± 6.85 | 37.19 ± 3.06 | 11.91 ± 0.74 | 11.54 ± 0.59 | 0.11 ± 0.01 | 76.87 ± 3.60 | 3.97 ± 0.09 | |
| 6 | 1.88 ± 0.15 | 54.78 ± 3.97 | 16.95 ± 0.19 | 1.11 ± 0.05 | 36.29 ± 2.90 | 0.22 ± 0.01 | 1.74 ± 0.06 | 4.67 ± 1.34 | |
| 7 | 6.10 ± 0.17 | 63.13 ± 2.55 | 22.40 ± 1.38 | 25.76 ± 0.91 | 9.68 ± 0.37 | 0.21 ± 0.02 | 67.83 ± 2.68 | 4.52 ± 0.15 | |
| 8 | 1.08 ± 0.03 | 7.66 ± 0.21 | <0.20 | <0.33 | 4.42 ± 0.16 | 0.28 ± 0.01 | 7.70 ± 0.45 | 3.51 ± 0.14 | |
| 9 | 1.54 ± 0.03 | 12.97 ± 0.26 | 3.34 ± 0.10 | 3.43 ± 0.14 | 4.94 ± 0.23 | 0.26 ± 0.02 | 10.43 ± 0.32 | 2.58 ± 0.12 | |
| Topolnita Cave | 10 | 1.23 ± 0.02 | 45.20 ± 0.72 | 19.44 ± 0.75 | 5.24 ± 0.24 | 6.18 ± 0.25 | 0.26 ± 0.01 | 12.46 ± 0.59 | 4.62 ± 0.18 |
| 11 | 0.81 ± 0.02 | 9.79 ± 0.26 | <0.20 | <0.33 | 2.75 ± 0.17 | 0.14 ± 0.01 | <0.13 | 2.10 ± 0.11 | |
| Aries River | 12 | 5.39 ± 0.13 | 129.51 ± 7.55 | <0.20 | 0.87 ± 0.03 | 65.92 ± 3.07 | 30.70 ± 0.79 | <0.13 | 1.64 ± 0.12 |
| 13 | 5.02 ± 0.22 | 135.98 ± 8.72 | 2.38 ± 0.06 | 2.09 ± 0.07 | <0.75 | 30.33 ± 1.55 | <0.13 | 2.00 ± 0.12 | |
| 14 | 1.74 ± 0.08 | 94.19 ± 4.36 | <0.20 | <0.33 | <0.75 | 15.45 ± 0.82 | <0.13 | 0.97 ± 0.03 | |
| RSD (%) 2 | 1.2–9.3 | 1.6–8.3 | 1.3–9.4 | 1.3–7.1 | 2.0–9.1 | 2.8–9.9 | 2.6–6.4 | 1.3–8.1 | |
| Component | Description |
|---|---|
| Plasma microtorch | Capacitively coupled; Mo tip microelectrode, 1.25 mm diameter (Goodfellow Cambridge, UK) mounted in a quartz tube (5 mm i.d, 25 mm length, 160 nm cut-off, H. Baumbach & Co. Ltd., Ipswich, UK); intake of Ar (100–200 mL min−1) transporting the vaporized microsample through four holes with a diameter of 750 µm drilled in the PTFE support around the microelectrode [63,69], (Babes-Bolyai University Cluj-Napoca, Romania) |
| Plasma power supply | Miniaturized r.f. generator (15 × 17 × 24 cm3), 13.56 MHz, 10–30 W (Technical University, Cluj-Napoca, Romania) |
| Small-sized electrothermal vaporizer | Rh filament (99.9% purity, 250 µm diameter, 4 turns with 1.5 mm diameter), mounted in a T-shaped vaporization chamber of quartz, sample volume: 10 µL, drying temperature: 80 °C for 180 s, vaporization temperature: 1300 °C for 10 s (Babes-Bolyai University Cluj-Napoca, Romania), 5–10% precision in temperature control [63,66,69]. |
| Power source for the Rh filament heating | Built in laboratory (Technical University, Cluj-Napoca, Romania), heating control of the Rh filament based on electrical resistance–temperature relationship [65,66]. |
| Control of Ar flow | Two-way valve SMCEVT307–5 DZ-01 F-Q supplied by the DC power source HY3003, Mastech, Premier Farnel (Leed, UK) [65]. |
| Optical detectors | Maya2000 Pro microspectrometer equipped with CCD (165–309 nm spectral range, 0.35 nm FWHM, detector chamber purged with Ar, Ocean Optics, Dunedin, USA).QE65 Pro microspectrometer equipped with CCD (190–380 nm spectral range, 0.4 nm FWHM, CCD cooled at −20 °C, Ocean Optics, Dunedin, USA). |
| Plasma viewing | Microspectrometer mounted on a 3D translator for targeting different heights by 100 µm increment, radial view through the collimating lens (10 mm focal length) without fiber optics [65,66]. |
| Spectrum recording/emission signal measurement | 70 episodic 3D spectra (signal–wavelength–time), 200 ms integration time per episode [67], measurement of the transient signal and peak height. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angyus, S.B.; Levei, E.; Petreus, D.; Etz, R.; Covaci, E.; Moldovan, O.T.; Ponta, M.; Darvasi, E.; Frentiu, T. Simultaneous Determination of As, Bi, Sb, Se, Te, Hg, Pb and Sn by Small-Sized Electrothermal Vaporization Capacitively Coupled Plasma Microtorch Optical Emission Spectrometry Using Direct Liquid Microsampling. Molecules 2021, 26, 2642. https://doi.org/10.3390/molecules26092642
Angyus SB, Levei E, Petreus D, Etz R, Covaci E, Moldovan OT, Ponta M, Darvasi E, Frentiu T. Simultaneous Determination of As, Bi, Sb, Se, Te, Hg, Pb and Sn by Small-Sized Electrothermal Vaporization Capacitively Coupled Plasma Microtorch Optical Emission Spectrometry Using Direct Liquid Microsampling. Molecules. 2021; 26(9):2642. https://doi.org/10.3390/molecules26092642
Chicago/Turabian StyleAngyus, Simion Bogdan, Erika Levei, Dorin Petreus, Radu Etz, Eniko Covaci, Oana Teodora Moldovan, Michaela Ponta, Eugen Darvasi, and Tiberiu Frentiu. 2021. "Simultaneous Determination of As, Bi, Sb, Se, Te, Hg, Pb and Sn by Small-Sized Electrothermal Vaporization Capacitively Coupled Plasma Microtorch Optical Emission Spectrometry Using Direct Liquid Microsampling" Molecules 26, no. 9: 2642. https://doi.org/10.3390/molecules26092642
APA StyleAngyus, S. B., Levei, E., Petreus, D., Etz, R., Covaci, E., Moldovan, O. T., Ponta, M., Darvasi, E., & Frentiu, T. (2021). Simultaneous Determination of As, Bi, Sb, Se, Te, Hg, Pb and Sn by Small-Sized Electrothermal Vaporization Capacitively Coupled Plasma Microtorch Optical Emission Spectrometry Using Direct Liquid Microsampling. Molecules, 26(9), 2642. https://doi.org/10.3390/molecules26092642