
The Structural Basis of Peptide Binding at Class A G Protein-Coupled Receptors

 , , ,
, , ,
Abstract
:
1. Introduction
1.1. G Protein-Coupled Receptors Are a Significant Target of Therapeutic Intervention
1.2. Peptide-Activated Receptors Are a Large Percentage of the GPCR Class A
1.3. Diversity of Peptide Ligands
1.4. Reducing the Flexibility of Peptide Ligands Is Crucial for Success in Co-Crystallization
1.5. Complexity of Peptide Ligand and Receptor Interactions
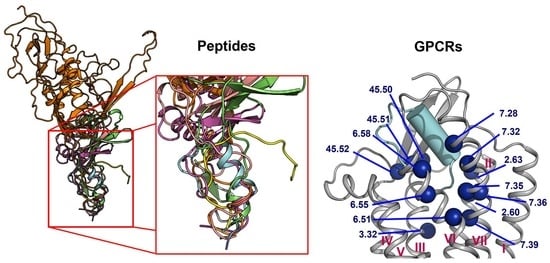
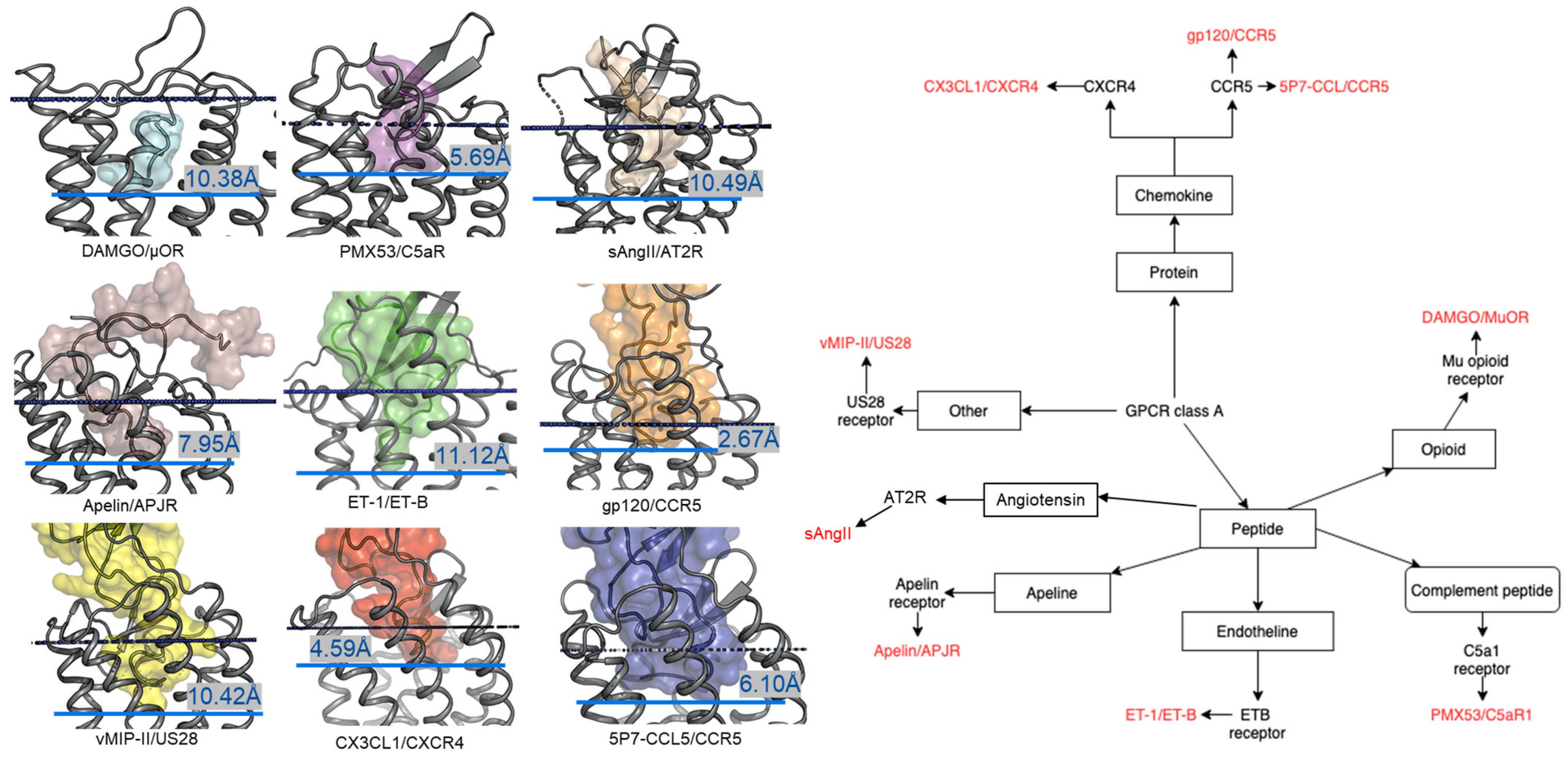
2. Comparison of Peptide Binding Modes across Class A GPCRs
2.1. Diversity in the Binding Modes of the Peptide Ligands to Class A GPCRs
2.2. Peptide Ligands Affect the Conformation of the Extracellular Surface
2.3. ECL1 and ECL2 Bound Conformation Have Conversed across Class A Peptide-GPCRs
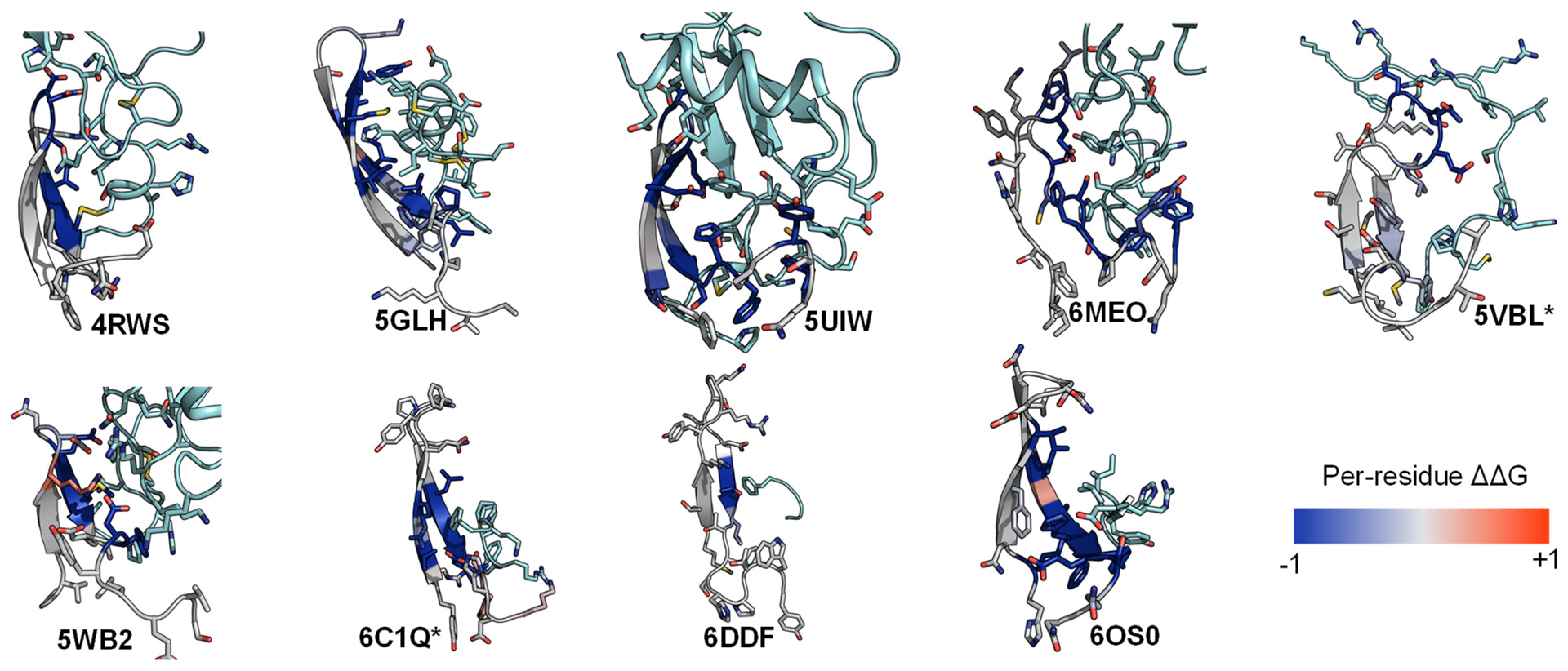
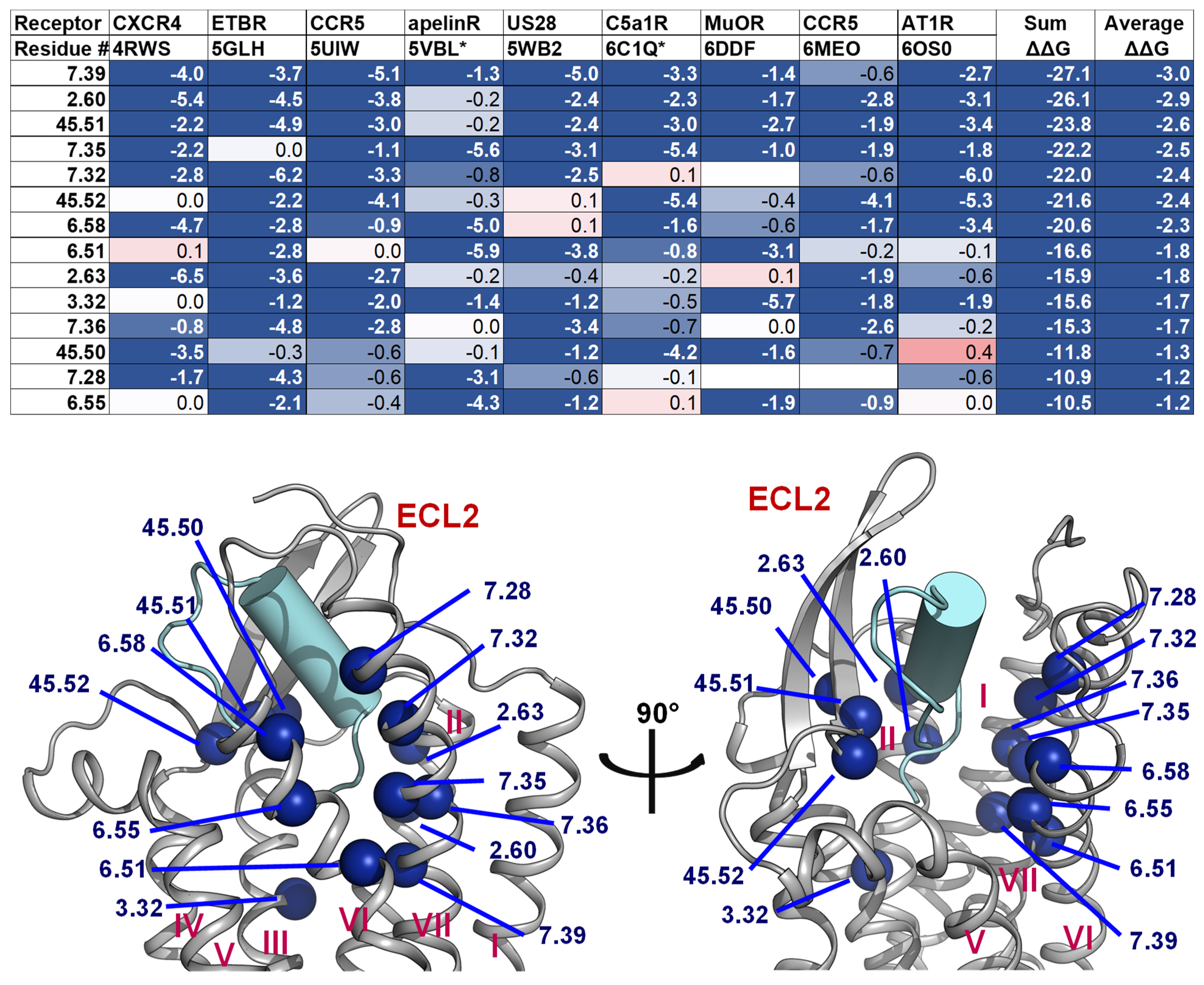
2.4. A List of 14 Common Interacting Residues Suggests a General Peptide Recognition and Binding Mechanism among Nine Class A GPCRs
3. Structural Changes in Peptides Induced by Receptors Are Critical for Binding
3.1. Neurotensin
3.2. Apelin
3.3. Endothelin
3.4. The Complement System Peptide Ligand C5a
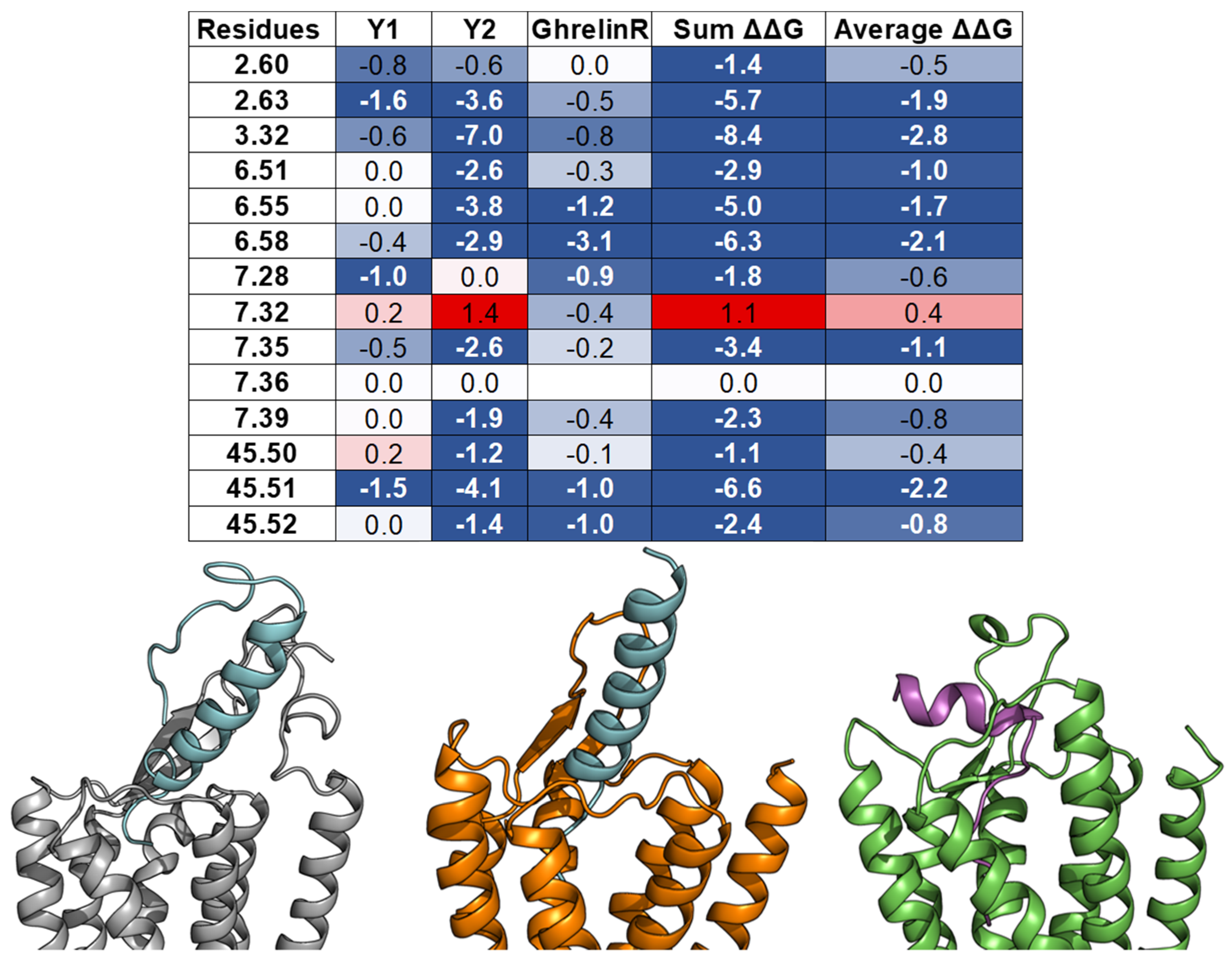
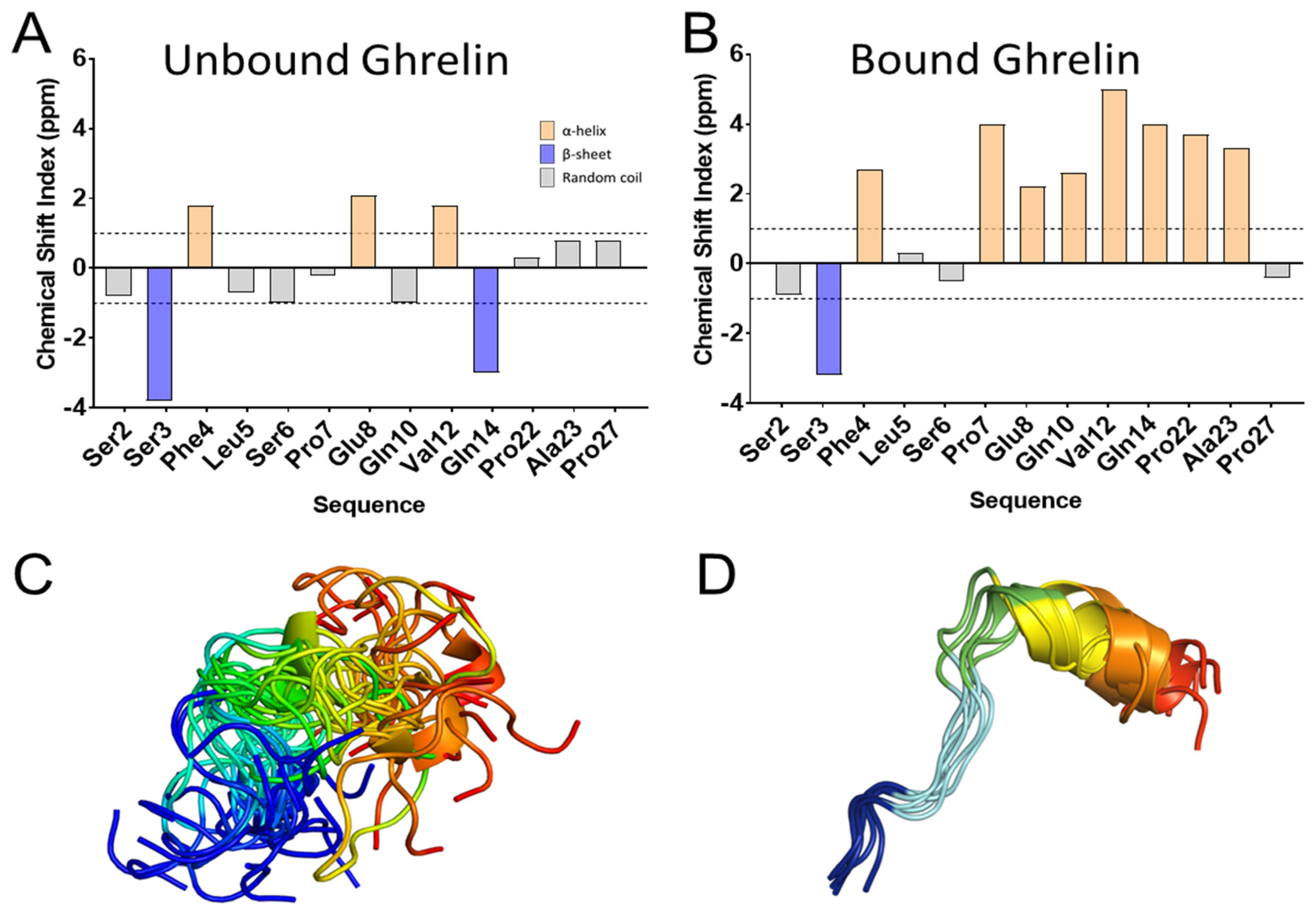
3.5. Ghrelin
3.6. Gonadotropin-Releasing Hormone
3.7. Neuropeptide Y
3.8. Opioid Peptides
4. Implications for Future Studies
4.1. Peptides Need to Be Characterized in Their Bound State
4.2. Mimetics of the Bound-State Conformations Can Aid in Structure Determination and Drug Discovery
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Flock, T.; Hauser, A.S.; Lund, N.; Gloriam, D.E.; Balaji, S.; Babu, M.M. Selectivity determinants of GPCR-G-protein binding. Nature 2017, 545, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef]
- Isberg, V.; Mordalski, S.; Munk, C.; Rataj, K.; Harpsoe, K.; Hauser, A.S.; Vroling, B.; Bojarski, A.J.; Vriend, G.; Gloriam, D.E. GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 2017, 45, 2936. [Google Scholar] [CrossRef] [Green Version]
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockaert, J.; Pin, J.P. Molecular tinkering of G protein-coupled receptors: An evolutionary success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J.; McCormack, P.L. Olmesartan Medoxomil. Drugs 2008, 68, 1239–1272. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Goa, K.L. Valsartan. Drugs 1997, 54, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Brogden, R.N.; Buckley, M.M.T.; Ward, A. Buserelin. Drugs 1990, 39, 399–437. [Google Scholar] [CrossRef]
- Davenport, A.P.; Scully, C.C.G.; de Graaf, C.; Brown, A.J.H.; Maguire, J.J. Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 2020, 19, 389–413. [Google Scholar] [CrossRef]
- Beck, A.; Bussat, M.C.; Klinguer-Hamour, C.; Goetsch, L.; Aubry, J.P.; Champion, T.; Julien, E.; Haeuw, J.F.; Bonnefoy, J.Y.; Corvaia, N. Stability and CTL activity of N-terminal glutamic acid containing peptides. J. Pept. Res. 2001, 57, 528–538. [Google Scholar] [CrossRef]
- Chance, R.E. Pancreatic Polypeptide. In Encyclopedia of Hormones; Henry, H.L., Norman, A.W., Eds.; Academic Press: New York, NY, USA, 2003; pp. 142–146. [Google Scholar]
- Kaiser, A.; Muller, P.; Zellmann, T.; Scheidt, H.A.; Thomas, L.; Bosse, M.; Meier, R.; Meiler, J.; Huster, D.; Beck-Sickinger, A.G.; et al. Unwinding of the C-Terminal Residues of Neuropeptide Y is critical for Y2 Receptor Binding and Activation. Angew. Chem. Int. Ed. Engl. 2015, 54, 7446–7449. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Vasile, S.; Østergaard, S.; Paulsson, J.F.; Pruner, J.; Åqvist, J.; Wulff, B.S.; Gutiérrez-de-Terán, H.; Larhammar, D. Elucidation of the Binding Mode of the Carboxyterminal Region of Peptide YY to the Human Y2 Receptor. Mol. Pharmacol. 2018, 93, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Bradbury, A.F.; Smyth, D.G. Peptide amidation. Trends Biochem. Sci. 1991, 16, 112–115. [Google Scholar] [CrossRef]
- Goren, H.J.; Bauce, L.G.; Vale, W. Forces and structural limitations of binding of thyrotrophin-releasing factor to the thyrotrophin-releasing receptor: The pyroglutamic acid moiety. Mol. Pharmacol. 1977, 13, 606–614. [Google Scholar]
- Morty, R.E.; Bulau, P.; Pellé, R.; Wilk, S.; Abe, K. Pyroglutamyl peptidase type I from Trypanosoma brucei: A new virulence factor from African trypanosomes that de-blocks regulatory peptides in the plasma of infected hosts. Biochem. J. 2006, 394, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef]
- Yang, J.; Brown, M.S.; Liang, G.; Grishin, N.V.; Goldstein, J.L. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 2008, 132, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Hewage, C.M.; Jiang, L.; Parkinson, J.A.; Ramage, R.; Sadler, I.H. Solution structure of a novel ETB receptor selective agonist ET1-21 [Cys(Acm)1,15, Aib3,11, Leu7] by nuclear magnetic resonance spectroscopy and molecular modelling. J. Pept. Res. 1999, 53, 223–233. [Google Scholar] [CrossRef]
- Hirata, Y.; Yoshimi, H.; Emori, T.; Shichiri, M.; Marumo, F.; Watanabe, T.X.; Kumagaye, S.; Nakajima, K.; Kimura, T.; Sakakibara, S. Receptor binding activity and cytosolic free calcium response by synthetic endothelin analogs in cultured rat vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1989, 160, 228–234. [Google Scholar] [CrossRef]
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.-R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, L.; Wolf, R.; Zeitschel, U.; Rossner, S.; Petersén, Å.; Leavitt, B.R.; Kästner, F.; Rothermundt, M.; Gärtner, U.-T.; Gündel, D.; et al. Proteolytic degradation of neuropeptide Y (NPY) from head to toe: Identification of novel NPY-cleaving peptidases and potential drug interactions in CNS and Periphery. J. Neurochem. 2015, 135, 1019–1037. [Google Scholar] [CrossRef] [Green Version]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the agonist-bound neurotensin receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, G.; Bondon, A.; Coutant, J.; Curmi, P.; Monti, J.P. Intermolecular interactions between the neurotensin and the third extracellular loop of human neurotensin 1 receptor. J. Biomol. Struct. Dyn. 2013, 31, 1381–1392. [Google Scholar] [CrossRef]
- Ma, Y.; Yue, Y.; Ma, Y.; Zhang, Q.; Zhou, Q.; Song, Y.; Shen, Y.; Li, X.; Ma, X.; Li, C.; et al. Structural Basis for Apelin Control of the Human Apelin Receptor. Structure 2017, 25, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Burg, J.S.; Ingram, J.R.; Venkatakrishnan, A.J.; Jude, K.M.; Dukkipati, A.; Feinberg, E.N.; Angelini, A.; Waghray, D.; Dror, R.O.; Ploegh, H.L.; et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science 2015, 347, 1113–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Han, G.W.; Abagyan, R.; Wu, B.; Stevens, R.C.; Cherezov, V.; Kufareva, I.; Handel, T.M. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 2017, 46, 1005–1017.e5. [Google Scholar] [CrossRef] [Green Version]
- Pedragosa-Badia, X.; Stichel, J.; Beck-Sickinger, A.G. Neuropeptide Y receptors: How to get subtype selectivity. Front. Endocrinol. (Lausanne) 2013, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joedicke, L.; Mao, J.; Kuenze, G.; Reinhart, C.; Kalavacherla, T.; Jonker, H.R.A.; Richter, C.; Schwalbe, H.; Meiler, J.; Preu, J.; et al. The molecular basis of subtype selectivity of human kinin G-protein-coupled receptors. Nat. Chem. Biol. 2018, 14, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Eisen, H.N.; Hou, X.H.; Shen, C.; Wang, K.; Tanguturi, V.K.; Smith, C.; Kozyrytska, K.; Nambiar, L.; McKinley, C.A.; Chen, J.; et al. Promiscuous binding of extracellular peptides to cell surface class I MHC protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4580–4585. [Google Scholar] [CrossRef] [Green Version]
- Ngo, T.; Ilatovskiy, A.V.; Stewart, A.G.; Coleman, J.L.; McRobb, F.M.; Riek, R.P.; Graham, R.M.; Abagyan, R.; Kufareva, I.; Smith, N.J. Orphan receptor ligand discovery by pickpocketing pharmacological neighbors. Nat. Chem. Biol. 2017, 13, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michino, M.; Beuming, T.; Donthamsetti, P.; Newman, A.H.; Javitch, J.A.; Shi, L. What can crystal structures of aminergic receptors tell us about designing subtype-selective ligands? Pharmacol. Rev. 2015, 67, 198–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooistra, A.J.; Kuhne, S.; de Esch, I.J.; Leurs, R.; de Graaf, C. A structural chemogenomics analysis of aminergic GPCRs: Lessons for histamine receptor ligand design. Br. J. Pharmacol. 2013, 170, 101–126. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Chien, E.Y.; Liu, W.; Zhao, Q.; Katritch, V.; Han, G.W.; Hanson, M.A.; Shi, L.; Newman, A.H.; Javitch, J.A.; Cherezov, V.; et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science 2010, 330, 1091–1095. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Song, G.; de Graaf, C.; Stevens, R.C. Structure and Function of Peptide-Binding G Protein-Coupled Receptors. J. Mol. Biol. 2017, 429, 2726–2745. [Google Scholar] [CrossRef]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the delta-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wacker, D.; Mileni, M.; Katritch, V.; Han, G.W.; Vardy, E.; Liu, W.; Thompson, A.A.; Huang, X.P.; Carroll, F.I.; et al. Structure of the human κ-opioid receptor in complex with JDTic. Nature 2012, 485, 327–332. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.A.; Liu, W.; Chun, E.; Katritch, V.; Wu, H.; Vardy, E.; Huang, X.P.; Trapella, C.; Guerrini, R.; Calo, G.; et al. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature 2012, 485, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 387–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Qin, L.; Zacarias, N.V.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Oswald, C.; Rappas, M.; Kean, J.; Dore, A.S.; Errey, J.C.; Bennett, K.; Deflorian, F.; Christopher, J.A.; Jazayeri, A.; Mason, J.S.; et al. Intracellular allosteric antagonism of the CCR9 receptor. Nature 2016, 540, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Babaoglu, K.; Brautigam, C.A.; Clark, L.; Shao, Z.; Scheuermann, T.H.; Harrell, C.M.; Gotter, A.L.; Roecker, A.J.; Winrow, C.J.; et al. Structure and ligand-binding mechanism of the human OX1 and OX2 orexin receptors. Nat. Struct. Mol. Biol. 2016, 23, 293–299. [Google Scholar] [CrossRef]
- Yin, J.; Mobarec, J.C.; Kolb, P.; Rosenbaum, D.M. Crystal structure of the human OX2 orexin receptor bound to the insomnia drug suvorexant. Nature 2015, 519, 247–250. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Desnoyer, R.; Han, G.W.; Patel, N.; Katritch, V.; Karnik, S.S.; Cherezov, V.; Stevens, R.C. Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [Green Version]
- Asada, H.; Horita, S.; Hirata, K.; Shiroishi, M.; Shiimura, Y.; Iwanari, H.; Hamakubo, T.; Shimamura, T.; Nomura, N.; Kusano-Arai, O.; et al. Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 2018, 25, 570–576. [Google Scholar] [CrossRef]
- Cheng, R.K.Y.; Fiez-Vandal, C.; Schlenker, O.; Edman, K.; Aggeler, B.; Brown, D.G.; Brown, G.A.; Cooke, R.M.; Dumelin, C.E.; Dore, A.S.; et al. Structural insight into allosteric modulation of protease-activated receptor 2. Nature 2017, 545, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Shihoya, W.; Nishizawa, T.; Okuta, A.; Tani, K.; Dohmae, N.; Fujiyoshi, Y.; Nureki, O.; Doi, T. Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 2016, 537, 363–368. [Google Scholar] [CrossRef]
- Yang, Z.; Han, S.; Keller, M.; Kaiser, A.; Bender, B.J.; Bosse, M.; Burkert, K.; Kogler, L.M.; Wifling, D.; Bernhardt, G.; et al. Structural basis of ligand binding modes at the neuropeptide Y Y1 receptor. Nature 2018, 556, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Chapman, K.; Clark, L.D.; Shao, Z.; Borek, D.; Xu, Q.; Wang, J.; Rosenbaum, D.M. Crystal structure of the human NK1 tachykinin receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 13264–13269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, N.; Rappas, M.; Dore, A.S.; Brown, J.; Bottegoni, G.; Koglin, M.; Cansfield, J.; Jazayeri, A.; Cooke, R.M.; Marshall, F.H. Structure of the complement C5a receptor bound to the extra-helical antagonist NDT9513727. Nature 2018, 553, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Yoshida, T.; Miyamoto, N.; Motoike, T.; Kurosu, H.; Shibata, K.; Yamanaka, A.; Williams, S.C.; Richardson, J.A.; Tsujino, N.; et al. Characterization of a family of endogenous neuropeptide ligands for the G protein-coupled receptors GPR7 and GPR8. Proc. Natl. Acad. Sci. USA 2003, 100, 6251–6256. [Google Scholar] [CrossRef] [Green Version]
- Shaik, M.M.; Peng, H.; Lu, J.; Rits-Volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2019, 565, 318–323. [Google Scholar] [CrossRef]
- Koehl, A.; Hu, H.; Maeda, S.; Zhang, Y.; Qu, Q.; Paggi, J.M.; Latorraca, N.R.; Hilger, D.; Dawson, R.; Matile, H.; et al. Structure of the micro-opioid receptor-Gi protein complex. Nature 2018, 558, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kim, H.R.; Deepak, R.; Wang, L.; Chung, K.Y.; Fan, H.; Wei, Z.; Zhang, C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018, 25, 472–481. [Google Scholar] [CrossRef]
- Krumm, B.E.; Grisshammer, R. Peptide ligand recognition by G protein-coupled receptors. Front. Pharmacol. 2015, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, I.G.; Gigoux, V.; Fourmy, D. Understanding peptide binding in Class A G protein-coupled receptors. Mol. Pharmacol. 2019, 96, 550–561. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef]
- Woolley, M.J.; Conner, A.C. Understanding the common themes and diverse roles of the second extracellular loop (ECL2) of the GPCR super-family. Mol. Cell. Endocrinol. 2017, 449, 3–11. [Google Scholar] [CrossRef]
- Isberg, V.; de Graaf, C.; Bortolato, A.; Cherezov, V.; Katritch, V.; Marshall, F.H.; Mordalski, S.; Pin, J.-P.; Stevens, R.C.; Vriend, G.; et al. Generic GPCR residue numbers-aligning topology maps while minding the gaps. Trends Pharmacol. Sci. 2015, 36, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Conway, P.; Tyka, M.D.; DiMaio, F.; Konerding, D.E.; Baker, D. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci. 2014, 23, 47–55. [Google Scholar] [CrossRef]
- Smith, C.A.; Kortemme, T. Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 2008, 380, 742–756. [Google Scholar] [CrossRef] [Green Version]
- Bender, B.J.; Vortmeier, G.; Ernicke, S.; Bosse, M.; Kaiser, A.; Els-Heindl, S.; Krug, U.; Beck-Sickinger, A.; Meiler, J.; Huster, D. Structural Model of Ghrelin Bound to its G Protein-Coupled Receptor. Structure 2019, 27, 537–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock ab-initio: Simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 20–41. [Google Scholar] [CrossRef] [Green Version]
- Lange, O.F.; Lakomek, N.A.; Fares, C.; Schroder, G.F.; Walter, K.F.; Becker, S.; Meiler, J.; Grubmuller, H.; Griesinger, C.; de Groot, B.L. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science 2008, 320, 1471–1475. [Google Scholar] [CrossRef] [Green Version]
- Tyndall, J.D.; Pfeiffer, B.; Abbenante, G.; Fairlie, D.P. Over one hundred peptide-activated G protein-coupled receptors recognize ligands with turn structure. Chem. Rev. 2005, 105, 793–826. [Google Scholar] [CrossRef]
- Tyndall, J.D.; Nall, T.; Fairlie, D.P. Proteases universally recognize beta strands in their active sites. Chem. Rev. 2005, 105, 973–999. [Google Scholar] [CrossRef]
- Siligardi, G.; Drake, A.F. The importance of extended conformations and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers 1995, 37, 281–292. [Google Scholar] [CrossRef]
- Carraway, R.; Leeman, S.E. The amino acid sequence of a hypothalamic peptide, neurotensin. J. Biol. Chem. 1975, 250, 1907–1911. [Google Scholar] [CrossRef]
- Henry, J.A.; Horwell, D.C.; Meecham, K.G.; Rees, D.C. A structure-affinity study of the amino acid side-chains in neurotensin: N and C terminal deletions and Ala-scan. Biorg. Med. Chem. Lett. 1993, 3, 949–952. [Google Scholar] [CrossRef]
- Xu, G.Y.; Deber, C.M. Conformations of neurotensin in solution and in membrane environments studied by 2-D NMR spectroscopy. Int. J. Pept. Protein Res. 1991, 37, 528–535. [Google Scholar] [CrossRef]
- Williamson, P.T.; Bains, S.; Chung, C.; Cooke, R.; Watts, A. Probing the environment of neurotensin whilst bound to the neurotensin receptor by solid state NMR. FEBS Lett. 2002, 518, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Luca, S.; White, J.F.; Sohal, A.K.; Filippov, D.V.; van Boom, J.H.; Grisshammer, R.; Baldus, M. The conformation of neurotensin bound to its G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 10706–10711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heise, H.; Luca, S.; de Groot, B.L.; Grubmuller, H.; Baldus, M. Probing conformational disorder in neurotensin by two-dimensional solid-state NMR and comparison to molecular dynamics simulations. Biophys. J. 2005, 89, 2113–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Kemmel, F.M.; Dubuc, I.; Bourdel, E.; Fehrentz, J.A.; Martinez, J.; Costentin, J. A C-terminal cyclic 8–13 neurotensin fragment analog appears less exposed to neprilysin when it crosses the blood-brain barrier than the cerebrospinal fluid-brain barrier in mice. Neurosci. Lett. 1996, 217, 58–60. [Google Scholar] [CrossRef]
- Fan, X.; Zhou, N.; Zhang, X.; Mukhtar, M.; Lu, Z.; Fang, J.; DuBois, G.C.; Pomerantz, R.J. Structural and functional study of the apelin-13 peptide, an endogenous ligand of the HIV-1 coreceptor, APJ. Biochemistry 2003, 42, 10163–10168. [Google Scholar] [CrossRef]
- Medhurst, A.D.; Jennings, C.A.; Robbins, M.J.; Davis, R.P.; Ellis, C.; Winborn, K.Y.; Lawrie, K.W.; Hervieu, G.; Riley, G.; Bolaky, J.E.; et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J. Neurochem. 2003, 84, 1162–1172. [Google Scholar] [CrossRef]
- Murza, A.; Parent, A.; Besserer-Offroy, E.; Tremblay, H.; Karadereye, F.; Beaudet, N.; Leduc, R.; Sarret, P.; Marsault, E. Elucidation of the structure-activity relationships of apelin: Influence of unnatural amino acids on binding, signaling, and plasma stability. ChemMedChem 2012, 7, 318–325. [Google Scholar] [CrossRef]
- Langelaan, D.N.; Bebbington, E.M.; Reddy, T.; Rainey, J.K. Structural insight into G-protein coupled receptor binding by apelin. Biochemistry 2009, 48, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.H.; Chen, C.P.; Marschner, T.M.; Krystek, S.R., Jr.; Bassolino, D.A. Conformational isomerism of endothelin in acidic aqueous media: A quantitative NOESY analysis. Biochemistry 1992, 31, 1280–1295. [Google Scholar] [CrossRef]
- Janes, R.W.; Peapus, D.H.; Wallace, B.A. The crystal structure of human endothelin. Nat. Struct. Biol. 1994, 1, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Aumelas, A.; Chiche, L.; Kubo, S.; Chino, N.; Tamaoki, H.; Kobayashi, Y. [Lys(−2)-Arg(−1)]endothelin-1 solution structure by two-dimensional 1H-NMR: Possible involvement of electrostatic interactions in native disulfide bridge formation and in biological activity decrease. Biochemistry 1995, 34, 4546–4561. [Google Scholar] [CrossRef]
- Atkins, A.R.; Martin, R.C.; Smith, R. 1H NMR studies of sarafotoxin SRTb, a nonselective endothelin receptor agonist, and IRL 1620, an ETB receptor-specific agonist. Biochemistry 1995, 34, 2026–2033. [Google Scholar] [CrossRef]
- Takashima, H.; Tamaoki, H.; Teno, N.; Nishi, Y.; Uchiyama, S.; Fukui, K.; Kobayashi, Y. Hydrophobic core around tyrosine for human endothelin-1 investigated by photochemically induced dynamic nuclear polarization nuclear magnetic resonance and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Biochemistry 2004, 43, 13932–13936. [Google Scholar] [CrossRef] [PubMed]
- Hoh, F.; Cerdan, R.; Kaas, Q.; Nishi, Y.; Chiche, L.; Kubo, S.; Chino, N.; Kobayashi, Y.; Dumas, C.; Aumelas, A. High-resolution X-ray structure of the unexpectedly stable dimer of the [Lys(−2)-Arg(−1)-des(17–21)]endothelin-1 peptide. Biochemistry 2004, 43, 15154–15168. [Google Scholar] [CrossRef]
- Lattig, J.; Oksche, A.; Beyermann, M.; Rosenthal, W.; Krause, G. Structural determinants for selective recognition of peptide ligands for endothelin receptor subtypes ETA and ETB. J. Pept. Sci. 2009, 15, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Hilal-Dandan, R.; Villegas, S.; Gonzalez, A.; Brunton, L.L. The quasi-irreversible nature of endothelin binding and G protein-linked signaling in cardiac myocytes. J. Pharmacol. Exp. Ther. 1997, 281, 267–273. [Google Scholar]
- Takasuka, T.; Sakurai, T.; Goto, K.; Furuichi, Y.; Watanabe, T. Human endothelin receptor ETB. Amino acid sequence requirements for super stable complex formation with its ligand. J. Biol. Chem. 1994, 269, 7509–7513. [Google Scholar] [CrossRef]
- Huber, R.; Scholze, H.; Paques, E.P.; Deisenhofer, J. Crystal structure analysis and molecular model of human C3a anaphylatoxin. Hoppe Seylers, Z. Physiol. Chem. 1980, 361, 1389–1399. [Google Scholar] [CrossRef]
- Nettesheim, D.G.; Edalji, R.P.; Mollison, K.W.; Greer, J.; Zuiderweg, E.R. Secondary structure of complement component C3a anaphylatoxin in solution as determined by NMR spectroscopy: Differences between crystal and solution conformations. Proc. Natl. Acad. Sci. USA 1988, 85, 5036–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuiderweg, E.R.; Nettesheim, D.G.; Mollison, K.W.; Carter, G.W. Tertiary structure of human complement component C5a in solution from nuclear magnetic resonance data. Biochemistry 1989, 28, 172–185. [Google Scholar] [CrossRef]
- Williamson, M.P.; Madison, V.S. Three-dimensional structure of porcine C5adesArg from 1H nuclear magnetic resonance data. Biochemistry 1990, 29, 2895–2905. [Google Scholar] [CrossRef]
- Fredslund, F.; Laursen, N.S.; Roversi, P.; Jenner, L.; Oliveira, C.L.; Pedersen, J.S.; Nunn, M.A.; Lea, S.M.; Discipio, R.; Sottrup-Jensen, L.; et al. Structure of and influence of a tick complement inhibitor on human complement component 5. Nat. Immunol. 2008, 9, 753–760. [Google Scholar] [CrossRef]
- Laursen, N.S.; Andersen, K.R.; Braren, I.; Spillner, E.; Sottrup-Jensen, L.; Andersen, G.R. Substrate recognition by complement convertases revealed in the C5-cobra venom factor complex. EMBO J. 2011, 30, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Bajic, G.; Yatime, L.; Klos, A.; Andersen, G.R. Human C3a and C3a desArg anaphylatoxins have conserved structures, in contrast to C5a and C5a desArg. Protein Sci. 2013, 22, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laursen, N.S.; Gordon, N.; Hermans, S.; Lorenz, N.; Jackson, N.; Wines, B.; Spillner, E.; Christensen, J.B.; Jensen, M.; Fredslund, F.; et al. Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2010, 107, 3681–3686. [Google Scholar] [CrossRef] [Green Version]
- Cook, W.J.; Galakatos, N.; Boyar, W.C.; Walter, R.L.; Ealick, S.E. Structure of human desArg-C5a. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Schatz-Jakobsen, J.A.; Yatime, L.; Larsen, C.; Petersen, S.V.; Klos, A.; Andersen, G.R. Structural and functional characterization of human and murine C5a anaphylatoxins. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1704–1717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Boyar, W.; Toth, M.J.; Wennogle, L.; Gonnella, N.C. Structural definition of the C5a C terminus by two-dimensional nuclear magnetic resonance spectroscopy. Proteins 1997, 28, 261–267. [Google Scholar] [CrossRef]
- Siciliano, S.J.; Rollins, T.E.; DeMartino, J.; Konteatis, Z.; Malkowitz, L.; Van Riper, G.; Bondy, S.; Rosen, H.; Springer, M.S. Two-site binding of C5a by its receptor: An alternative binding paradigm for G protein-coupled receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 1214–1218. [Google Scholar] [CrossRef] [Green Version]
- Nikiforovich, G.V.; Marshall, G.R.; Baranski, T.J. Modeling molecular mechanisms of binding of the anaphylatoxin C5a to the C5a receptor. Biochemistry 2008, 47, 3117–3130. [Google Scholar] [CrossRef]
- Rana, S.; Sahoo, A.R. Model structures of inactive and peptide agonist bound C5aR: Insights into agonist binding, selectivity and activation. Biochem. Biophys. Rep. 2015, 1, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, A.R.; Mishra, R.; Rana, S. The Model Structures of the Complement Component 5a Receptor (C5aR) Bound to the Native and Engineered (h)C5a. Sci. Rep. 2018, 8, 2955. [Google Scholar] [CrossRef] [Green Version]
- Bednarek, M.A.; Feighner, S.D.; Pong, S.S.; McKee, K.K.; Hreniuk, D.L.; Silva, M.V.; Warren, V.A.; Howard, A.D.; Van Der Ploeg, L.H.; Heck, J.V. Structure-function studies on the new growth hormone-releasing peptide, ghrelin: Minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J. Med. Chem. 2000, 43, 4370–4376. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hosoda, H.; Kitajima, Y.; Morozumi, N.; Minamitake, Y.; Tanaka, S.; Matsuo, H.; Kojima, M.; Hayashi, Y.; Kangawa, K. Structure-activity relationship of ghrelin: Pharmacological study of ghrelin peptides. Biochem. Biophys. Res. Commun. 2001, 287, 142–146. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kitajima, Y.; Iwanami, T.; Hayashi, Y.; Tanaka, S.; Minamitake, Y.; Hosoda, H.; Kojima, M.; Matsuo, H.; Kangawa, K. Structural similarity of ghrelin derivatives to peptidyl growth hormone secretagogues. Biochem. Biophys. Res. Commun. 2001, 284, 655–659. [Google Scholar] [CrossRef]
- Van Craenenbroeck, M.; Gregoire, F.; De Neef, P.; Robberecht, P.; Perret, J. Ala-scan of ghrelin (1–14): Interaction with the recombinant human ghrelin receptor. Peptides 2004, 25, 959–965. [Google Scholar] [CrossRef]
- Silva Elipe, M.V.; Bednarek, M.A.; Gao, Y.D. 1H NMR structural analysis of human ghrelin and its six truncated analogs. Biopolymers 2001, 59, 489–501. [Google Scholar] [CrossRef]
- Martin-Pastor, M.; De Capua, A.; Alvarez, C.J.; Diaz-Hernandez, M.D.; Jimenez-Barbero, J.; Casanueva, F.F.; Pazos, Y. Interaction between ghrelin and the ghrelin receptor (GHS-R1a), a NMR study using living cells. Bioorg. Med. Chem. 2010, 18, 1583–1590. [Google Scholar] [CrossRef]
- Staes, E.; Absil, P.A.; Lins, L.; Brasseur, R.; Deleu, M.; Lecouturier, N.; Fievez, V.; Rieux, A.; Mingeot-Leclercq, M.P.; Raussens, V.; et al. Acylated and unacylated ghrelin binding to membranes and to ghrelin receptor: Towards a better understanding of the underlying mechanisms. Biochim. Biophys. Acta 2010, 1798, 2102–2113. [Google Scholar] [CrossRef]
- De Ricco, R.; Valensin, D.; Gaggelli, E.; Valensin, G. Conformation propensities of des-acyl-ghrelin as probed by CD and NMR. Peptides 2013, 43, 62–67. [Google Scholar] [CrossRef]
- Vortmeier, G.; DeLuca, S.H.; Els-Heindl, S.; Chollet, C.; Scheidt, H.A.; Beck-Sickinger, A.G.; Meiler, J.; Huster, D. Integrating solid-state NMR and computational modeling to investigate the structure and dynamics of membrane-associated ghrelin. PLoS ONE 2015, 10, e0122444. [Google Scholar] [CrossRef] [PubMed]
- Ferré, G.; Louet, M.; Saurel, O.; Delort, B.; Czaplicki, G.; M’Kadmi, C.; Damian, M.; Renault, P.; Cantel, S.; Gavara, L.; et al. Structure and dynamics of G protein-coupled receptor–bound ghrelin reveal the critical role of the octanoyl chain. Proc. Natl. Acad. Sci. USA 2019, 116, 17525–17530. [Google Scholar] [CrossRef] [Green Version]
- Millar, R.P. GnRHs and GnRH receptors. Anim. Reprod. Sci. 2005, 88, 5–28. [Google Scholar] [CrossRef]
- Sealfon, S.C.; Weinstein, H.; Millar, R.P. Molecular mechanisms of ligand interaction with the gonadotropin-releasing hormone receptor. Endocr. Rev. 1997, 18, 180–205. [Google Scholar] [CrossRef]
- Chary, K.V.; Srivastava, S.; Hosur, R.V.; Roy, K.B.; Govil, G. Molecular conformation of gonadoliberin using two-dimensional NMR spectroscopy. Eur. J. Biochem. 1986, 158, 323–332. [Google Scholar] [CrossRef]
- Grant, G.; Vale, W. Speculations on structural relationships between the hypothalamic releasing factors of pituitary hormones. Nat. New Biol. 1972, 237, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.K.; Williams, R.H.; Humphries, A.J.; Johansson, N.G.; Folkers, K.; Bowers, C.Y. Luteinizing releasing hormone, synthesis and arg 8 -analogs, and conformation-sequence-activity relationships. Biochem. Biophys. Res. Commun. 1972, 47, 727–732. [Google Scholar] [CrossRef]
- Monahan, M.W.; Amoss, M.S.; Anderson, H.A.; Vale, W. Synthetic analogs of the hypothalamic luteinizing hormone releasing factor with increased agonist or antagonist properties. Biochemistry 1973, 12, 4616–4620. [Google Scholar] [CrossRef]
- Pincus, M.R.; Woo, J.; Monaco, R.; Lubowsky, J.; Avitable, M.J.; Carty, R.P. The low-energy conformations of gonadotropin-releasing hormone in aqueous solution. Protein J. 2014, 33, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Pincus, M.R.; Woo, J.; Monaco, R.; Lubowsky, J.; Carty, R.P. Low energy conformations for gonadotropin-releasing hormone with D- and L-amino acid substitutions for Gly 6: Possible receptor-bound conformations. Protein J. 2014, 33, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Millar, R.P.; Flanagan, C.A.; Milton, R.C.; King, J.A. Chimeric analogues of vertebrate gonadotropin-releasing hormones comprising substitutions of the variant amino acids in positions 5, 7, and 8. Characterization of requirements for receptor binding and gonadotropin release in mammalian and avian pituitary gonadotropes. J. Biol. Chem. 1989, 264, 21007–21013. [Google Scholar]
- Kumar, P.; Sharma, A. Gonadotropin-releasing hormone analogs: Understanding advantages and limitations. J. Hum. Reprod. Sci. 2014, 7, 170–174. [Google Scholar] [CrossRef]
- Andersen, N.H.; Hammen, P.K. A conformation-preference/potency correlation for GnRH analogs: NMR evidence. Biorg. Med. Chem. Lett. 1991, 1, 263–266. [Google Scholar] [CrossRef]
- Laimou, D.K.; Katsara, M.; Matsoukas, M.T.; Apostolopoulos, V.; Troganis, A.N.; Tselios, T.V. Structural elucidation of Leuprolide and its analogues in solution: Insight into their bioactive conformation. Amino Acids 2010, 39, 1147–1160. [Google Scholar] [CrossRef]
- Beck-Sickinger, A.G.; Jung, G. Structure-activity relationships of neuropeptide Y analogues with respect to Y1 and Y2 receptors. Biopolymers 1995, 37, 123–142. [Google Scholar] [CrossRef]
- Blundell, T.L.; Pitts, J.E.; Tickle, I.J.; Wood, S.P.; Wu, C.W. X-ray analysis (1. 4-A resolution) of avian pancreatic polypeptide: Small globular protein hormone. Proc. Natl. Acad. Sci. USA 1981, 78, 4175–4179. [Google Scholar] [CrossRef] [Green Version]
- Saudek, V.; Pelton, J.T. Sequence-specific 1H NMR assignment and secondary structure of neuropeptide Y in aqueous solution. Biochemistry 1990, 29, 4509–4515. [Google Scholar] [CrossRef]
- Schüß, C.; Vu, O.; Schubert, M.; Du, Y.; Mishra, N.M.; Tough, I.R.; Stichel, J.; Weaver, C.D.; Emmitte, K.A.; Cox, H.M.; et al. Highly Selective Y4 Receptor Antagonist Binds in an Allosteric Binding Pocket. J. Med. Chem. 2021, 64, 2801–2814. [Google Scholar] [CrossRef]
- Thomas, L.; Scheidt, H.A.; Bettio, A.; Huster, D.; Beck-Sickinger, A.G.; Arnold, K.; Zschornig, O. Membrane interaction of neuropeptide Y detected by EPR and NMR spectroscopy. Biochim. Biophys. Acta 2005, 1714, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.; Bettio, A.; Beck-Sickinger, A.G.; Zerbe, O. Structure and dynamics of micelle-bound neuropeptide Y: Comparison with unligated NPY and implications for receptor selection. J. Mol. Biol. 2001, 305, 307–329. [Google Scholar] [CrossRef]
- Portoghese, P.S. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends Pharmacol. Sci. 1989, 10, 230–235. [Google Scholar] [CrossRef]
- O’Connor, C.; White, K.L.; Doncescu, N.; Didenko, T.; Roth, B.L.; Czaplicki, G.; Stevens, R.C.; Wuthrich, K.; Milon, A. NMR structure and dynamics of the agonist dynorphin peptide bound to the human kappa opioid receptor. Proc. Natl. Acad. Sci. USA 2015, 112, 11852–11857. [Google Scholar] [CrossRef] [Green Version]
- Andersson, A.; Maler, L. NMR solution structure and dynamics of motilin in isotropic phospholipid bicellar solution. J. Biomol. NMR 2002, 24, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Rathmann, D.; Lindner, D.; DeLuca, S.H.; Kaufmann, K.W.; Meiler, J.; Beck-Sickinger, A.G. Ligand-mimicking receptor variant discloses binding and activation mode of prolactin-releasing peptide. J. Biol. Chem. 2012, 287, 32181–32194. [Google Scholar] [CrossRef] [Green Version]
- Deluca, S.H.; Rathmann, D.; Beck-Sickinger, A.G.; Meiler, J. The activity of prolactin releasing peptide correlates with its helicity. Biopolymers 2013, 99, 314–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubecka, E.A.; Sikorska, E.; Sobolewski, D.; Prahl, A.; Slaninova, J.; Ciarkowski, J. Arginine-, D-arginine-vasopressin, and their inverso analogues in micellar and liposomic models of cell membrane: CD, NMR, and molecular dynamics studies. Eur. Biophys. J. 2015, 44, 727–743. [Google Scholar] [CrossRef] [Green Version]
- Haugaard-Kedstrom, L.M.; Hossain, M.A.; Daly, N.L.; Bathgate, R.A.; Rinderknecht, E.; Wade, J.D.; Craik, D.J.; Rosengren, K.J. Solution structure, aggregation behavior, and flexibility of human relaxin-2. ACS Chem. Biol. 2015, 10, 891–900. [Google Scholar] [CrossRef]
- Rosengren, K.J.; Lin, F.; Bathgate, R.A.; Tregear, G.W.; Daly, N.L.; Wade, J.D.; Craik, D.J. Solution structure and novel insights into the determinants of the receptor specificity of human relaxin-3. J. Biol. Chem. 2006, 281, 5845–5851. [Google Scholar] [CrossRef] [Green Version]
- Grace, C.R.; Erchegyi, J.; Reubi, J.C.; Rivier, J.E.; Riek, R. Three-dimensional consensus structure of sst2-selective somatostatin (SRIF) antagonists by NMR. Biopolymers 2008, 89, 1077–1087. [Google Scholar] [CrossRef] [Green Version]
- Grace, C.R.; Erchegyi, J.; Koerber, S.C.; Reubi, J.C.; Rivier, J.; Riek, R. Novel sst2-selective somatostatin agonists. Three-dimensional consensus structure by NMR. J. Med. Chem. 2006, 49, 4487–4496. [Google Scholar] [CrossRef] [Green Version]
- Grace, C.R.; Durrer, L.; Koerber, S.C.; Erchegyi, J.; Reubi, J.C.; Rivier, J.E.; Riek, R. Somatostatin receptor 1 selective analogues: 4. Three-dimensional consensus structure by NMR. J. Med. Chem. 2005, 48, 523–533. [Google Scholar] [CrossRef]
- Grace, C.R.; Koerber, S.C.; Erchegyi, J.; Reubi, J.C.; Rivier, J.; Riek, R. Novel sst4-selective somatostatin (SRIF) agonists. 4. Three-dimensional consensus structure by NMR. J. Med. Chem. 2003, 46, 5606–5618. [Google Scholar] [CrossRef]
- Denys, L.; Bothner-By, A.A.; Fisher, G.H.; Ryan, J.W. Conformational diversity of bradykinin in aqueous solution. Biochemistry 1982, 21, 6531–6536. [Google Scholar] [CrossRef]
- Lopez, J.J.; Shukla, A.K.; Reinhart, C.; Schwalbe, H.; Michel, H.; Glaubitz, C. The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2008, 47, 1668–1671. [Google Scholar] [CrossRef]
- Kaiser, A.; Coin, I. Capturing Peptide–GPCR Interactions and Their Dynamics. Molecules 2020, 25, 4724. [Google Scholar] [CrossRef]
- Singh, K.D.; Unal, H.; Desnoyer, R.; Karnik, S.S. Mechanism of Hormone Peptide Activation of a GPCR: Angiotensin II Activated State of AT1R Initiated by van der Waals Attraction. J. Chem. Inf. Model. 2019, 59, 373–385. [Google Scholar] [CrossRef]
- Lane, J.R.; May, L.T.; Parton, R.G.; Sexton, P.M.; Christopoulos, A. A kinetic view of GPCR allostery and biased agonism. Nat. Chem. Biol. 2017, 13, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S.; Kaufman, B. Machine learning and AI-based approaches for bioactive ligand discovery and GPCR-ligand recognition. Methods 2020, 180, 89–110. [Google Scholar] [CrossRef]
- Bandholtz, S.; Wichard, J.; Kühne, R.; Grötzinger, C. Molecular Evolution of a Peptide GPCR Ligand Driven by Artificial Neural Networks. PLoS ONE 2012, 7, e36948. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Modification | Example | Function |
|---|---|---|
| C-terminal amidation | Neuropeptide Y (NPY), neuromedin B | C-terminal amidation reduces the overall charge of a peptide, forms key hydrogen interactions that are important for the potency of the peptide [15], and increases the metabolic stability of peptides as well as their ability to resist enzymatic degradation [16] |
| N-terminal pyroglutamic acid | Thyroid stimulating hormone (TSH), gonadotropin-releasing hormone I (GnRHI), regulated upon activation, Normal T cell expressed, and presumably secreted (RANTES)/chemokine ligand 5 (CCL5) | The pyroglutamic acid is often involved in peptide-receptor recognition and potency [17], and provides stability against N-terminal degradation [18] |
| Bromination | Neuropeptides B and W (NPBW) | Bromination on N-terminal tryptophan might protect the peptide from amino-peptidases’ degradation [19] |
| Lipidation | Ghrelin | The attached lipid group (e.g., octanoyl group) is essential to the activity of the peptide [19] and affects the hydrophobicity of the peptide [20] |
| Disulfide bridge formation | Endothelin, vasopressin | The disulfide bonds stabilize the defined secondary structure [21], stabilizing the bound conformation of the peptide [22] |
| Differential proteolysis | Bradykinin, angiotensin, NPY/NPY3-36, apelin (Ape)-13/Ape-17/Ape 36, adrenocorticotropic hormone (ACTH), pro-opiomelanocortin (POMC) cleavage yielding α-, β-, and γ- melanocyte-stimulating hormone (MSH), and endorphins | Proteolysis can switch the activity of the peptides on and off [23] or differentiates the binding selectivity and the biological responses of the peptides [24] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, O.; Bender, B.J.; Pankewitz, L.; Huster, D.; Beck-Sickinger, A.G.; Meiler, J. The Structural Basis of Peptide Binding at Class A G Protein-Coupled Receptors. Molecules 2022, 27, 210. https://doi.org/10.3390/molecules27010210
Vu O, Bender BJ, Pankewitz L, Huster D, Beck-Sickinger AG, Meiler J. The Structural Basis of Peptide Binding at Class A G Protein-Coupled Receptors. Molecules. 2022; 27(1):210. https://doi.org/10.3390/molecules27010210
Chicago/Turabian StyleVu, Oanh, Brian Joseph Bender, Lisa Pankewitz, Daniel Huster, Annette G. Beck-Sickinger, and Jens Meiler. 2022. "The Structural Basis of Peptide Binding at Class A G Protein-Coupled Receptors" Molecules 27, no. 1: 210. https://doi.org/10.3390/molecules27010210
APA StyleVu, O., Bender, B. J., Pankewitz, L., Huster, D., Beck-Sickinger, A. G., & Meiler, J. (2022). The Structural Basis of Peptide Binding at Class A G Protein-Coupled Receptors. Molecules, 27(1), 210. https://doi.org/10.3390/molecules27010210