
“On-The-Fly” Non-Adiabatic Dynamics Simulations on Photoinduced Ring-Closing Reaction of a Nucleoside-Based Diarylethene Photoswitch



Abstract
:
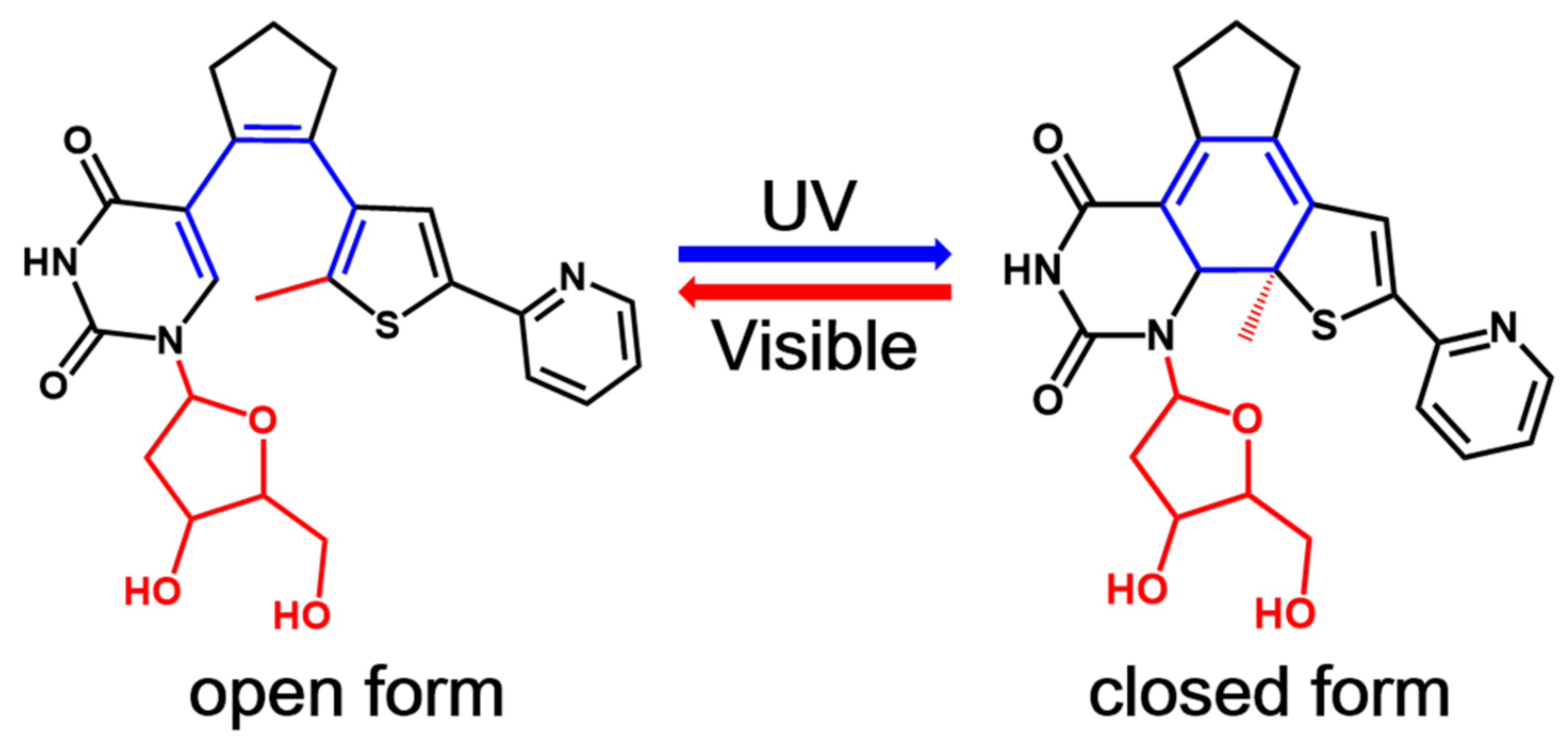
1. Introduction
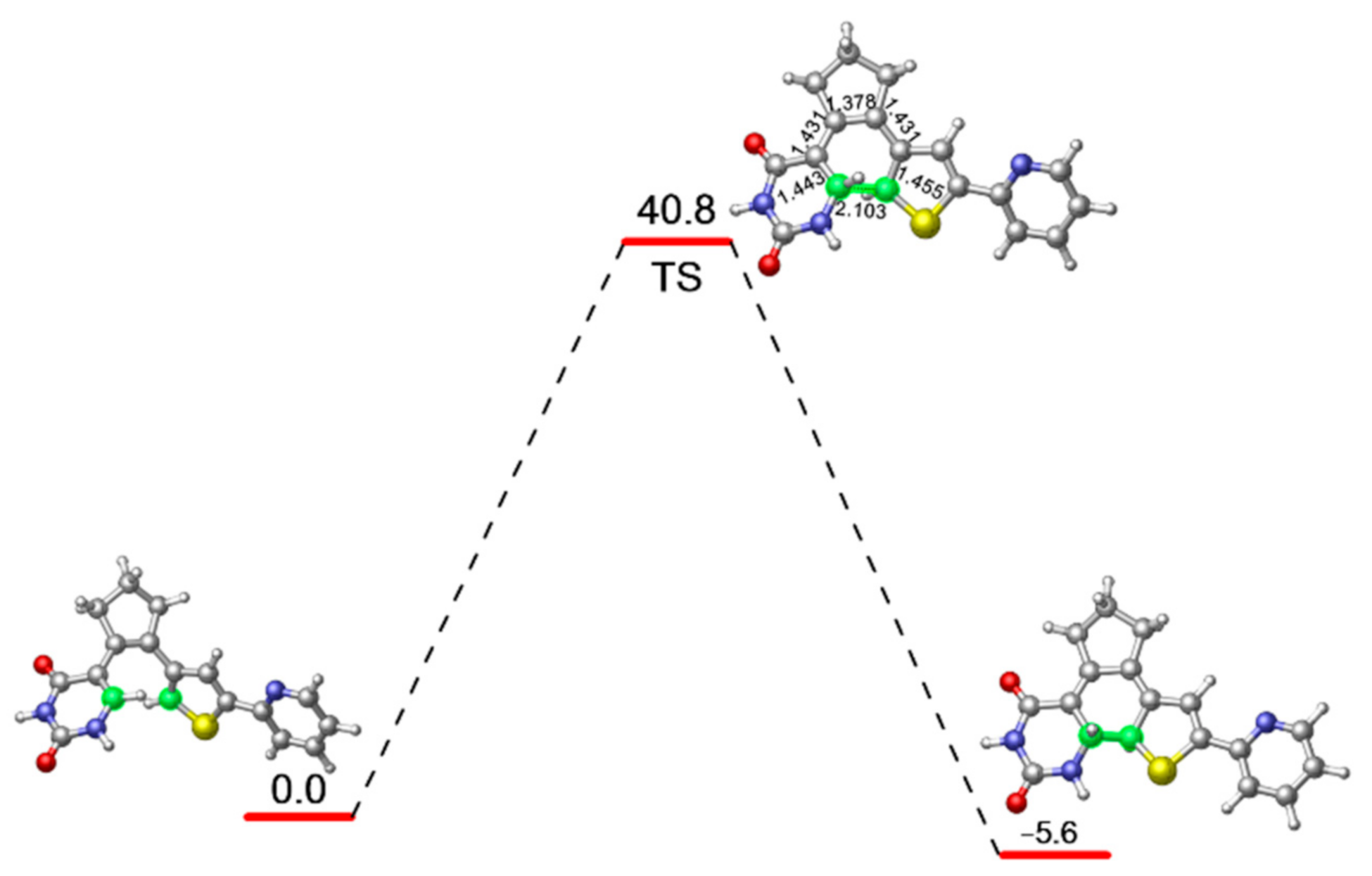
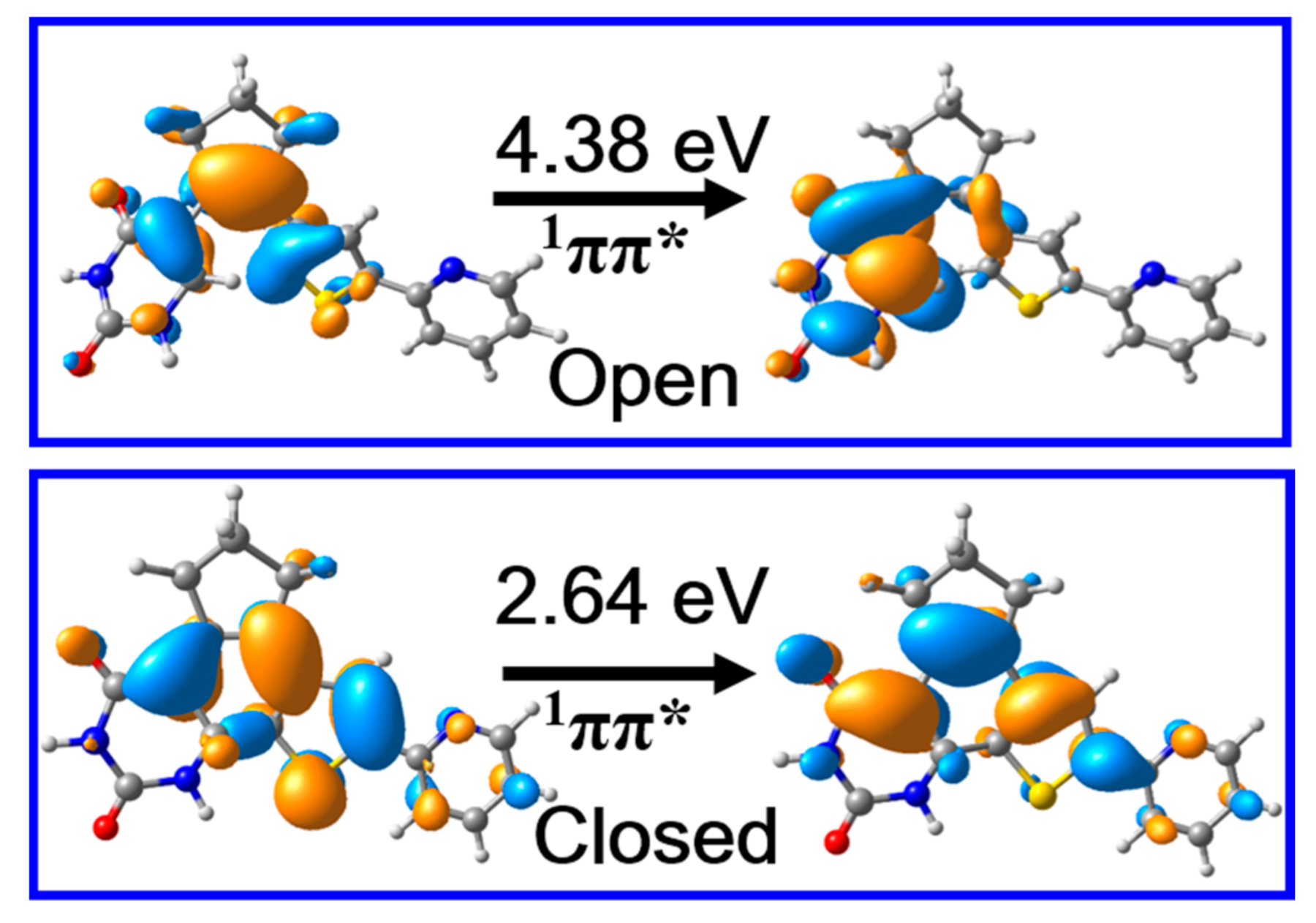
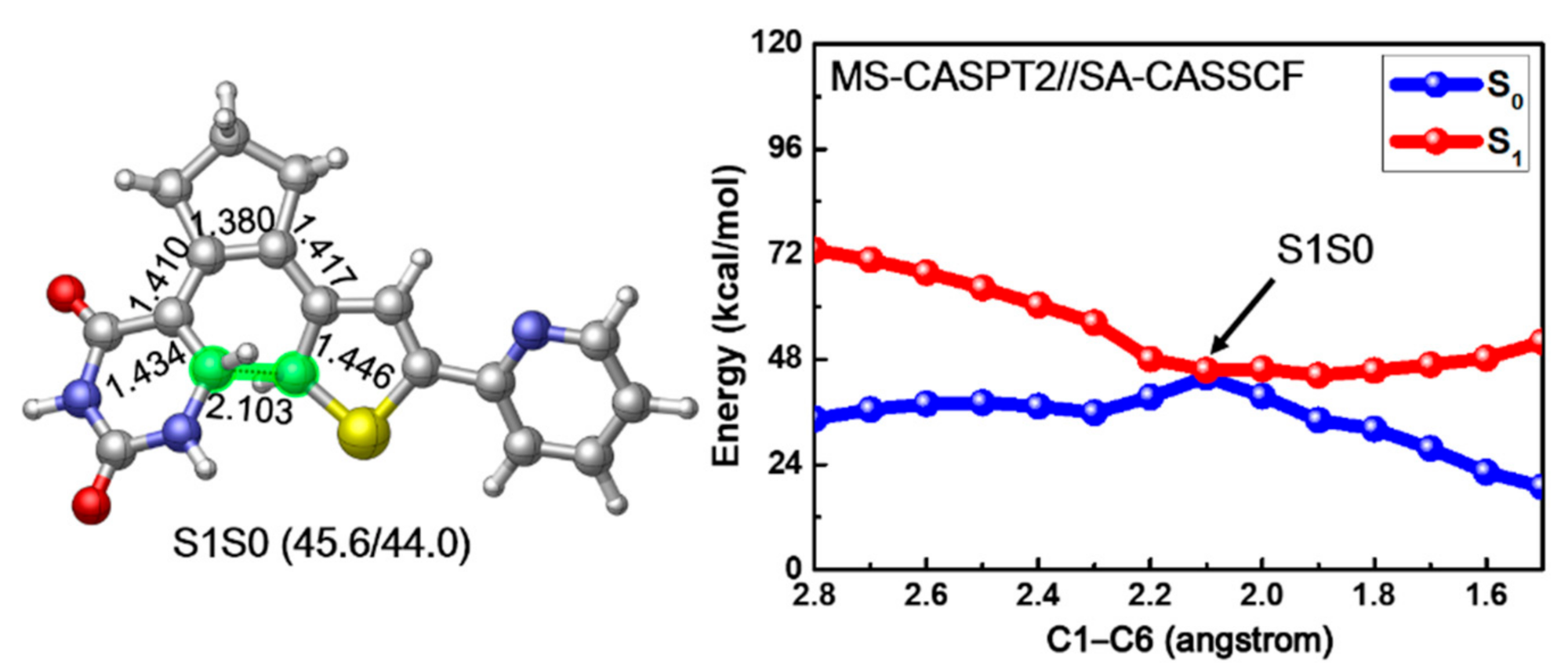
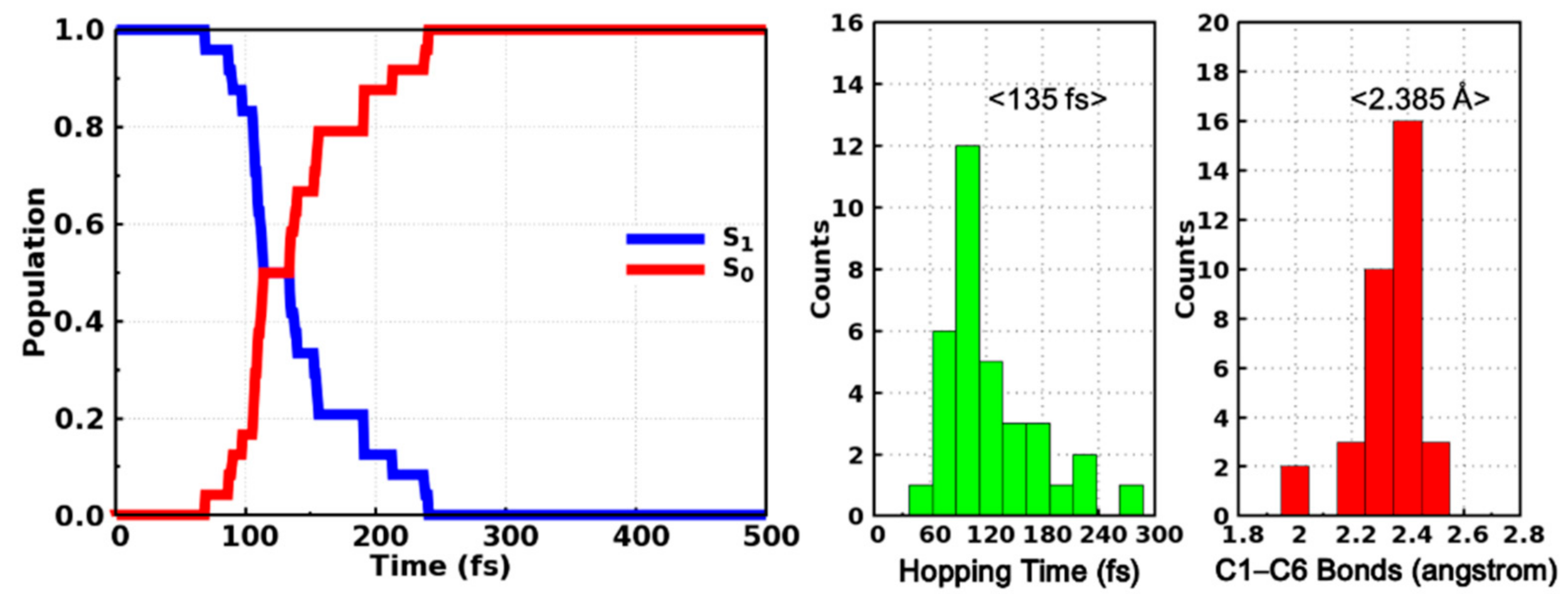
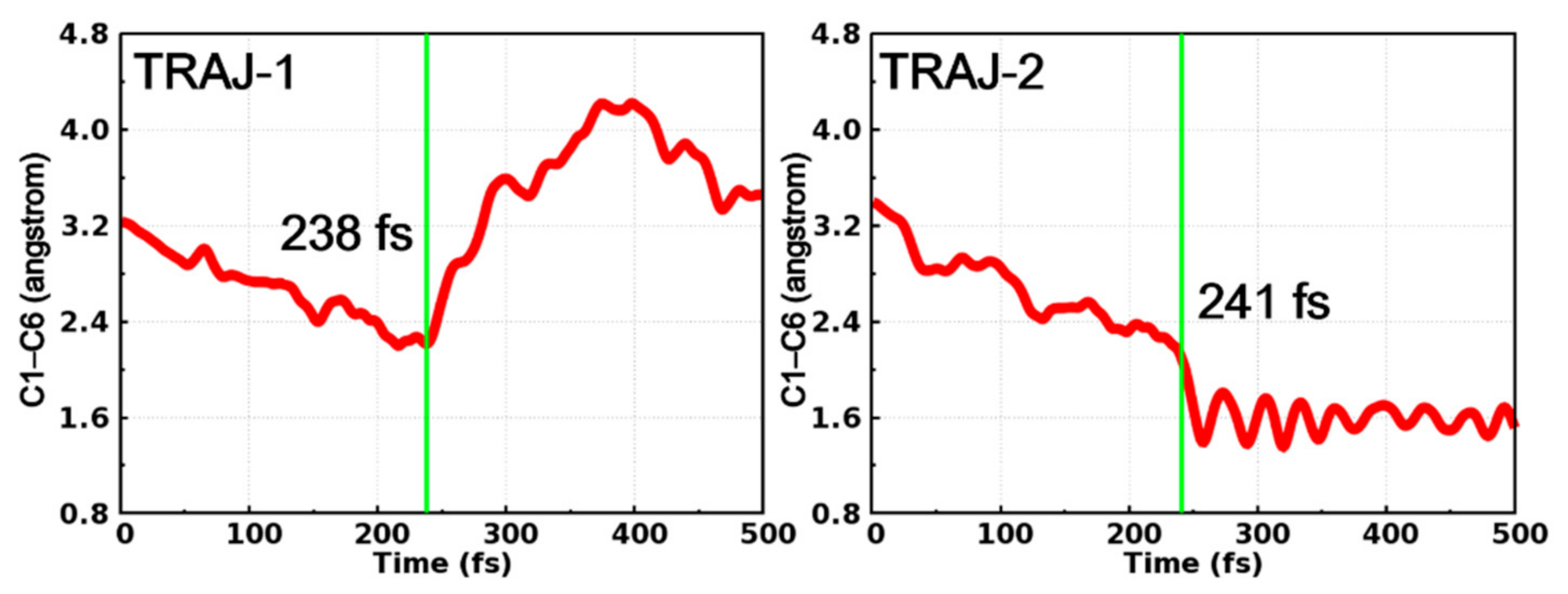
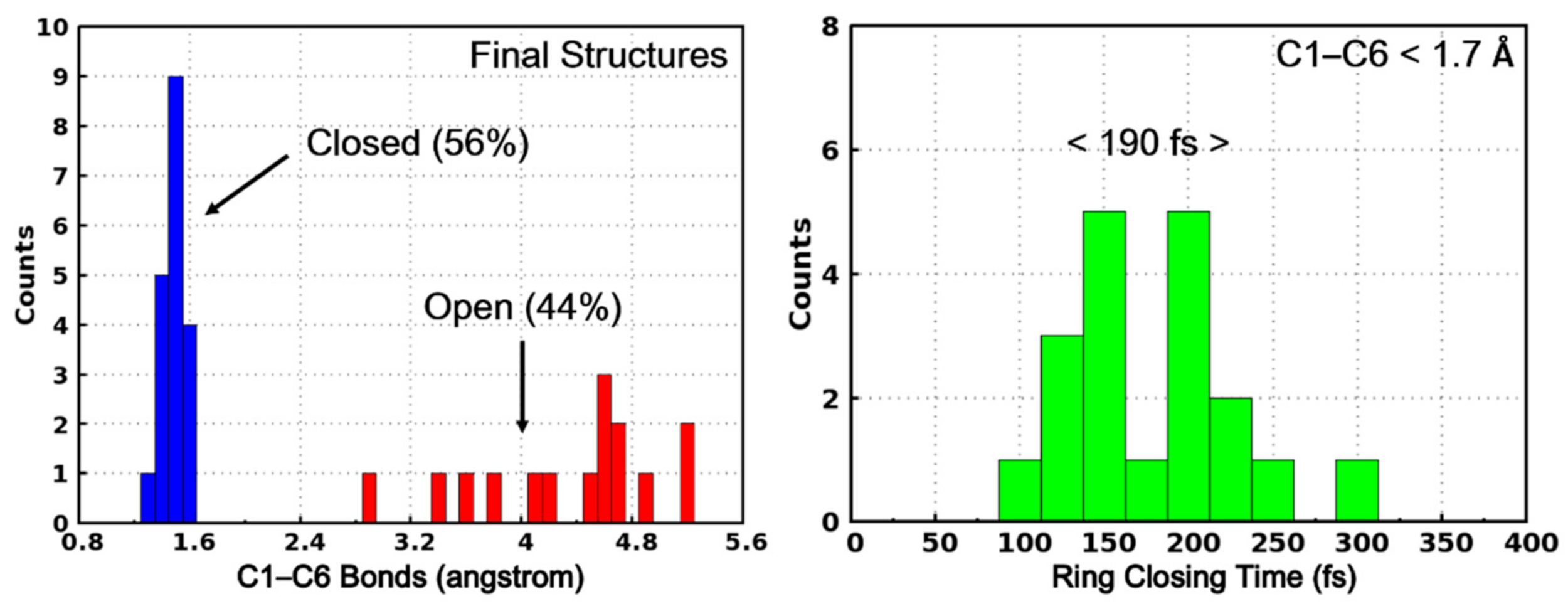
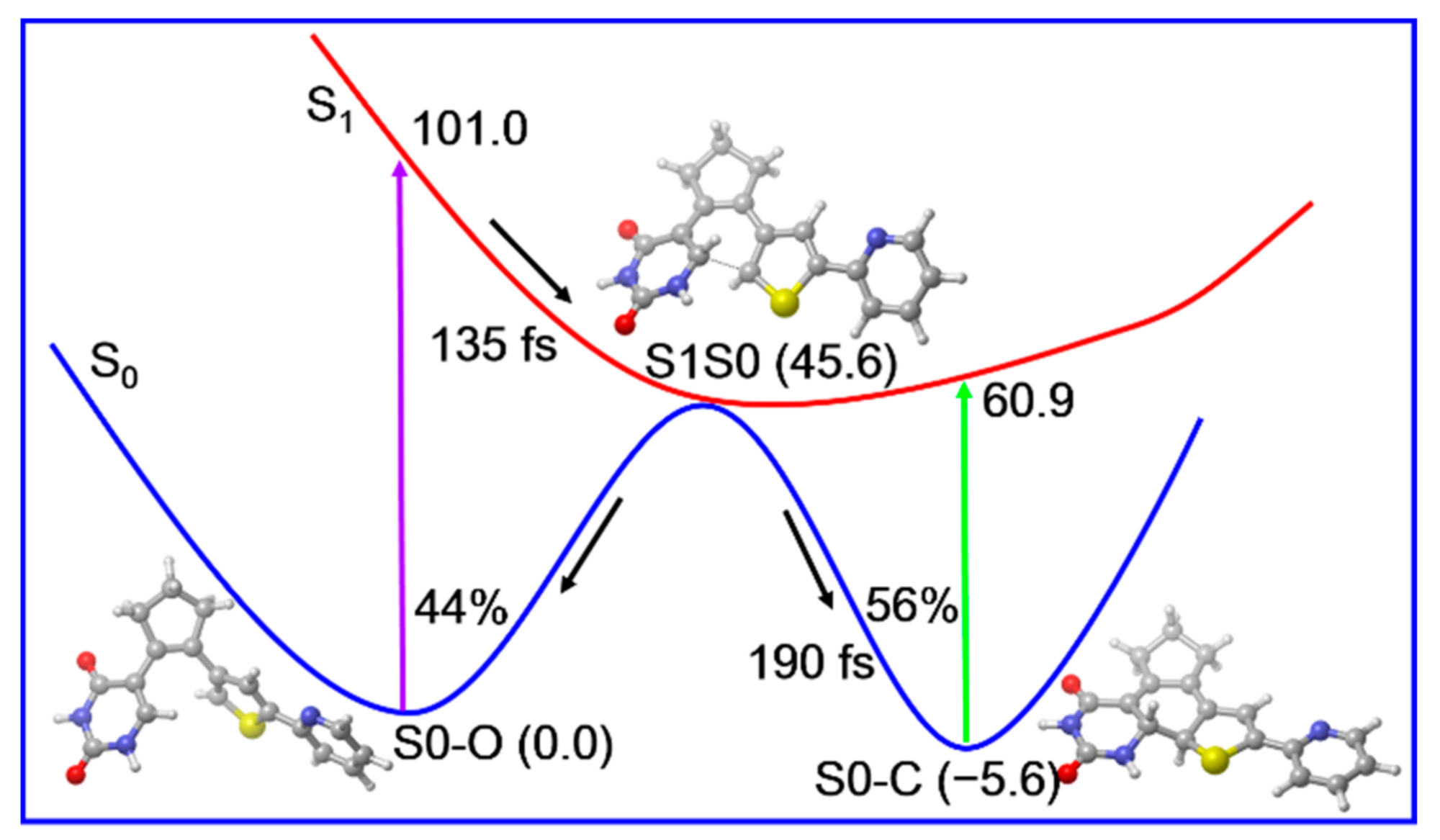
2. Results and Discussion
3. Materials and Methods
3.1. Electronic Structure Calculations
3.2. Non-Adiabatic Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Natali, M.; Giordani, S. Molecular Switches as Photocontrollable “Smart” Receptors. Chem. Soc. Rev. 2012, 41, 4010–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szymański, W.; Beierle, J.M.; Kistemaker, H.A.V.; Velema, W.A.; Feringa, B.L. Reversible Photocontrol of Biological Systems by the Incorporation of Molecular Photoswitches. Chem. Rev. 2013, 113, 6114–6178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamiya, Y.; Asanuma, H. Light-Driven DNA Nanomachine with a Photoresponsive Molecular Engine. Accounts Chem. Res. 2014, 47, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y. Design and Applications of Metastable-State Photoacids. Acc. Chem. Res. 2017, 50, 1956–1964. [Google Scholar] [CrossRef]
- Li, Z.; Wang, G.; Ye, Y.; Li, B.; Li, H.; Chen, B. Loading Photochromic Molecules into a Luminescent Metal–Organic Framework for Information Anticounterfeiting. Angew. Chem. Int. Ed. 2019, 58, 18025–18031. [Google Scholar] [CrossRef]
- Huang, X.; Li, T. Recent progress in the development of molecular-scale electronics based on photoswitchable molecules. J. Mater. Chem. C 2020, 8, 821–848. [Google Scholar] [CrossRef]
- Wakayama, Y.; Hayakawa, R.; Higashiguchi, K.; Matsuda, K. Photochromism for optically functionalized organic field-effect transistors: A comprehensive review. J. Mater. Chem. C 2020, 8, 10956–10974. [Google Scholar] [CrossRef]
- Andréasson, J.; Pischel, U. Light-stimulated molecular and supramolecular systems for information processing and beyond. Co-ord. Chem. Rev. 2021, 429, 213695. [Google Scholar] [CrossRef]
- Zhang, J.; Zou, Q.; Tian, H. Photochromic Materials: More Than Meets The Eye. Adv. Mater. 2013, 25, 378–399. [Google Scholar] [CrossRef]
- Orgiu, E.; Samori, P. 25th Anniversary Article: Organic Electronics Marries Photochromism: Generation of Multifunctional Interfaces, Materials, and Devices. Adv. Mater. 2014, 26, 1827–1845. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Tian, H. Taking orders from light: Progress in photochromic bio-materials. Mater. Horizons 2014, 1, 169–184. [Google Scholar] [CrossRef]
- Pianowski, Z.L. Recent Implementations of Molecular Photoswitches into Smart Materials and Biological Systems. Chem. Eur. J. 2019, 25, 5128–5144. [Google Scholar] [CrossRef]
- Jia, S.; Fong, W.-K.; Graham, B.; Boyd, B.J. Photoswitchable Molecules in Long-Wavelength Light-Responsive Drug Delivery: From Molecular Design to Applications. Chem. Mater. 2018, 30, 2873–2887. [Google Scholar] [CrossRef]
- Bléger, D.; Hecht, S. Visible-Light-Activated Molecular Switches. Angew. Chem. Int. Ed. 2015, 54, 11338–11349. [Google Scholar] [CrossRef]
- Wang, L.; Li, Q. Photochromism into Nanosystems: Towards Lighting up the Future Nanoworld. Chem. Soc. Rev. 2018, 47, 1044–1097. [Google Scholar] [CrossRef]
- Boelke, J.; Hecht, S. Designing Molecular Photoswitches for Soft Materials Applications. Adv. Opt. Mater. 2019, 7, 1900404. [Google Scholar] [CrossRef]
- Goulet-Hanssens, A.; Eisenreich, F.; Hecht, S. Enlightening Materials with Photoswitches. Adv. Mater. 2020, 32, e1905966. [Google Scholar] [CrossRef]
- Rice, A.M.; Martin, C.R.; Galitskiy, V.A.; Berseneva, A.A.; Leith, G.A.; Shustova, N.B. Photophysics Modulation in Photoswitchable Metal-Organic Frameworks. Chem. Rev. 2020, 120, 8790–8813. [Google Scholar] [CrossRef]
- Irie, M. Diarylethenes for Memories and Switches. Chem. Rev. 2000, 100, 1685–1716. [Google Scholar] [CrossRef]
- Matsuda, K.; Irie, M. Diarylethene as a Photo Switching Unit. J. Photoch. Photobio. C 2004, 5, 169–182. [Google Scholar] [CrossRef]
- Tian, H.; Yang, S. Recent progresses on diarylethene based photochromic switches. Chem. Soc. Rev. 2004, 33, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Irie, M. Photochromism of diarylethene single molecules and single crystals. Photochem. Photobiol. Sci. 2010, 9, 1535–1542. [Google Scholar] [CrossRef] [PubMed]
- Pu, S.-Z.; Sun, Q.; Fan, C.-B.; Wang, R.-J.; Liu, G. Recent advances in diarylethene-based multi-responsive molecular switches. J. Mater. Chem. C 2016, 4, 3075–3093. [Google Scholar] [CrossRef]
- Uchida, K.; Nishimura, R.; Hatano, E.; Mayama, H.; Yokojima, S. Photochromic Crystalline Systems Mimicking Bio-Functions. Chem. Eur. J. 2018, 24, 8491–8506. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, H. The Endeavor of Diarylethenes: New Structures, High Performance, and Bright Future. Adv. Opt. Mater. 2018, 6, 1701278. [Google Scholar] [CrossRef]
- Lin, S.Y.; Gutierrez-Cuevas, K.G.; Zhang, X.F.; Guo, J.B.; Li, Q. Fluorescent Photochromic Alpha-Cyanodiarylethene Molecular Switches: An Emerging and Promising Class of Functional Diarylethene. Adv. Funct. Mater. 2021, 31, 2007957. [Google Scholar] [CrossRef]
- Komarov, I.V.; Afonin, S.; Babii, O.; Schober, T.; Ulrich, A.S. Efficiently Photocontrollable or Not? Biological Activity of Photoisomerizable Diarylethenes. Chem. Eur. J. 2018, 24, 11245–11254. [Google Scholar] [CrossRef]
- Babii, O.; Afonin, S.; Berditsch, M.; Reiβer, S.; Mykhailiuk, P.K.; Kubyshkin, V.S.; Steinbrecher, T.; Ulrich, A.S.; Komarov, I.V. Controlling Biological Activity with Light: Diarylethene-Containing Cyclic Peptidomimetics. Angew. Chem. Int. Ed. 2014, 53, 3392–3395. [Google Scholar] [CrossRef]
- Lubbe, A.S.; Szymanski, W.; Feringa, B.L. Recent Developments in Reversible Photoregulation of Oligonucleotide Structure and Function. Chem. Soc. Rev. 2017, 46, 1052–1079. [Google Scholar] [CrossRef]
- Schweigert, C.; Babii, O.; Afonin, S.; Schober, T.; Leier, J.; Michenfelder, N.C.; Komarov, I.V.; Ulrich, A.S.; Unterreiner, A.N. Real-Time Observation of Diarylethene-Based Photoswitches in a Cyclic Peptide Environment. ChemPhotoChem 2019, 3, 403–410. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, L. Photoregulation between small DNAs and reversible photochromic molecules. Biomater. Sci. 2019, 7, 4944–4962. [Google Scholar] [CrossRef]
- Singer, M.; Jäschke, A. Reversibly Photoswitchable Nucleosides: Synthesis and Photochromic Properties of Diarylethene-Functionalized 7-Deazaadenosine Derivatives. J. Am. Chem. Soc. 2010, 132, 8372–8377. [Google Scholar] [CrossRef]
- Singer, M.; Nierth, A.; Jäschke, A. Photochromism of Diarylethene-Functionalized 7-Deazaguanosines. Eur. J. Org. Chem. 2013, 2013, 2766–2769. [Google Scholar] [CrossRef]
- Cahová, H.; Jäschke, A. Nucleoside-Based Diarylethene Photoswitches and Their Facile Incorporation into Photoswitchable DNA. Angew. Chem. Int. Ed. 2013, 52, 3186–3190. [Google Scholar] [CrossRef]
- Sarter, C.; Dey, S.; Jäschke, A. Photoswitchable Oligonucleotides Containing Different Diarylethene-Modified Nucleotides. ACS Omega 2019, 4, 12125–12129. [Google Scholar] [CrossRef] [Green Version]
- Buckup, T.; Sarter, C.; Volpp, H.-R.; Jäschke, A.; Motzkus, M. Ultrafast Time-Resolved Spectroscopy of Diarylethene-Based Photoswitchable Deoxyuridine Nucleosides. J. Phys. Chem. Lett. 2015, 6, 4717–4721. [Google Scholar] [CrossRef]
- Li, Y.; Lustres, J.L.P.; Volpp, H.-R.; Buckup, T.; Kolmar, T.; Jäschke, A.; Motzkus, M. Ultrafast Ring Closing of a Diarylethene-Based Photoswitchable Nucleoside. Phys. Chem. Chem. Phys. 2018, 20, 22867–22876. [Google Scholar] [CrossRef]
- Hania, P.R.; Pugžlys, A.; Lucas, L.N.; De Jong, J.J.D.; Feringa, B.L.; Van Esch, J.H.; Jonkman, H.T.; Duppen, K. Ring Closure Dynamics of BTE-Based Photochromic Switches: Perfluoro- versus Perhydrocyclopentene Derivatives. J. Phys. Chem. A 2005, 109, 9437–9442. [Google Scholar] [CrossRef]
- Shim, S.; Eom, I.; Joo, T.; Kim, E.; Kim, K.S. Ring Closure Reaction Dynamics of Diarylethene Derivatives in Solution. J. Phys. Chem. A 2007, 111, 8910–8917. [Google Scholar] [CrossRef]
- Aloïse, S.; Sliwa, M.; Pawlowska, Z.; Réhault, J.; Dubois, J.; Poizat, O.; Buntinx, G.; Perrier, A.; Maurel, F.; Yamaguchi, S.; et al. Bridged Photochromic Diarylethenes Investigated by Ultrafast Absorption Spectroscopy: Evidence for Two Distinct Photocy-clization Pathways. J. Am. Chem. Soc. 2010, 132, 7379–7390. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Fujiwara, M.; Umesato, T.; Saito, H.; Kobatake, S.; Irie, M.; Miyasaka, H. Cyclization Reaction Dynamics of a Photochromic Diarylethene Derivative as Revealed by Femtosecond to Microsecond Time-Resolved Spectroscopy. J. Phys. Chem. C 2011, 115, 4265–4272. [Google Scholar] [CrossRef]
- Jean-Ruel, H.; Cooney, R.R.; Gao, M.; Lu, C.; Kochman, M.A.; Morrison, C.A.; Miller, R.J.D. Femtosecond Dynamics of the Ring Closing Process of Diarylethene: A Case Study of Electrocyclic Reactions in Photochromic Single Crystals. J. Phys. Chem. A 2011, 115, 13158–13168. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.L.; Elles, C.G. Controlling the Excited-State Reaction Dynamics of a Photochromic Molecular Switch with Sequential Two-Photon Excitation. J. Phys. Chem. Lett. 2012, 3, 2995–3000. [Google Scholar] [CrossRef]
- Jean-Ruel, H.; Gao, M.; Kochman, M.A.; Lu, C.; Liu, L.C.; Cooney, R.R.; Morrison, C.A.; Miller, R.J.D. Ring-Closing Reaction in Diarylethene Captured by Femtosecond Electron Crystallography. J. Phys. Chem. B 2013, 117, 15894–15902. [Google Scholar] [CrossRef] [Green Version]
- Sumi, T.; Takagi, Y.; Yagi, A.; Morimoto, M.; Irie, M. Photoirradiation wavelength dependence of cycloreversion quantum yields of diarylethenes. Chem. Commun. 2014, 50, 3928–3930. [Google Scholar] [CrossRef]
- Ward, C.L.; Elles, C.G. Cycloreversion Dynamics of a Photochromic Molecular Switch via One-Photon and Sequential Two-Photon Excitation. J. Phys. Chem. A 2014, 118, 10011–10019. [Google Scholar] [CrossRef]
- Hamdi, I.; Buntinx, G.; Perrier, A.; Devos, O.; Jaïdane, N.; Delbaere, S.; Tiwari, A.K.; Dubois, J.; Takeshita, M.; Wada, Y.; et al. New insights into the photoswitching mechanisms of normal dithienylethenes. Phys. Chem. Chem. Phys. 2016, 18, 28091–28100. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Umesato, T.; Fujiwara, M.; Une, K.; Yoneda, Y.; Sotome, H.; Katayama, T.; Kobatake, S.; Asahi, T.; Irie, M.; et al. Solvent Polarity Dependence of Photochromic Reactions of a Diarylethene Derivative as Revealed by Steady-State and Tran-sient Spectroscopies. J. Phys. Chem. C 2016, 120, 1170–1177. [Google Scholar] [CrossRef]
- Schweighöfer, F.; Moreno, J.; Bobone, S.; Chiantia, S.; Herrmann, A.; Hecht, S.; Wachtveitl, J. Connectivity pattern modifies excited state relaxation dynamics of fluorophore–photoswitch molecular dyads. Phys. Chem. Chem. Phys. 2017, 19, 4010–4018. [Google Scholar] [CrossRef]
- Sotome, H.; Nagasaka, T.; Une, K.; Okui, C.; Ishibashi, Y.; Kamada, K.; Kobatake, S.; Irie, M.; Miyasaka, H. Efficient Cycloreversion Reaction of a Diarylethene Derivative in Higher Excited States Attained by Off-Resonant Simultaneous Two-Photon Absorption. J. Phys. Chem. Lett. 2017, 8, 3272–3276. [Google Scholar] [CrossRef]
- Barrez, E.; Laurent, G.; Pavageau, C.; Sliwa, M.; Metivier, R. Comparative photophysical investigation of doubly-emissive photochromic-fluorescent diarylethenes. Phys. Chem. Chem. Phys. 2017, 20, 2470–2479. [Google Scholar] [CrossRef] [PubMed]
- Sotome, H.; Kitagawa, D.; Nakahama, T.; Ito, S.; Kobatake, S.; Irie, M.; Miyasaka, H. Cyclization reaction dynamics of an inverse type diarylethene derivative as revealed by time-resolved absorption and fluorescence spectroscopies. Phys. Chem. Chem. Phys. 2019, 21, 8623–8632. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, I.; Buntinx, G.; Tiwari, A.K.; Delbaere, S.; Takeshita, M.; Aloïse, S. Cyclization Dynamics and Competitive Processes of Photochromic Perfluorocyclopentene Dithienylethylene in Solution. ChemPhysChem 2020, 21, 2223–2229. [Google Scholar] [CrossRef] [PubMed]
- Oplachko, M.V.; Smolentsev, A.B.; Magin, I.M.; Pozdnyakov, I.P.; Nichiporenko, V.A.; Grivin, V.P.; Plyusnin, V.F.; Vyazovkin, V.V.; Yanshole, V.V.; Parkhats, M.V.; et al. Mechanism of photochromic transformations and photodegradation of an asymmetrical 2,3-diarylcyclopentenone. Phys. Chem. Chem. Phys. 2020, 22, 5220–5228. [Google Scholar] [CrossRef]
- Sotome, H.; Okajima, H.; Nagasaka, T.; Tachii, Y.; Sakamoto, A.; Kobatake, S.; Irie, M.; Miyasaka, H. Geometrical Evolution and Formation of the Photoproduct in the Cycloreversion Reaction of a Diarylethene Derivative Probed by Vibrational Spectroscopy. ChemPhysChem. 2020, 21, 1524–1530. [Google Scholar] [CrossRef]
- Sotome, H.; Une, K.; Nagasaka, T.; Kobatake, S.; Irie, M.; Miyasaka, H. A dominant factor of the cycloreversion reactivity of diarylethene derivatives as revealed by femtosecond time-resolved absorption spectroscopy. J. Chem. Phys. 2020, 152, 034301. [Google Scholar] [CrossRef]
- Clark, A.E. Time-Dependent Density Functional Theory Studies of the Photoswitching of the Two-Photon Absorption Spectra in Stilbene, Metacyclophenadiene, and Diarylethene Chromophores. J. Phys. Chem. A 2006, 110, 3790–3796. [Google Scholar] [CrossRef]
- Perpète, E.A.; Maurel, F.; Jacquemin, D. TD−DFT Investigation of Diarylethene Dyes with Cyclopentene, Dihydrothiophene, and Dihydropyrrole Bridges. J. Phys. Chem. A 2007, 111, 5528–5535. [Google Scholar] [CrossRef]
- Indelli, M.T.; Carli, S.; Ghirotti, M.; Chiorboli, C.; Ravaglia, M.; Garavelli, M.; Scandola, F. Triplet Pathways in Diarylethene Photochromism: Photophysical and Computational Study of Dyads Containing Ruthenium(II) Polypyridine and 1,2-Bis(2-methylbenzothiophene-3-yl)maleimide Units. J. Am. Chem. Soc. 2008, 130, 7286–7299. [Google Scholar] [CrossRef]
- Patel, P.D.; Masunov, A.E. Theoretical Study of Photochromic Compounds. 1. Bond Length Alternation and Absorption Spectra for the Open and Closed Forms of 29 Diarylethene Derivatives. J. Phys. Chem. A 2009, 113, 8409–8414. [Google Scholar] [CrossRef]
- Tsuji, Y.; Staykov, A.; Yoshizawa, K. Orbital Control of the Conductance Photoswitching in Diarylethene. J. Phys. Chem. C 2009, 113, 21477–21483. [Google Scholar] [CrossRef]
- Chan, J.C.-H.; Lam, W.H.; Wong, H.-L.; Zhu, N.; Wong, W.-T.; Yam, V.W.-W. Diarylethene-Containing Cyclometalated Platinum(II) Complexes: Tunable Photochromism via Metal Coordination and Rational Ligand Design. J. Am. Chem. Soc. 2011, 133, 12690–12705. [Google Scholar] [CrossRef]
- Liu, S.; Sun, S.; Wang, C.-M.; Zhao, Q.; Sun, H.; Li, F.; Fan, Q.; Huang, W. DFT/TDDFT Investigation of the Modulation of Photochromic Properties in an Organoboron-Based Diarylethene by Fluoride Ions. ChemPhysChem 2010, 12, 313–321. [Google Scholar] [CrossRef]
- Patel, P.D.; Masunov, A.E. Theoretical Study of Photochromic Compounds: Part 3. Prediction of Thermal Stability. J. Phys. Chem. C 2011, 115, 10292–10297. [Google Scholar] [CrossRef]
- Perrier, A.; Maurel, F.; Jacquemin, D. Diarylethene–dihydroazulene multimode photochrome: A theoretical spectroscopic investigation. Phys. Chem. Chem. Phys. 2011, 13, 13791–13799. [Google Scholar] [CrossRef]
- Okuno, K.; Shigeta, Y.; Kishi, R.; Miyasaka, H.; Nakano, M. Tuned CAM-B3LYP functional in the time-dependent density functional theory scheme for excitation energies and properties of diarylethene derivatives. J. Photochem. Photobiol. A Chem. 2012, 235, 29–34. [Google Scholar] [CrossRef]
- Barone, V.; Cacelli, I.; Ferretti, A.; Visciarelli, M. Electron Transport Properties of Diarylethene Photoswitches by a Simplified NEGF-DFT Approach. J. Phys. Chem. B 2014, 118, 4976–4981. [Google Scholar] [CrossRef]
- Chantzis, A.; Cerezo, J.; Perrier, A.; Santoro, F.; Jacquemin, D. Optical Properties of Diarylethenes with TD-DFT: 0–0 Energies, Fluorescence, Stokes Shifts, and Vibronic Shapes. J. Chem. Theory Comput. 2014, 10, 3944–3957. [Google Scholar] [CrossRef]
- Fihey, A.; Kloss, B.; Perrier, A.; Maurel, F. Density Functional Theory Study of the Conformation and Optical Properties of Hybrid Au-N-Dithienylethene Systems (N = 3, 19, 25). J. Phys. Chem. A 2014, 118, 4695–4706. [Google Scholar] [CrossRef]
- Fihey, A.; Jacquemin, D. Designing efficient photochromic dithienylethene dyads. Chem. Sci. 2015, 6, 3495–3504. [Google Scholar] [CrossRef] [Green Version]
- Lasorne, B.; Fihey, A.; Mendive-Tapia, D.; Jacquemin, D. A curve-crossing model to rationalize and optimize diarylethene dyads. Chem. Sci. 2015, 6, 5695–5702. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, S.; Hasegawa, J.-Y.; Matsuda, K. Computational Investigation into Photoswitching Efficiency of Diarylethene Derivatives: An Insight Based on the Decay Constant of Electron Tunneling. J. Phys. Chem. C 2015, 119, 20169–20178. [Google Scholar] [CrossRef]
- Okuno, K.; Shigeta, Y.; Kishi, R.; Nakano, M. Theoretical design of solvatochromism switching by photochromic reactions using donor–acceptor disubstituted diarylethene derivatives with oxidized thiophene rings. Phys. Chem. Chem. Phys. 2015, 17, 6484–6494. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Bai, F.-Q.; Li, L.; Zhang, H.-X. Theoretical investigation on a series of novel S,S-dioxide diarylethenes with abnormal photochromic properties and design of new dyads. New J. Chem. 2014, 39, 1634–1642. [Google Scholar] [CrossRef]
- Fihey, A.; Russo, R.; Cupellini, L.; Jacquemin, D.; Mennucci, B. Is energy transfer limiting multiphotochromism? answers from ab initio quantifications. Phys. Chem. Chem. Phys. 2016, 19, 2044–2052. [Google Scholar] [CrossRef]
- Hamdi, I.; Buntinx, G.; Poizat, O.; Delbaere, S.; Perrier, A.; Yamashita, R.; Muraoka, K.-I.; Takeshita, M.; Aloïse, S. Unraveling ultrafast dynamics of the photoswitchable bridged dithienylethenes under structural constraints. Phys. Chem. Chem. Phys. 2019, 21, 6407–6414. [Google Scholar] [CrossRef]
- Avramopoulos, A.; Zaleśny, R.; Reis, H.; Papadopoulos, M.G. A Computational Strategy for the Design of Photochromic Derivatives Based on Diarylethene and Nickel Dithiolene with Large Contrast in Nonlinear Optical Properties. J. Phys. Chem. C 2020, 124, 4221–4241. [Google Scholar] [CrossRef]
- Le Bras, L.; Berthin, R.; Hamdi, I.; Louati, M.; Aloïse, S.; Takeshita, M.; Adamo, C.; Perrier, A. Understanding the properties of dithienylethenes functionalized for supramolecular self-assembly: A molecular modeling study. Phys. Chem. Chem. Phys. 2020, 22, 6942–6952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; He, L.; Feng, J.; Liu, X.; Zhou, L.; Zhang, H. Unveiling the Relationship between Energy Transfer and the Triplet Energy Level by Tuning Diarylethene within Europium(III) Complexes. Inorg. Chem. 2020, 59, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Guillaumont, D.; Kobayashi, T.; Kanda, K.; Miyasaka, H.; Uchida, K.; Kobatake, S.; Shibata, K.; Nakamura, S.; Irie, M. An ab Initio MO Study of the Photochromic Reaction of Dithienylethenes. J. Phys. Chem. A 2002, 106, 7222–7227. [Google Scholar] [CrossRef]
- Boggio-Pasqua, M.; Ravaglia, M.; Bearpark, M.J.; Garavelli, M.; Robb, M.A. Can Diarylethene Photochromism Be Explained by a Reaction Path Alone? A CASSCF Study with Model MMVB Dynamics. J. Phys. Chem. A 2003, 107, 11139–11152. [Google Scholar] [CrossRef]
- Asano, Y.; Murakami, A.; Kobayashi, T.; Goldberg, A.; Guillaumont, D.; Yabushita, S.; Irie, M.; Nakamura, S. Theoretical Study on the Photochromic Cycloreversion Reactions of Dithienylethenes; on the Role of the Conical Intersections. J. Am. Chem. Soc. 2004, 126, 12112–12120. [Google Scholar] [CrossRef]
- Nakamura, S.; Yokojima, S.; Uchida, K.; Tsujioka, T.; Goldberg, A.; Murakami, A.; Shinoda, K.; Mikami, M.; Kobayashi, T.; Kobatake, S.; et al. Theoretical investigation on photochromic diarylethene: A short review. J. Photochem. Photobiol. A Chem. 2008, 200, 10–18. [Google Scholar] [CrossRef]
- Perrier, A.; Aloise, S.; Olivucci, M.; Jacquemin, D. Inverse versus Normal Dithienylethenes: Computational Investigation of the Photocyclization Reaction. J. Phys. Chem. Lett. 2013, 4, 2190–2196. [Google Scholar] [CrossRef] [Green Version]
- Mendive-Tapia, D.; Perrier, A.; Bearpark, M.J.; Robb, M.A.; Lasorne, B.; Jacquemin, D. New insights into the by-product fatigue mechanism of the photo-induced ring-opening in diarylethenes. Phys. Chem. Chem. Phys. 2014, 16, 18463–18471. [Google Scholar] [CrossRef]
- Wiebeler, C.; Bader, C.A.; Meier, C.; Schumacher, S. Optical spectrum, perceived color, refractive index, and non-adiabatic dynamics of the photochromic diarylethene CMTE. Phys. Chem. Chem. Phys. 2014, 16, 14531–14538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-T.; Gao, Y.-J.; Wang, Q.; Cui, G.L. Photochromic Mechanism of a Bridged Diarylethene: Combined Electronic Structure Calculations and Non-adiabatic Dynamics Simulations. J. Phys. Chem. A 2017, 121, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Chiariello, M.G.; Raucci, U.; Coppola, F.; Rega, N. Unveiling anharmonic coupling by means of excited state ab initio dynamics: Application to diarylethene photoreactivity. Phys. Chem. Chem. Phys. 2018, 21, 3606–3614. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W.T. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef] [Green Version]
- Francés-Monerris, A.; Holub, J.; Roca-Sanjuán, D.; Hnyk, D.; Lang, K.; Oliva-Enrich, J.M. Photochromic System among Boron Hydrides: The Hawthorne Rearrangement. J. Phys. Chem. Lett. 2019, 10, 6202–6207. [Google Scholar] [CrossRef]
- Karlström, G.; Lindh, R.; Malmqvist, P.-Å.; Roos, O.B.; Ryde, U.; Veryazov, V.; Widmark, P.-O.; Cossi, M.; Schimmelpfennig, B.; Neogrady, P.; et al. MOLCAS: A program package for computational chemistry. Comput. Mater. Sci. 2003, 28, 222–239. [Google Scholar] [CrossRef]
- Nelson, T.; Fernandez-Alberti, S.; Roitberg, A.E.; Tretiak, S. Non-adiabatic Excited-State Molecular Dynamics: Modeling Photophysics in Organic Conjugated Materials. Acc. Chem. Res. 2014, 47, 1155–1164. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S.; Tully, J.C. Proton transfer in solution: Molecular dynamics with quantum transitions. J. Chem. Phys. 1994, 101, 4657–4667. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Xu, C.; Zhu, C. Probing the π → π* photoisomerization mechanism of cis-azobenzene by multi-state ab initio on-the-fly trajectory dynamics simulation. Phys. Chem. Chem. Phys. 2015, 17, 17646–17660. [Google Scholar] [CrossRef]
- Cui, G.; Thiel, W. Generalized trajectory surface-hopping method for internal conversion and intersystem crossing. J. Chem. Phys. 2014, 141, 124101. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.-Y.; Fang, Y.-G.; Xie, B.-B.; Fang, W.-H.; Cui, G.L. QM/MM Non-adiabatic Dynamics Simulations on Photoinduced Wolff Rearrangements of 1,2,3-Thiadiazole. J. Chem. Phys. 2017, 146, 224302. [Google Scholar] [CrossRef]
- Wigner, E. On the Quantum Correction for Thermodynamic Equilibrium. Phys. Rev. 1932, 40, 749–759. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Aquilante, F.; Lindh, R.; Pedersen, T.B. Unbiased auxiliary basis sets for accurate two-electron integral approximations. J. Chem. Phys. 2007, 127, 114107. [Google Scholar] [CrossRef] [PubMed]
- Ghigo, G.; Roos, B.O.; Malmqvist, P.-Å. A modified definition of the zeroth-order Hamiltonian in multiconfigurational perturbation theory (CASPT2). Chem. Phys. Lett. 2004, 396, 142–149. [Google Scholar] [CrossRef]
- Forsberg, N.; Malmqvist, P.-Å. Multiconfiguration perturbation theory with imaginary level shift. Chem. Phys. Lett. 1997, 274, 196–204. [Google Scholar] [CrossRef]
- Chang, X.-P.; Li, C.-X.; Xie, B.-B.; Cui, G.L. Photoprotection Mechanism of P-Methoxy Methylcinnamate: A CASPT2 Study. J. Phys. Chem. A 2015, 119, 11488–11497. [Google Scholar] [CrossRef]
- Fang, Y.-G.; Li, C.-X.; Chang, X.-P.; Cui, G. Photophysics of a UV-B Filter 4-Methylbenzylidene Camphor: Intersystem Crossing Plays an Important Role. ChemPhysChem 2018, 19, 744–752. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Chang, X.-P.; Xia, S.-H.; Cui, G.L.; Thiel, W. Excited-State Proton-Transfer-Induced Trapping Enhances the Fluo-rescence Emission of a Locked Gfp Chromophore. J. Chem. Theory Comput. 2016, 12, 753–764. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheesem, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Aquilante, F.; De Vico, L.; Ferré, N.; Ghigo, G.; Malmqvist, P.-Å.; Neogrády, P.; Pedersen, T.B.; Pitoňák, M.; Reiher, M.; Roos, B.O.; et al. MOLCAS 7: The Next Generation. J. Comput. Chem. 2010, 31, 224–247. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Barbatti, M.; Aquino, A.J.A.; Szymczak, J.J.; Nachtigallová, D.; Hobza, P.; Lischka, H. Relaxation mechanisms of UV-photoexcited DNA and RNA nucleobases. Proc. Natl. Acad. Sci. USA 2010, 107, 21453–21458. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Lan, Z.; Thiel, W. Hydrogen Bonding Regulates the Monomeric Nonradiative Decay of Adenine in DNA Strands. Angew. Chem. Int. Ed. 2011, 50, 6864–6867. [Google Scholar] [CrossRef]
- Cui, G.; Lan, Z.; Thiel, W. Intramolecular Hydrogen Bonding Plays a Crucial Role in the Photophysics and Photochemistry of the GFP Chromophore. J. Am. Chem. Soc. 2012, 134, 1662–1672. [Google Scholar] [CrossRef]
- Long, R.; English, N.J.; Prezhdo, O.V. Photo-induced Charge Separation across the Graphene–TiO2 Interface Is Faster than Energy Losses: A Time-Domain ab Initio Analysis. J. Am. Chem. Soc. 2012, 134, 14238–14248. [Google Scholar] [CrossRef]
- Fischer, S.A.; Chapman, C.T.; Li, X. Surface hopping with Ehrenfest excited potential. J. Chem. Phys. 2011, 135, 144102. [Google Scholar] [CrossRef]
- Fischer, S.A.; Lingerfelt, D.B.; May, J.W.; Li, X. Non-adiabatic molecular dynamics investigation of photoionization state formation and lifetime in Mn2+-doped ZnO quantum dots. Phys. Chem. Chem. Phys. 2014, 16, 17507–17514. [Google Scholar] [CrossRef]
- Richter, M.; Marquetand, P.; González-Vázquez, J.; Sola, I.; González, L. SHARC: Ab Initio Molecular Dynamics with Surface Hopping in the Adiabatic Representation Including Arbitrary Couplings. J. Chem. Theory Comput. 2011, 7, 1253–1258. [Google Scholar] [CrossRef]
- Richter, M.; Marquetand, P.; González-Vázquez, J.; Sola, I.; González, L. Femtosecond Intersystem Crossing in the DNA Nu-cleobase Cytosine. J. Phys. Chem. Lett. 2012, 3, 3090–3095. [Google Scholar] [CrossRef]
- Wang, Y.-T.; Liu, X.-Y.; Cui, G.; Fang, W.-H.; Thiel, W. Photoisomerization of Arylazopyrazole Photoswitches: Stereospecific Excited-State Relaxation. Angew. Chem. Int. Ed. 2016, 55, 14009–14013. [Google Scholar] [CrossRef]
- Xia, S.-H.; Cui, G.; Fang, W.-H.; Thiel, W. How Photoisomerization Drives Peptide Folding and Unfolding: Insights from QM/MM and MM Dynamics Simulations. Angew. Chem. Int. Ed. 2016, 55, 2067–2072. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.-J.; Chang, X.-P.; Liu, X.-Y.; Li, Q.-S.; Cui, G.; Thiel, W. Excited-State Decay Paths in Tetraphenylethene Derivatives. J. Phys. Chem. A 2017, 121, 2572–2579. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Sun, X.W.; Zhang, T.S.; Liu, X.Y.; Cui, G.L. Non-adiabatic Dynamics Simulations on Early-Time Photochemistry of Spirobenzopyran. J. Phys. Chem. A 2020, 124, 2547–2559. [Google Scholar] [CrossRef]
- Tully, J.C.; Preston, R.K. Trajectory Surface Hopping Approach to Non-adiabatic Molecular Collisions: Reaction of H+ with D2. J. Chem. Phys. 1971, 55, 562–572. [Google Scholar] [CrossRef]
- Shen, L.; Xie, B.; Li, Z.; Liu, L.; Cui, G.; Fang, W.-H. Role of Multistate Intersections in Photochemistry. J. Phys. Chem. Lett. 2020, 11, 8490–8501. [Google Scholar] [CrossRef]
- Martínez, T.J. Insights for Light-Driven Molecular Devices from Ab Initio Multiple Spawning Excited-State Dynamics of Or-ganic and Biological Chromophores. Acc. Chem. Res. 2006, 39, 119–126. [Google Scholar] [CrossRef]
- Yu, L.; Xu, C.; Lei, Y.; Zhu, C.; Wen, Z. Trajectory-Based Non-adiabatic Molecular Dynamics without Calculating Non-adiabatic Coupling in the Avoided Crossing Case: Trans Cis Photoisomerization in Azobenzene. Phys. Chem. Chem. Phys. 2014, 16, 25883–25895. [Google Scholar] [CrossRef]
- Zhu, C.; Nakamura, H. Theory of Non-adiabatic Transition for General Two-State Curve Crossing Problems. I. Non-adiabatic Tunneling Case. J. Chem. Phys. 1994, 101, 10630–10647. [Google Scholar] [CrossRef]
- Zhu, C.; Nakamura, H. Theory of Non-adiabatic Transition for General Two-State Curve Crossing Problems. II. Landau-Zener Case. J. Chem. Phys. 1995, 102, 7448–7461. [Google Scholar] [CrossRef]
- Zhu, C.; Nobusada, K.; Nakamura, H. New implementation of the trajectory surface hopping method with use of the Zhu–Nakamura theory. J. Chem. Phys. 2001, 115, 3031–3044. [Google Scholar] [CrossRef]
- Barbatti, M.; Aquino, A.J.A.; Lischka, H.; Schriever, C.; Lochbrunner, S.; Riedle, E. Ultrafast internal conversion pathway and mechanism in 2-(2′-hydroxyphenyl)benzothiazole: A case study for excited-state intramolecular proton transfer systems. Phys. Chem. Chem. Phys. 2009, 11, 1406–1415. [Google Scholar] [CrossRef]
- Li, C.-X.; Guo, W.-W.; Xie, B.-B.; Cui, G.L. Photodynamics of Oxybenzone Sunscreen: Non-adiabatic Dynamics Simulations. J. Chem. Phys. 2016, 145, 074308. [Google Scholar] [CrossRef]
- Freitag, L.; González, L. Theoretical Spectroscopy and Photodynamics of a Ruthenium Nitrosyl Complex. Inorg. Chem. 2014, 53, 6415–6426. [Google Scholar] [CrossRef] [PubMed]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond Orders | C1–C2 | C2–C3 | C3–C4 | C4–C5 | C5–C6 | C1–C6 |
|---|---|---|---|---|---|---|
| S0-O | 1.663 | 1.026 | 1.840 | 1.000 | 1.596 | 0.004 |
| S0-C | 0.939 | 1.710 | 1.141 | 1.683 | 0.964 | 0.972 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, D.-H.; Li, L.; Liu, X.-Y.; Cui, G. “On-The-Fly” Non-Adiabatic Dynamics Simulations on Photoinduced Ring-Closing Reaction of a Nucleoside-Based Diarylethene Photoswitch. Molecules 2021, 26, 2724. https://doi.org/10.3390/molecules26092724
Xu D-H, Li L, Liu X-Y, Cui G. “On-The-Fly” Non-Adiabatic Dynamics Simulations on Photoinduced Ring-Closing Reaction of a Nucleoside-Based Diarylethene Photoswitch. Molecules. 2021; 26(9):2724. https://doi.org/10.3390/molecules26092724
Chicago/Turabian StyleXu, Dong-Hui, Laicai Li, Xiang-Yang Liu, and Ganglong Cui. 2021. "“On-The-Fly” Non-Adiabatic Dynamics Simulations on Photoinduced Ring-Closing Reaction of a Nucleoside-Based Diarylethene Photoswitch" Molecules 26, no. 9: 2724. https://doi.org/10.3390/molecules26092724