
Evaluation of Virtual Screening Strategies for the Identification of γ-Secretase Inhibitors and Modulators


Abstract
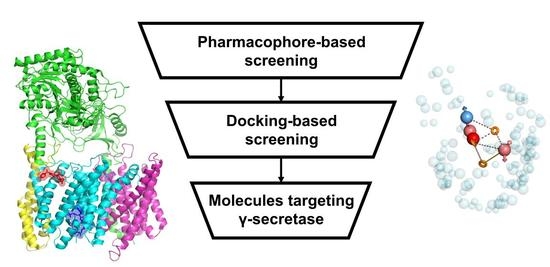
1. Introduction
2. Results
2.1. Evaluation of Docking and Pharmacophore Modelling for Pose Prediction of γ-Secretase Inhibitors and Modulators
2.2. Derivation of Virtual Screening Strategies for γ-Secretase Inhibitors and Modulators
2.3. Application of Optimal Virtual Screening Strategies to the ZINC15 Investigational Set
3. Discussion
4. Materials and Methods
4.1. Selection and Preparation of γ-Secretase Structures
4.2. Molecular Docking
4.3. Pharmacophore Modelling and Screening
4.4. Validation of Pose Prediction
4.5. Optimisation of Virtual Screening for Identifying γ-Secretase Inhibitors and γ-Secretase Modulators
4.6. Evaluation of Virtual Screening Performance
4.7. Application of Optimised Virtual Screening Procedures to an External Test Set
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lichtenthaler, S.F.; Steiner, H. Sheddases and intramembrane-cleaving proteases: RIPpers of the membrane. Symposium on regulated intramembrane proteolysis. EMBO J. 2007, 8, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimers Dis. 2011, 25, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A. Activity of γ-secretase on substrates other than APP. Curr. Top. Med. Chem. 2008, 8, 9–16. [Google Scholar] [CrossRef]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Yu, G.; Nishimura, M.; Arawaka, S.; Levitan, D.; Zhang, L.; Tandon, A.; Song, Y.Q.; Rogaeva, E.; Chen, F.; Kawarai, T.; et al. Nicastrin modulates presenilin-mediated Notch/glp-1 signal transduction and βAPP processing. Nature 2000, 407, 48–54. [Google Scholar] [CrossRef]
- Goutte, C.; Tsunozaki, M.; Hale, V.A.; Priess, J.R. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc. Natl. Acad. Sci. USA 2002, 99, 775–779. [Google Scholar] [CrossRef]
- Francis, R.; McGrath, G.; Zhang, J.; Ruddy, D.A.; Sym, M.; Apfeld, J.; Nicoll, M.; Maxwell, M.; Hai, B.; Ellis, M.C.; et al. Aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev. Cell 2002, 3, 85–97. [Google Scholar] [CrossRef]
- Kimberly, W.T.; Xia, W.; Rahmati, T.; Wolfe, M.S.; Selkoe, D.J. The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J. Biol. Chem. 2000, 275, 3173–3178. [Google Scholar] [CrossRef]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Golde, T.E.; Koo, E.H.; Felsenstein, K.M.; Osborne, B.A.; Miele, L. γ-Secretase inhibitors and modulators. Biochim. Biophys. Acta 2013, 1828, 2898–2907. [Google Scholar] [CrossRef]
- Borgegård, T.; Gustavsson, S.; Nilsson, C.; Parpal, S.; Klintenberg, R.; Berg, A.-L.; Rosqvist, S.; Serneels, L.; Svensson, S.; Olsson, F.; et al. Alzheimer’s disease: Presenilin 2-sparing γ-secretase inhibition is a tolerable Aβ peptide-lowering strategy. J. Neurosci. 2012, 32, 17297–17305. [Google Scholar] [CrossRef]
- Beher, D.; Clarke, E.E.; Wrigley, J.D.; Martin, A.C.; Nadin, A.; Churcher, I.; Shearman, M.S. Selected non-steroidal anti-inflammatory drugs and their derivatives target γ-secretase at a novel site: Evidence for an allosteric mechanism. J. Biol. Chem. 2004, 279, 43419–43426. [Google Scholar] [CrossRef]
- Gillman, K.W.; Starrett, J.E., Jr.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.E.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. NEJM 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Coric, V.; van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and tolerability of the γ-secretase inhibitor avagacestat in a phase 2 study of mild to moderate Alzheimer disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef]
- Barthet, G.; Georgakopoulos, A.; Robakis, N.K. Cellular mechanisms of γ-secretase substrate selection, processing and toxicity. Prog. Neurobiol. 2012, 98, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Shearman, M.S.; Beher, D.; Clarke, E.E.; Lewis, H.D.; Harrison, T.; Hunt, P.; Nadin, A.; Smith, A.L.; Stevenson, G.; Castro, J.L. L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 2000, 39, 8698–8704. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Y.; Li, J.M.; Xiao, L.; Mou, L.; Cai, Y.; Huang, H.; Luo, X.G.; Yan, X.X. [(3) H]-L685,458 binding sites are abundant in multiple peripheral organs in rats: Implications for safety assessment of putative γ-secretase targeting drugs. Basic Clin. Pharmacol. Toxicol. 2014, 115, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Esler, W.P.; Kimberly, W.T.; Ostaszewski, B.L.; Ye, W.; Diehl, T.S.; Selkoe, D.J.; Wolfe, M.S. Activity-dependent isolation of the presenilin-γ-secretase complex reveals nicastrin and a γ substrate. Proc. Natl. Acad. Sci. USA 2002, 99, 2720–2725. [Google Scholar] [CrossRef] [PubMed]
- Morohashi, Y.; Kan, T.; Tominari, Y.; Fuwa, H.; Okamura, Y.; Watanabe, N.; Sato, C.; Natsugari, H.; Fukuyama, T.; Iwatsubo, T.; et al. C-terminal fragment of presenilin is the molecular target of a dipeptidic γ-secretase-specific inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester). J. Biol. Chem. 2006, 281, 14670–14676. [Google Scholar] [CrossRef]
- May, P.; Yang, Z.; Li, W.; Hyslop, P.; Siemers, E.; Boggs, L. Multi-compartmental pharmacodynamic assessment of the functional γ-secretase inhibitor LY450139 in PDAPP transgenic mice and non-transgenic mice. Neurobiol. Aging 2004, 25, S65. [Google Scholar] [CrossRef]
- In’T Veld, B.A.; Ruitenberg, A.; Hofman, A.; Launer, L.J.; van Duijn, C.M.; Stijnen, T.; Breteler, M.M.; Stricker, B.H. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N. Engl. J. Med. 2001, 345, 1515–1521. [Google Scholar] [CrossRef] [PubMed]
- Etminan, M.; Gill, S.; Samii, A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: Systematic review and meta-analysis of observational studies. BMJ 2003, 327, 128–131. [Google Scholar] [CrossRef]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef]
- Kounnas, M.Z.; Danks, A.M.; Cheng, S.; Tyree, C.; Ackerman, E.; Zhang, X.; Ahn, K.; Nguyen, P.; Comer, D.; Mao, L.; et al. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron 2010, 67, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Kounnas, M.Z.; Lane-Donovan, C.; Nowakowski, D.W.; Herz, J.; Comer, W.T. NGP 555, a γ-secretase modulator, lowers the amyloid biomarker, Aβ42, in cerebrospinal fluid while preventing Alzheimer’s disease cognitive decline in rodents. Alzheimers Dement. 2017, 3, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Oehlrich, D.; Berthelot, D.J.; Gijsen, H.J. γ-Secretase modulators as potential disease modifying anti-Alzheimer’s drugs. J. Med. Chem. 2011, 54, 669–698. [Google Scholar] [CrossRef]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 2015, 4, e11182. [Google Scholar] [CrossRef]
- Bai, X.C.; Yan, C.; Yang, G.; Lu, P.; Ma, D.; Sun, L.; Zhou, R.; Scheres, S.H.W.; Shi, Y. An atomic structure of human γ-secretase. Nature 2015, 525, 212–217. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human γ-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human γ-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef]
- Yang, G.; Zhou, R.; Guo, X.; Yan, C.; Lei, J.; Shi, Y. Structural basis of γ-secretase inhibition and modulation by small molecule drugs. Cell 2021, 184, 521–533.e514. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free Energy Methods in Drug Design: Prospects of “Alchemical Perturbation” in Medicinal Chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef]
- Kimura, T.; Kawano, K.; Doi, E.; Kitazawa, N.; Takaishi, M.; Ito, K.; Kaneko, T.; Sasaki, T.; Sato, N.; Miyagawa, T.; et al. Morpholine Type Cinnamide. Patent No. US20070117798A1, 17 November 2009. [Google Scholar]
- Kimura, T.; Kawano, K.; Doi, E.; Kitazawa, N.; Takaishi, M.; Ito, K.; Kaneko, T.; Sasaki, T.; Miyagawa, T.; Hagiwara, H.; et al. Two Cyclic Cinnamide Compound. Patent No. US20070117839, 8 November 2006. [Google Scholar]
- Fischer, C.; Munoz, B.; Zultanski, S.; Fischer, C.; Zhou, H.; Brown, W.C. Triazole Derivatives for Treating Alzheimer’s Disease and Related Conditions. Patent No. WO2008156580, 9 June 2008. [Google Scholar]
- Huang, X.; Aslanian, R.; Zhou, W.; Zhu, X.; Qin, J.; Greenlee, W.; Zhu, Z.; Zhang, L.; Hyde, L.; Chu, I.; et al. The Discovery of Pyridone and Pyridazone Heterocycles as γ-Secretase Modulators. ACS Med. Chem. Lett. 2010, 1, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K.; Flohr, A.; Jacobsen, H.; Jolidon, S.; Luebbers, T. Hetarylanilines as Modulators for Amyloid Beta. Patent no. WO2008138753, 30 April 2008. [Google Scholar]
- Goetschi, E.; Jolidon, S.; Luebbers, T. Modulators for Amyloid Beta. Patent No. WO2010040661, 29 September 2009. [Google Scholar]
- Fischer, C.; Methot, J.; Zhou, H.; Schell, A.J.; Munoz, B.; Rivkin, A.A.; Ahearn, S.P.; Chichetti, S.; Maccoss, R.N.; Kattar, S.D.; et al. Triazole Derivatives for Treatment of Alzheimer’s Disease. Patent No. WO2010071741, 7 December 2009. [Google Scholar]
- Kimura, T.; Doi, E.; Doko, T.; Shinmyo, D.; Ito, K.; Sato, N.; Hasegawa, D.; Uemera, T.; Watanabe, T. Aryl Imidazole Compounds and Their Use as Beta Amyloid Production Inhibitors. Patent No. WO2010098488, 24 February 2010. [Google Scholar]
- Kitazawa, N.; Shinmyo, D.; Ito, K.; Sato, N.; Hasegawa, D.; Uemera, T.; Watanabe, T. Nitrogen Containing Fused Heterocyclic Compounds and Their Use as Beta Amyloid Production Inhibitors. Patent No. WO2010098487, 24 February 2010. [Google Scholar]
- Gu, K.; Li, Q.; Lin, H.; Zhu, J.; Mo, J.; He, S.; Lu, X.; Jiang, X.; Sun, H. Gamma secretase inhibitors: A patent review (2013–2015). Expert Opin. Ther. Pat. 2017, 27, 851–866. [Google Scholar] [CrossRef]
- Lan, X.; Kiyota, T.; Hanamsagar, R.; Huang, Y.; Andrews, S.; Peng, H.; Zheng, J.C.; Swindells, S.; Carlson, G.A.; Ikezu, T. The effect of HIV protease inhibitors on amyloid-β peptide degradation and synthesis in human cells and Alzheimer’s disease animal model. J. Neuroimmune Pharmacol. 2012, 7, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Höttecke, N.; Liebeck, M.; Baumann, K.; Schubenel, R.; Winkler, E.; Steiner, H.; Schmidt, B. Inhibition of γ-secretase by the CK1 inhibitor IC261 does not depend on CK1δ. Bioorg. Med. Chem. Lett. 2010, 20, 2958–2963. [Google Scholar] [CrossRef] [PubMed]
- Amombo, G.M.O.; Kramer, T.; Lo Monte, F.; Göring, S.; Fach, M.; Smith, S.; Kolb, S.; Schubenel, R.; Baumann, K.; Schmidt, B. Modification of a promiscuous inhibitor shifts the inhibition from γ-secretase to FLT-3. Bioorg. Med. Chem. Lett. 2012, 22, 7634–7640. [Google Scholar] [CrossRef] [PubMed]
- Narlawar, R.; Baumann, K.; Czech, C.; Schmidt, B. Conversion of the LXR-agonist TO-901317—From inverse to normal modulation of γ-secretase by addition of a carboxylic acid and a lipophilic anchor. Bioorg. Med. Chem. Lett. 2007, 17, 5428–5431. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Webber, S.E.; Marakovits, J.T.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Ford, C.E.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J. Med. Chem. 1999, 42, 1213–1224. [Google Scholar] [CrossRef]
- Greer, J.; Erickson, J.W.; Baldwin, J.J.; Varney, M.D. Application of the three-dimensional structures of protein target molecules in structure-based drug design. J. Med. Chem. 1994, 37, 1035–1054. [Google Scholar] [CrossRef] [PubMed]
- Kale, A.J.; Moore, B.S. Molecular mechanisms of acquired proteasome inhibitor resistance. J. Med. Chem. 2012, 55, 10317–10327. [Google Scholar] [CrossRef]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef]
- Säfholm, A.; Tuomela, J.; Rosenkvist, J.; Dejmek, J.; Härkönen, P.; Andersson, T. The Wnt-5a–Derived Hexapeptide Foxy-5 Inhibits Breast Cancer Metastasis In Vivo by Targeting Cell Motility. Clin. Cancer Res. 2008, 14, 6556–6563. [Google Scholar] [CrossRef] [PubMed]
- Millan, D.S.; Bunnage, M.E.; Burrows, J.L.; Butcher, K.J.; Dodd, P.G.; Evans, T.J.; Fairman, D.A.; Hughes, S.J.; Kilty, I.C.; Lemaitre, A.; et al. Design and synthesis of inhaled p38 inhibitors for the treatment of chronic obstructive pulmonary disease. J. Med. Chem. 2011, 54, 7797–7814. [Google Scholar] [CrossRef]
- Araya, E.; Rodriguez, A.; Rubio, J.; Spada, A.; Joglar, J.; Llebaria, A.; Lagunas, C.; Fernandez, A.G.; Spisani, S.; Perez, J.J. Synthesis and evaluation of diverse analogs of amygdalin as potential peptidomimetics of peptide T. Bioorg. Med. Chem. Lett. 2005, 15, 1493–1496. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Inhibition of Notch Pathway Signaling: A One Compound Mission to Treat Cancer. ACS Med. Chem. Lett. 2013, 4, 373–374. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chin, N.X.; Gu, J.W.; Neu, H.C. In vitro activity of cefcanel versus other oral cephalosporins. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 676–682. [Google Scholar] [CrossRef]
- Delong, M.A.; Sznaidman, M.L.; Oakley, R.H.; Eckhardt, A.E.; Hudson, C.; Yingling, J.D.; Peel, M.; Richardson, T.E.; Murray, C.L.; Rao, B.N.N.; et al. Isoquinoline Compounds. Patent No. US20070142429, 30 December 2008. [Google Scholar]
- Smidt, M.L.; Potts, K.E.; Tucker, S.P.; Blystone, L.; Stiebel, T.R.; Stallings, W.C.; McDonald, J.J.; Pillay, D.; Richman, D.D.; Bryant, M.L. A mutation in human immunodeficiency virus type 1 protease at position 88, located outside the active site, confers resistance to the hydroxyethylurea inhibitor SC-55389A. Antimicrob. Agents Chemother. 1997, 41, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Cooper, K.; Fray, M.J.; Parry, M.J.; Richardson, K.; Steele, J. 1,4-Dihydropyridines as antagonists of platelet activating factor. 1. Synthesis and structure-activity relationships of 2-(4-heterocyclyl)phenyl derivatives. J. Med. Chem. 1992, 35, 3115–3129. [Google Scholar] [CrossRef] [PubMed]
- Yotsuji, A.; Mitsuyama, J.; Hori, R.; Yasuda, T.; Saikawa, I.; Inoue, M.; Mitsuhashi, S. Mechanism of action of cephalosporins and resistance caused by decreased affinity for penicillin-binding proteins in Bacteroides fragilis. Antimicrob. Agents Chemother. 1988, 32, 1848–1853. [Google Scholar] [CrossRef] [PubMed]
- Bühlmayer, P.; Caselli, A.; Fuhrer, W.; Göschke, R.; Rasetti, V.; Rüeger, H.; Stanton, J.L.; Criscione, L.; Wood, J.M. Synthesis and biological activity of some transition-state inhibitors of human renin. J. Med. Chem. 1988, 31, 1839–1846. [Google Scholar] [CrossRef]
- Repine, J.T.; Himmelsbach, R.J.; Hodges, J.C.; Kaltenbronn, J.S.; Sircar, I.; Skeean, R.W.; Brennan, S.T.; Hurley, T.R.; Lunney, E.; Humblet, C.C.; et al. Renin inhibitors containing esters at the P2-position. Oral activity in a derivative of methyl aminomalonate. J. Med. Chem. 1991, 34, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Daines, R.A.; Chambers, P.A.; Foley, J.J.; Griswold, D.E.; Kingsbury, W.D.; Martin, L.D.; Schmidt, D.B.; Sham, K.K.; Sarau, H.M. (E)-3-[6-[[(2,6-dichlorophenyl)thio]methyl]-3-(2-phenylethoxy)-2- pyridinyl]-2-propenoic acid: A high-affinity leukotriene B4 receptor antagonist with oral antiinflammatory activity. J. Med. Chem. 1996, 39, 3837–3841. [Google Scholar] [CrossRef]
- Deng, G.; Liu, Z.; Ye, F.; Luo, X.; Zhu, W.; Shen, X.; Liu, H.; Jiang, H. Tryptophan-containing dipeptide derivatives as potent PPARγ antagonists: Design, synthesis, biological evaluation, and molecular modeling. Eur. J. Med. Chem. 2008, 43, 2699–2716. [Google Scholar] [CrossRef]
- Albrecht, H.P.; Hofman, H.P.; Klebe, G.; Kreiskott, H. L-dopa potentiating analogs of Pro-Leu-Gly-NH2 with oral efficacy. Boll. Chim. Farm. 1991, 130, 55–59. [Google Scholar] [PubMed]
- Yu, K.L.; Rajakumar, G.; Srivastava, L.K.; Mishra, R.K.; Johnson, R.L. Dopamine receptor modulation by conformationally constrained analogues of Pro-Leu-Gly-NH2. J. Med. Chem. 1988, 31, 1430–1436. [Google Scholar] [CrossRef]
- Doebber, T.W.; Kelly, L.J.; Zhou, G.; Meurer, R.; Biswas, C.; Li, Y.; Wu, M.S.; Ippolito, M.C.; Chao, Y.S.; Wang, P.R.; et al. MK-0767, a novel dual PPARalpha/gamma agonist, displays robust antihyperglycemic and hypolipidemic activities. BioChem. Biophys. Res. Commun. 2004, 318, 323–328. [Google Scholar] [CrossRef]
- Getman, D.P.; DeCrescenzo, G.A.; Heintz, R.M.; Reed, K.L.; Talley, J.J.; Bryant, M.L.; Clare, M.; Houseman, K.A.; Marr, J.J.; Mueller, R.A.; et al. Discovery of a novel class of potent HIV-1 protease inhibitors containing the (R)-(hydroxyethyl)urea isostere. J. Med. Chem. 1993, 36, 288–291. [Google Scholar] [CrossRef]
- Dwyer, M.P.; Yu, Y.; Chao, J.; Aki, C.; Chao, J.; Biju, P.; Girijavallabhan, V.; Rindgen, D.; Bond, R.; Mayer-Ezel, R.; et al. Discovery of 2-hydroxy-N,N-dimethyl-3-{2-[[(R)-1-(5- methylfuran-2-yl)propyl]amino]-3,4-dioxocyclobut-1-enylamino}benzamide (SCH 527123): A potent, orally bioavailable CXCR2/CXCR1 receptor antagonist. J. Med. Chem. 2006, 49, 7603–7606. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Xue, C.-B.; Li, H.-Y.; Li, Q. Piperidin-4-yl Azetidine Derivatives as JAK1 Inhibitors. Patent No. US-201113043986-A, 1 July 2014. [Google Scholar]
- Doherty, A.M.; Kaltenbronn, J.S.; Hudspeth, J.P.; Repine, J.T.; Roark, W.H.; Sircar, I.; Tinney, F.J.; Connolly, C.J.; Hodges, J.C.; Taylor, M.D.; et al. New inhibitors of human renin that contain novel replacements at the P2 site. J. Med. Chem. 1991, 34, 1258–1271. [Google Scholar] [CrossRef]
- Reiter, L.A.; Koch, K.; Piscopio, A.D.; Showell, H.J.; Alpert, R.; Biggers, M.S.; Chambers, R.J.; Conklyn, M.J.; Cooper, K.; Cortina, S.R.; et al. trans-3-Benzyl-4-hydroxy-7-chromanylbenzoic acid derivatives as antagonists of the leukotriene B4 (LTB4) receptor. Bioorg. Med. Chem. Lett. 1998, 8, 1781–1786. [Google Scholar] [CrossRef]
- Albright, J.D.; Reich, M.F.; Delos Santos, E.G.; Dusza, J.P.; Sum, F.W.; Venkatesan, A.M.; Coupet, J.; Chan, P.S.; Ru, X.; Mazandarani, H.; et al. 5-Fluoro-2-methyl-N-[4-(5H-pyrrolo [2,1-c]-[1,4]benzodiazepin-10(11H)-ylcarbonyl)-3-chlorophenyl]benzamide (VPA-985): An orally active arginine vasopressin antagonist with selectivity for V2 receptors. J. Med. Chem. 1998, 41, 2442–2444. [Google Scholar] [CrossRef]
- Molinari, A.J.; Trybulski, E.J.; Bagli, J.; Croce, S.; Considine, J.; Qi, J.; Ali, K.; Demaio, W.; Lihotz, L.; Cochran, D. Identification and synthesis of major metabolites of Vasopressin V2-receptor agonist WAY-151932, and antagonist, Lixivaptan. Bioorg. Med. Chem. Lett. 2007, 17, 5796–5800. [Google Scholar] [CrossRef] [PubMed]
- Portevin, B.; Benoist, A.; Rémond, G.; Hervé, Y.; Vincent, M.; Lepagnol, J.; De Nanteuil, G. New prolyl endopeptidase inhibitors: In vitro and in vivo activities of azabicyclo[2.2.2]octane, azabicyclo[2.2.1]heptane, and perhydroindole derivatives. J. Med. Chem. 1996, 39, 2379–2391. [Google Scholar] [CrossRef]
- Williamson, R.; Hakenbeck, R.; Tomasz, A. In vivo interaction of beta-lactam antibiotics with the penicillin-binding proteins of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 1980, 18, 629–637. [Google Scholar] [CrossRef]
- Cheng, H.; Hoffman, J.E.; Le, P.T.; Pairish, M.; Kania, R.; Farrell, W.; Bagrodia, S.; Yuan, J.; Sun, S.; Zhang, E.; et al. Structure-based design, SAR analysis and antitumor activity of PI3K/mTOR dual inhibitors from 4-methylpyridopyrimidinone series. Bioorg. Med. Chem. Lett. 2013, 23, 2787–2792. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Bagrodia, S.; Bailey, S.; Edwards, M.; Hoffman, J.; Hu, Q.; Kania, R.; Knighton, D.R.; Marx, M.A.; Ninkovic, S.; et al. Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04691502 through structure based drug design. MedChemComm 2010, 1, 139–144. [Google Scholar] [CrossRef]
- Albrecht, B.K.; Harmange, J.C.; Bauer, D.; Berry, L.; Bode, C.; Boezio, A.A.; Chen, A.; Choquette, D.; Dussault, I.; Fridrich, C.; et al. Discovery and optimization of triazolopyridazines as potent and selective inhibitors of the c-Met kinase. J. Med. Chem. 2008, 51, 2879–2882. [Google Scholar] [CrossRef]
- Kimura, T.; Kawano, K.; Doi, E.; Kitazawa, N.; Shin, K.; Miyagawa, T.; Kaneko, T.; Ito, K.; Takaishi, M.; Sasaki, T.; et al. Cinnamide compound. Patent No. WO2005115990, 25 May 2005. [Google Scholar]
- Kuzikov, M.; Costanzi, E.; Reinshagen, J.; Esposito, F.; Vangeel, L.; Wolf, M.; Ellinger, B.; Claussen, C.; Geisslinger, G.; Corona, A.; et al. Identification of Inhibitors of SARS-CoV-2 3CL-Pro Enzymatic Activity Using a Small Molecule in Vitro Repurposing Screen. ACS Pharmacol. Translat. Sci. 2021, 4, 1096–1110. [Google Scholar] [CrossRef]
- Arun Prasad, P.; Gautham, N. A new peptide docking strategy using a mean field technique with mutually orthogonal Latin square sampling. J. Comput. Aided Mol. Des. 2008, 22, 815–829. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—High resolution modeling of peptide–protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein–peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef]
- Mehra, R.; Kepp, K.P. Computational prediction and molecular mechanism of γ-secretase modulators. Eur. J. Pharm. Sci. 2021, 157, 105626. [Google Scholar] [CrossRef]
- Santiago, Á.; Guzmán-Ocampo, D.C.; Aguayo-Ortiz, R.; Dominguez, L. Characterizing the Chemical Space of γ-Secretase Inhibitors and Modulators. ACS Chem. Neurosci. 2021, 12, 2765–2775. [Google Scholar] [CrossRef]
- Mancarella, S.; Serino, G.; Dituri, F.; Cigliano, A.; Ribback, S.; Wang, J.; Chen, X.; Calvisi, D.F.; Giannelli, G. Crenigacestat, a selective NOTCH1 inhibitor, reduces intrahepatic cholangiocarcinoma progression by blocking VEGFA/DLL4/MMP13 axis. Cell Death Diff. 2020, 27, 2330–2343. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A Novel Approach to Pharmacophore Modeling and 3D Database Searching. Chem. Biol Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15 -Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2016, 45, D945–D954. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Sastry, G.M.; Dixon, S.L.; Sherman, W. Rapid shape-based alignment and virtual screening method based on atom/feature-pair similiarities and volume overlap scoring. J. Chem. Inf. Model. 2011, 51, 2455–2466. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Modulators | |||
|---|---|---|---|---|
| Glide HTVS | ePharmacophore | Glide HTVS | ePharmacophore | |
| 6IYC | 0.67 (0.16, 7%) | 0.35 (0.06, 15%) | 0.65 (0.04, 20%) | 0.80 (0.11, 19%) |
| 6IDF | 0.56 (0.06, 5%) | 0.45 (0.03, 5%) | 0.64 (0.05, 10%) | 0.54 (0.02, 23%) |
| 6LQG | 0.56 (0.06, 24%) | 0.60 (0.12, 12%) | 0.51 (0.02, 10%) | 0.76 (0.06, 13%) |
| 6LR4 | 0.50 (0.02, 9%) | 0.45 (0.11, 12%) | 0.58 (0.02, 6%) | 0.72 (0.07, 12%) |
| 7C9I | 0.45 (0.10, 7%) | 0.63 (0.17, 6%) | 0.56 (0.08, 12%) | 0.57 (0.00, 5%) |
| 7D8X | 0.48 (0.07, 5%) | 0.38 (0.07, 5%) | 0.63 (0.06, 23%) | 0.73 (0.16, 7%) |
| Inhibitors 2 | Modulators 3 | |||
|---|---|---|---|---|
| SP Flexible | SP Refine Only | SP Flexible | SP Refine Only | |
| 6IYC | 0.99 (0.93, 11%) | 0.97 (0.75, 9%) | 0.65 (0.19, 15%) | 0.54 (0.28, 12%) |
| 6IDF | 0.95 (0.74, 6%) | 0.71 (0.83, 7%) | 0.61 (0.15, 10%) | - 4 |
| 6LQG | 0.74 (0.27, 23%) | 0.77 (0.74, 5%) | 0.56 (0.10, 6%) | 0.36 (−0.06, 5%) |
| 6LR4 | 0.97 (0.83, 7%) | 0.63 (0.74, 5%) | 0.50 (0.07, 8%) | 0.44 (−0.03, 5%) |
| 7C9I | 0.80 (0.65, 7%) | 0.82 (0.83, 7%) | 0.64 (0.26, 10%) | 0.35 (0.00, 5%) |
| 7D8X | 0.94 (0.74, 6%) | 0.83 (0.83, 7%) | 0.57 (0.00, 17%) | 0.70 (0.39, 9%) |
| Performance 1 | |
|---|---|
| Inhibitors 2 | 0.14 (0.14, 5%) |
| Modulators 3 | 0.81 (0.47, 24%) |
| ZINC ID | Generic Name | Target Class(es) | Specific Target(s) |
|---|---|---|---|
| ZINC000003919807 | AG7088 | Protease | Human rhinovirus A protease [50] |
| ZINC000085548251 | A-77003 | Protease, lyase | HIV-1 protease [51], carbonic anhydrase II [52] |
| ZINC000043202141 | Oprozomib | Protease | Proteasome subunits beta type 5 and 8 [52] |
| ZINC000068077856 | Foxy-5 | Class F GPCR, kinase, co-receptor 1 | Frizzleds, Ryk, RORs, LRP 1 [53,54] |
| ZINC000082138051 | PF-03715455 | Kinase | VEGFR1 [54], MAP kinase p38 beta [55], misshapen-like kinase 1 [55] |
| ZINC000169345692 | Peptide T | Surface antigen | CD4 [56] |
| ZINC000095586643 | Crenigacestat | Protease | γ-secretase [57] |
| ZINC000049694463 | Cefcanel daloxate | Transferase 2 | Penicillin-binding proteins 2 [58] |
| ZINC000090636091 | - 3 | Kinase | G protein-coupled receptor kinases [59] |
| ZINC000003935423 | Droxinavir | Protease | HIV-1 protease [60] |
| ZINC000027657184 | Modipafant | Class A GPCR, voltage-gated ion channel | Platelet-activating factor receptor [61], voltage-dependent L-type calcium channel subunit α1D [61] |
| ZINC000003830407 | Cefazolin | Transferase | Penicillin-binding proteins [62] |
| ZINC000003917787 | - 3 | Protease | Renin [63], cathepsin D [64] |
| ZINC000001541366 | Ticolubant | Class A GPCR, reductase | Leukotriene B4 receptor 1 [65], arachidonate 5-lipoxygenase [65] |
| ZINC000002012859 | Halofenate | Transcription factor | PPARγ [66] |
| ZINC000005599165 | Doreptide | Class A GPCR 2 | Dopamine receptors2 [67,68] |
| ZINC000200259560 | MK-0767 | Transcription factor | PPARα [69], PPARγ [69] |
| ZINC000003915259 | Telinavir | Protease | Human rhinovirus A protease [70], HIV-1 protease [70] |
| ZINC000206178236 | Navarixin | Class A GPCR | CXCR2 [71], CXCR1 [71] |
| ZINC000118795962 | Itacitinib | Kinase | JAK1 [72], JAK2 [72] |
| ZINC000028257302 | - 3 | Protease | Renin [73] |
| ZINC000004392972 | CP-195543 | Class A GPCR | Leukotriene B4 receptor [74] |
| ZINC000000600399 | Lixivaptan | Class A GPCR | Vasopressin receptors (V1a, V2) [75], oxytocin receptor [76] |
| ZINC000003807687 | JTP-4819 | Protease | Prolyl endopeptidase [77] |
| ZINC000003831243 | Oxacillin | Transferase | Penicillin-binding protein [78] |
| ZINC ID | Generic Name | Target Class(es) | Specific Target(s) |
|---|---|---|---|
| ZINC000117704832 | PF-04691502 | Kinase | AKT [79], PI3Kα [80], mTOR [79,80] |
| ZINC000003919807 | AG7088 | Protease | Human rhinovirus A protease [50] |
| ZINC000034285235 | AMG-208 | Kinase | MET [81] |
| ZINC000067172224 | E2012 | Protease | γ-secretase [82] |
| ZINC000000005014 | Ocinaplon | Protease | SARS-CoV-2 main protease [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ioppolo, A.; Eccles, M.; Groth, D.; Verdile, G.; Agostino, M. Evaluation of Virtual Screening Strategies for the Identification of γ-Secretase Inhibitors and Modulators. Molecules 2022, 27, 176. https://doi.org/10.3390/molecules27010176
Ioppolo A, Eccles M, Groth D, Verdile G, Agostino M. Evaluation of Virtual Screening Strategies for the Identification of γ-Secretase Inhibitors and Modulators. Molecules. 2022; 27(1):176. https://doi.org/10.3390/molecules27010176
Chicago/Turabian StyleIoppolo, Alicia, Melissa Eccles, David Groth, Giuseppe Verdile, and Mark Agostino. 2022. "Evaluation of Virtual Screening Strategies for the Identification of γ-Secretase Inhibitors and Modulators" Molecules 27, no. 1: 176. https://doi.org/10.3390/molecules27010176
APA StyleIoppolo, A., Eccles, M., Groth, D., Verdile, G., & Agostino, M. (2022). Evaluation of Virtual Screening Strategies for the Identification of γ-Secretase Inhibitors and Modulators. Molecules, 27(1), 176. https://doi.org/10.3390/molecules27010176