
Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold


Abstract
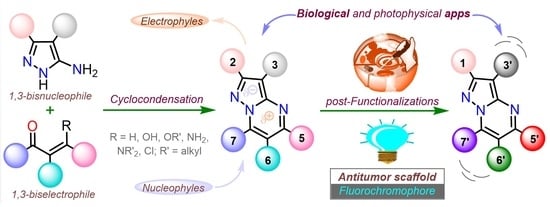
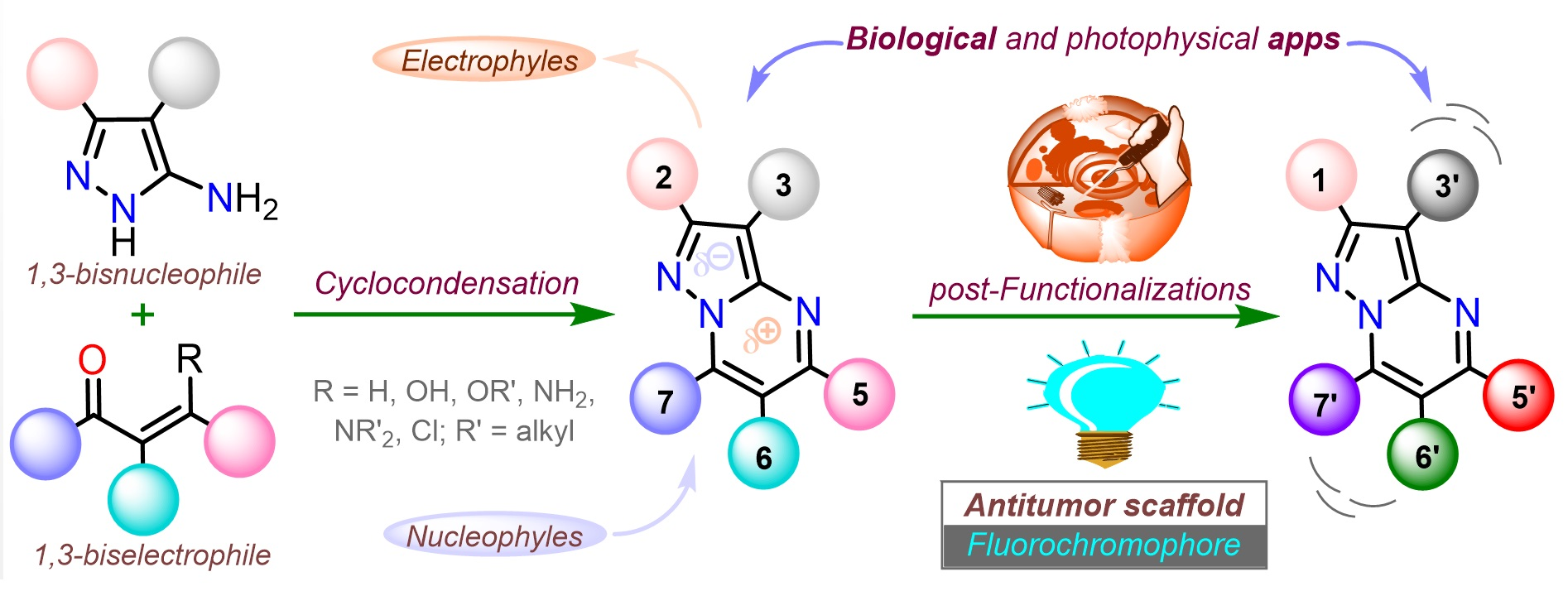
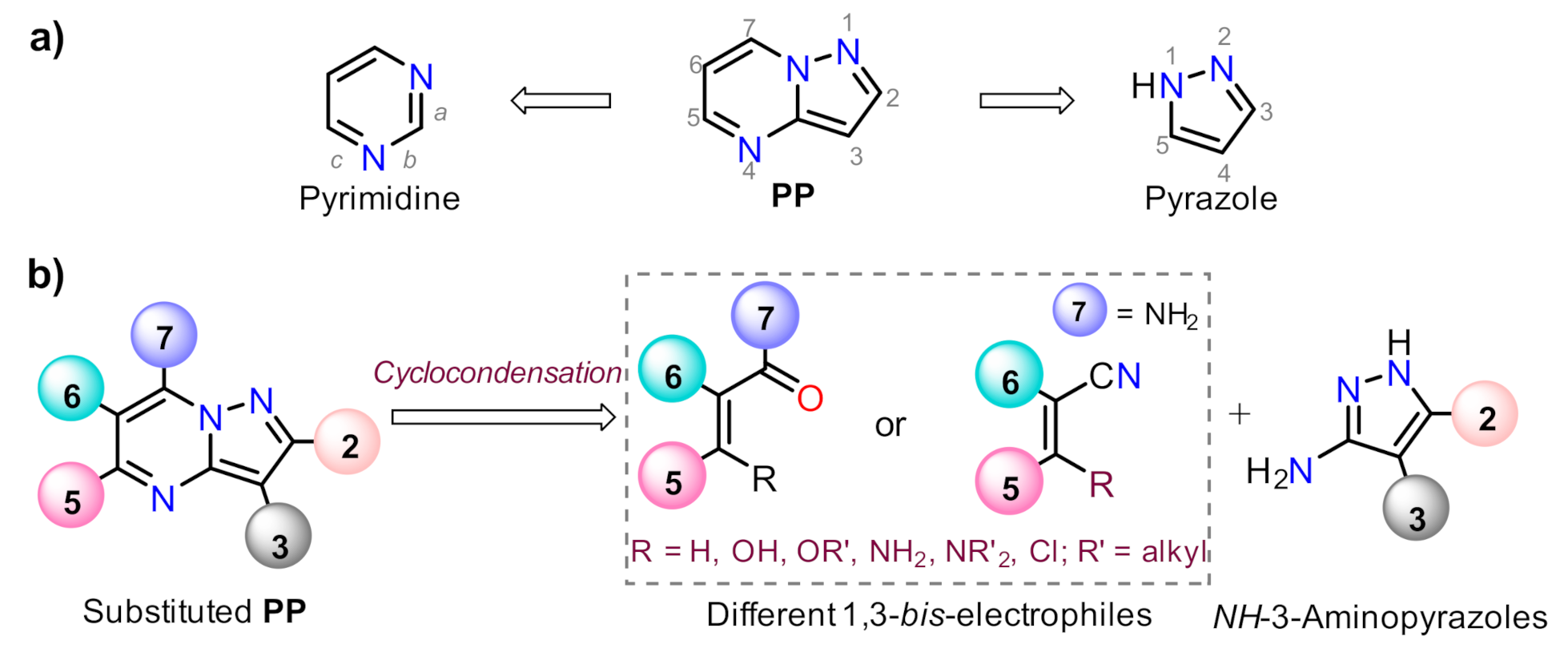
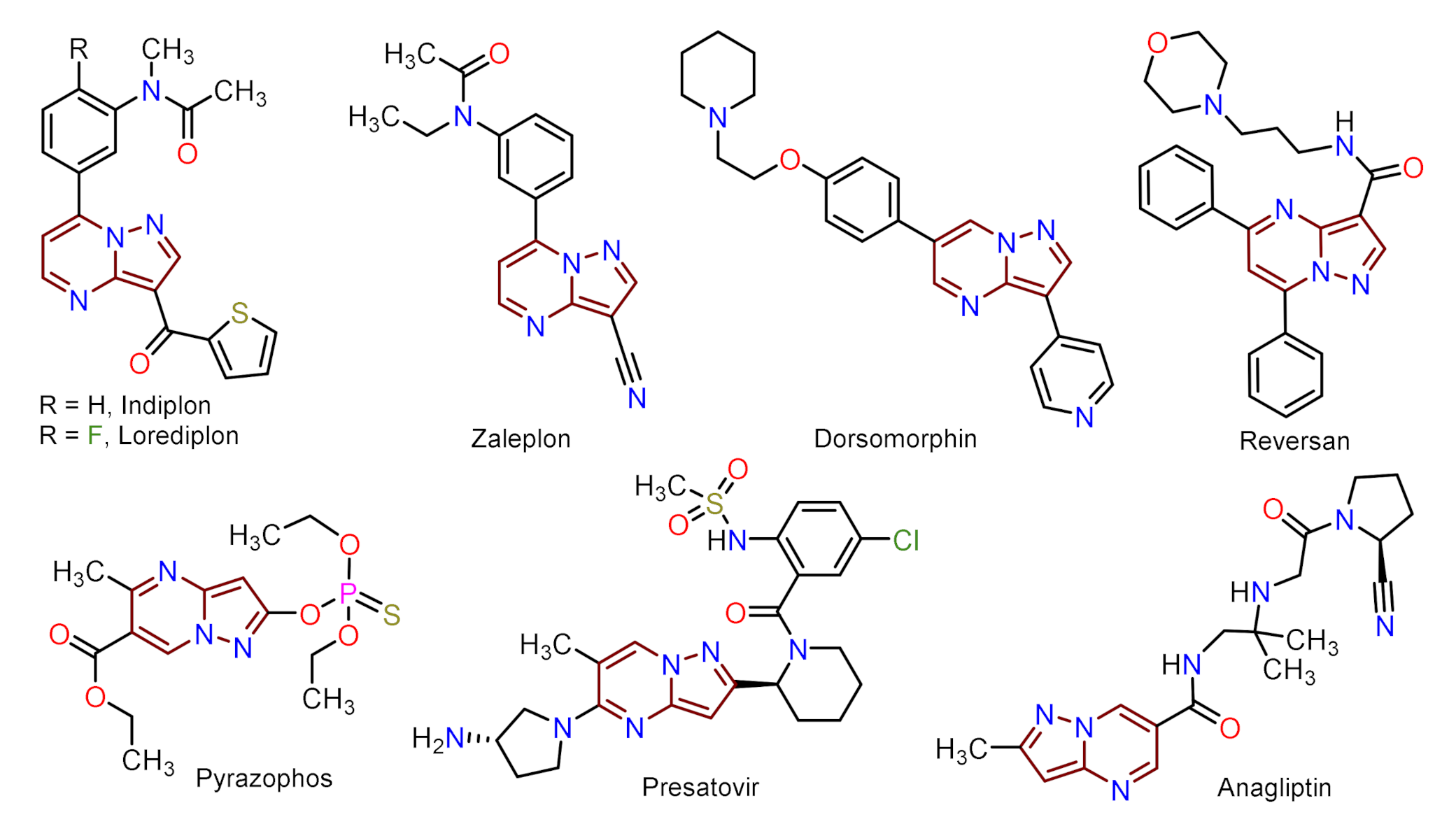
1. Introduction
2. Synthesis and Functionalization
2.1. Synthesis
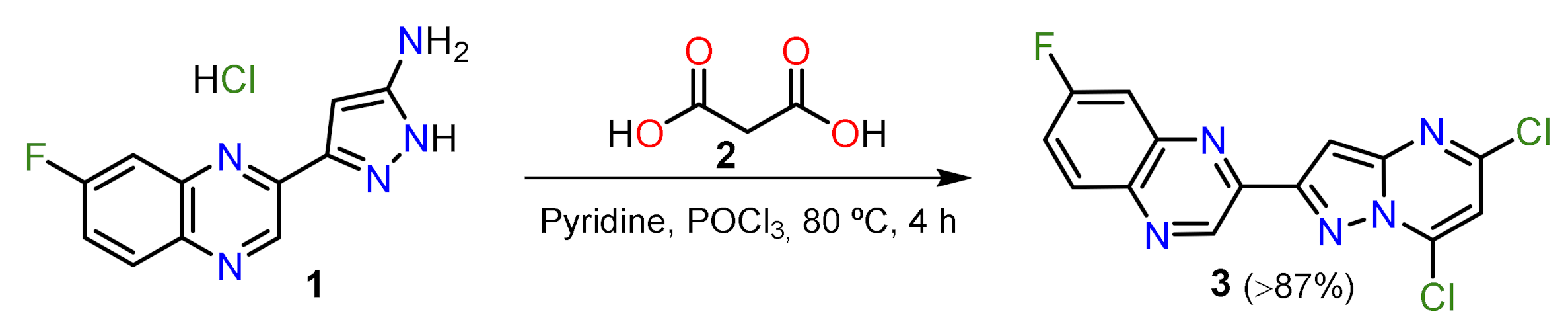
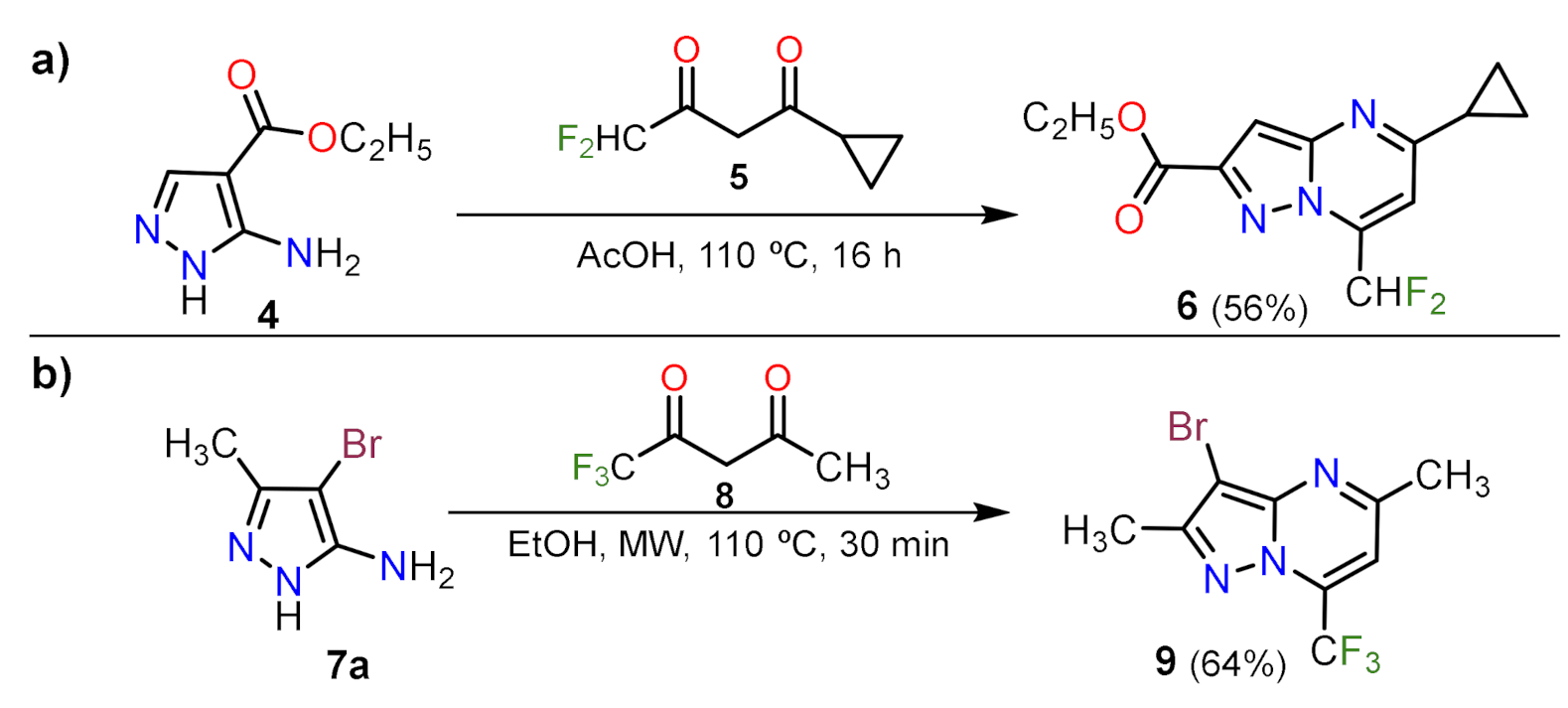
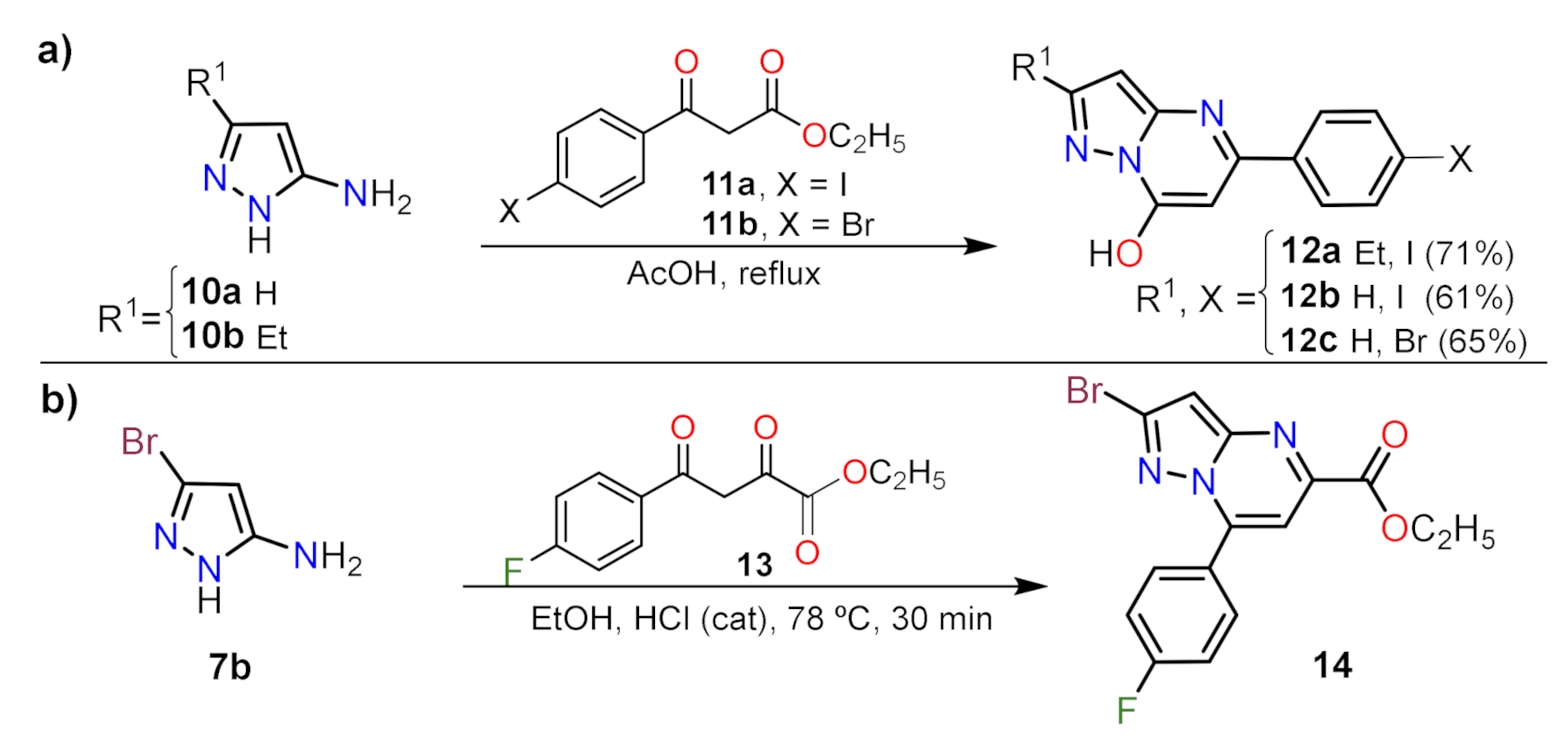
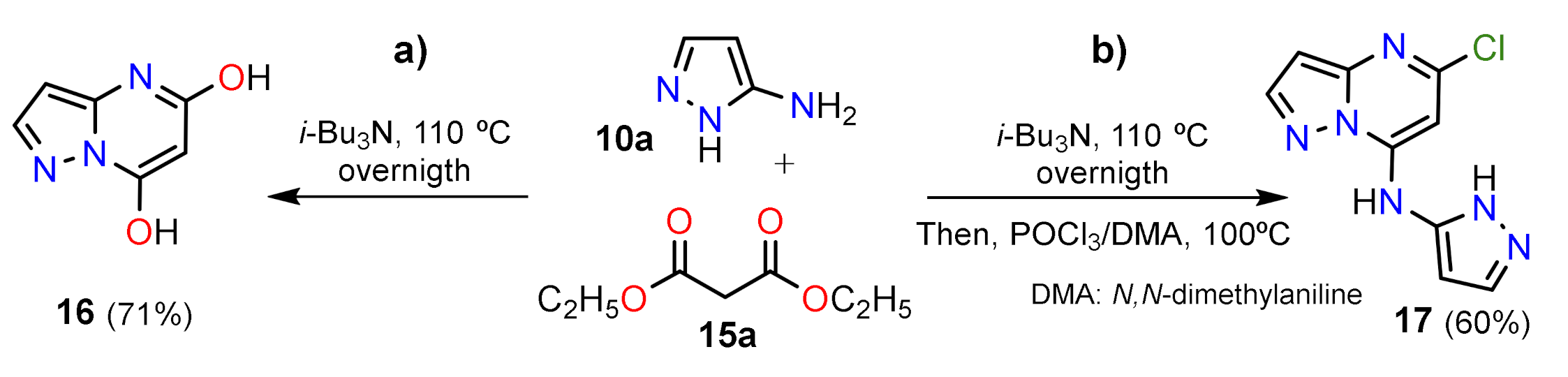
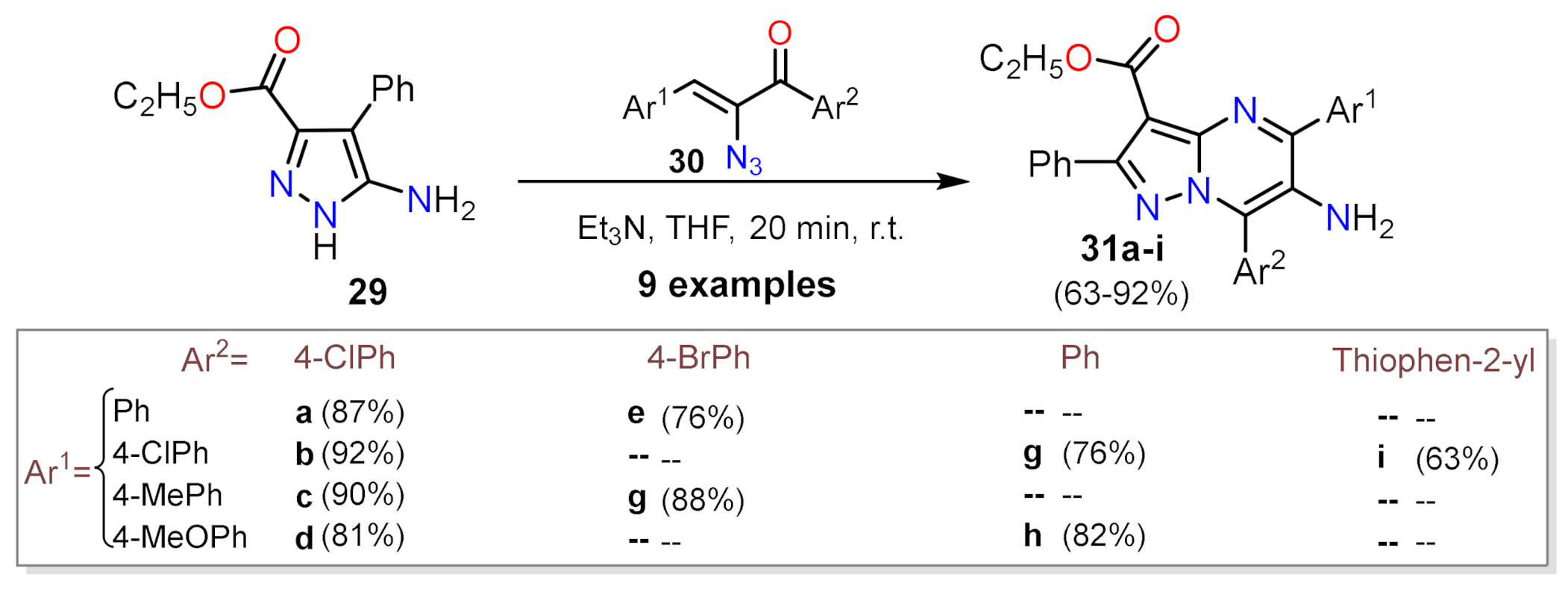
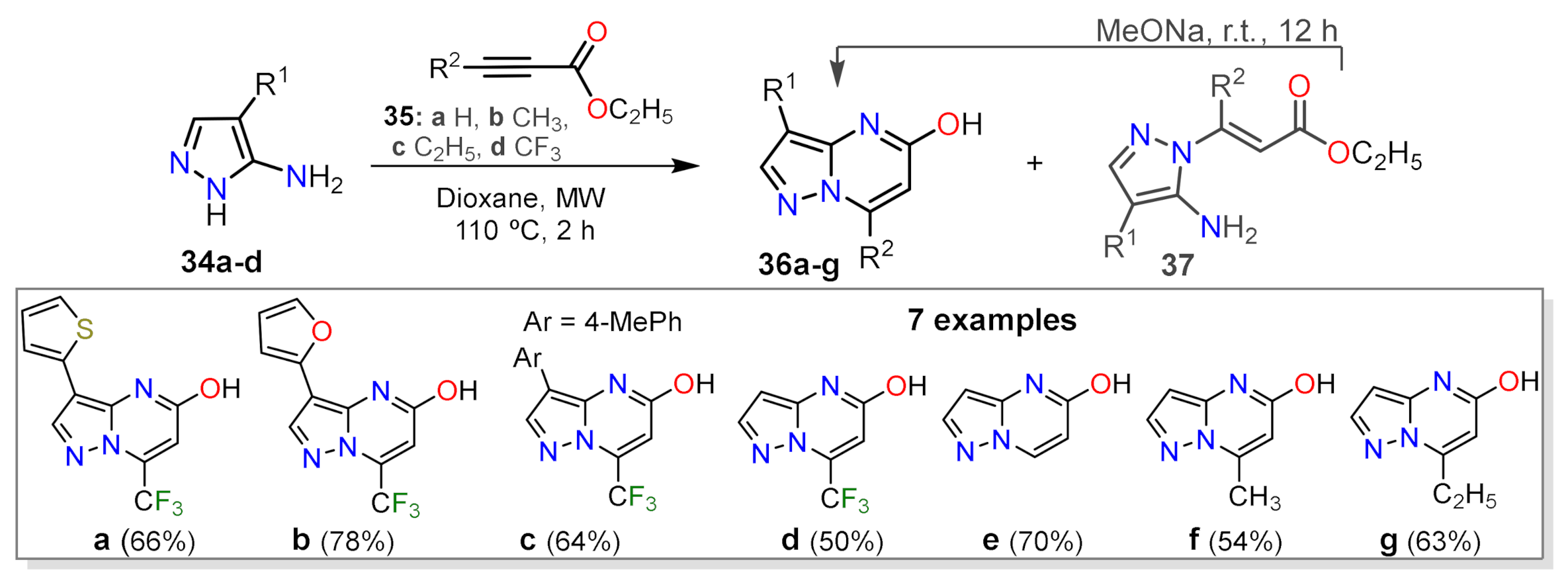
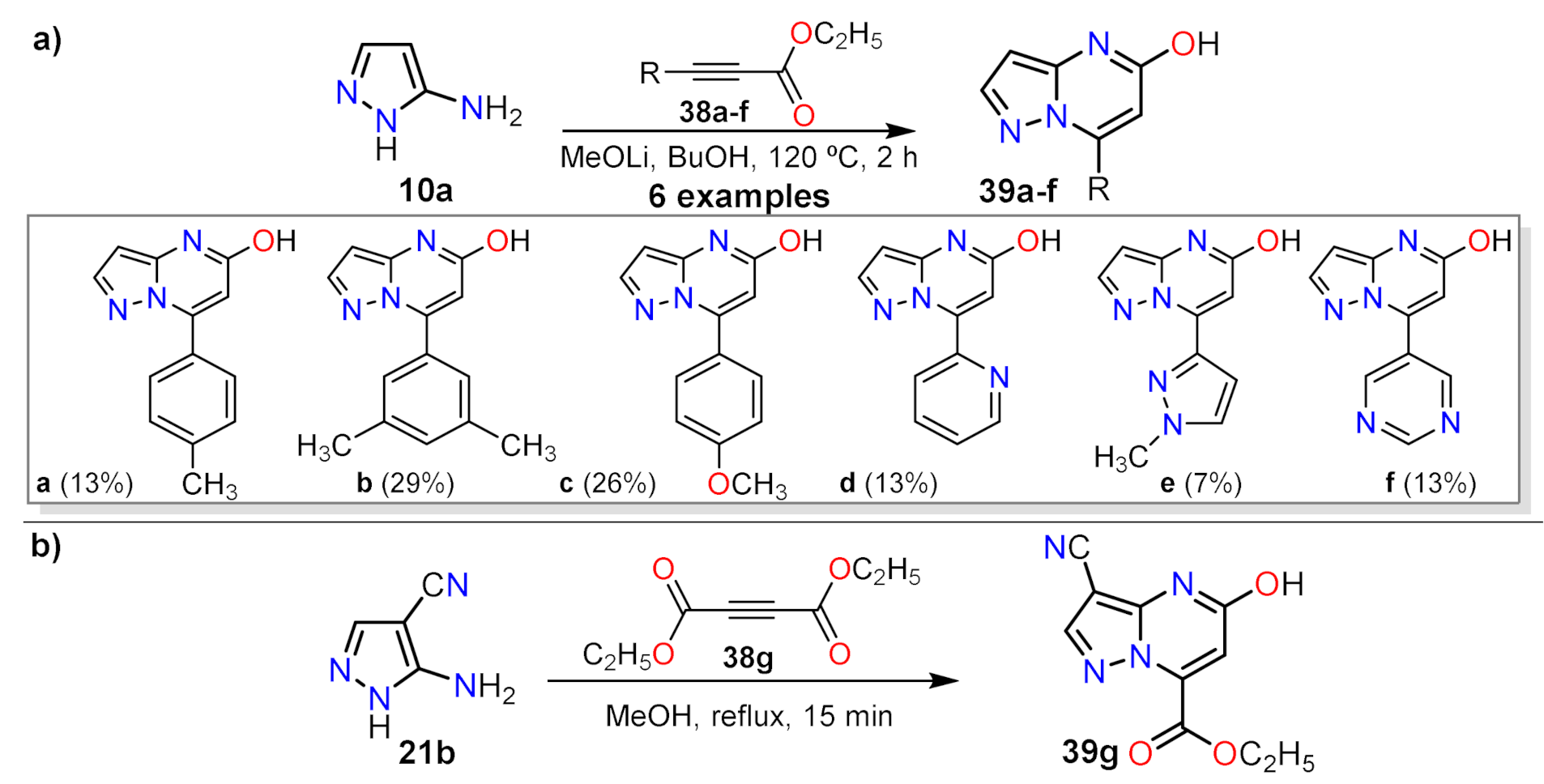
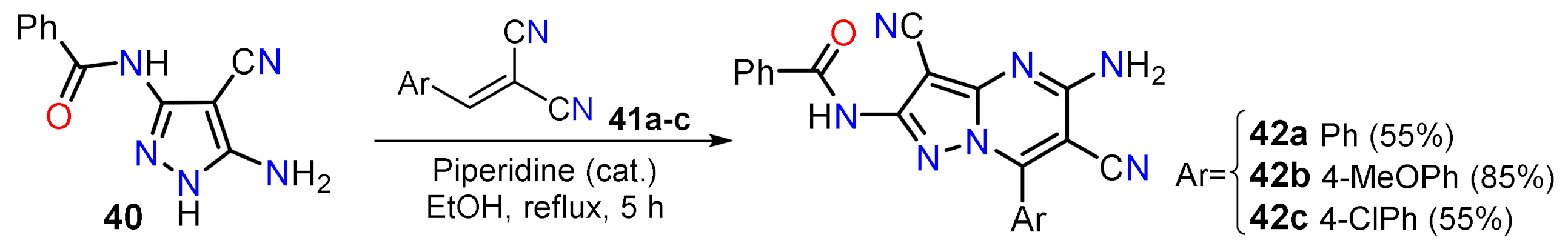
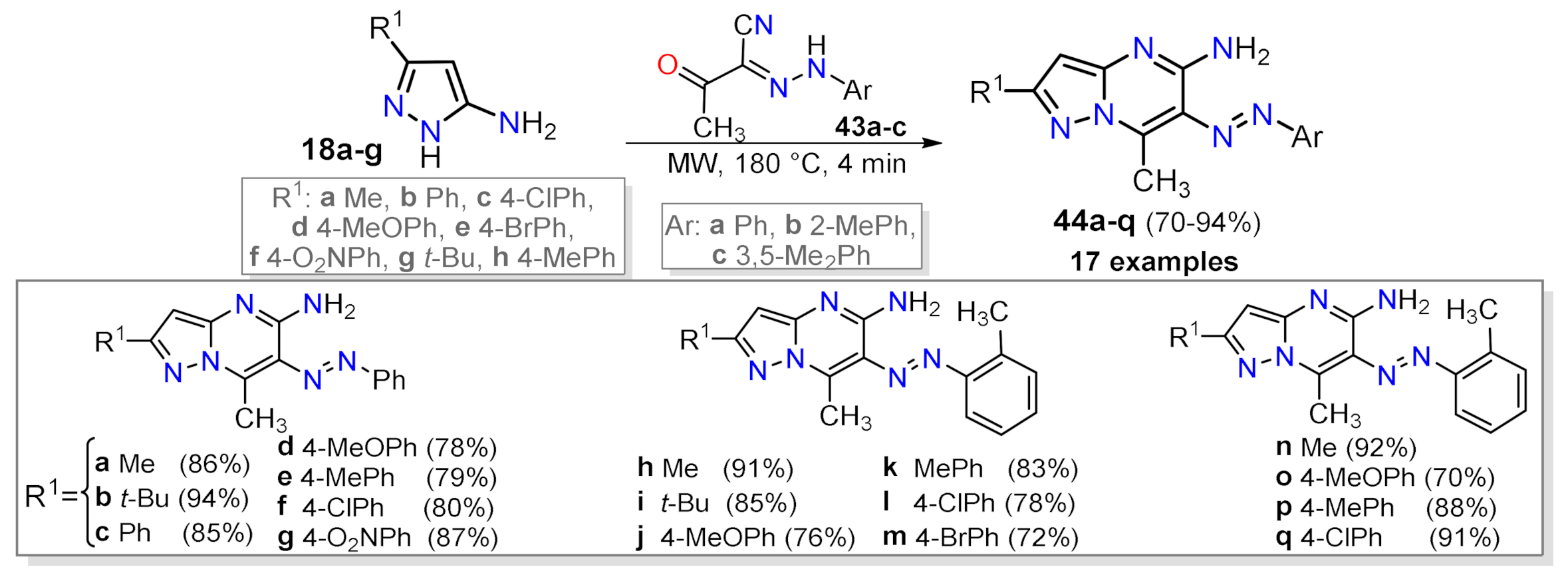
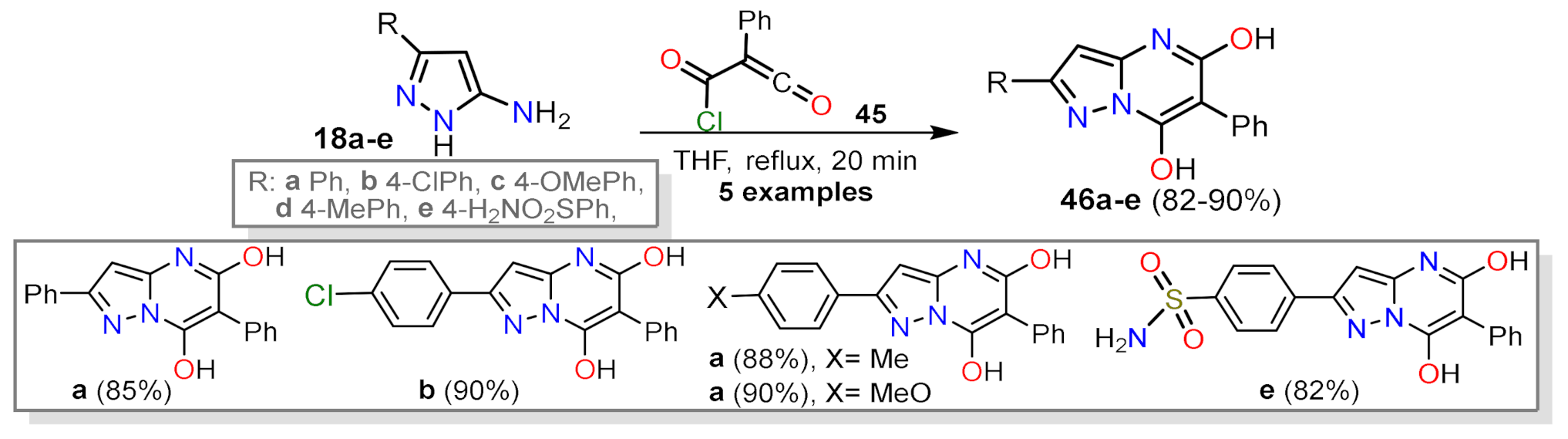
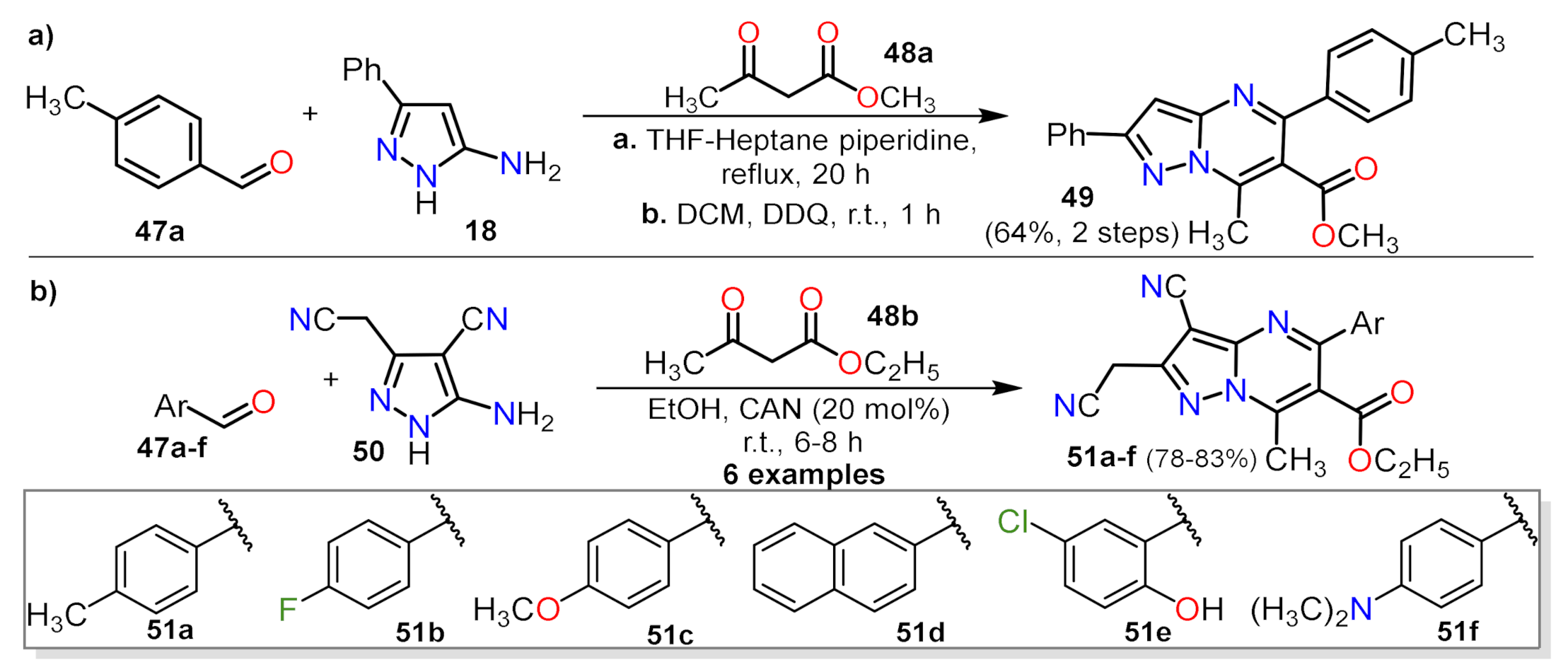
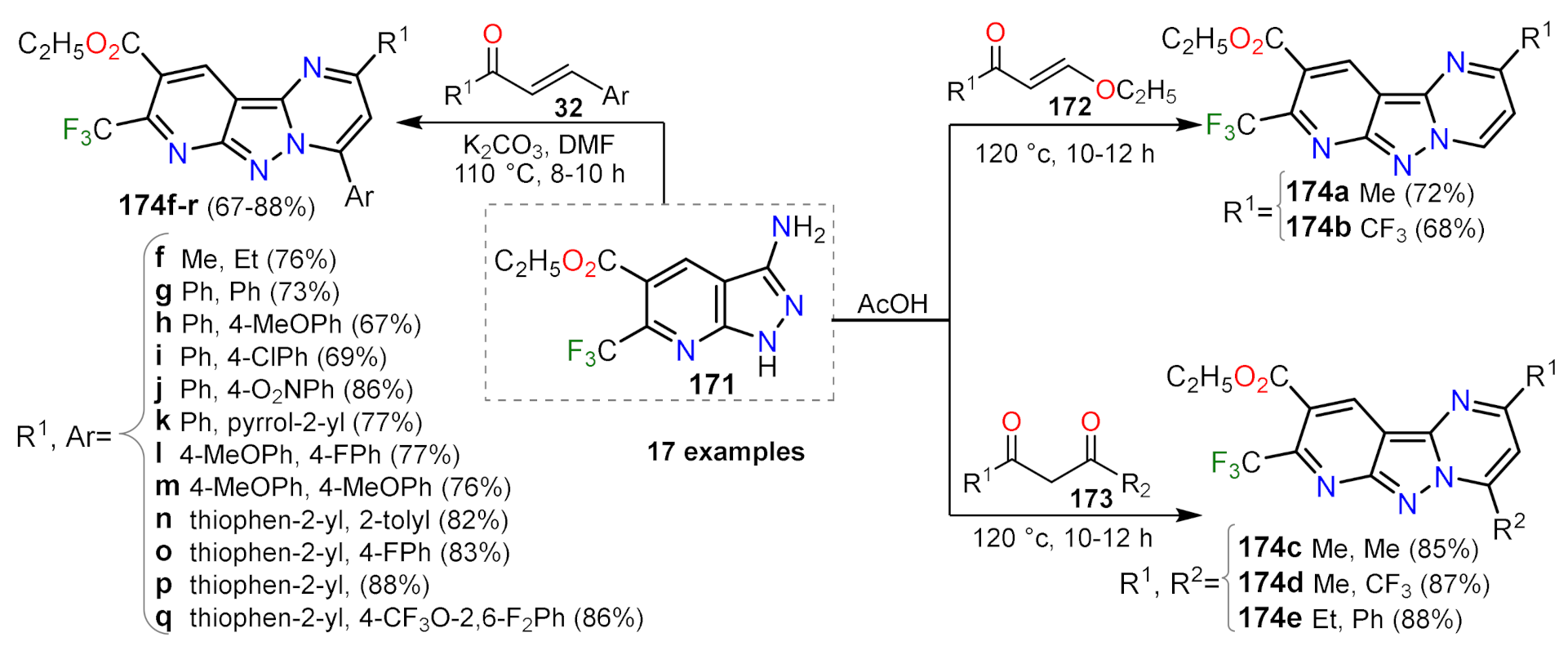
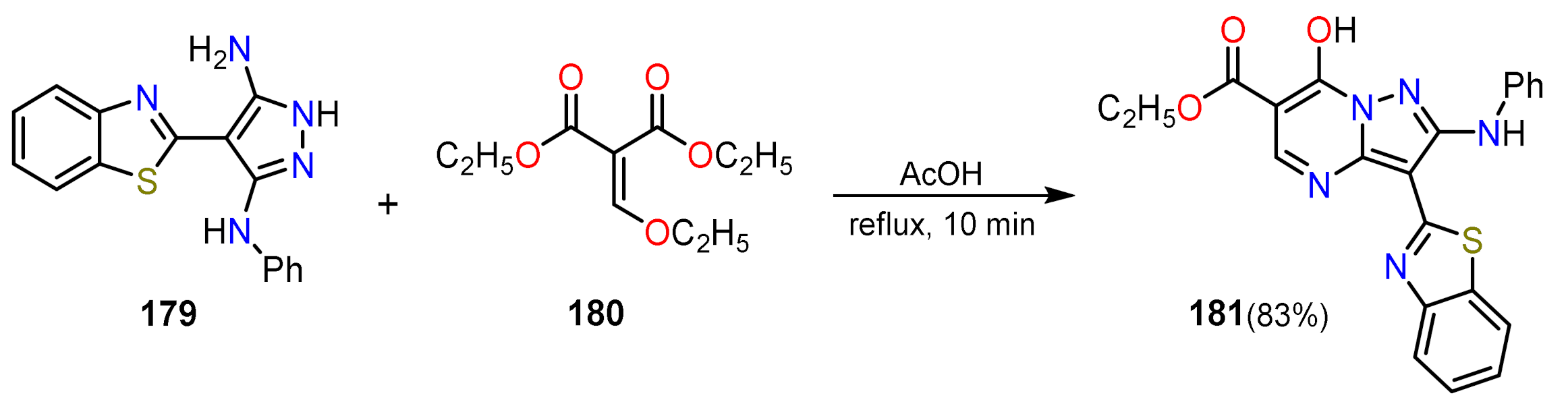
2.1.1. Biselectrophillic Systems
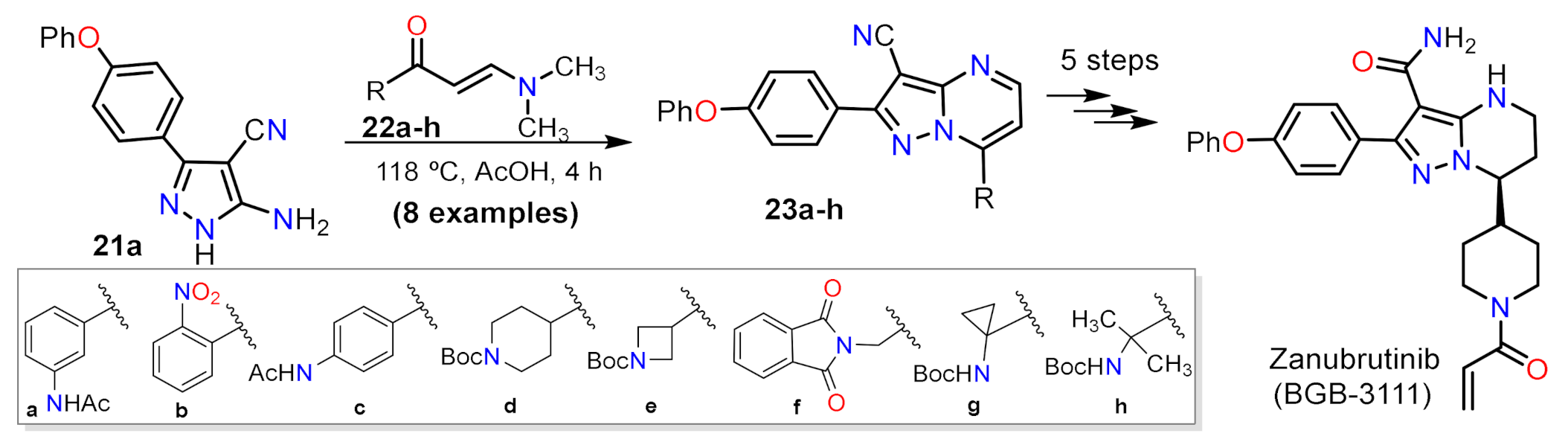
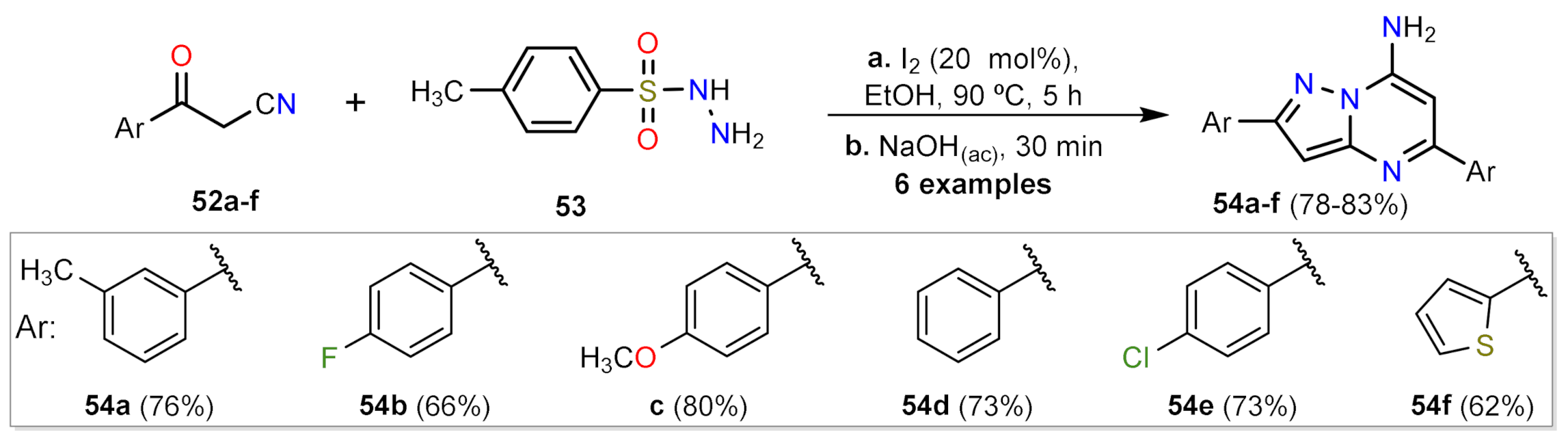
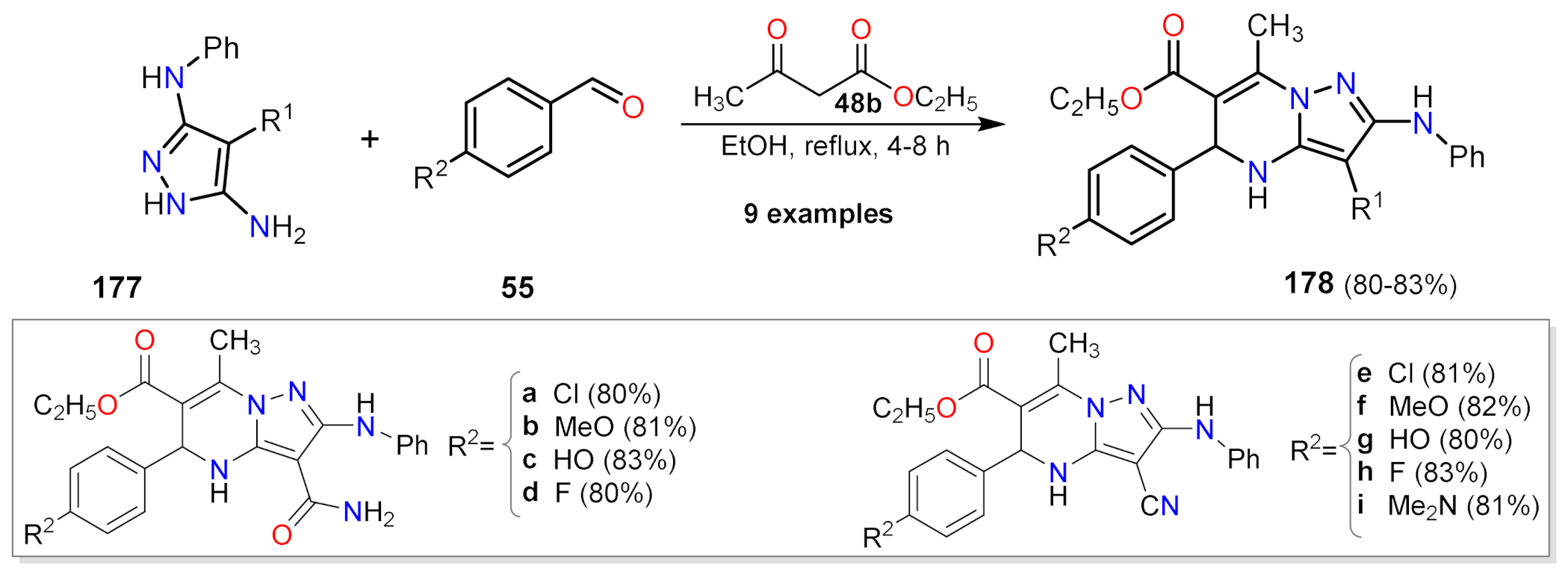
2.1.2. Multicomponent Reactions
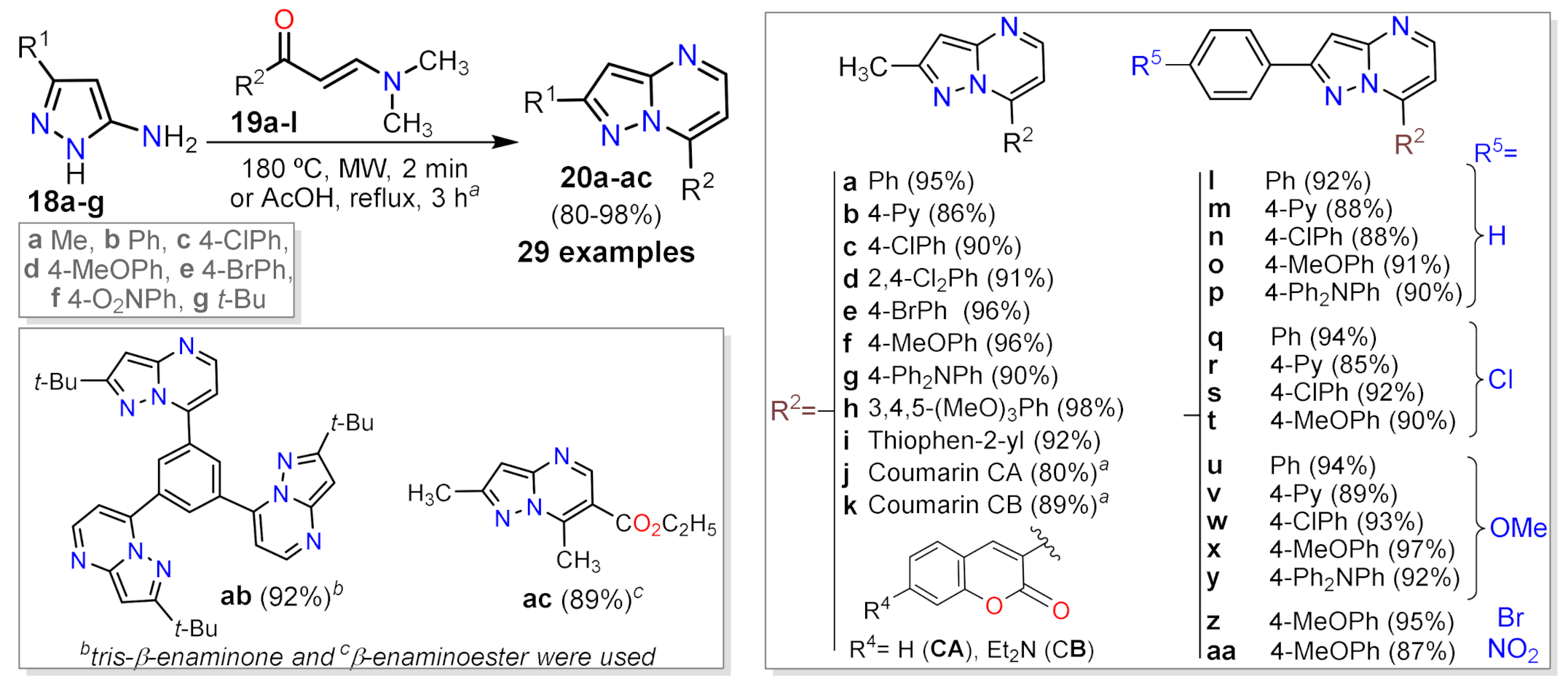
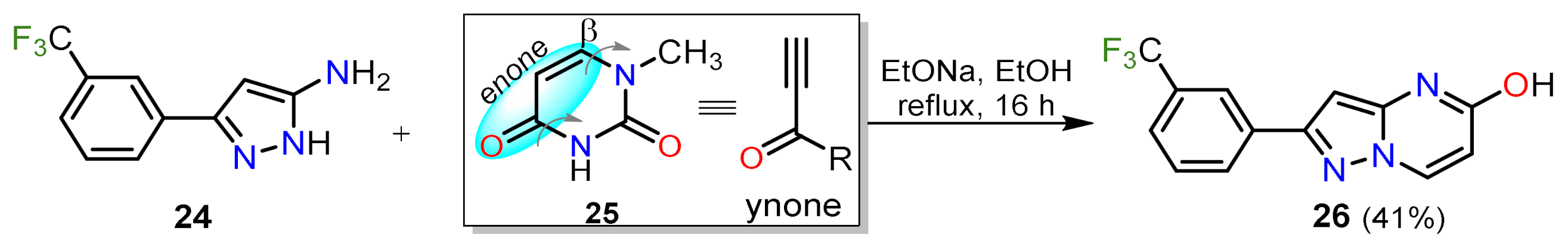
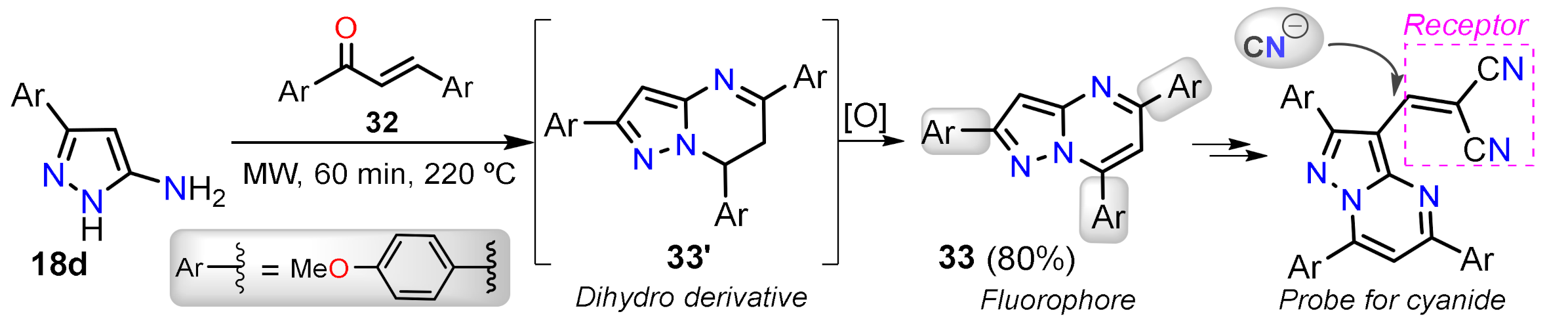
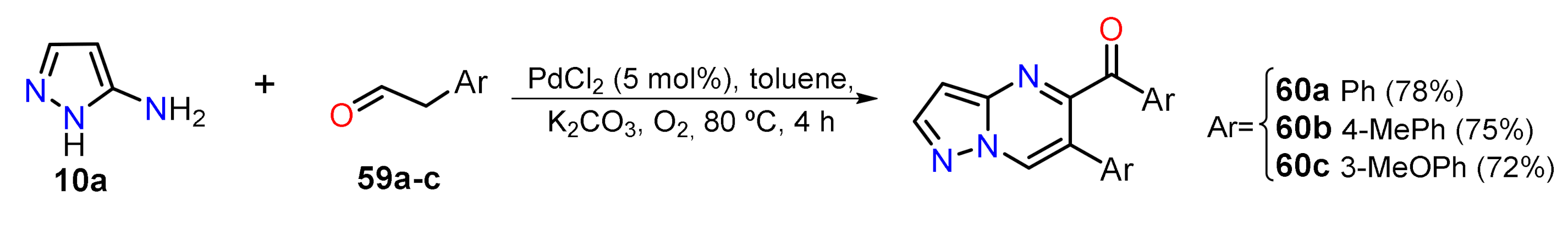
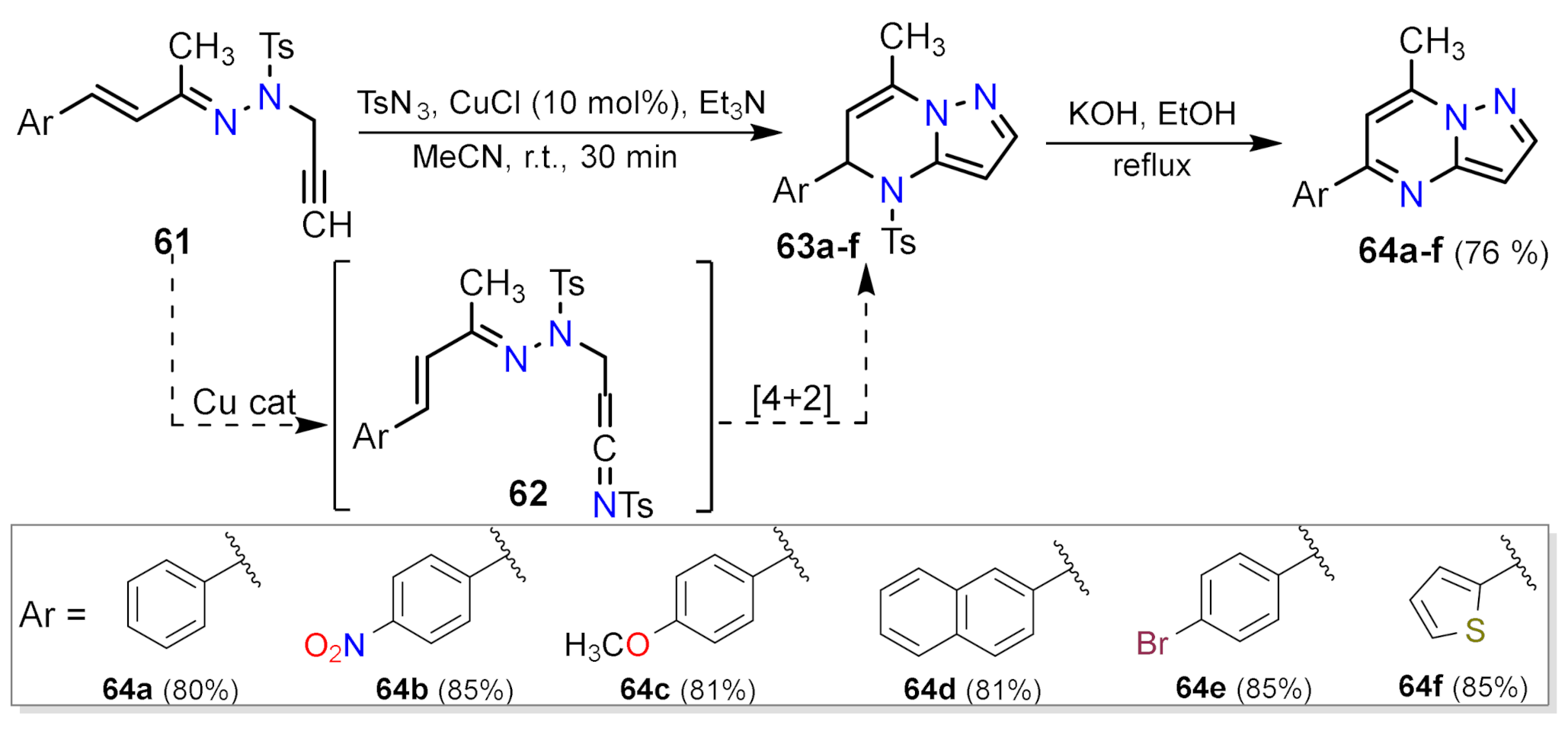
2.1.3. Synthesis by Pericyclic Reactions
2.1.4. Synthesis with Fused Cores
2.2. Functionalization
2.2.1. Metal Catalyzed Reactions
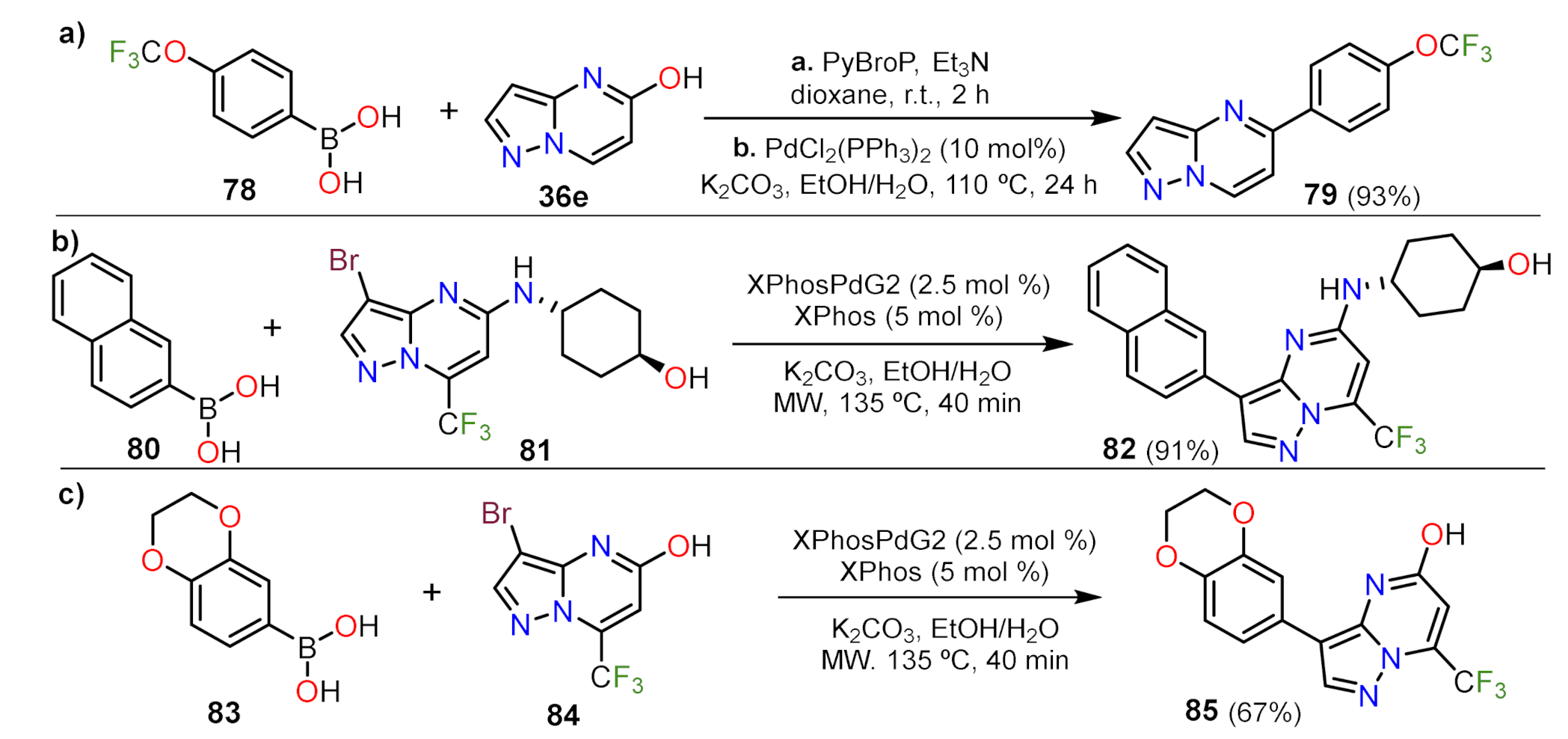
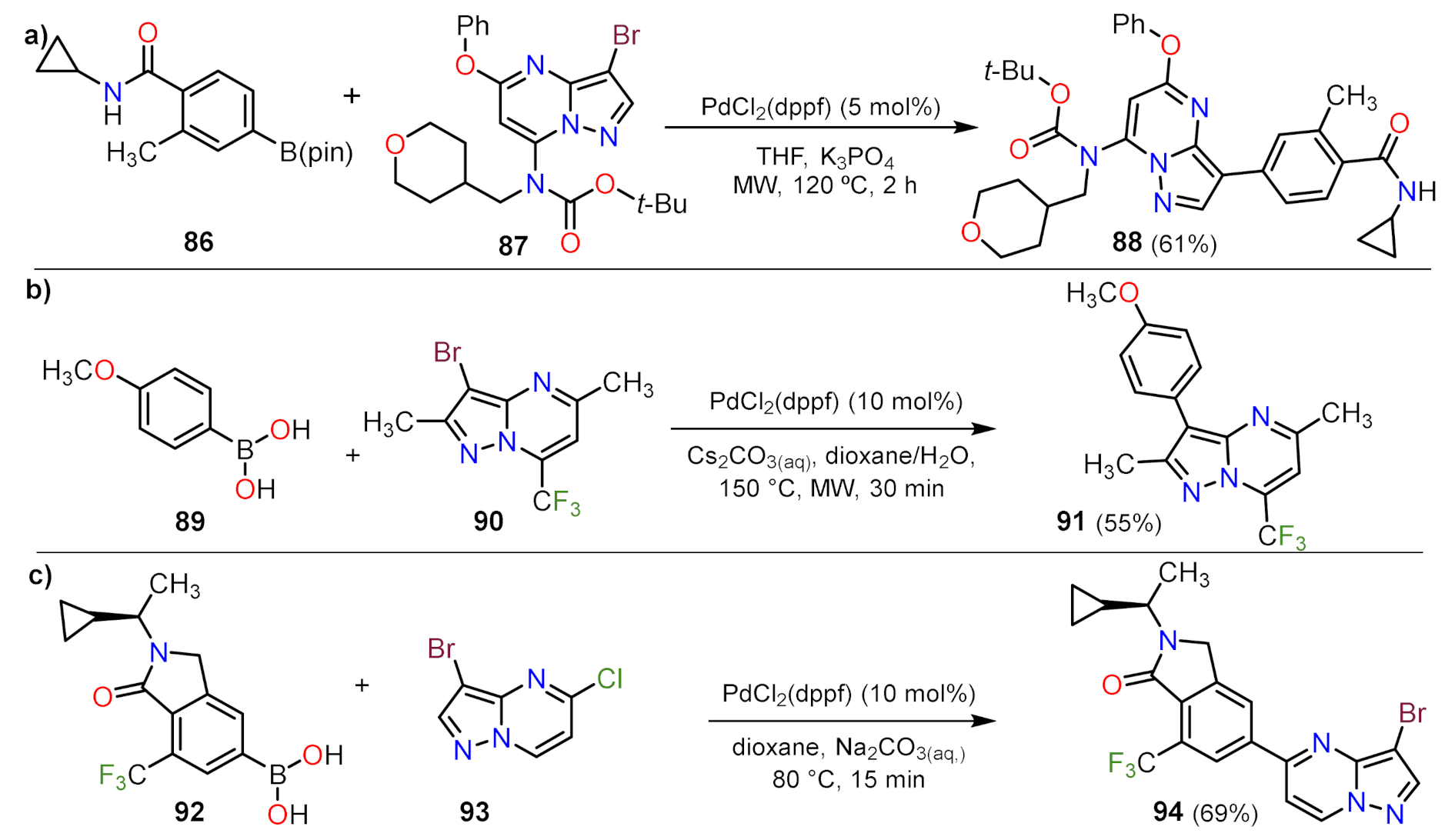
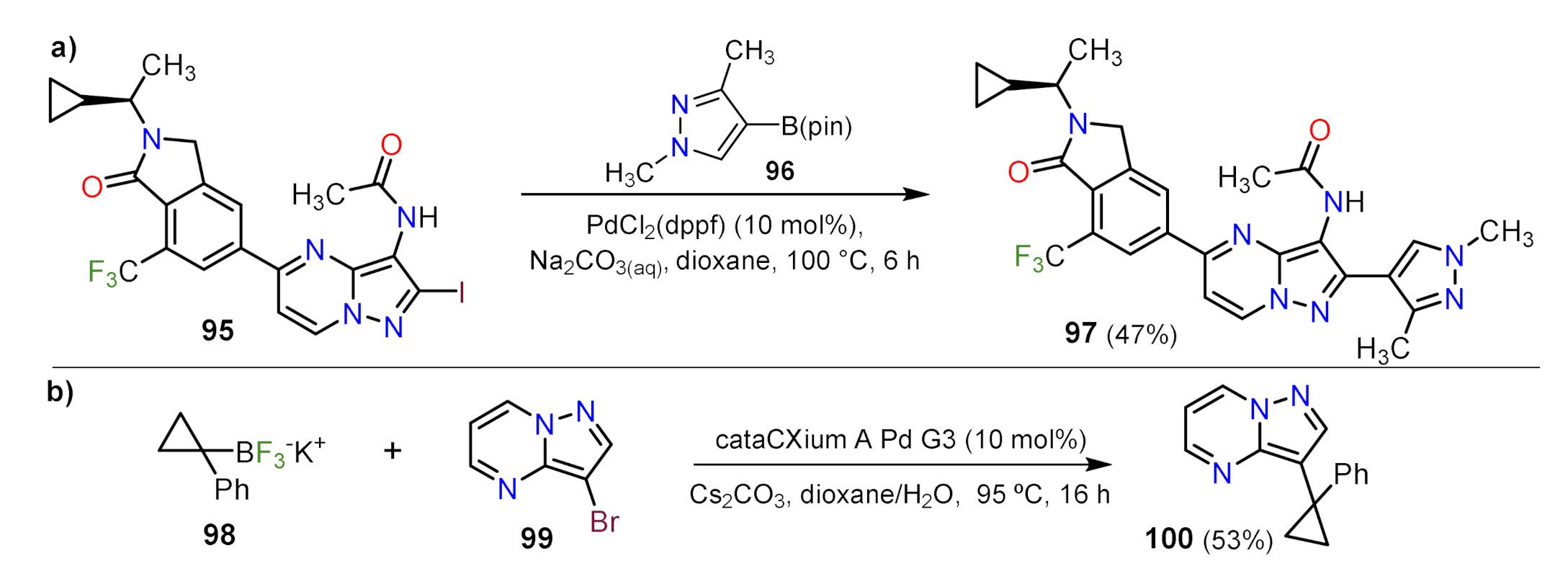
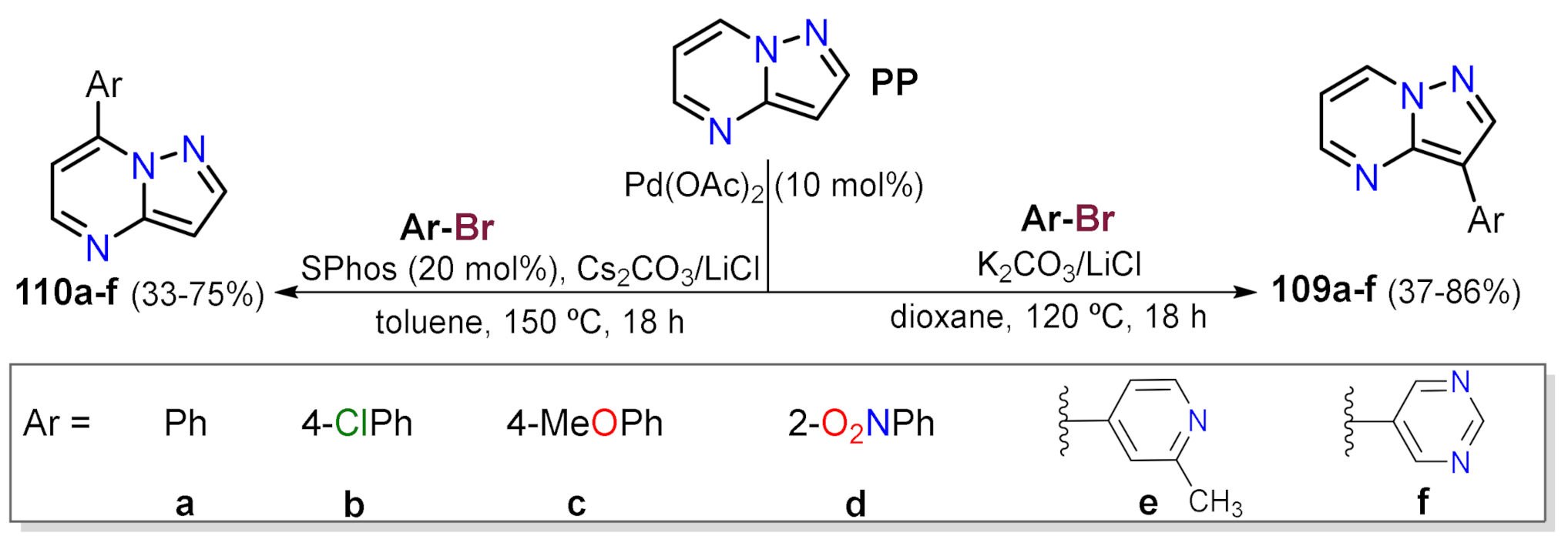
Suzuki Couplings
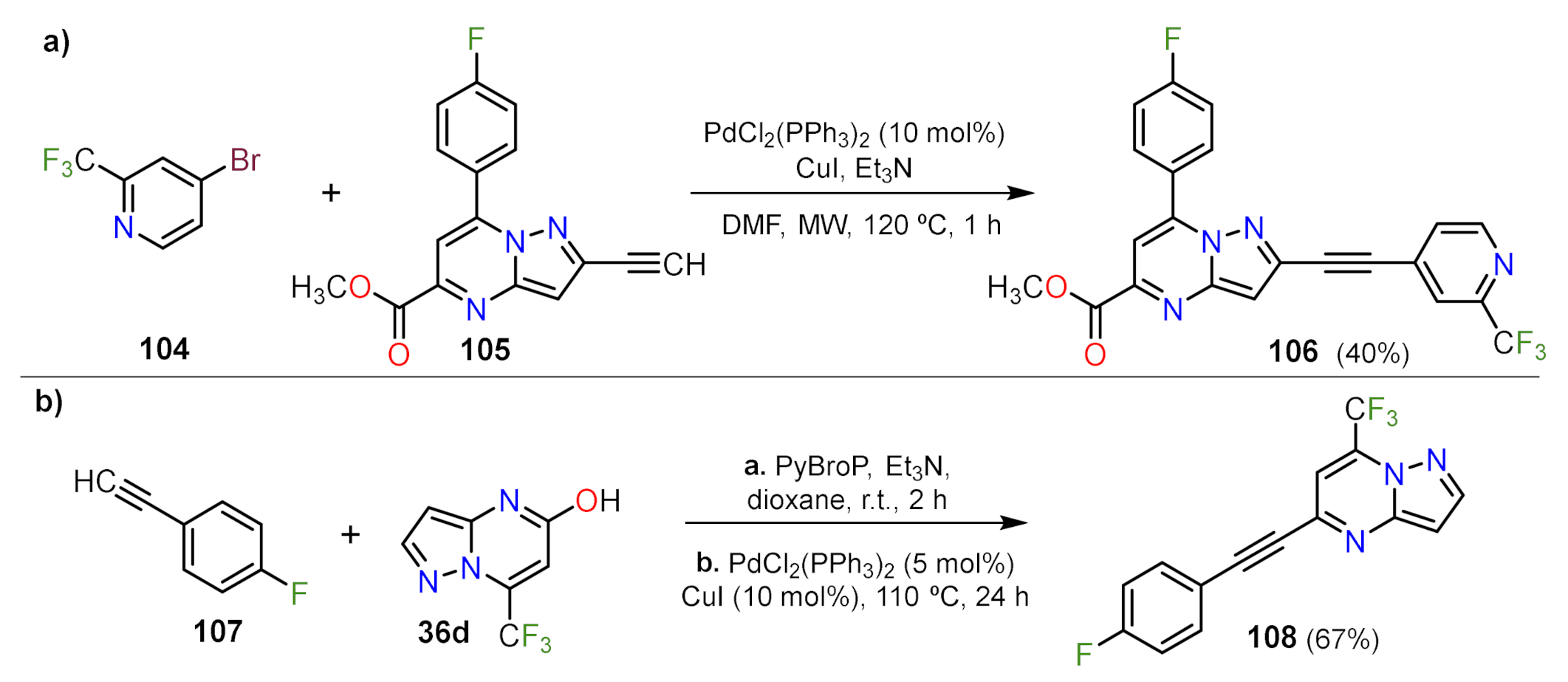
Sonogashira Couplings
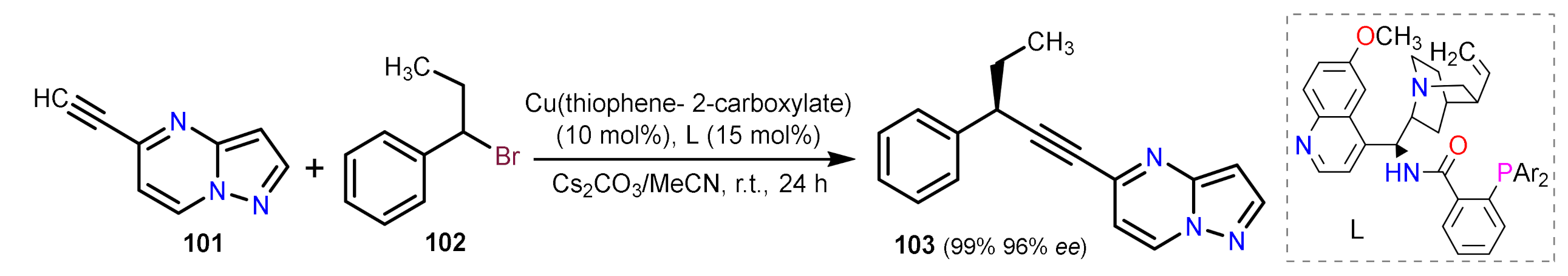
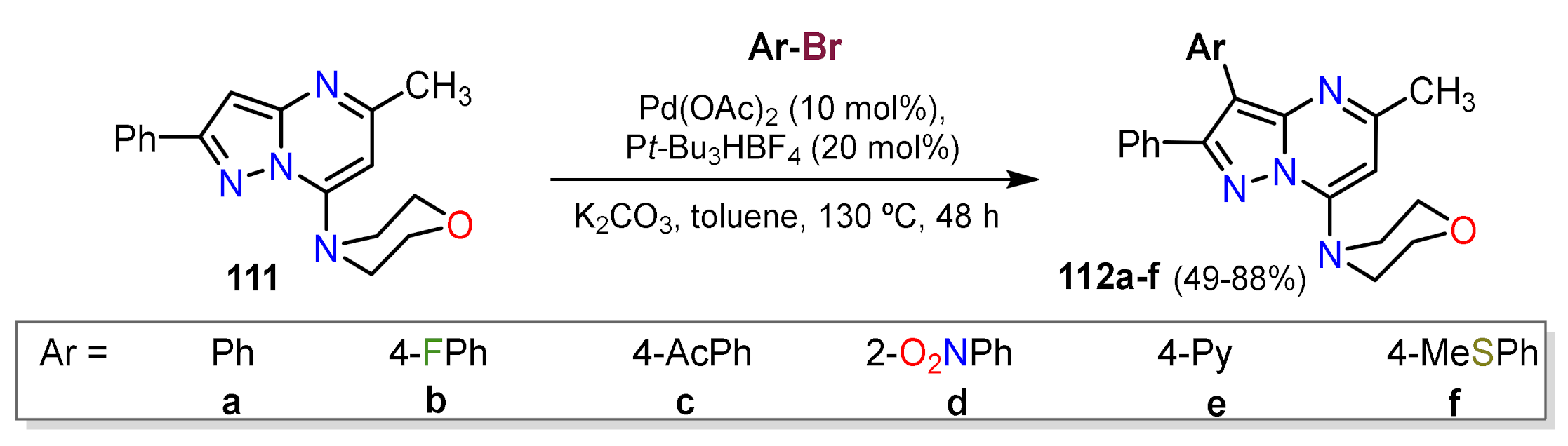
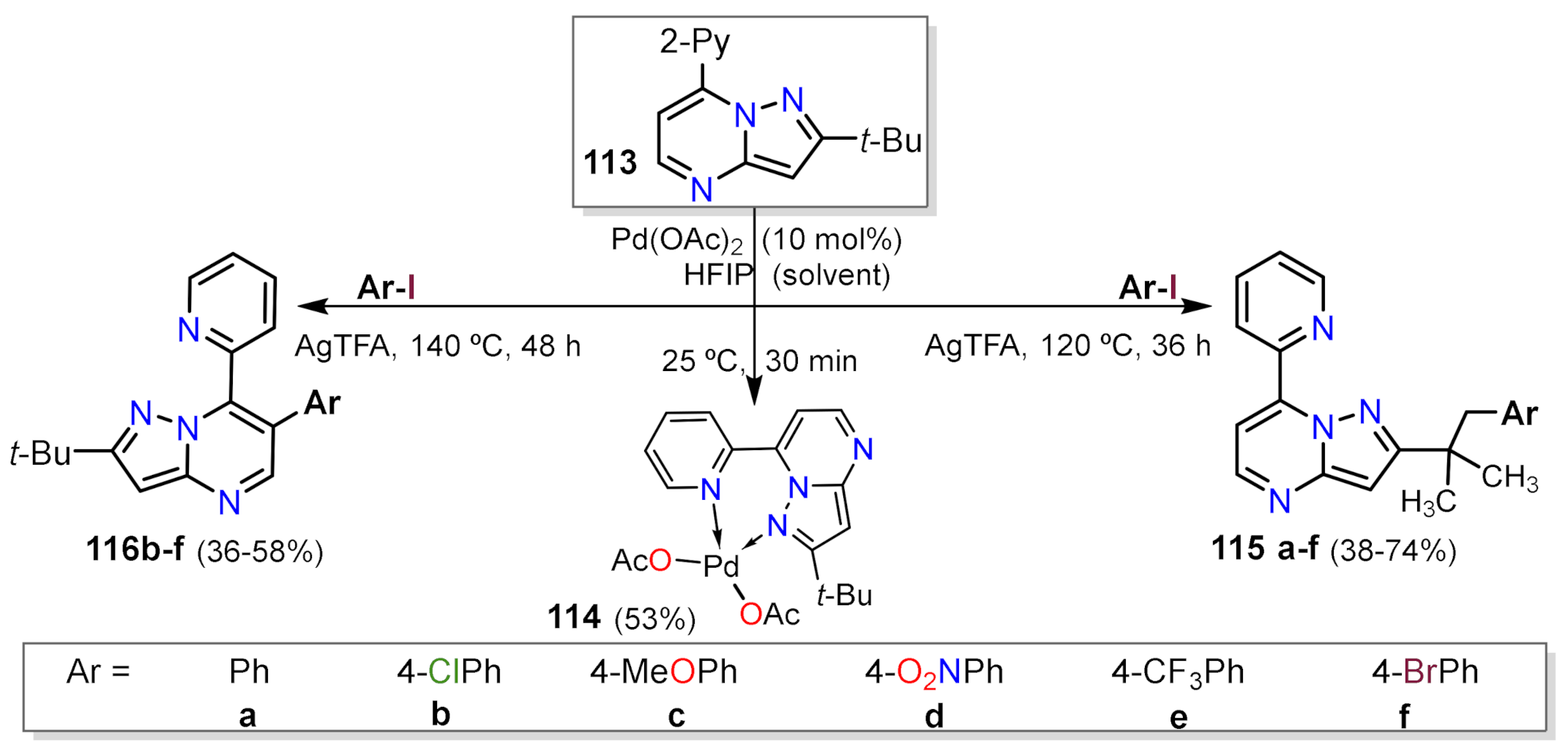
Other Metal-Catalyzed Reactions
2.2.2. Nucleophilic Aromatic Substitution Reactions
2.2.3. Other Functionalization Reactions
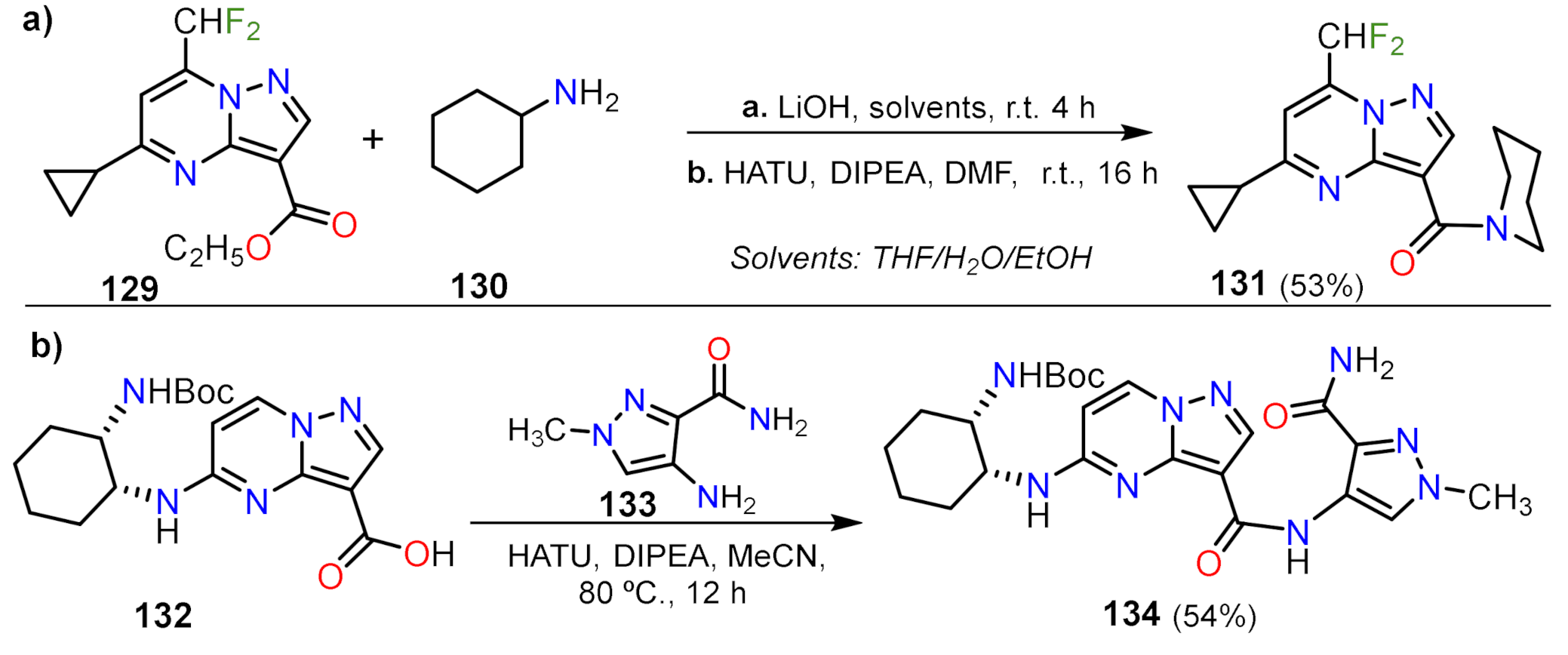
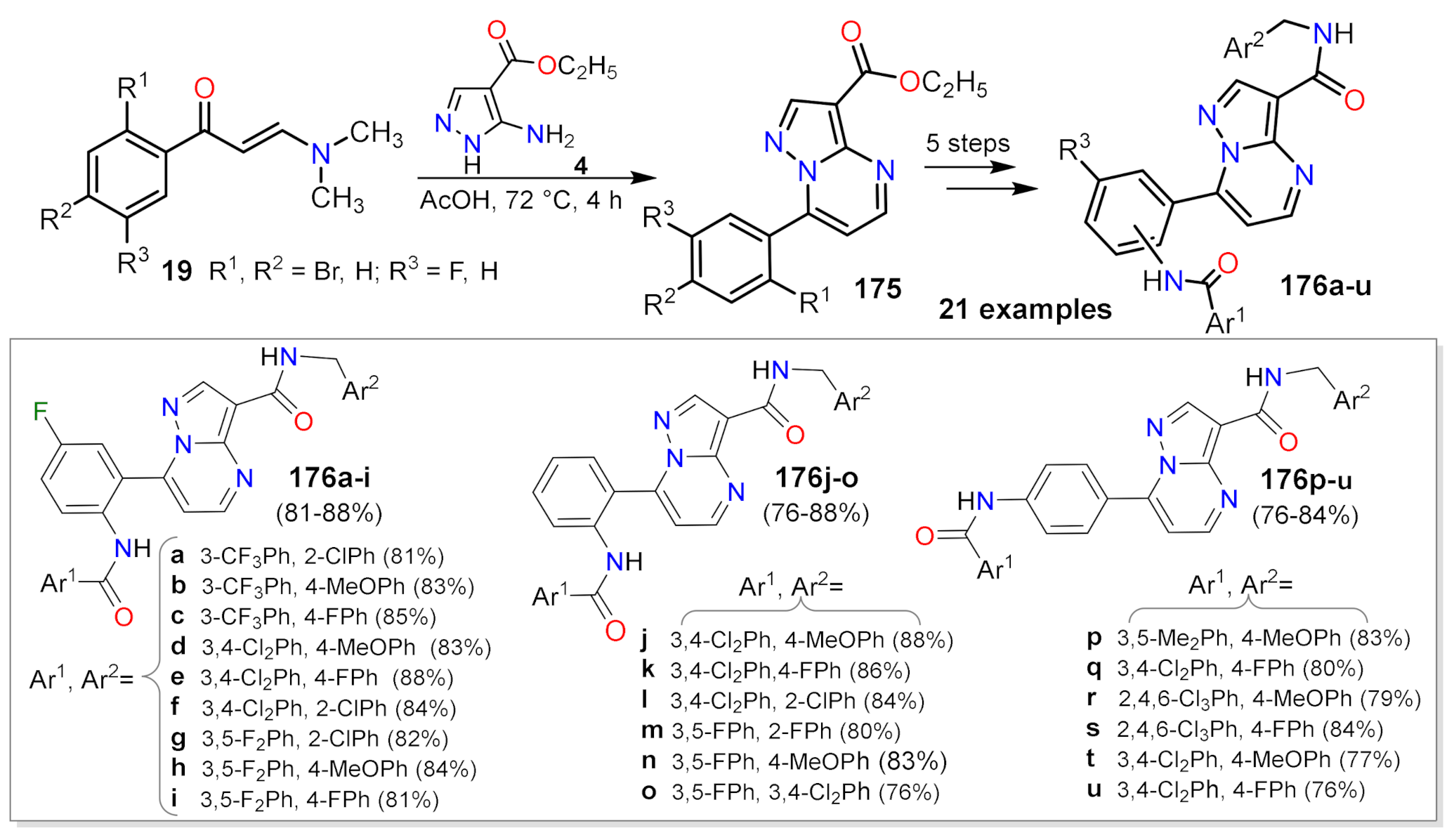
Carboxamide Synthesis
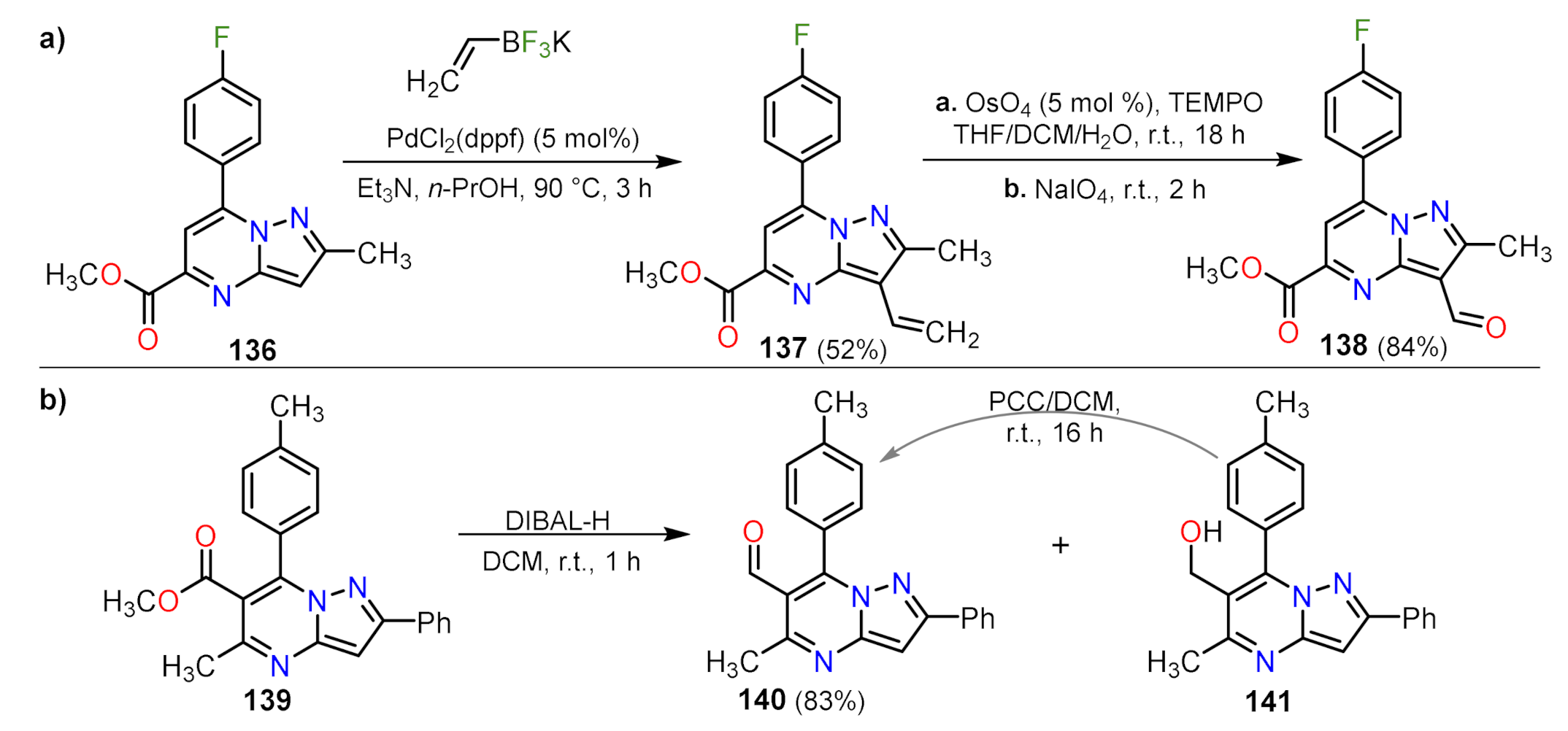
Formylation Reactions
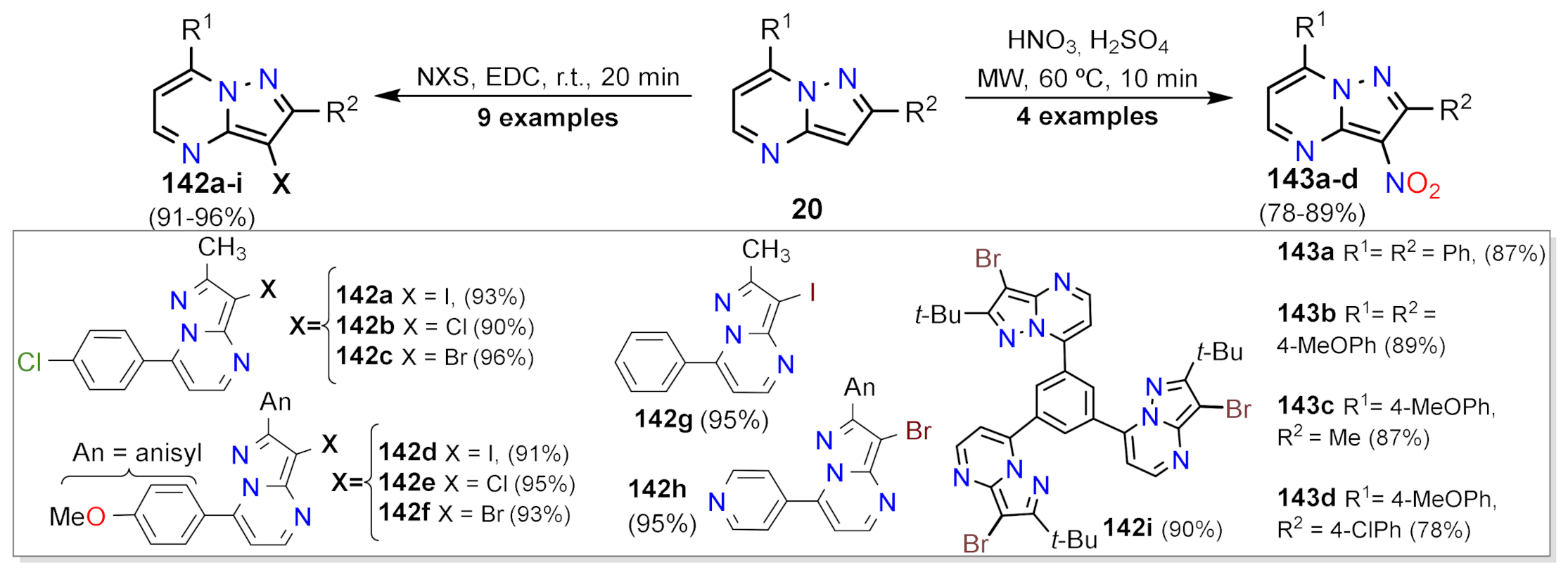
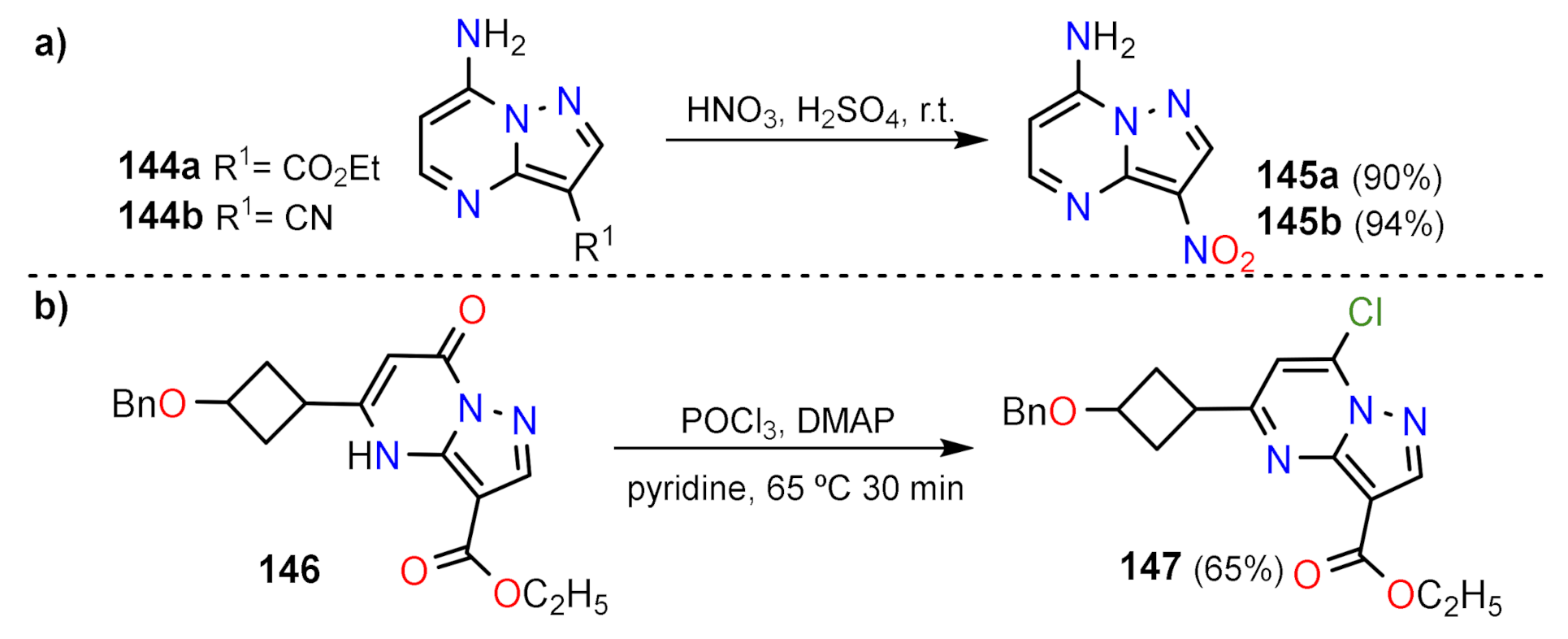
Nitration and Halogenation Reactions
Reduction Reactions
3. Antitumor Activity
Antiproliferative Activity
4. Enzymatic Inhibition
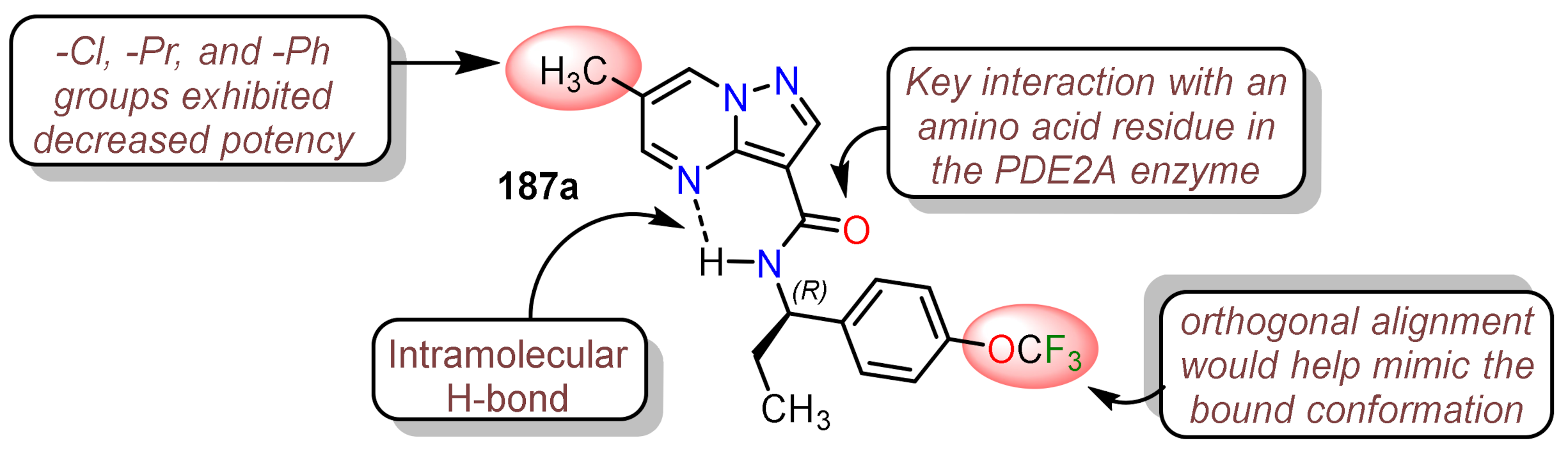
- 1.
- Pazolo[1,5-a]pyrimidine core that makes π − π and CH − π interactions with the residues in the enzyme.
- 2.
- The central amide linker generated the hydrogen bond with the more polar residues stabilizing the PDE2A enzyme, while the amide NH plays a crucial role in constraining the binding conformation via intramolecular hydrogen bonding to the pyrimidine nitrogen atom.
- 3.
- α-Branched benzylamine portion and the p-CF3O group fits into the sits in the hydrophobic cavity in an energetically favorable orthogonal orientation.
- 4.
- The substitution at position 6 combines two effects, the Van der Walls interactions with the residues and the smallest possible size.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Castillo, J.C.; Portilla, J. Recent advances in the synthesis of new pyrazole derivatives. Targets Heterocycl. Syst. 2018, 22, 194–223. [Google Scholar] [CrossRef]
- Salem, M.A.; Helal, M.H.; Gouda, M.A.; Abd EL-Gawad, H.H.; Shehab, M.A.M.; El-Khalafawy, A. Recent synthetic methodologies for pyrazolo[1,5-a]pyrimidine. Synth. Commun. 2019, 49, 1750–1776. [Google Scholar] [CrossRef]
- Al-Azmi, A. Pyrazolo[1,5-a]pyrimidines: A Close Look into their Synthesis and Applications. Curr. Org. Chem. 2019, 23, 721–743. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Karpoormath, R.; Chandrasekaran, B.; Hampannavar, G.A.; Thapliyal, N.; Palakollu, V.N. An insight on synthetic and medicinal aspects of pyrazolo[1,5-a]pyrimidine scaffold. Eur. J. Med. Chem. 2017, 126, 298–352. [Google Scholar] [CrossRef]
- Eftekhari-Sis, B.; Zirak, M. Chemistry of α-oxoesters: A powerful tool for the synthesis of heterocycles. Chem. Rev. 2015, 115, 151–264. [Google Scholar] [CrossRef] [PubMed]
- Abu Elmaati, T.M.; El-Taweel, F.M. New Trends in the Chemistry of 5-Aminopyrazoles. J. Heterocycl. Chem. 2004, 41, 109–134. [Google Scholar] [CrossRef]
- Asano, T.; Yamazaki, H.; Kasahara, C.; Kubota, H.; Kontani, T.; Harayama, Y.; Ohno, K.; Mizuhara, H.; Yokomoto, M.; Misumi, K.; et al. Identification, Synthesis, and Biological Evaluation of 6-[(6R)-2-(4-Fluorophenyl)-6-(hydroxymethyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidin-3-yl]-2-(2-methylphenyl)pyridazin-3(2H)-one (AS1940477), a Potent p38 MAP Kinase Inhibitor. J. Med. Chem. 2012, 55, 7772–7785. [Google Scholar] [CrossRef] [PubMed]
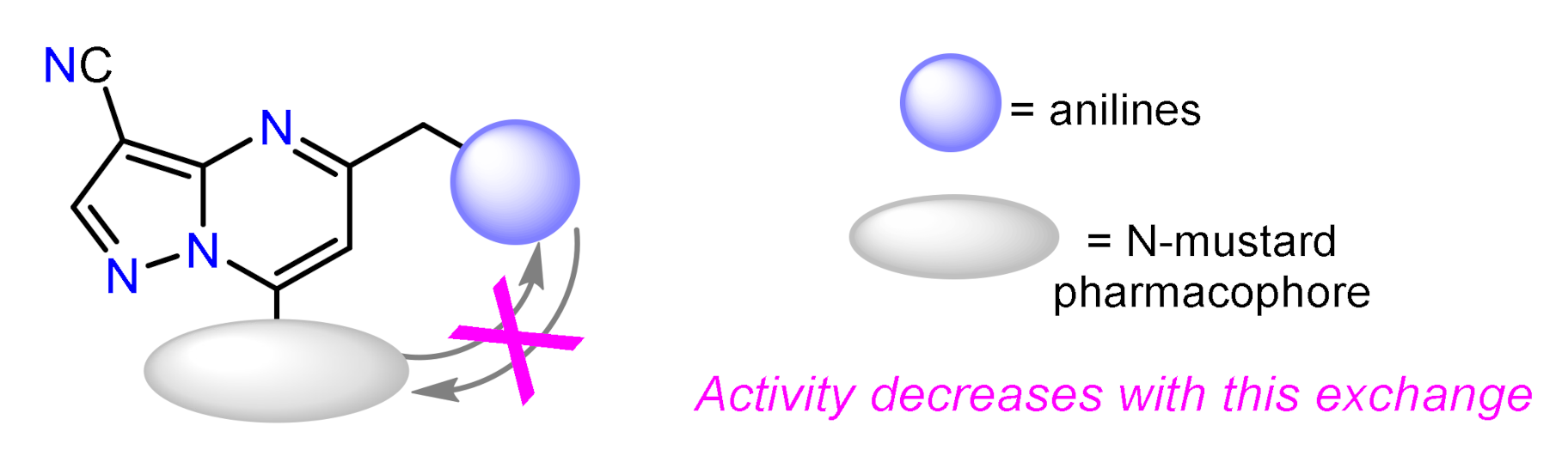
- Zhao, M.; Ren, H.; Chang, J.; Zhang, D.; Yang, Y.; He, Y.; Qi, C.; Zhang, H. Design and synthesis of novel pyrazolo[1,5-a]pyrimidine derivatives bearing nitrogen mustard moiety and evaluation of their antitumor activity in vitro and in vivo. Eur. J. Med. Chem. 2016, 119, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.J.; Attkins, N.J.; Fish, R.; van der Graaf, P.H. Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. Br. J. Pharmacol. 2011, 164, 992–1007. [Google Scholar] [CrossRef]
- Babaoglu, K.; Boojamra, C.G.; Eisenberg, E.J.; Hui, H.C.; Mackman, R.L.; Parrish, J.P.; Sangi, M.; Saunders, O.L.; Siegel, D.; Sperandio, D.; et al. Pyrazolo[1,5-a]pyrimidines as antiviral agents. Patent WO2011163518A1, 29 December 2011. [Google Scholar]
- Naidu, B.N.; Patel, M.; D’andrea, S.; Zheng, Z.B.; Connolly, T.P.; Langley, D.R.; Peese, K.; Wang, Z.; Walker, M.A.; Kadow, J.F. Inhibitors of Human Immunodeficiency Virus Replication. Patent WO2014028384A1, 20 February 2014. [Google Scholar]
- Yang, X.Z.; Sun, R.; Guo, X.; Wei, X.R.; Gao, J.; Xu, Y.J.; Ge, J.F. The application of bioactive pyrazolopyrimidine unit for the construction of fluorescent biomarkers. Dye. Pigment. 2020, 173, 107878. [Google Scholar] [CrossRef]
- Singsardar, M.; Sarkar, R.; Majhi, K.; Sinha, S.; Hajra, A. Brønsted Acidic Ionic Liquid-Catalyzed Regioselective Synthesis of Pyrazolopyrimidines and Their Photophysical Properties. ChemistrySelect 2018, 3, 1404–1410. [Google Scholar] [CrossRef]
- Golubev, P.; Karpova, E.A.; Pankova, A.S.; Sorokina, M.; Kuznetsov, M.A. Regioselective Synthesis of 7-(Trimethylsilylethynyl)pyrazolo[1,5-a]pyrimidines via Reaction of Pyrazolamines with Enynones. J. Org. Chem. 2016, 81, 11268–11275. [Google Scholar] [CrossRef] [PubMed]
- Tigreros, A.; Portilla, J. Fluorescent Pyrazole Derivatives: An Attractive Scaffold for Biological Imaging Applications. Curr. Chinese Sci. 2021, 1, 197–206. [Google Scholar] [CrossRef]
- Tigreros, A.; Portilla, J. Recent progress in chemosensors based on pyrazole derivatives. RSC Adv. 2020, 10, 19693–19712. [Google Scholar] [CrossRef]
- Tigreros, A.; Macías, M.; Portilla, J. Photophysical and crystallographic study of three integrated pyrazolo[1,5-a]pyrimidine–triphenylamine systems. Dye. Pigment. 2021, 184, 108730. [Google Scholar] [CrossRef]
- Tigreros, A.; Aranzazu, S.L.; Bravo, N.F.; Zapata-Rivera, J.; Portilla, J. Pyrazolo[1,5-a]pyrimidines-based fluorophores: A comprehensive theoretical-experimental study. RSC Adv. 2020, 10, 39542–39552. [Google Scholar] [CrossRef]
- Tigreros, A.; Castillo, J.C.; Portilla, J. Cyanide chemosensors based on 3-dicyanovinylpyrazolo[1,5-a]pyrimidines: Effects of peripheral 4-anisyl group substitution on the photophysical properties. Talanta 2020, 215, 120905. [Google Scholar] [CrossRef]
- Tigreros, A.; Rosero, H.A.; Castillo, J.C.; Portilla, J. Integrated pyrazolo[1,5-a]pyrimidine–hemicyanine system as a colorimetric and fluorometric chemosensor for cyanide recognition in water. Talanta 2019, 196, 395–401. [Google Scholar] [CrossRef]
- Castillo, J.C.; Tigreros, A.; Portilla, J. 3-Formylpyrazolo[1,5-a]pyrimidines as Key Intermediates for the Preparation of Functional Fluorophores. J. Org. Chem. 2018, 83, 10887–10897. [Google Scholar] [CrossRef] [PubMed]
- Portilla, J.; Quiroga, J.; Cobo, J.; Low, J.N.; Glidewell, C. Hydrogen-bonded chains in isostruo tural 5-methyl-2-(4-methylphenyl)-7,8-dihydro-6H-cyclopenta[g]pyrazolo[1,5-a]pyrimidine, 2-(4-chloro-phenyl)-5-methyl-7,8-dihydro-6H-cyclopenta[g]pyrazolo[1,5-a]pyrimidine and 2-(4-bromophenyl)-5-methyl-7,8-dihydro-6H-. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2005, 61, 452–456. [Google Scholar] [CrossRef]
- Portilla, J.; Quiroga, J.; Cobo, J.; Low, J.N.; Glidewell, C. 7-Amino-2,5-dimethylpyrazolo[1,5-a]pyrimidine hemihydrate redetermined at 120 K: A three-dimensional hydrogen-bonded framework. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2006, 62, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Regan, A.C. 11.12- Bicyclic 5-6 Systems with One Bridgehead (Ring Junction) Nitrogen Atom: Two Extra Heteroatoms 1:1. In Comprehensive Heterocyclic Chemistry III; Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; pp. 551–587. ISBN 978-0-08-044992-0. [Google Scholar]
- Candeias, N.R.; Branco, L.C.; Gois, P.M.P.; Afonso, C.A.M.; Trindade, A.F. More sustainable approaches for the synthesis of n-based heterocycles. Chem. Rev. 2009, 109, 2703–2802. [Google Scholar] [CrossRef]
- Bondock, S.; Alqahtani, S.; Fouda, A.M. Synthesis and anticancer evaluation of some new pyrazolo[3,4-d][1,2,3]triazin-4-ones, pyrazolo[1,5-a]pyrimidines, and imidazo[1,2-b]pyrazoles clubbed with carbazole. J. Heterocycl. Chem. 2021, 58, 56–73. [Google Scholar] [CrossRef]
- Hebishy, A.M.S.; Salama, H.T.; Elgemeie, G.H. New Route to the Synthesis of Benzamide-Based 5-Aminopyrazoles and Their Fused Heterocycles Showing Remarkable Antiavian Influenza Virus Activity. ACS Omega 2020, 5, 25104–25112. [Google Scholar] [CrossRef]
- Metwally, N.H.; Mohamed, M.S.; Ragb, E.A. Design, synthesis, anticancer evaluation, molecular docking and cell cycle analysis of 3-methyl-4,7-dihydropyrazolo[1,5-a]pyrimidine derivatives as potent histone lysine demethylases (KDM) inhibitors and apoptosis inducers. Bioorg. Chem. 2019, 88, 102929. [Google Scholar] [CrossRef]
- Yamaguchi-Sasaki, T.; Tokura, S.; Ogata, Y.; Kawaguchi, T.; Sugaya, Y.; Takahashi, R.; Iwakiri, K.; Abe-Kumasaka, T.; Yoshida, I.; Arikawa, K.; et al. Discovery of a Potent Dual Inhibitor of Wild-Type and Mutant Respiratory Syncytial Virus Fusion Proteins. ACS Med. Chem. Lett. 2020, 11, 1145–1151. [Google Scholar] [CrossRef]
- Yamagami, T.; Kobayashi, R.; Moriyama, N.; Horiuchi, H.; Toyofuku, E.; Kadoh, Y.; Kawanishi, E.; Izumoto, S.; Hiramatsu, H.; Nanjo, T.; et al. Scalable Process Design for a PDE10A Inhibitor Consisting of Pyrazolopyrimidine and Quinoxaline as Key Units. Org. Process Res. Dev. 2019, 23, 578–587. [Google Scholar] [CrossRef]
- Manetti, F.; Stecca, B.; Santini, R.; Maresca, L.; Giannini, G.; Taddei, M.; Petricci, E. Pharmacophore-Based Virtual Screening for Identification of Negative Modulators of GLI1 as Potential Anticancer Agents. ACS Med. Chem. Lett. 2020, 11, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Abe, M.; Seto, M.; Gogliotti, R.G.; Loch, M.T.; Bollinger, K.A.; Chang, S.; Engelberg, E.M.; Luscombe, V.B.; Harp, J.M.; Bubser, M.; et al. Discovery of VU6005649, a CNS Penetrant mGlu7/8 Receptor PAM Derived from a Series of Pyrazolo[1,5-a]pyrimidines. ACS Med. Chem. Lett. 2017, 8, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Hylsová, M.; Carbain, B.; Fanfrlík, J.; Musilová, L.; Haldar, S.; Köprülüoğlu, C.; Ajani, H.; Brahmkshatriya, P.S.; Jorda, R.; Kryštof, V.; et al. Explicit treatment of active-site waters enhances quantum mechanical/implicit solvent scoring: Inhibition of CDK2 by new pyrazolo[1,5-a]pyrimidines. Eur. J. Med. Chem. 2017, 126, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Silveira, F.F.; de Souza, J.O.; Hoelz, L.V.B.; Campos, V.R.; Jabor, V.A.P.; Aguiar, A.C.C.; Nonato, M.C.; Albuquerque, M.G.; Guido, R.V.C.; Boechat, N.; et al. Comparative study between the anti-P. falciparum activity of triazolopyrimidine, pyrazolopyrimidine and quinoline derivatives and the identification of new PfDHODH inhibitors. Eur. J. Med. Chem. 2021, 209. [Google Scholar] [CrossRef]
- Childress, E.S.; Wieting, J.M.; Felts, A.S.; Breiner, M.M.; Long, M.F.; Luscombe, V.B.; Rodriguez, A.L.; Cho, H.P.; Blobaum, A.L.; Niswender, C.M.; et al. Discovery of Novel Central Nervous System Penetrant Metabotropic Glutamate Receptor Subtype 2 (mGlu 2 ) Negative Allosteric Modulators (NAMs) Based on Functionalized Pyrazolo[1,5-a]pyrimidine-5-carboxamide and Thieno[3,2-b]pyridine-5-carboxamide Cores. J. Med. Chem. 2019, 62, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Deng, X.; Sun, R.; Tavallaie, M.S.; Wang, J.; Cai, Q.; Lam, C.; Lei, S.; Fu, L.; Jiang, F. Structural optimization of pyrazolo[1,5-a]pyrimidine derivatives as potent and highly selective DPP-4 inhibitors. Eur. J. Med. Chem. 2020, 208, 112850. [Google Scholar] [CrossRef] [PubMed]
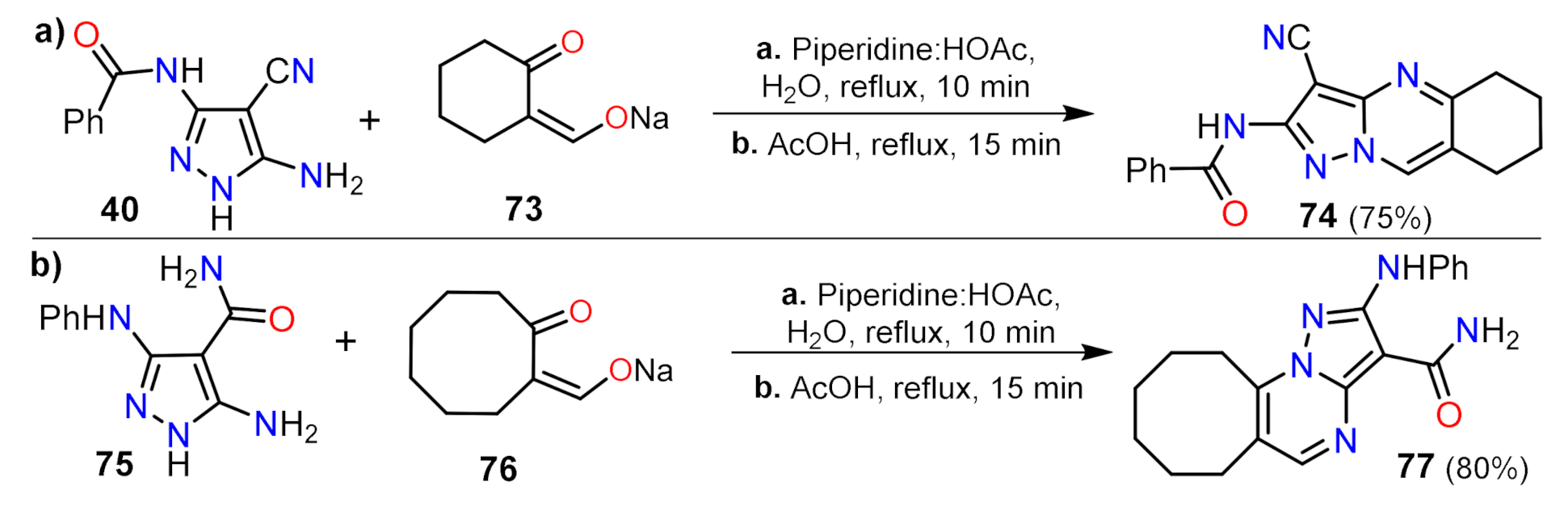
- Portilla, J.; Quiroga, J.; Nogueras, M.; Cobo, J. Regioselective synthesis of fused pyrazolo[1,5-a]pyrimidines by reaction of 5-amino-1H-pyrazoles and β-dicarbonyl compounds containing five-membered rings. Tetrahedron 2012, 68, 988–994. [Google Scholar] [CrossRef]
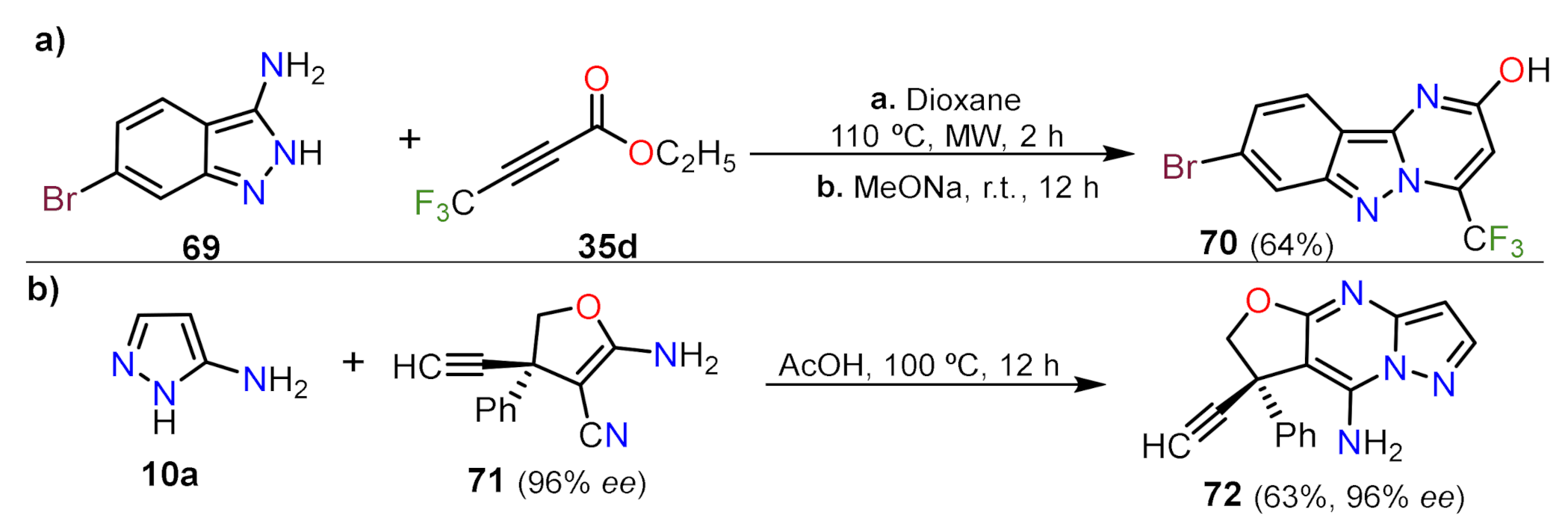
- Castillo, J.C.; Rosero, H.A.; Portilla, J. Simple access toward 3-halo- and 3-nitro-pyrazolo[1,5-a] pyrimidines through a one-pot sequence. RSC Adv. 2017, 7, 28483–28488. [Google Scholar] [CrossRef]
- Guo, Y.; Liu, Y.; Hu, N.; Yu, D.; Zhou, C.; Shi, G.; Zhang, B.; Wei, M.; Liu, J.; Luo, L.; et al. Discovery of Zanubrutinib (BGB-3111), a Novel, Potent, and Selective Covalent Inhibitor of Bruton’s Tyrosine Kinase. J. Med. Chem. 2019, 62, 7923–7940. [Google Scholar] [CrossRef] [PubMed]
- Attia, M.H.; Elrazaz, E.Z.; El-Emam, S.Z.; Taher, A.T.; Abdel-Aziz, H.A.; Abouzid, K.A.M. Synthesis and in-vitro anti-proliferative evaluation of some pyrazolo[1,5-a]pyrimidines as novel larotrectinib analogs. Bioorg. Chem. 2020, 94, 103458. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, Y.; Li, L.; Ren, Y.; Deng, X.; Liu, J.; Wang, W.; Luo, M.; Liu, S.; Chen, J. Design, synthesis, and bioevaluation of pyrazolo[1,5-a]pyrimidine derivatives as tubulin polymerization inhibitors targeting the colchicine binding site with potent anticancer activities. Eur. J. Med. Chem. 2020, 202, 112519. [Google Scholar] [CrossRef]
- Ajeesh Kumar, A.K.; Bodke, Y.D.; Lakra, P.S.; Sambasivam, G.; Bhat, K.G. Design, synthesis and anti-cancer evaluation of a novel series of pyrazolo [1, 5-a] pyrimidine substituted diamide derivatives. Med. Chem. Res. 2017, 26, 714–744. [Google Scholar] [CrossRef]
- Xu, Y.; Brenning, B.G.; Kultgen, S.G.; Foulks, J.M.; Clifford, A.; Lai, S.; Chan, A.; Merx, S.; McCullar, M.V.; Kanner, S.B.; et al. Synthesis and Biological Evaluation of Pyrazolo[1,5-a ]pyrimidine Compounds as Potent and Selective Pim-1 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 63–67. [Google Scholar] [CrossRef]
- Peytam, F.; Adib, M.; Shourgeshty, R.; Firoozpour, L.; Rahmanian-Jazi, M.; Jahani, M.; Moghimi, S.; Divsalar, K.; Faramarzi, M.A.; Mojtabavi, S.; et al. An efficient and targeted synthetic approach towards new highly substituted 6-amino-pyrazolo[1,5-a]pyrimidines with α-glucosidase inhibitory activity. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Xing, S.; Zheng, K.; Sadeghzadeh, S.M. Synthesis of pyrazolopyrimidines in mild conditions by gold nanoparticles supported on magnetic ionic gelation in aqueous solution. Appl. Organomet. Chem. 2020, 34, 1–9. [Google Scholar] [CrossRef]
- Quiroga, J.; Trilleras, J.; Insuasty, B.; Abonía, R.; Nogueras, M.; Cobo, J. Regioselective formylation of pyrazolo[3,4-b]pyridine and pyrazolo[1,5-a]pyrimidine systems using Vilsmeier-Haack conditions. Tetrahedron Lett. 2008, 49, 2689–2691. [Google Scholar] [CrossRef]
- Jismy, B.; Tikad, A.; Akssira, M.; Guillaumet, G. Efficient Access to 3,5-Disubstituted 7-(Trifluoromethyl)pyrazolo[1,5-a]pyrimidines Involving SNAr and Suzuki Cross-Coupling Reactions. Molecules 2020, 25, 2062. [Google Scholar] [CrossRef] [PubMed]
- Jismy, B.; Guillaumet, G.; Allouchi, H.; Akssira, M.; Abarbri, M. Concise and Efficient Access to 5,7-Disubstituted Pyrazolo[1,5-a]pyrimidines by Pd-Catalyzed Sequential Arylation, Alkynylation and SNAr Reaction. Eur. J. Org. Chem. 2017, 2017, 6168–6178. [Google Scholar] [CrossRef]
- Jismy, B.; Allouchi, H.; Guillaumet, G.; Akssira, M.; Abarbri, M. An Efficient Synthesis of New 7-Trifluoromethyl-2,5-disubstituted Pyrazolo[1,5-a]pyrimidines. Synth. 2018, 50, 1675–1686. [Google Scholar] [CrossRef]
- Schmitt, D.C.; Niljianskul, N.; Sach, N.W.; Trujillo, J.I. A Parallel Approach to 7-(Hetero)arylpyrazolo[1,5- a]pyrimidin-5-ones. ACS Comb. Sci. 2018, 20, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Akrami, S.; Karami, B.; Farahi, M. A novel protocol for catalyst-free synthesis of fused six-member rings to triazole and pyrazole. Mol. Divers. 2020, 24, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Fouda, A.M.; Abbas, H.A.S.; Ahmed, E.H.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Synthesis, in vitro antimicrobial and cytotoxic activities of some new pyrazolo[1,5-a]pyrimidine derivatives. Molecules 2019, 24, 1080. [Google Scholar] [CrossRef]
- Castillo, J.C.; Estupiñan, D.; Nogueras, M.; Cobo, J.; Portilla, J. 6-(Aryldiazenyl)pyrazolo[1,5-a]pyrimidines as Strategic Intermediates for the Synthesis of Pyrazolo[5,1-b]purines. J. Org. Chem. 2016, 81, 12364–12373. [Google Scholar] [CrossRef]
- Zahedifar, M.; Razavi, R.; Sheibani, H. Reaction of (chloro carbonyl) phenyl ketene with 5-amino pyrazolones: Synthesis, characterization and theoretical studies of 7-hydroxy-6-phenyl-3-(phenyldiazenyl)pyrazolo[1,5-a]pyrimidine-2,5(1H,4H)-dione derivatives. J. Mol. Struct. 2016, 1125, 730–735. [Google Scholar] [CrossRef]
- Li, G.; Meanwell, N.A.; Krystal, M.R.; Langley, D.R.; Naidu, B.N.; Sivaprakasam, P.; Lewis, H.; Kish, K.; Khan, J.A.; Ng, A.; et al. Discovery and Optimization of Novel Pyrazolopyrimidines as Potent and Orally Bioavailable Allosteric HIV-1 Integrase Inhibitors. J. Med. Chem. 2020, 63, 2620–2637. [Google Scholar] [CrossRef] [PubMed]
- Naik, N.S.; Shastri, L.A.; Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Joshi, S.D.; Sunagar, V. Synthesis of novel aryl and coumarin substituted pyrazolo[1,5-a]pyrimidine derivatives as potent anti-inflammatory and anticancer agents. Chem. Data Collect. 2020, 30, 100550. [Google Scholar] [CrossRef]
- Ali, G.M.E.; Ibrahim, D.A.; Elmetwali, A.M.; Ismail, N.S.M. Design, synthesis and biological evaluation of certain CDK2 inhibitors based on pyrazole and pyrazolo[1,5-a] pyrimidine scaffold with apoptotic activity. Bioorg. Chem. 2019, 86, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Qiu, J.K.; Jiang, B.; Hao, W.J.; Guo, C.; Tu, S.J. I2-Catalyzed Multicomponent Reactions for Accessing Densely Functionalized Pyrazolo[1,5-a]pyrimidines and Their Disulphenylated Derivatives. J. Org. Chem. 2016, 81, 3321–3328. [Google Scholar] [CrossRef] [PubMed]
- Hoang, G.L.; Streit, A.D.; Ellman, J.A. Three-Component Coupling of Aldehydes, Aminopyrazoles, and Sulfoxonium Ylides via Rhodium(III)-Catalyzed Imidoyl C–H Activation: Synthesis of Pyrazolo[1,5- a ]pyrimidines. J. Org. Chem. 2018, 83, 15347–15360. [Google Scholar] [CrossRef] [PubMed]
- Reddy Kotla, S.K.; Vandavasi, J.K.; Wang, J.J.; Parang, K.; Tiwari, R.K. Palladium-Catalyzed Intramolecular Cross-Dehydrogenative Coupling: Synthesis of Fused Imidazo[1,2-a]pyrimidines and Pyrazolo[1,5-a]pyrimidines. ACS Omega 2017, 2, 11–19. [Google Scholar] [CrossRef]
- Ding, Z.C.; An, X.M.; Zeng, J.H.; Tang, Z.N.; Zhan, Z.P. Copper(I)-Catalyzed Synthesis of 4,5-Dihydropyrazolo[1,5-a]pyrimidines via Cascade Transformation of N-Propargylic Sulfonylhydrazones with Sulfonyl Azides. Adv. Synth. Catal. 2017, 359, 3319–3324. [Google Scholar] [CrossRef]
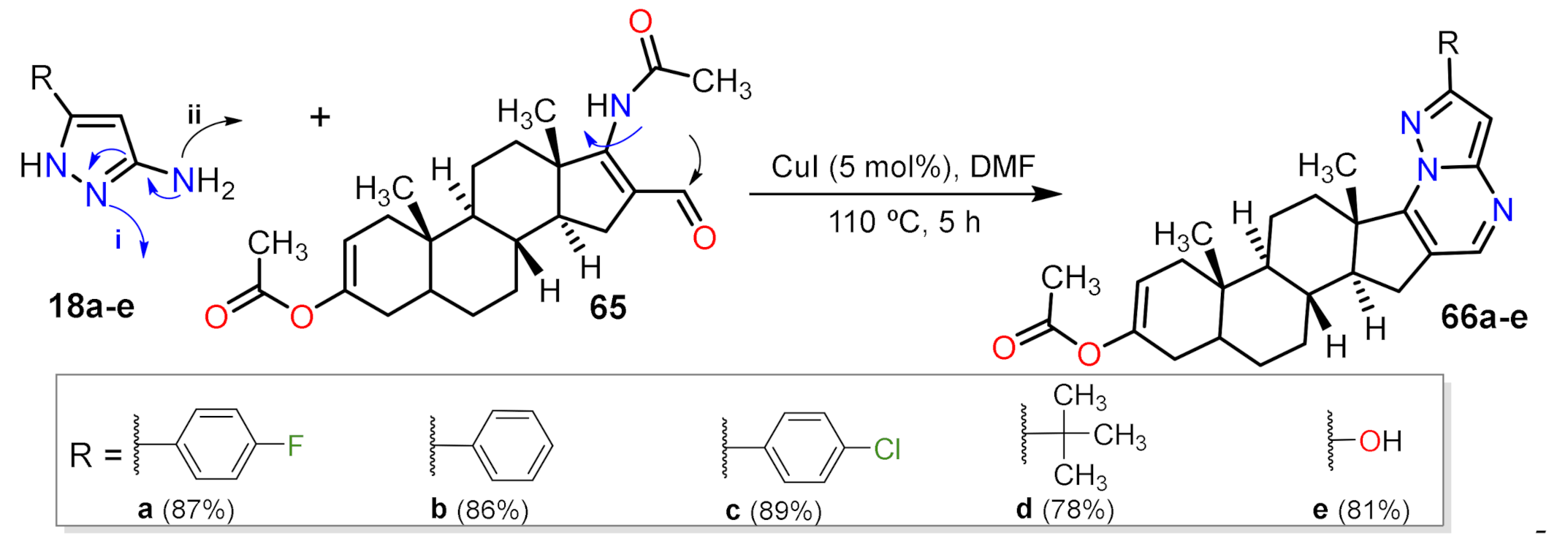
- Nongthombam, G.S.; Boruah, R.C. Augmentation of steroidal β-formylenamide with pyrazolo and benzimidazo moieties: A tandem approach to highly fluorescent steroidal heterocycles. Tetrahedron Lett. 2021, 152893. [Google Scholar] [CrossRef]
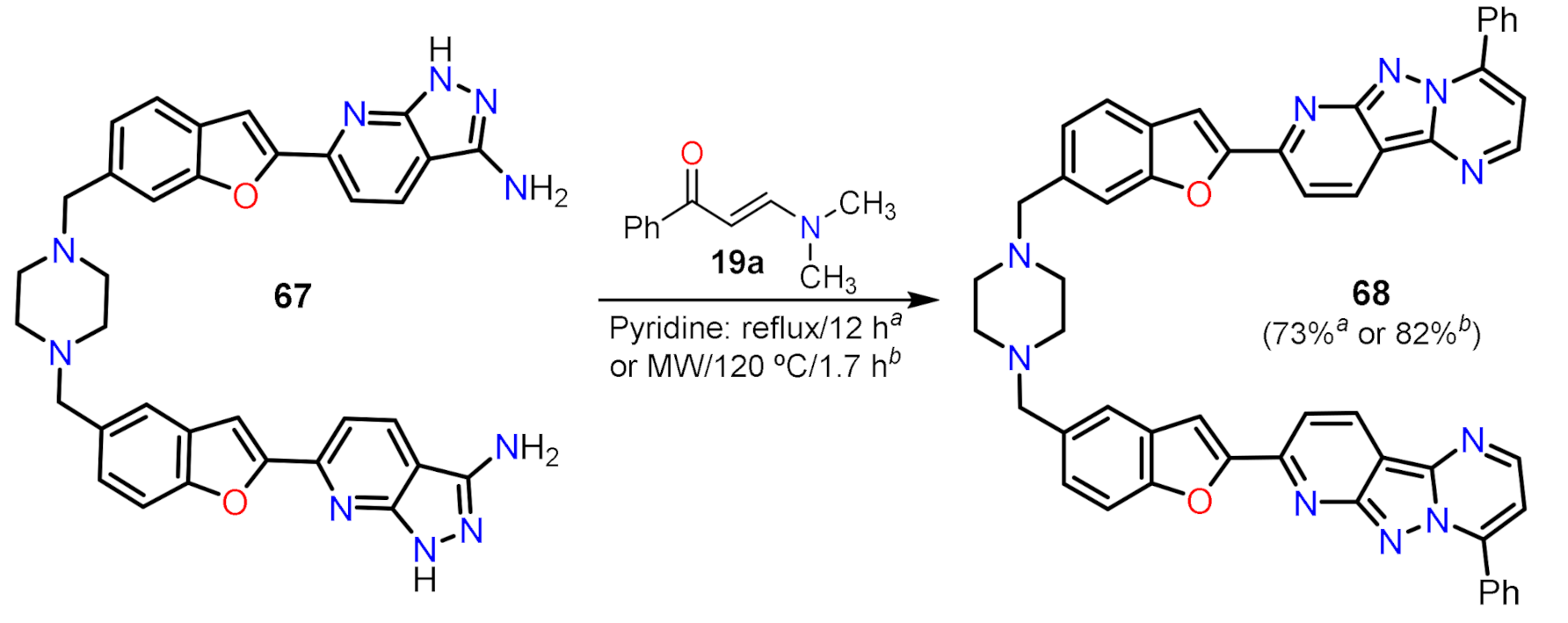
- Mekky, A.E.M.; Ahmed, M.S.M.; Sanad, S.M.H.; Abdallah, Z.A. Bis(benzofuran-enaminone) hybrid possessing piperazine linker: Versatile precursor for microwave assisted synthesis of bis(pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidines). Synth. Commun. 2021, 51, 1–19. [Google Scholar] [CrossRef]
- Jismy, B.; Guillaumet, G.; Akssira, M.; Tikad, A.; Abarbri, M. Efficient microwave-assisted Suzuki-Miyaura cross-coupling reaction of 3-bromo pyrazolo[1,5-a] pyrimidin-5(4 H)-one: Towards a new access to 3,5-diarylated 7-(trifluoromethyl)pyrazolo[1,5-a] pyrimidine derivatives. RSC Adv. 2021, 11, 1287–1302. [Google Scholar] [CrossRef]
- Zhang, Y.C.; Zhang, B.W.; Geng, R.L.; Song, J. Enantioselective [3 + 2] Cycloaddition Reaction of Ethynylethylene Carbonates with Malononitrile Enabled by Organo/Metal Cooperative Catalysis. Org. Lett. 2018, 20, 7907–7911. [Google Scholar] [CrossRef]
- Abdallah, A.E.M.; Elgemeie, G.H. Design, synthesis, docking, and antimicrobial evaluation of some novel pyrazolo[1,5-a] pyrimidines and their corresponding cycloalkane ring-fused derivatives as purine analogs. Drug Des. Devel. Ther. 2018, 12, 1785–1798. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Laufer, R.; Patel, N.K.; Ng, G.; Sampson, P.B.; Li, S.-W.; Lang, Y.; Feher, M.; Brokx, R.; Beletskaya, I.; et al. Discovery of Pyrazolo[1,5-a]pyrimidine TTK Inhibitors: CFI-402257 is a Potent, Selective, Bioavailable Anticancer Agent. ACS Med. Chem. Lett. 2016, 7, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Drew, S.L.; Thomas-Tran, R.; Beatty, J.W.; Fournier, J.; Lawson, K.V.; Miles, D.H.; Mata, G.; Sharif, E.U.; Yan, X.; Mailyan, A.K.; et al. Discovery of Potent and Selective PI3KγInhibitors. J. Med. Chem. 2020, 63, 11235–11257. [Google Scholar] [CrossRef]
- Harris, M.R.; Wisniewska, H.M.; Jiao, W.; Wang, X.; Bradow, J.N. A Modular Approach to the Synthesis of gem-Disubstituted Cyclopropanes. Org. Lett. 2018, 20, 2867–2871. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.Y.; Zhang, Y.F.; Ma, C.L.; Gu, Q.S.; Wang, F.L.; Li, Z.L.; Jiang, S.P.; Liu, X.Y. A general asymmetric copper-catalysed Sonogashira C(sp 3)–C(sp) coupling. Nat. Chem. 2019, 11, 1158–1166. [Google Scholar] [CrossRef]
- Bedford, R.B.; Durrant, S.J.; Montgomery, M. Catalyst-Switchable Regiocontrol in the Direct Arylation of Remote C-H Groups in Pyrazolo[1,5-a]pyrimidines. Angew. Chemie Int. Ed. 2015, 54, 8787–8790. [Google Scholar] [CrossRef] [PubMed]
- Loubidi, M.; Manga, C.; Tber, Z.; Bassoude, I.; Essassi, E.M.; Berteina-Raboin, S. One-Pot SNAr/Direct Pd-Catalyzed CH Arylation Functionalization of Pyrazolo[1,5-a]pyrimidine at the C3 and C7 Positions. Eur. J. Org. Chem. 2018, 2018, 3936–3942. [Google Scholar] [CrossRef]
- Gogula, T.; Zhang, J.; Lonka, M.R.; Zhang, S.; Zou, H. Temperature-modulated selective C(sp3)-H or C(sp2)-H arylation through palladium catalysis. Chem. Sci. 2020, 11, 11461–11467. [Google Scholar] [CrossRef]
- Colomer, I.; Chamberlain, A.E.R.; Haughey, M.B.; Donohoe, T.J. Fc12 Hfip. Nat. Rev. Chem. 2017, 1, 0088. [Google Scholar] [CrossRef]
- McCoull, W.; Abrams, R.D.; Anderson, E.; Blades, K.; Barton, P.; Box, M.; Burgess, J.; Byth, K.; Cao, Q.; Chuaqui, C.; et al. Discovery of Pyrazolo[1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors. J. Med. Chem. 2017, 60, 4386–4402. [Google Scholar] [CrossRef] [PubMed]
- Mackman, R.L.; Sangi, M.; Sperandio, D.; Parrish, J.P.; Eisenberg, E.; Perron, M.; Hui, H.; Zhang, L.; Siegel, D.; Yang, H.; et al. Discovery of an oral respiratory syncytial virus (RSV) fusion inhibitor (GS-5806) and clinical proof of concept in a human RSV challenge study. J. Med. Chem. 2015, 58, 1630–1643. [Google Scholar] [CrossRef]
- Mathison, C.J.N.; Chianelli, D.; Rucker, P.V.; Nelson, J.; Roland, J.; Huang, Z.; Yang, Y.; Jiang, J.; Xie, Y.F.; Epple, R.; et al. Efficacy and Tolerability of Pyrazolo[1,5-a]pyrimidine RET Kinase Inhibitors for the Treatment of Lung Adenocarcinoma. ACS Med. Chem. Lett. 2020, 11, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Sasaki, T.; Kawaguchi, T.; Okada, A.; Tokura, S.; Tanaka-Yamamoto, N.; Takeuchi, T.; Ogata, Y.; Takahashi, R.; Kurimoto-Tsuruta, R.; Tamaoki, T.; et al. Discovery of a potent dual inhibitor of wild-type and mutant respiratory syncytial virus fusion proteins through the modulation of atropisomer interconversion properties. Bioorg. Med. Chem. 2020, 28, 115818. [Google Scholar] [CrossRef]
- Levy, J.N.; Alegre-Requena, J.V.; Liu, R.; Paton, R.S.; McNally, A. Selective Halogenation of Pyridines Using Designed Phosphine Reagents. J. Am. Chem. Soc. 2020, 142, 11295–11305. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Altman, M.D.; Baker, J.; Brubaker, J.D.; Chen, H.; Chen, Y.; Fischmann, T.; Gibeau, C.; Kleinschek, M.A.; Leccese, E.; et al. Discovery of 5-Amino- N -(1 H -pyrazol-4-yl)pyrazolo[1,5-a]pyrimidine-3-carboxamide Inhibitors of IRAK4. ACS Med. Chem. Lett. 2015, 6, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Bryan, M.C.; Drobnick, J.; Gobbi, A.; Kolesnikov, A.; Chen, Y.; Rajapaksa, N.; Ndubaku, C.; Feng, J.; Chang, W.; Francis, R.; et al. Development of Potent and Selective Pyrazolopyrimidine IRAK4 Inhibitors. J. Med. Chem. 2019, 62, 6223–6240. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Sasaki, S.; Asano, Y.; Ujikawa, O.; Fukumoto, S.; Nakashima, K.; Oki, H.; Kamiguchi, N.; Imada, H.; Iwashita, H.; et al. Discovery of an Orally Bioavailable, Brain-Penetrating, in Vivo Active Phosphodiesterase 2A Inhibitor Lead Series for the Treatment of Cognitive Disorders. J. Med. Chem. 2017, 60, 7658–7676. [Google Scholar] [CrossRef] [PubMed]
- Gazizov, D.A.; Gorbunov, E.B.; Rusinov, G.L.; Ulomsky, E.N.; Charushin, V.N. A New Family of Fused Azolo[1,5-a]pteridines and Azolo[5,1-b]purines. ACS Omega 2020, 5, 18226–18233. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Utecht, G.; Fruziński, A.; Jasiński, M. Polysubstituted 3-trifluoromethylpyrazoles: Regioselective (3 + 2)-cycloaddition of trifluoroacetonitrile imines with enol ethers and functional group transformations. Org. Biomol. Chem. 2018, 16, 1252–1257. [Google Scholar] [CrossRef]
- Li, M.; Zhao, B.X. Progress of the synthesis of condensed pyrazole derivatives (from 2010 to mid-2013). Eur. J. Med. Chem. 2014, 85, 311–340. [Google Scholar] [CrossRef]
- Scott, L.J. Larotrectinib: First Global Approval. Drugs 2019, 79, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Gana, C.C.; Hanssen, K.M.; Yu, D.M.T.; Flemming, C.L.; Wheatley, M.S.; Conseil, G.; Cole, S.P.C.; Norris, M.D.; Haber, M.; Fletcher, J.I. MRP1 modulators synergize with buthionine sulfoximine to exploit collateral sensitivity and selectively kill MRP1-expressing cancer cells. Biochem. Pharmacol. 2019, 168, 237–248. [Google Scholar] [CrossRef]
- McCoull, W.; Abrams, R.D.; Anderson, E.; Blades, K.; Barton, P.; Box, M.; Burgess, J.; Byth, K.; Cao, Q.; Chuaqui, C.; et al. Correction to Discovery of Pyrazolo[1,5-a]pyrimidine B-Cell Lymphoma 6 (BCL6) Binders and Optimization to High Affinity Macrocyclic Inhibitors. J. Med. Chem. 2017, 60, 6459. [Google Scholar] [CrossRef] [PubMed]
- Bou Karroum, N.; Moarbess, G.; Guichou, J.F.; Bonnet, P.A.; Patinote, C.; Bouharoun-Tayoun, H.; Chamat, S.; Cuq, P.; Diab-Assaf, M.; Kassab, I.; et al. Novel and selective TLR7 antagonists among the imidazo[1,2-a]pyrazines, Imidazo[1,5-a]quinoxalines, and Pyrazolo[1,5-a]quinoxalines Series. J. Med. Chem. 2019, 62, 7015–7031. [Google Scholar] [CrossRef]
- Philoppes, J.N.; Khedr, M.A.; Hassan, M.H.A.; Kamel, G.; Lamie, P.F. New pyrazolopyrimidine derivatives with anticancer activity: Design, synthesis, PIM-1 inhibition, molecular docking study and molecular dynamics. Bioorg. Chem. 2020, 100, 103944. [Google Scholar] [CrossRef]
- Ravi Kumar, N.; Poornachandra, Y.; Krishna Swaroop, D.; Jitender Dev, G.; Ganesh Kumar, C.; Narsaiah, B. Synthesis of novel ethyl 2,4-disubstituted 8-(trifluoromethyl)pyrido[2′,3′:3,4]pyrazolo[1,5-a]pyrimidine-9-carboxylate derivatives as promising anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 5203–5206. [Google Scholar] [CrossRef]
- Henderson, S.H.; Sorrell, F.; Bennett, J.; Hanley, M.T.; Robinson, S.; Hopkins Navratilova, I.; Elkins, J.M.; Ward, S.E. Mining Public Domain Data to Develop Selective DYRK1A Inhibitors. ACS Med. Chem. Lett. 2020, 11, 1620–1626. [Google Scholar] [CrossRef]
- Dowling, J.E.; Alimzhanov, M.; Bao, L.; Chuaqui, C.; Denz, C.R.; Jenkins, E.; Larsen, N.A.; Lyne, P.D.; Pontz, T.; Ye, Q.; et al. Potent and Selective CK2 Kinase Inhibitors with Effects on Wnt Pathway Signaling in Vivo. ACS Med. Chem. Lett. 2016, 7, 300–305. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, M.T.; Hussein, H.A.R.; Elebiary, N.M.; Hassan, G.S.; Elmessery, S.M.; Elsheakh, A.R.; Nayel, M.; Abdel-Aziz, H.A. Tyrosine kinase inhibition effects of novel Pyrazolo[1,5-a]pyrimidines and Pyrido[2,3-d]pyrimidines ligand: Synthesis, biological screening and molecular modeling studies. Bioorg. Chem. 2018, 78, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, B.; Kovvuri, J.; Kumar, C.G.; Routhu, S.R.; Shareef, M.A.; Kadagathur, M.; Adiyala, P.R.; Alavala, S.; Nagesh, N.; Kamal, A. Synthesis and biological evaluation of pyrazole linked benzothiazole-β-naphthol derivatives as topoisomerase I inhibitors with DNA binding ability. Bioorg. Med. Chem. 2019, 27, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Husseiny, E.M. Synthesis, cytotoxicity of some pyrazoles and pyrazolo[1,5-a]pyrimidines bearing benzothiazole moiety and investigation of their mechanism of action. Bioorg. Chem. 2020, 102, 104053. [Google Scholar] [CrossRef] [PubMed]
















































| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| Hela | A549 | HCT-116 | B16-F10 | |
| 162 | 0.019 ± 0.001 | 0.015 ± 0.001 | 0.039 ± 0.003 | 0.048 ± 0.004 |
| 163 | 0.021 ± 0.001 | 0.003 ± 0.001 | 0.048 ± 0.005 | 0.021 ± 0.002 |
| Colchicine | 0.081 ± 0.006 | 0.107 ± 0.009 | 0.042 ± 0.004 | 0.087 ± 0.006 |



| Compound | IC50 (µM) | |||
|---|---|---|---|---|
| Huh-7 | HeLa | MCF7 | MDA-MB231 | |
| 170e | 6.3 | 48.8 | 118.4 | 74.25 |
| 170j | 54.1 | 7.8 | 106 | 5.74 |
| 170k | 95.0 | 21.6 | 3.0 | 4.32 |
| DOX | 3.2 | 8.1 | 5.9 | 6.0 |







| Compound | GI50 (µM) | |
|---|---|---|
| HCT-116 | SW620 | |
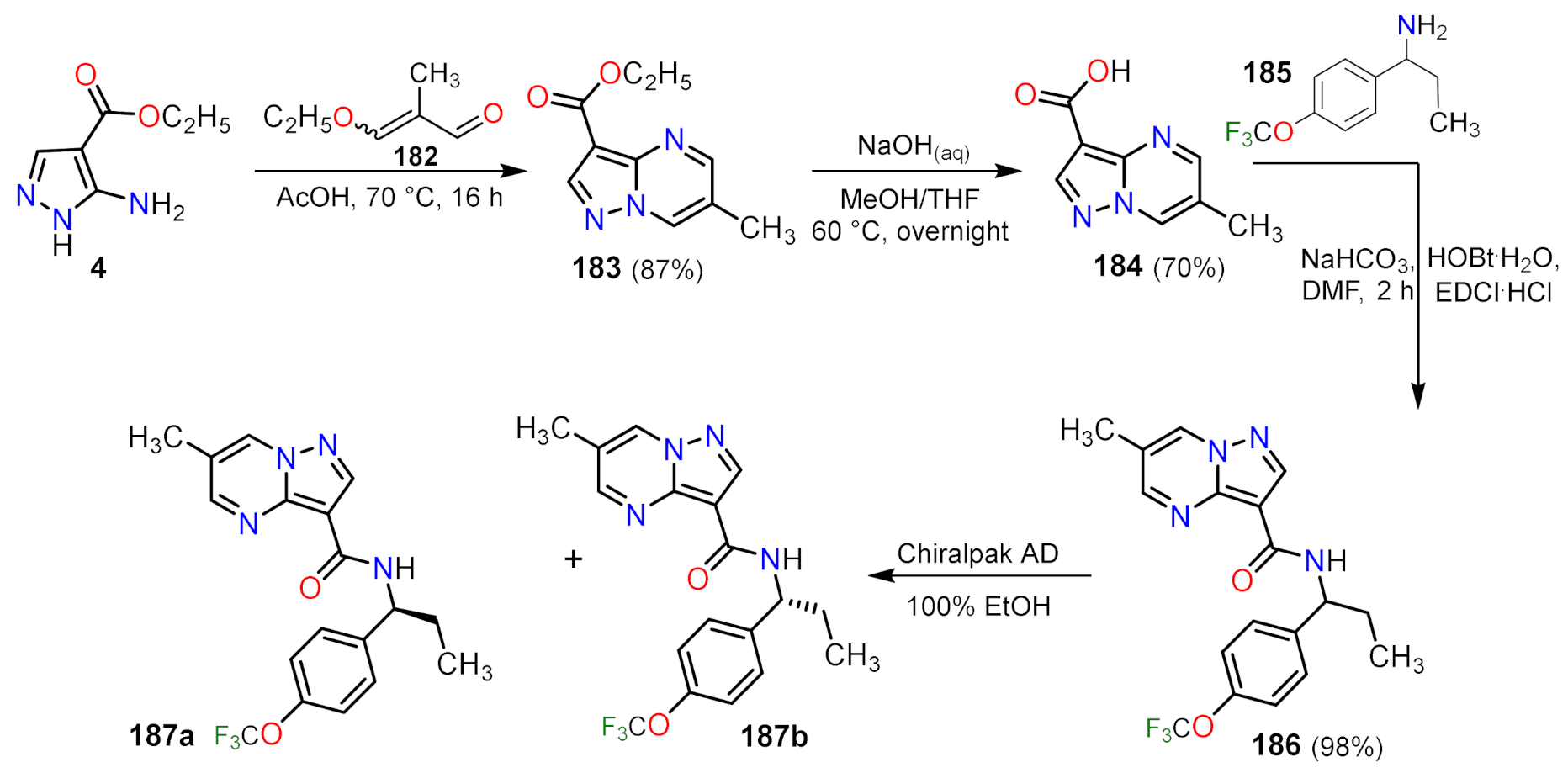
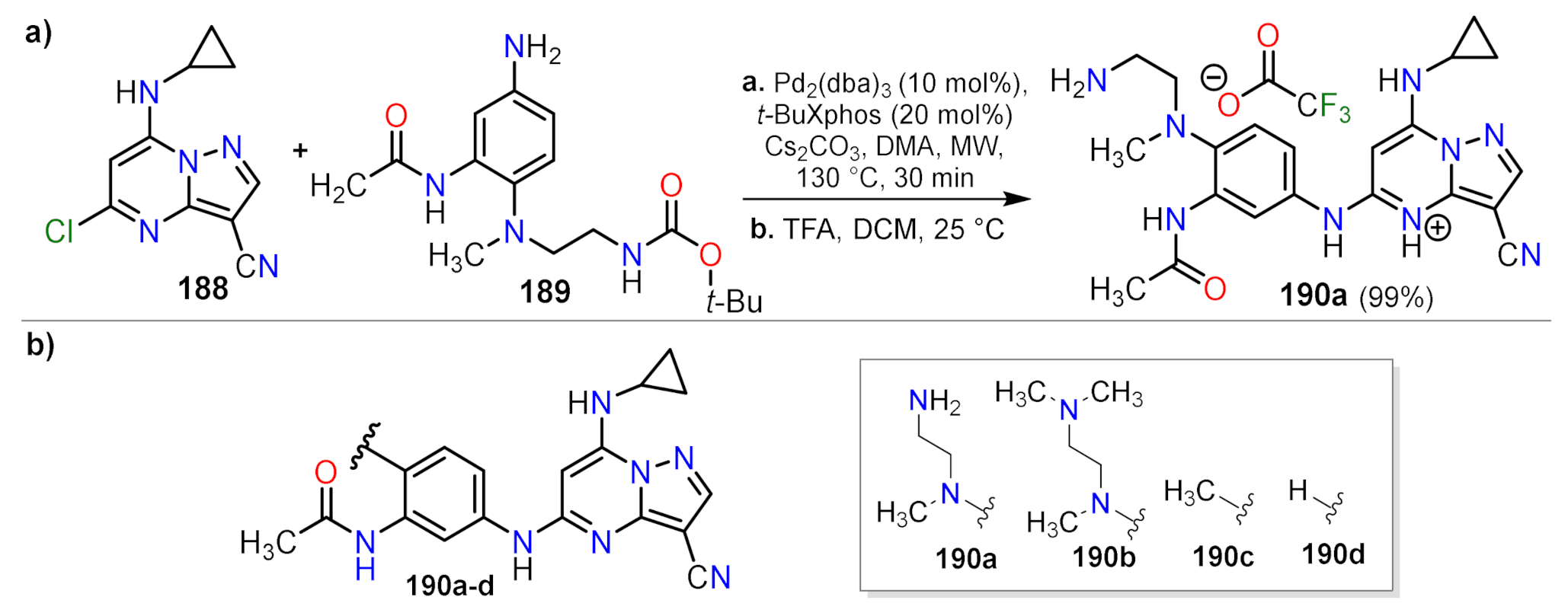
| 190a | 0.7 | 0.7 |
| 190b | 0.08 | 0.1 |
| 190c | 0.03 | 0.03 |
| 190d | 0.01 | 0.005 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthetic Methods | Substituted PP | Functionalization Reactions | ||
|---|---|---|---|---|
| Section 2.1.1 | 1,3-Biselectrophillic systems: β-dicarbonyls, etc. |  | Metal catalyzed reactions: Suzuki, etc. | Section 2.2.1 |
| Section 2.1.2 | Multicomponent reactions | Nucleophilic aromatic substitution. | Section 2.2.2 | |
| Section 2.1.3 | Synthesis by a pericyclic reaction | Other functionalization reactions. | Section 2.2.3 | |
| [a] General examples of some relevant synthetic transformations (synthesis and functionalizations) are showed. | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias-Gómez, A.; Godoy, A.; Portilla, J. Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold. Molecules 2021, 26, 2708. https://doi.org/10.3390/molecules26092708
Arias-Gómez A, Godoy A, Portilla J. Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold. Molecules. 2021; 26(9):2708. https://doi.org/10.3390/molecules26092708
Chicago/Turabian StyleArias-Gómez, Andres, Andrés Godoy, and Jaime Portilla. 2021. "Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold" Molecules 26, no. 9: 2708. https://doi.org/10.3390/molecules26092708
APA StyleArias-Gómez, A., Godoy, A., & Portilla, J. (2021). Functional Pyrazolo[1,5-a]pyrimidines: Current Approaches in Synthetic Transformations and Uses As an Antitumor Scaffold. Molecules, 26(9), 2708. https://doi.org/10.3390/molecules26092708