
Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis



{kind=link}
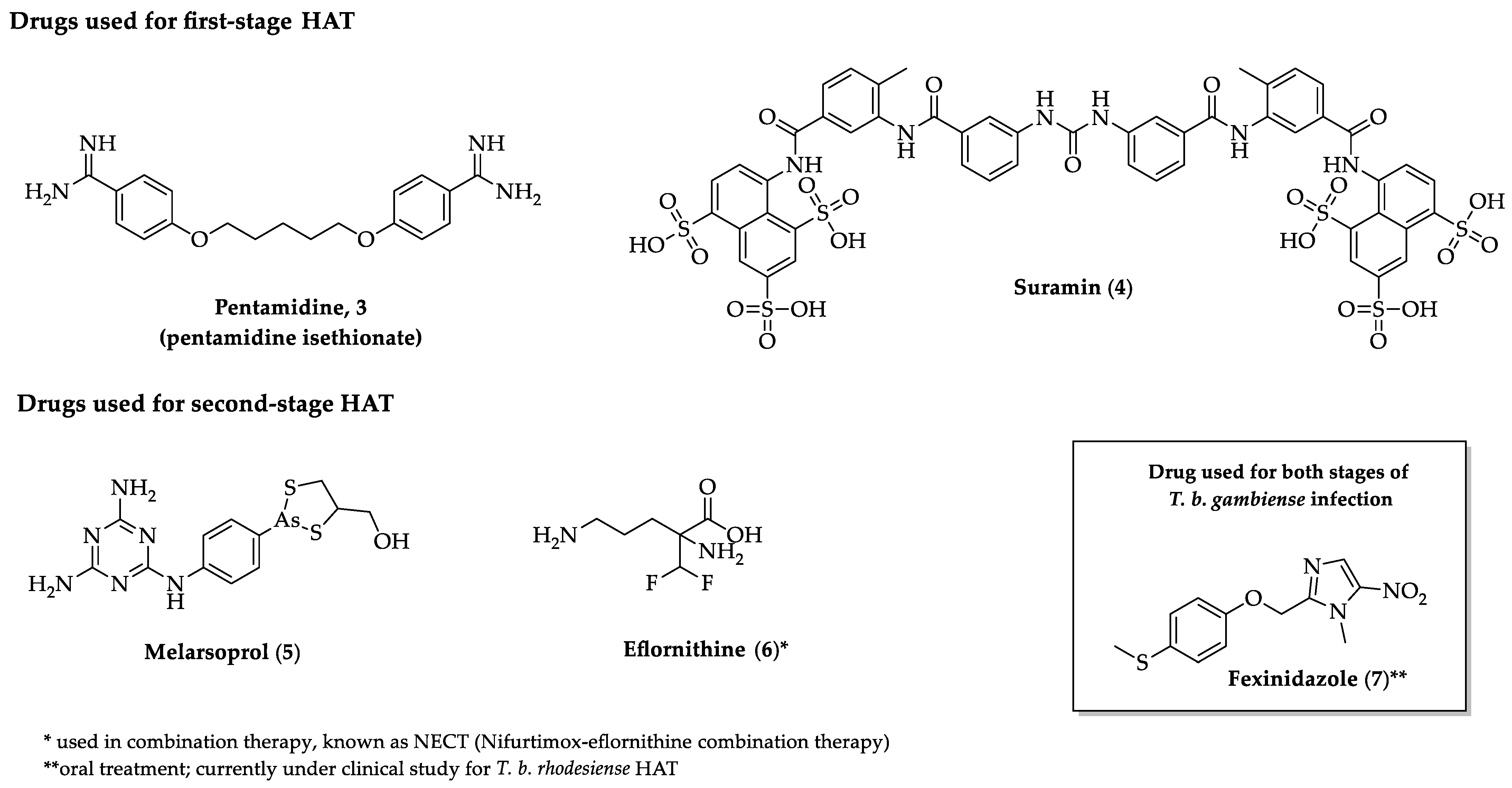{kind=link}
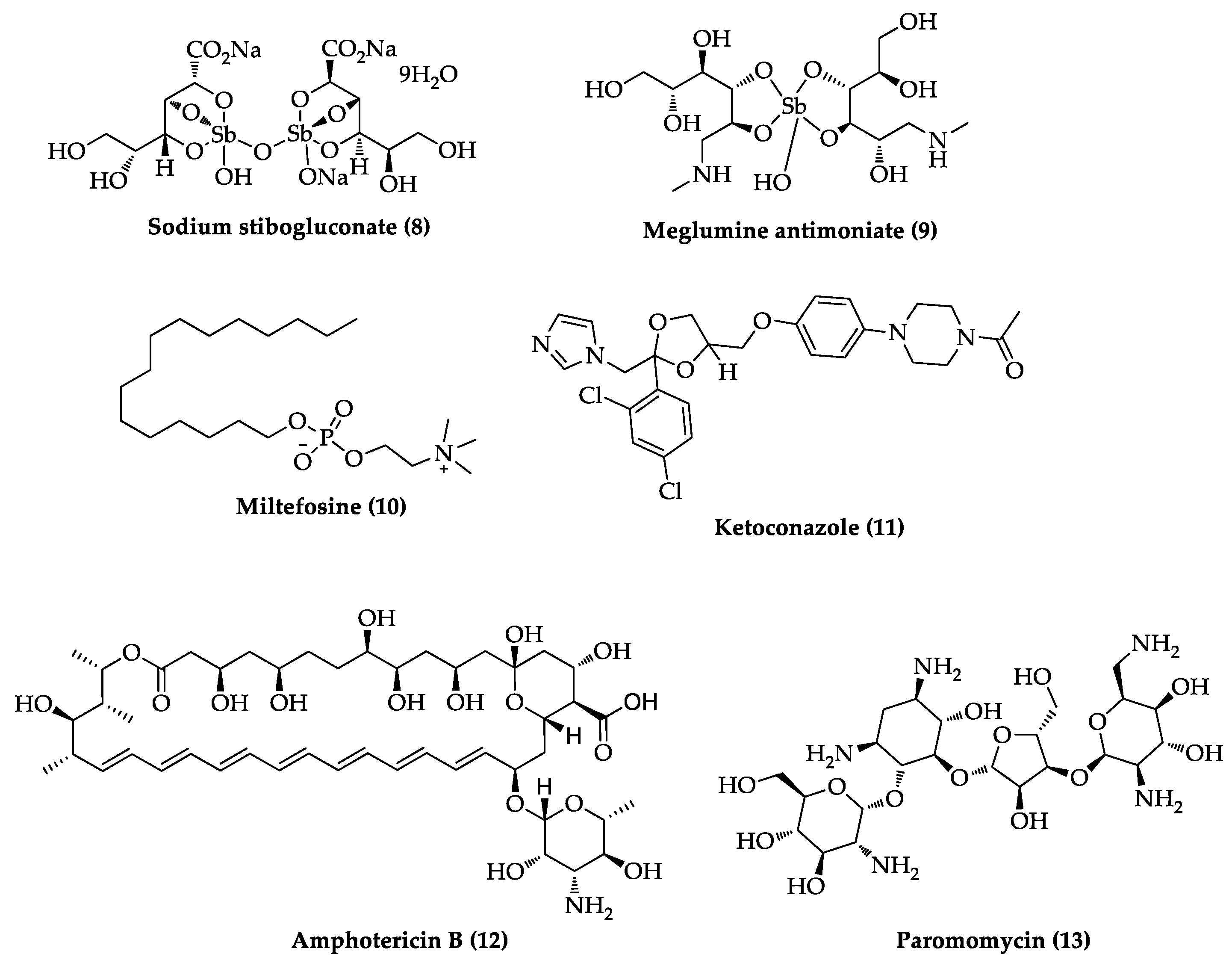{kind=link}
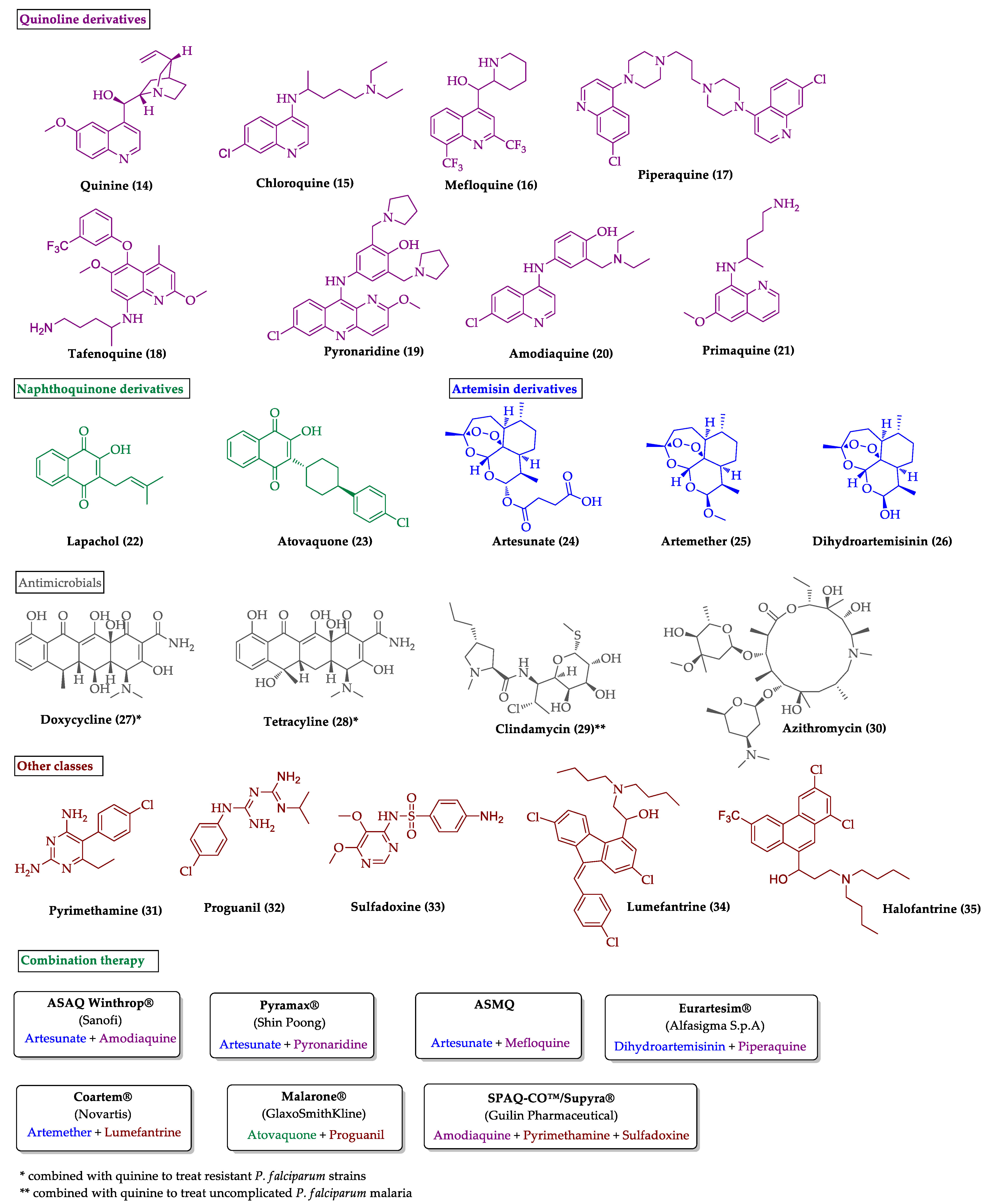{kind=link}
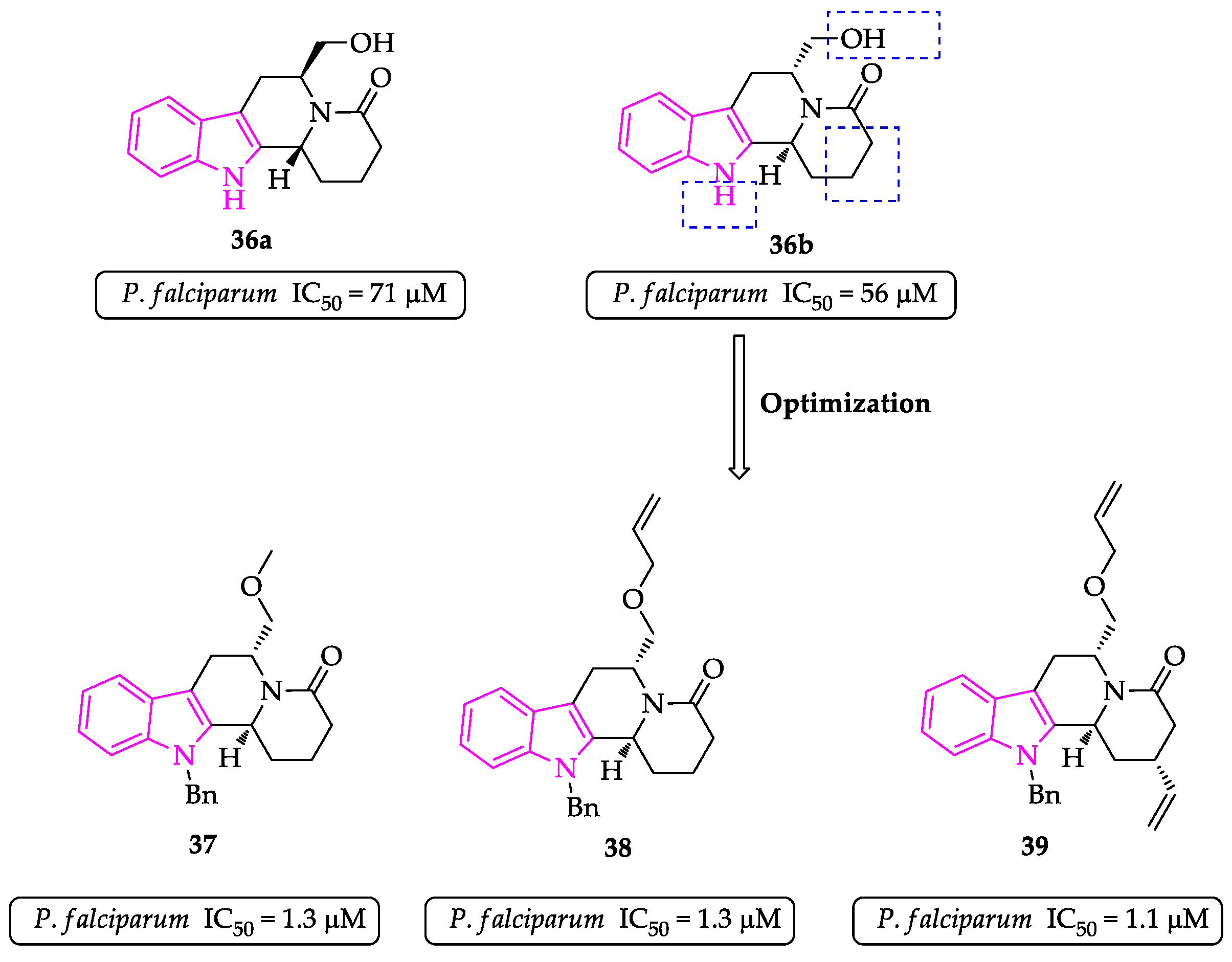{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
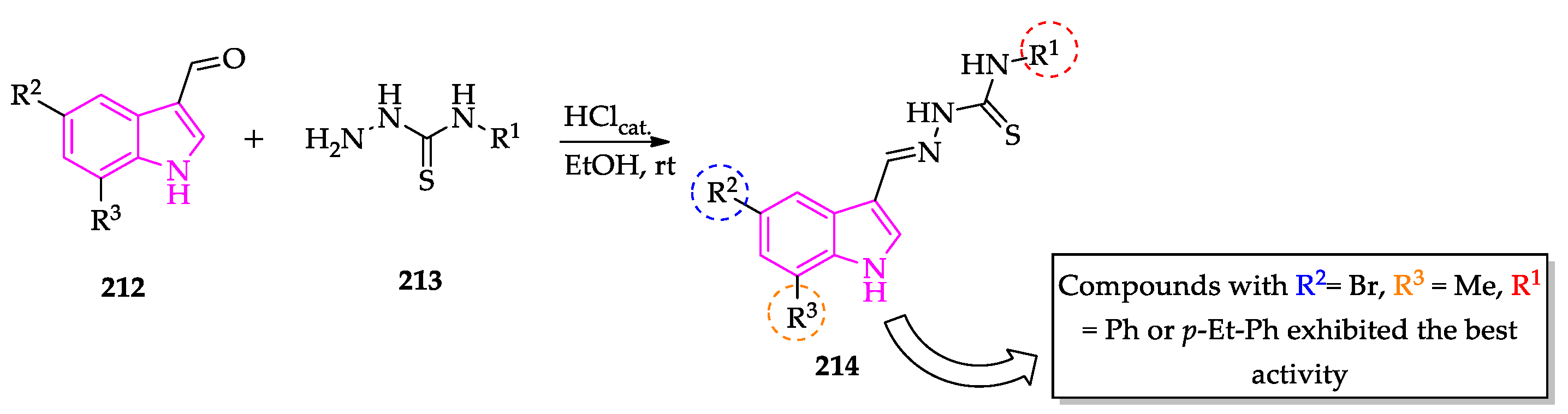{kind=link}
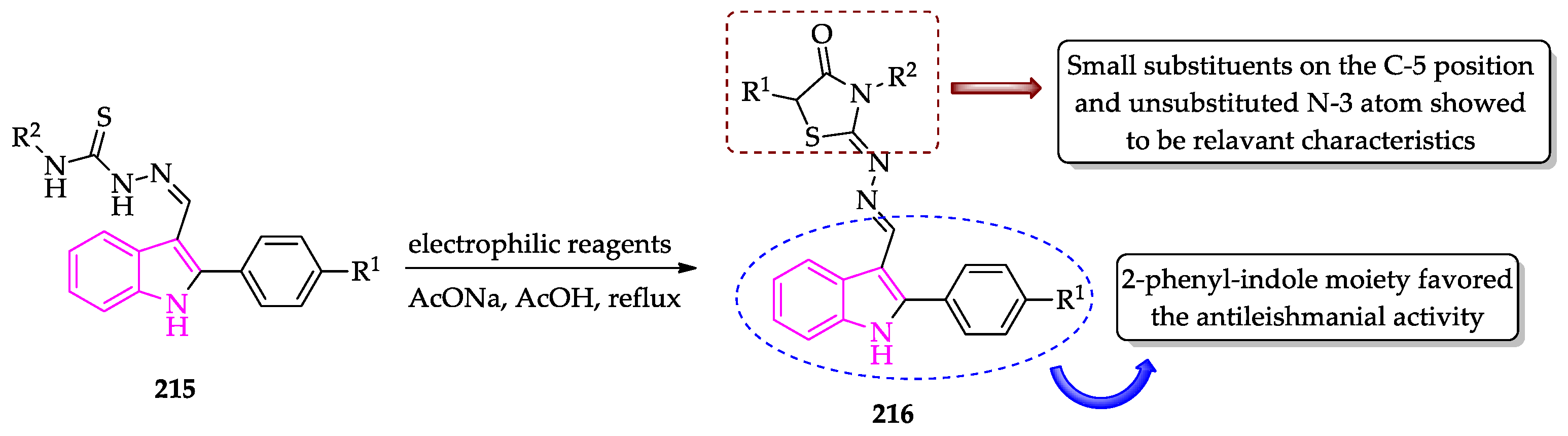{kind=link}
Abstract
:1. Introduction
2. Recent Development of Indole-Based Small Molecules Targeting Malaria, Trypanosomiasis and Leishmaniasis
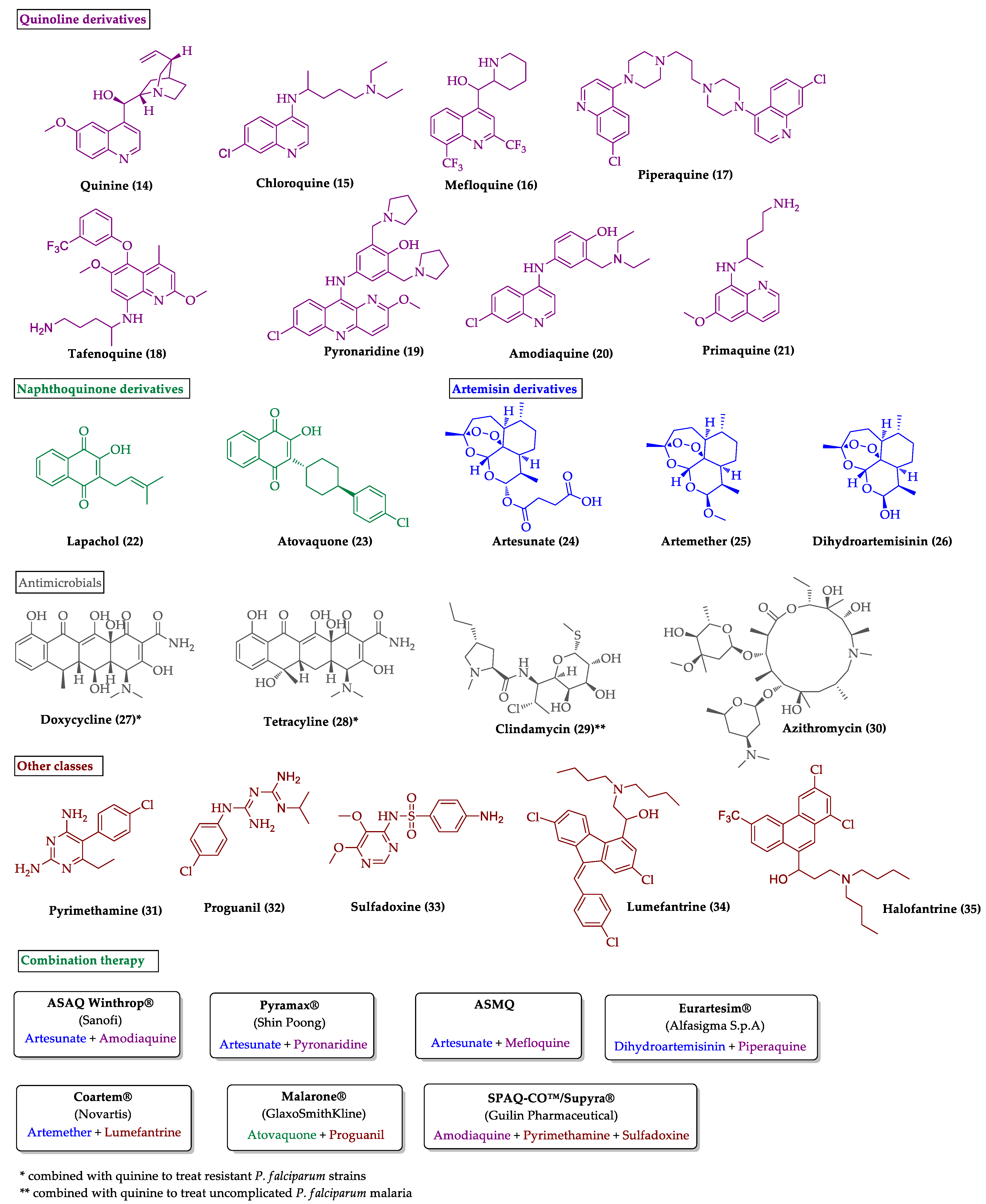
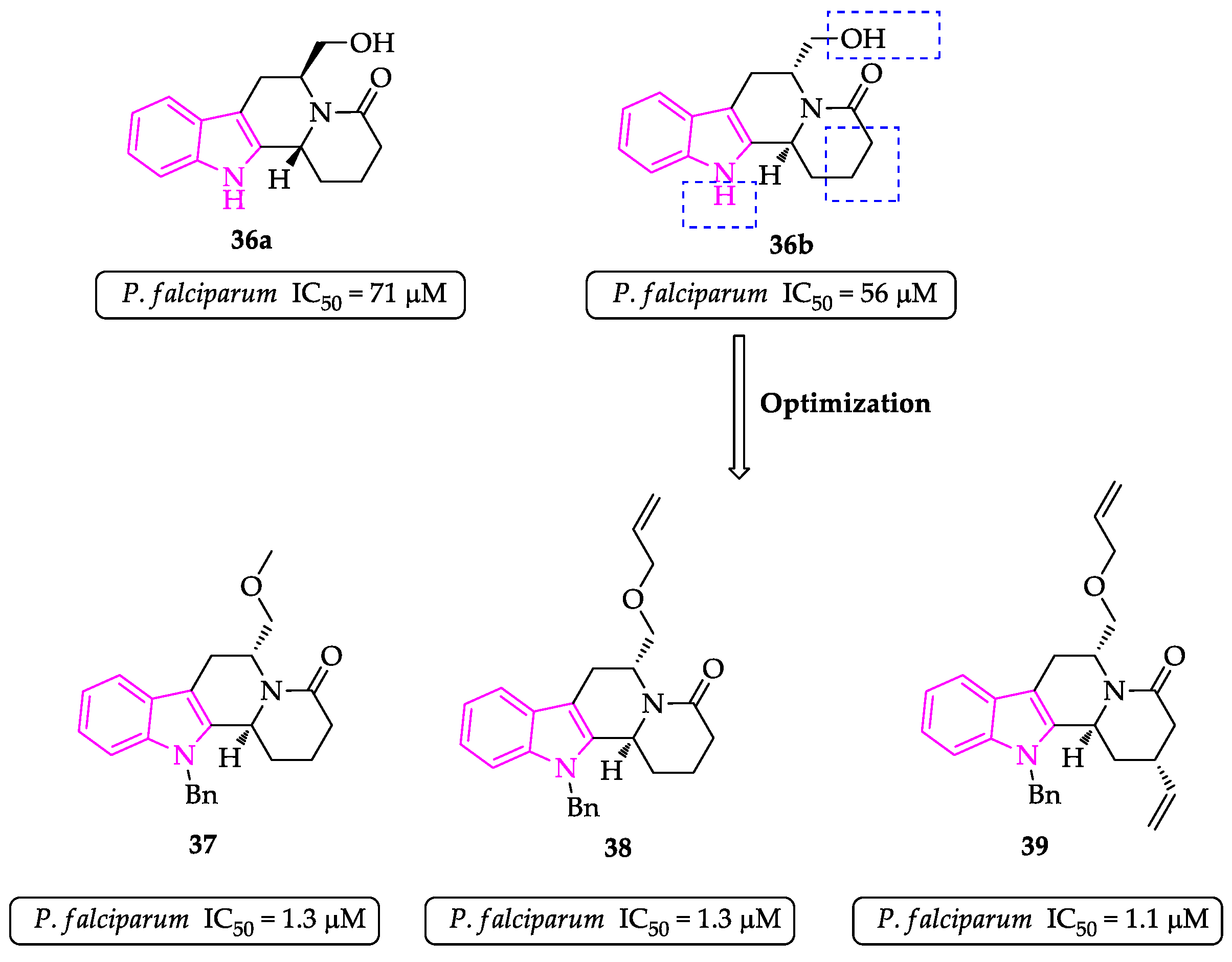
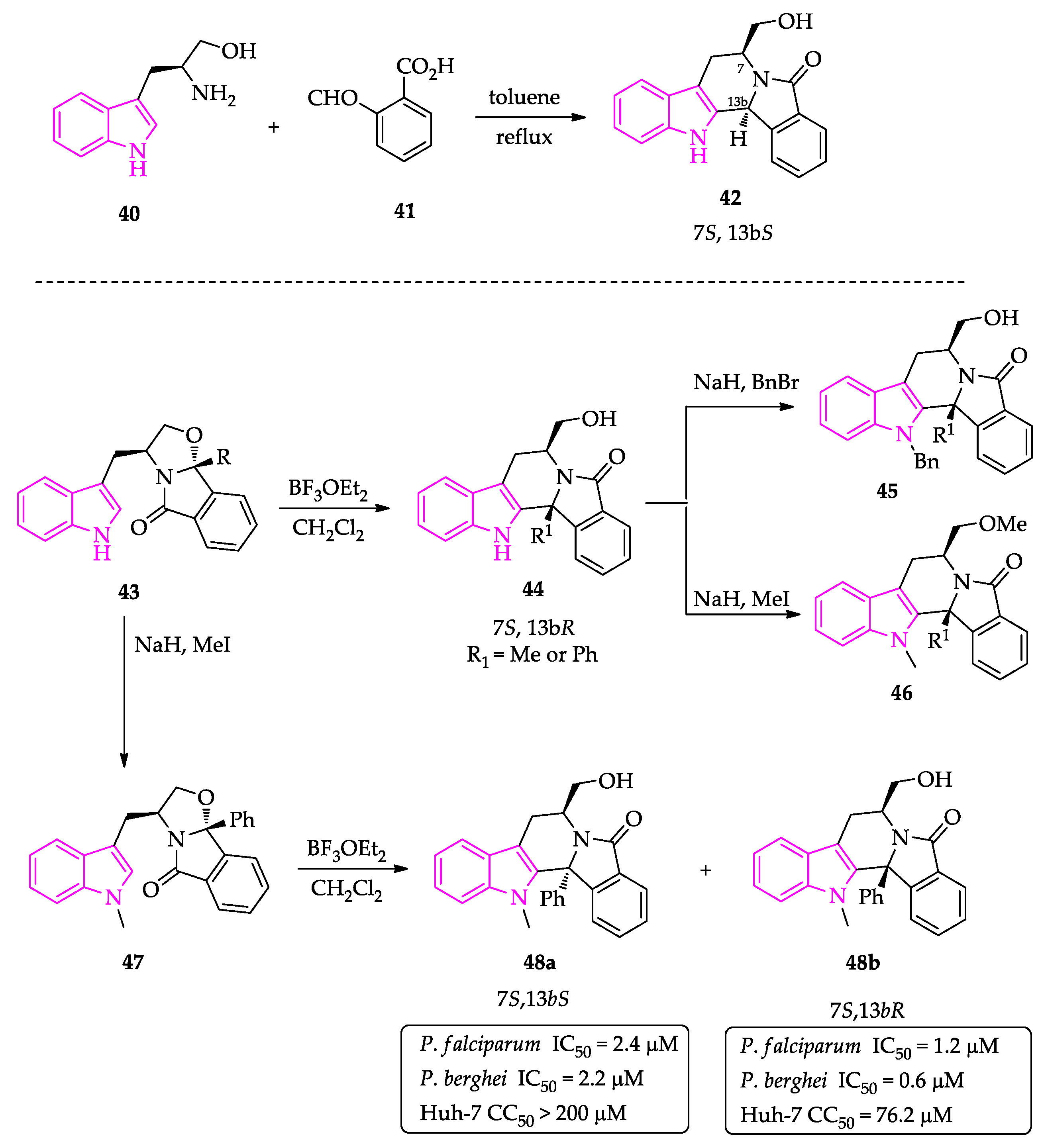
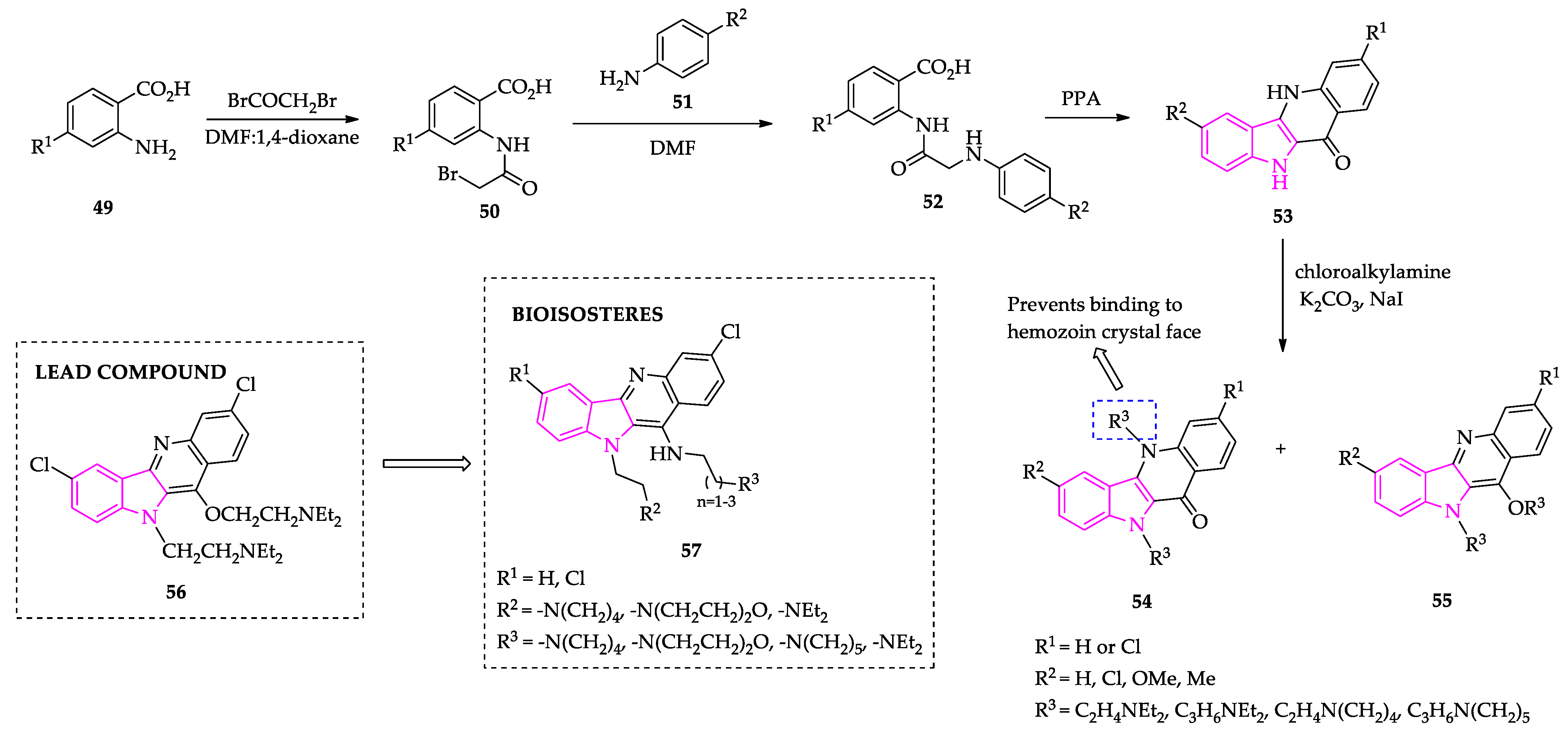
2.1. Indole Derivatives with Antiplasmodial Activity
2.2. Indole Derivatives with Antitrypanosomal Activity
2.3. Indole Derivatives with Antileishmanial Activity
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Molyneux, D.H.; Savioli, L.; Engels, D. Neglected tropical diseases: Progress towards addressing the chronic pandemic. Lancet 2017, 389, 312–325. [Google Scholar] [CrossRef]
- Pastrana, N.A.; Beran, D.; Somerville, C.; Heller, O.; Correia, J.C.; Suggs, L.S. The process of building the priority of neglected tropical diseases: A global policy analysis. PLoS Negl. Trop. Dis. 2020, 14, e0008498. [Google Scholar] [CrossRef]
- DESA, UN. Transforming Our World: The 2030 Agenda for Sustainable Development. UN General Assembly. Available online: https://sdgs.un.org/2030agenda (accessed on 12 October 2021).
- Institute for Health Metrics and Evaluation (IHME). GBD 2019 Cause and Risk Summary: Neglected Diseases and Malaria; IHME, University of Washington: Seattle, WA, USA, 2020; Available online: https://www.healthdata.org/results/gbd_summaries/2019/neglected-tropical-diseases-and-malaria-level-2-cause (accessed on 12 October 2021).
- Aagaard-Hansen, J.; Nombela, N.; Alvar, J. Population movement: A key factor in the epidemiology of neglected tropical diseases. Trop. Med. Int. Health 2010, 15, 1281–1288. [Google Scholar] [CrossRef]
- Tidman, R.; Abela-Ridder, B.; De Castañeda, R.R. The impact of climate change on neglected tropical diseases: A systematic review. Trans. R. Soc. Trop. Med. Hyg. 2021, 115, 147–168. [Google Scholar] [CrossRef]
- Altamura, F.; Rajesh, R.; Catta-Preta, C.M.C.; Moretti, N.S.; Cestari, I. The current drug discovery landscape for trypanosomiasis and leishmaniasis: Challenges and strategies to identify drug targets. Drug Dev. Res. 2020, 1–28. [Google Scholar] [CrossRef]
- Lukeš, J.; Skalický, T.; Týč, J.; Votýpka, J.; Yurchenko, V. Evolution of parasitism in kinetoplastid flagellates. Mol. Biochem. Parasitol. 2014, 195, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Büscher, P.; Cecchi, G.; Jamonneau, V.; Priotto, G. Human African Trypanosomiasis. Lancet 2017, 390, 2397–2409. [Google Scholar] [CrossRef]
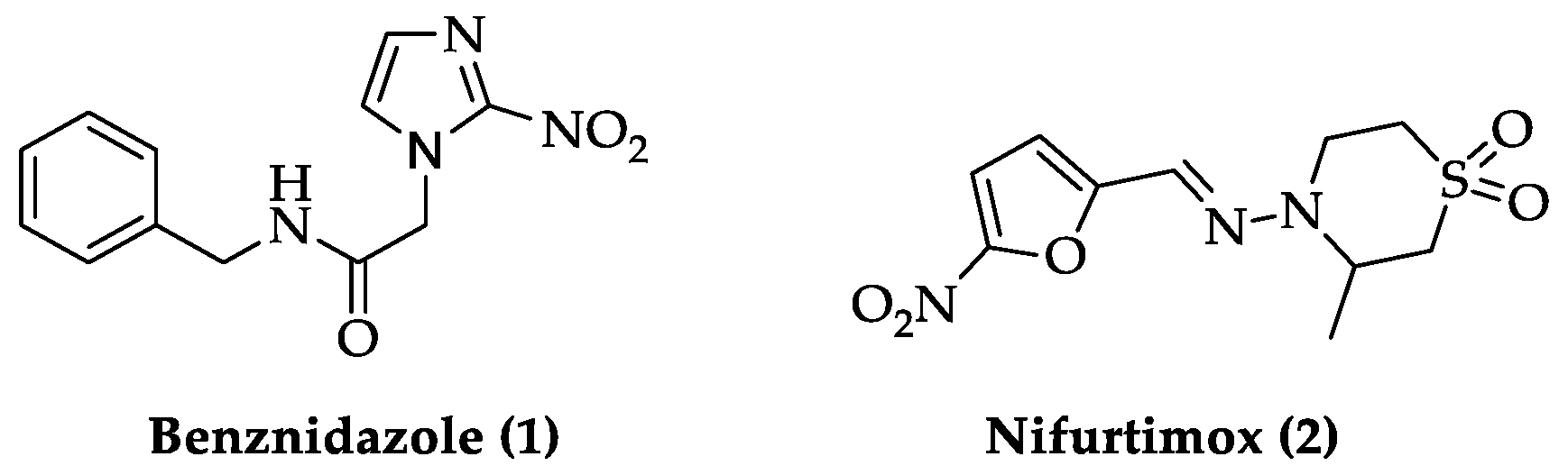
- Echeverria, L.E.; Morillo, C.A. American trypanosomiasis (Chagas Disease). Infect. Dis. Clin. N. Am. 2019, 33, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.C.P.; Dones, W.; Morillo, C.A.; Encina, J.J.; Ribeiro, A.L. Chagas disease: An overview of clinical and epidemiological aspects. J. Am. Coll. Cardiol. 2013, 62, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Coura, J.R. The main sceneries of Chagas disease transmission. The vectors, blood and oral transmissions—A comprehensive review. Mem. Inst. Oswaldo Cruz 2015, 110, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Coura, J.R.; Vĩas, P.A. Chagas disease: A new worldwide challenge. Nature 2010, 465, S06–S07. [Google Scholar] [CrossRef] [PubMed]
- Chagas Disease (American Trypanosomiasis). Available online: https://www.who.int/health-topics/chagas-disease#tab=tab_1 (accessed on 12 October 2021).
- Lee, B.Y.; Bacon, K.M.; Bottazzi, M.E.; Hotez, P.J. Global economic burden of Chagas disease: A computational simulation model. Lancet Infect. Dis. 2013, 13, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Steverding, D. The history of Chagas disease. Parasites Vectors 2014, 7, 317. [Google Scholar] [CrossRef]
- Lascano, F.; García Bournissen, F.; Altcheh, J. Review of pharmacological options for the treatment of Chagas disease. Br. J. Clin. Pharmacol. 2020, 1–20. [Google Scholar] [CrossRef]
- Lidani, K.C.F.; Andrade, F.A.; Bavia, L.; Damasceno, F.S.; Beltrame, M.H.; Messias-Reason, I.J.; Sandri, T.L. Chagas disease: From discovery to a worldwide health problem. J. Phys. Oceanogr. 2019, 49, 166. [Google Scholar] [CrossRef] [PubMed]
- García-Huertas, P.; Cardona-Castro, N. Advances in the treatment of Chagas disease: Promising new drugs, plants and targets. Biomed. Pharmacother. 2021, 142, 112020. [Google Scholar] [CrossRef]
- Santos, S.S.; de Araújo, R.V.; Giarolla, J.; El Seoud, O.; Ferreira, E.I. Searching for drugs for Chagas disease, leishmaniasis and schistosomiasis: A review. Int. J. Antimicrob. Agents 2020, 55, 105906. [Google Scholar] [CrossRef]
- Bermudez, J.; Davies, C.; Simonazzi, A.; Pablo Real, J.; Palma, S. Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Trop. 2016, 156, 1–16. [Google Scholar] [CrossRef]
- Hide, G. History of sleeping sickness in East Africa. Clin. Microbiol. Rev. 1999, 12, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Malvy, D.; Chappuis, F. Sleeping sickness. Clin. Microbiol. Infect. 2011, 17, 986–995. [Google Scholar] [CrossRef] [Green Version]
- WHO. Trypanosomiasis, Human African (Sleeping Sickness). 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 13 October 2021).
- Wamwiri, F.N.; Changasi, R.E. Tsetse Flies (Glossina) as vectors of human African Trypanosomiasis: A review. Biomed. Res. Int. 2016, 2016, 6201350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, P.G.E. Update on human African Trypanosomiasis (sleeping sickness). J. Neurol. 2019, 266, 2334–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, C.S.; Stone, C.M.; Steinmann, P.; Tanner, M.; Tediosi, F. Seeing beyond 2020: An economic evaluation of contemporary and emerging strategies for elimination of Trypanosoma brucei gambiense. Lancet Glob. Health 2017, 5, e69–e79. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.G.E.; Rodgers, J. Clinical and neuropathogenetic aspects of human African Trypanosomiasis. Front. Immunol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human African Trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, C.S.; Yukich, J.; Goeree, R.; Tediosi, F. A Literature Review of Economic Evaluations for a Neglected Tropical Disease: Human African Trypanosomiasis (“Sleeping Sickness”). PLoS Negl. Trop. Dis. 2015, 9, e0003397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martínez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A Review of Leishmaniasis: Current Knowledge and Future Directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votýpka, J.; Marty, P.; Delaunay, P.; Sereno, D. A Historical Overview of the Classification, Evolution, and Dispersion of Leishmania Parasites and Sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- Karunaweera, N.D.; Ferreira, M.U. Leishmaniasis: Current challenges and prospects for elimination with special focus on the South Asian region. Parasitology 2018, 145, 425–429. [Google Scholar] [CrossRef] [Green Version]
- Alemayehu, B.; Alemayehu, M. Leishmaniasis: A Review on Parasite, Vector and Reservoir Host. Health Sci. J. 2017, 11, 1. [Google Scholar] [CrossRef]
- WHO. Leishmaniasis. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 13 October 2021).
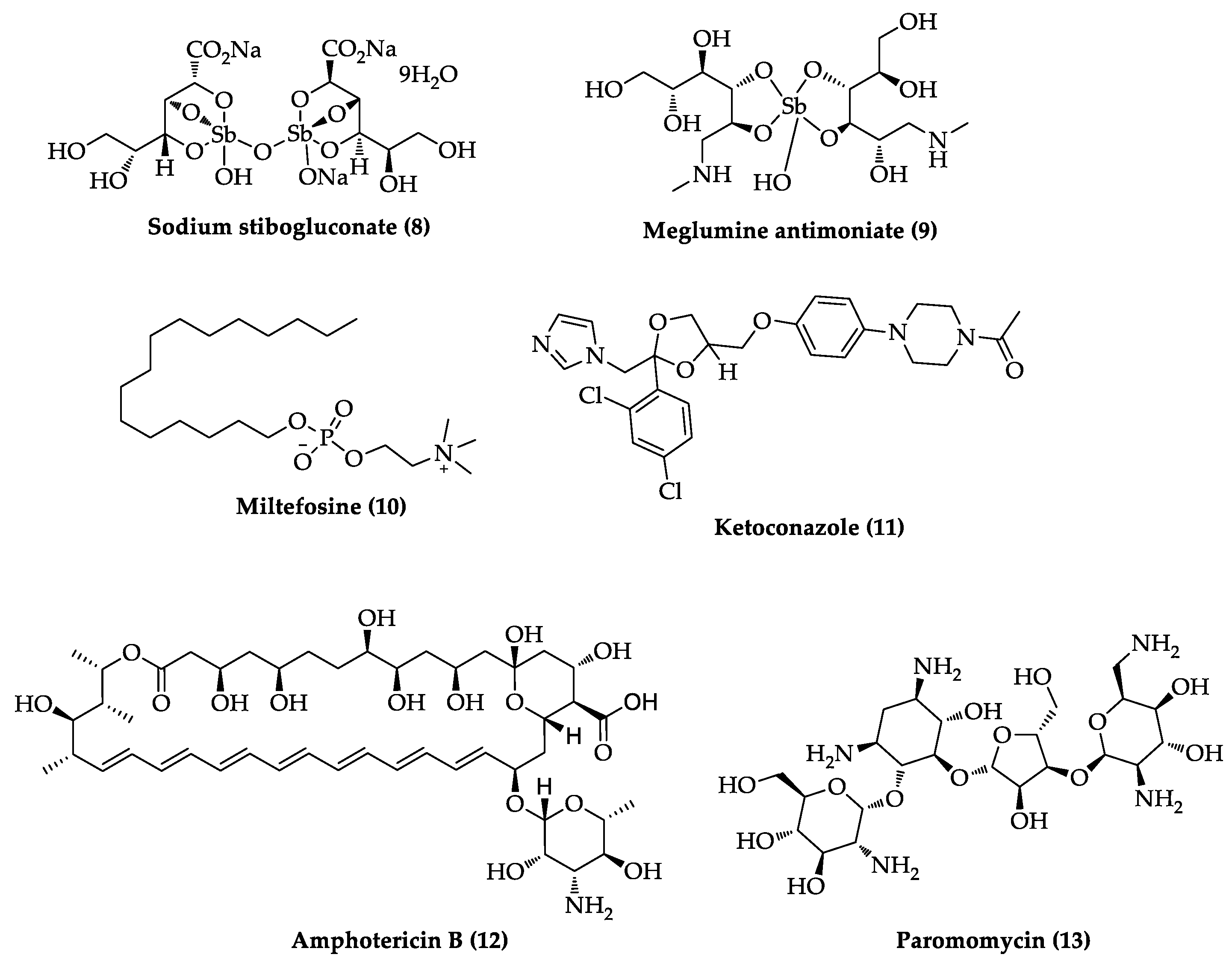
- Croft, S.L.; Olliaro, P. Leishmaniasis chemotherapy-challenges and opportunities. Clin. Microbiol. Infect. 2011, 17, 1478–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burza, S.; Crof, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Salman, S.M.; Rubeiz, N.G.; Kibbi, A.G. Cutaneous leishmaniasis: Clinical features and diagnosis. Clin. Dermatol. 1999, 17, 291–296. [Google Scholar] [CrossRef]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, S7–S16. [Google Scholar] [CrossRef]
- Santos, D.O.; Coutinho, C.E.R.; Madeira, M.F.; Bottino, C.G.; Vieira, R.T.; Nascimento, S.B.; Bernardino, A.; Bourguignon, S.C.; Corte-Real, S.; Pinho, R.T.; et al. Leishmaniasis treatment—A challenge that remains: A review. Parasitol. Res. 2008, 103, 1–10. [Google Scholar] [CrossRef]
- Burrows, J.N.; Elliott, R.L.; Kaneko, T.; Mowbray, C.E.; Waterson, D. The role of modern drug discovery in the fight against neglected and tropical diseases. Med. Chem. Comm. 2014, 5, 688–700. [Google Scholar] [CrossRef]
- Bethony, J.M.; Cole, R.N.; Guo, X.; Kamhawi, S.; Lightowlers, M.W.; Loukas, A.; Petri, W.; Reed, S.; Valenzuela, J.G.; Hotez, P.J. Vaccines to combat the neglected tropical diseases. Immunol. Rev. 2011, 239, 237–270. [Google Scholar] [CrossRef] [Green Version]
- Boutayeb, A. Developing countries and neglected diseases: Challenges and perspectives. Int. J. Equity Health 2007, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Laurens, M.B. RTS,S/AS01 vaccine (MosquirixTM): An overview. Hum. Vaccines Immunother. 2019, 16, 480–489. [Google Scholar] [CrossRef]
- Scientists Hail Historic Malaria Vaccine Approval—But Point to Challenges Ahead. Available online: https://www.nature.com/articles/d41586-021-02755-5 (accessed on 15 October 2021).
- WHO. World Malaria Report 2020: 20 Years of Global Progress and Challenges. 2020. Available online: https://apps.who.int/iris/handle/10665/337660 (accessed on 15 October 2021).
- Lalremruata, A.; Jeyaraj, S.; Engleitner, T.; Joanny, F.; Lang, A.; Bélard, S.; Mombo-Ngoma, G.; Ramharter, M.; Kremsner, P.G.; Mordmüller, B.; et al. Species and genotype diversity of Plasmodium in malaria patients from Gabon analysed by next generation sequencing. Malar. J. 2017, 16, 398. [Google Scholar] [CrossRef]
- Plewes, K.; Leopold, S.J.; Kingston, H.W.F.; Dondorp, A.M. Malaria: What’s New in the Management of Malaria? Infect. Dis. Clin. N. Am. 2019, 33, 39–60. [Google Scholar] [CrossRef] [PubMed]
- Bousema, T.; Drakeley, C. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin. Microbiol. Rev. 2011, 24, 377–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Mharakurwa, S.; Ndiaye, D.; Rathod, P.K.; Rosenthal, P.J. Antimalarial drug resistance: Literature review and activities and findings of the ICEMR network. Am. J. Trop. Med. Hyg. 2015, 93, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Mita, T.; Tanabe, K. Evolution of Plasmodium falciparum drug resistance: Implications for the development and containment of artemisinin resistance. Jpn. J. Infect. Dis. 2012, 65, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Price, R.N.; von Seidlein, L.; Valecha, N.; Nosten, F.; Baird, J.K.; White, N.J. Global extent of chloroquine-resistant Plasmodium vivax: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 982–991. [Google Scholar] [CrossRef] [Green Version]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belete, T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Dev. Ther. 2020, 14, 3875–38892. [Google Scholar] [CrossRef]
- WHO. Artemisinin Resistance and Artemisinin-Based Combination Therapy Efficacy. 2018. Available online: https://apps.who.int/iris/handle/10665/274362 (accessed on 15 October 2021).
- Thanikachalam, P.V.; Maurya, R.K.; Garg, V.; Monga, V. An insight into the medicinal perspective of synthetic analogs of indole: A review. Eur. J. Med. Chem. 2019, 180, 562–612. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef]
- Chauhan, M.; Saxena, A.; Saha, B. An insight in anti-malarial potential of indole scaffold: A review. Eur. J. Med. Chem. 2021, 218, 113400. [Google Scholar] [CrossRef]
- Surur, A.S.; Huluka, S.A.; Mitku, M.L.; Asres, K. Indole: The after next scaffold of antiplasmodial agents? Drug Des. Dev. Ther. 2020, 14, 4855–4867. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sharma, S.; Kalra, S.; Singh, G.; Monga, V.; Kumar, B. Medicinal Perspective of Indole Derivatives: Recent Developments and Structure-Activity Relationship Studies. Curr. Drug Targets 2020, 21, 864–891. [Google Scholar] [CrossRef] [PubMed]
- Lopes, E.A.; Santos, M.M.M.; Mori, M. Pharmacological treatment of malaria. In Antiprotozoal Drug Development and Delivery; Springer, 2021; accepted; Available online: https://link.springer.com/chapter/10.1007/7355_2021_125 (accessed on 27 August 2021).
- Horrocks, P.; Fallon, S.; Denman, L.; Devine, O.; Duffy, L.J.; Harper, A.; Meredith, E.L.; Hasenkamp, S.; Sidaway, A.; Monnery, D.; et al. Synthesis and evaluation of a novel series of indoloisoquinolines as small molecule anti-malarial leads. Bioorg. Med. Chem. Lett. 2012, 22, 1770–1773. [Google Scholar] [CrossRef]
- Pereira, N.A.L.; Monteiro, Â.; Machado, M.; Gut, J.; Molins, E.; Perry, M.J.; Dourado, J.; Moreira, R.; Rosenthal, P.J.; Prudêncio, M.; et al. Enantiopure Indolizinoindolones with in vitro Activity against Blood- and Liver-Stage Malaria Parasites. ChemMedChem 2015, 10, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Paulo, A.; Figueiras, M.; MacHado, M.; Charneira, C.; Lavrado, J.; Santos, S.A.; Lopes, D.; Gut, J.; Rosenthal, P.J.; Nogueira, F.; et al. Bis-alkylamine indolo[3,2-b]quinolines as hemozoin ligands: Implications for antimalarial cytostatic and cytocidal activities. J. Med. Chem. 2014, 57, 3295–3313. [Google Scholar] [CrossRef]
- Figueiras, M.; Coelho, L.; Wicht, K.J.; Santos, S.A.; Lavrado, J.; Gut, J.; Rosenthal, P.J.; Nogueira, F.; Egan, T.J.; Moreira, R.; et al. N10,N11-di-alkylamine indolo[3,2-b]quinolines as hemozoin inhibitors: Design, synthesis and antiplasmodial activity. Bioorg. Med. Chem. 2015, 23, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.A.; Lukens, A.K.; Coelho, L.; Nogueira, F.; Wirth, D.F.; Mazitschek, R.; Moreira, R.; Paulo, A. Exploring the 3-piperidin-4-yl-1H-indole scaffold as a novel antimalarial chemotype. Eur. J. Med. Chem. 2015, 102, 320–333. [Google Scholar] [CrossRef]
- Schuck, D.C.; Jordão, A.K.; Nakabashi, M.; Cunha, A.C.; Ferreira, V.F.; Garcia, C.R.S. Synthetic indole and melatonin derivatives exhibit antimalarial activity on the cell cycle of the human malaria parasite Plasmodium falciparum. Eur. J. Med. Chem. 2014, 78, 375–382. [Google Scholar] [CrossRef] [PubMed]
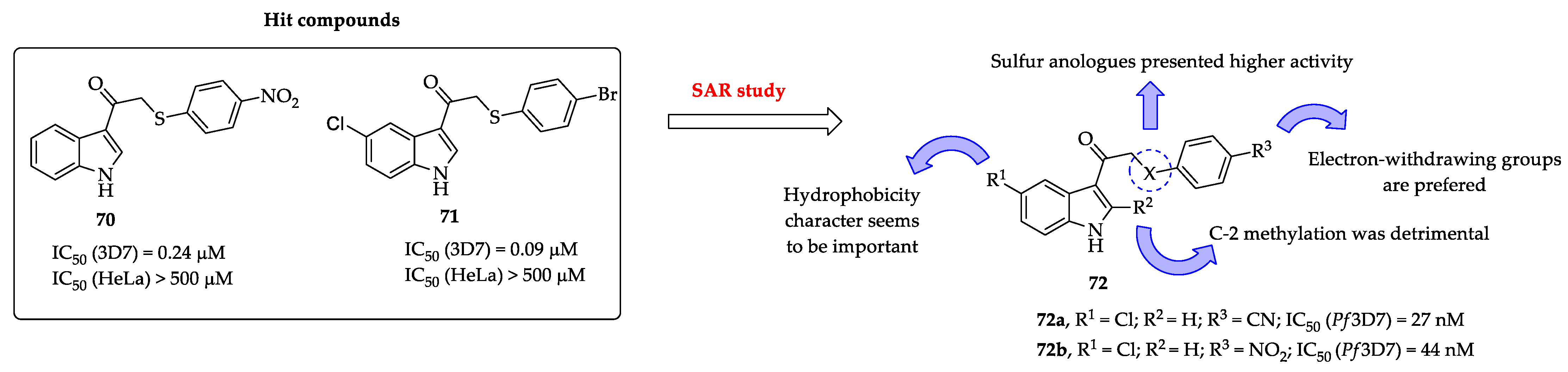
- Lunga, M.J.; Chisango, R.L.; Weyers, C.; Isaacs, M.; Taylor, D.; Edkins, A.L.; Khanye, S.D.; Hoppe, H.C.; Veale, C.G.L. Expanding the SAR of Nontoxic Antiplasmodial Indolyl-3-ethanone Ethers and Thioethers. ChemMedChem 2018, 13, 1353–1362. [Google Scholar] [CrossRef]
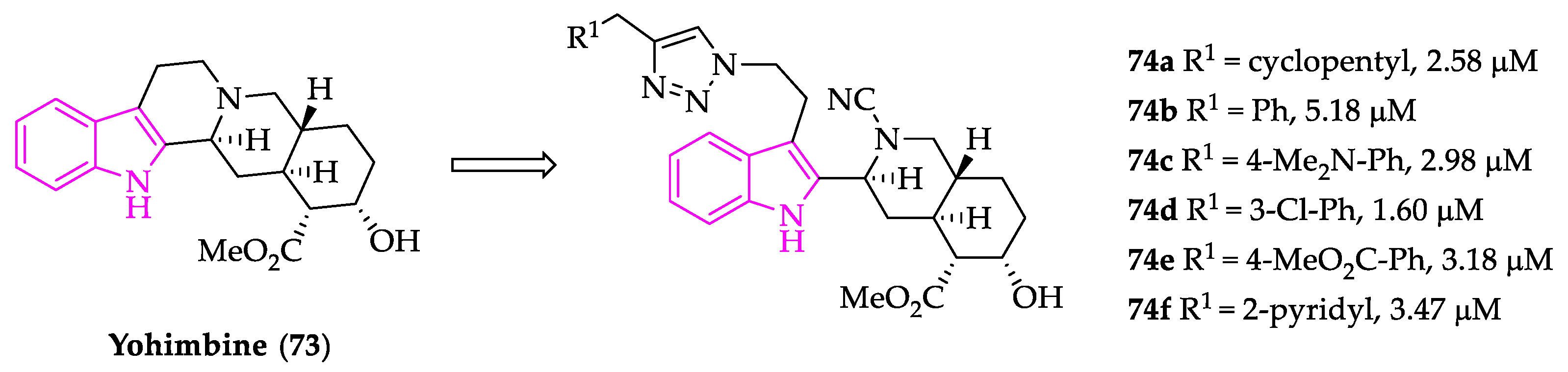
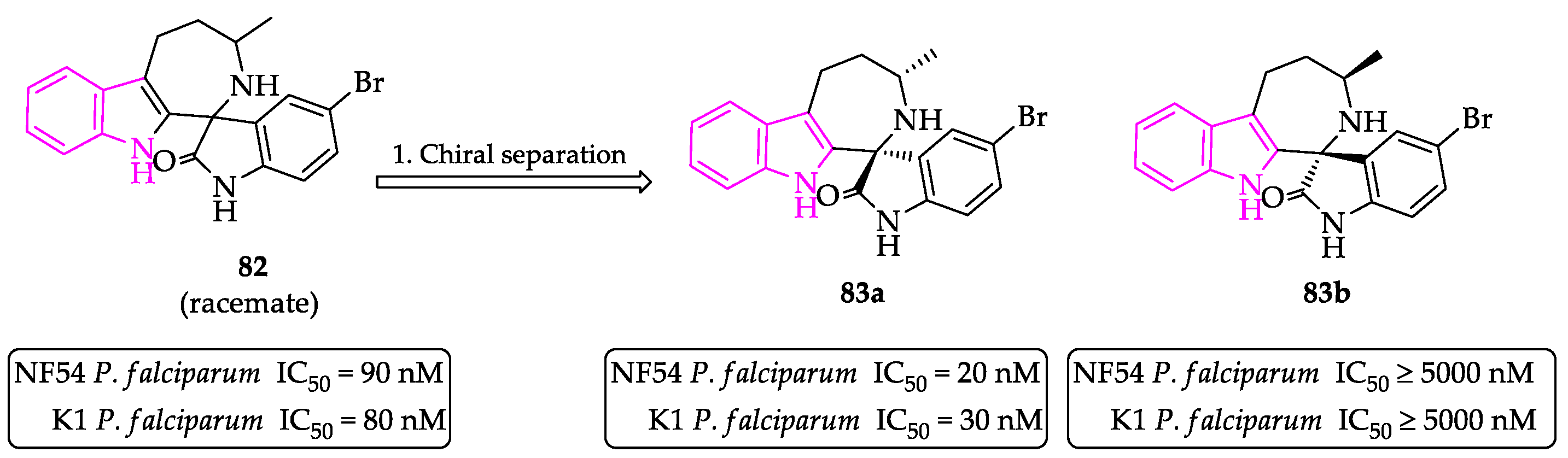
- Paciaroni, N.G.; Perry, D.L.; Norwood, V.M.; Murillo-Solano, C.; Collins, J.; Tenneti, S.; Chakrabarti, D.; Huigens, R.W. Re-Engineering of Yohimbine’s Biological Activity through Ring Distortion: Identification and Structure-Activity Relationships of a New Class of Antiplasmodial Agents. ACS Infect. Dis. 2020, 6, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Lande, D.H.; Nasereddin, A.; Alder, A.; Gilberger, T.W.; Dzikowski, R.; Grünefeld, J.; Kunick, C. Synthesis and antiplasmodial activity of bisindolylcyclobutenediones. Molecules 2021, 26, 4739. [Google Scholar] [CrossRef] [PubMed]
- Plouffe, D.; Brinker, A.; McNamara, C.; Henson, K.; Kato, N.; Kuhen, K.; Nagle, A.; Adrián, F.; Matzen, J.T.; Anderson, P.; et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. USA 2008, 105, 9059–9064. [Google Scholar] [CrossRef] [Green Version]
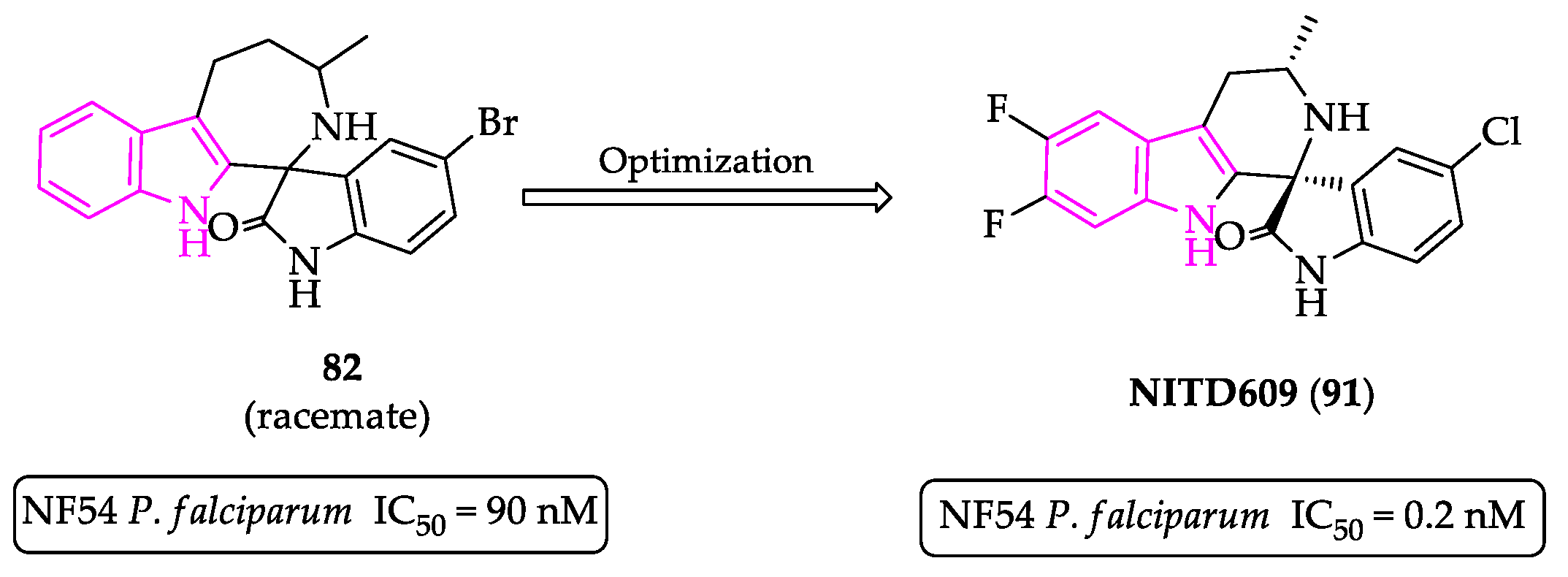
- Yeung, B.K.S.; Zou, B.; Rottmann, M.; Lakshminarayana, S.B.; Ang, S.H.; Leong, S.Y.; Tan, J.; Wong, J.; Keller-Maerki, S.; Fischli, C.; et al. Spirotetrahydro β-carbolines (spiroindolones): A new class of potent and orally efficacious compounds for the treatment of malaria. J. Med. Chem. 2010, 53, 5155–5164. [Google Scholar] [CrossRef]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillman, N.J.; Allen, R.J.W.; McNamara, C.W.; Yeung, B.K.S.; Winzeler, E.A.; Diagana, T.T.; Kirk, K. Na+ regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 2013, 13, 227–237. [Google Scholar] [CrossRef] [Green Version]
- A Study to Assess Efficacy, Safety of KAE609 in Adult Patients with Acute Malaria Mono-Infection, Identifier: NCT01860989. Available online: https://clinicaltrials.gov/ct2/show/NCT01860989 (accessed on 10 November 2021).
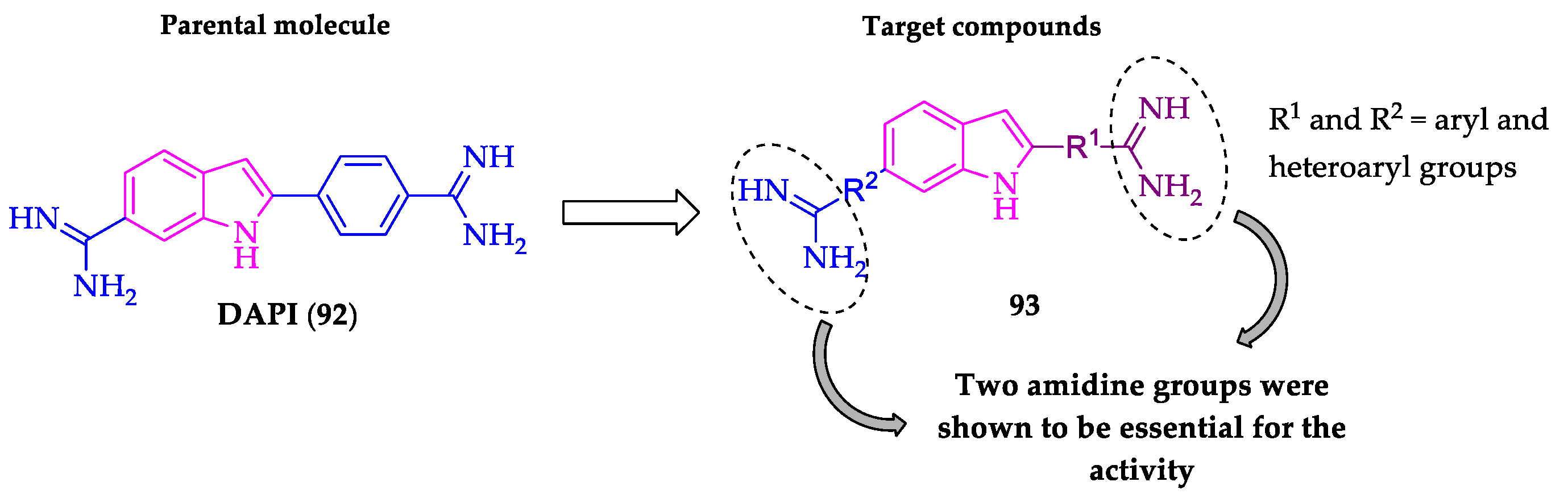
- Farahat, A.A.; Kumar, A.; Say, M.; Barghash, A.E.D.M.; Goda, F.E.; Eisa, H.M.; Wenzler, T.; Brun, R.; Liu, Y.; Mickelson, L.; et al. Synthesis, DNA binding, fluorescence measurements and antiparasitic activity of DAPI related diamidines. Bioorg. Med. Chem. 2010, 18, 557–566. [Google Scholar] [CrossRef]
- Farahat, A.A.; Kumar, A.; Say, M.; Wenzler, T.; Brun, R.; Paul, A.; Wilson, W.D.; Boykin, D.W. Exploration of DAPI analogues: Synthesis, antitrypanosomal activity, DNA binding and fluorescence properties. Eur. J. Med. Chem. 2017, 128, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Farahat, A.A.; Ismail, M.A.; Kumar, A.; Wenzler, T.; Brun, R.; Paul, A.; Wilson, W.D.; Boykin, D.W. Indole and Benzimidazole Bichalcophenes: Synthesis, DNA Binding and Antiparasitic Activity. Eur. J. Med. Chem. 2018, 143, 1590–1596. [Google Scholar] [CrossRef]
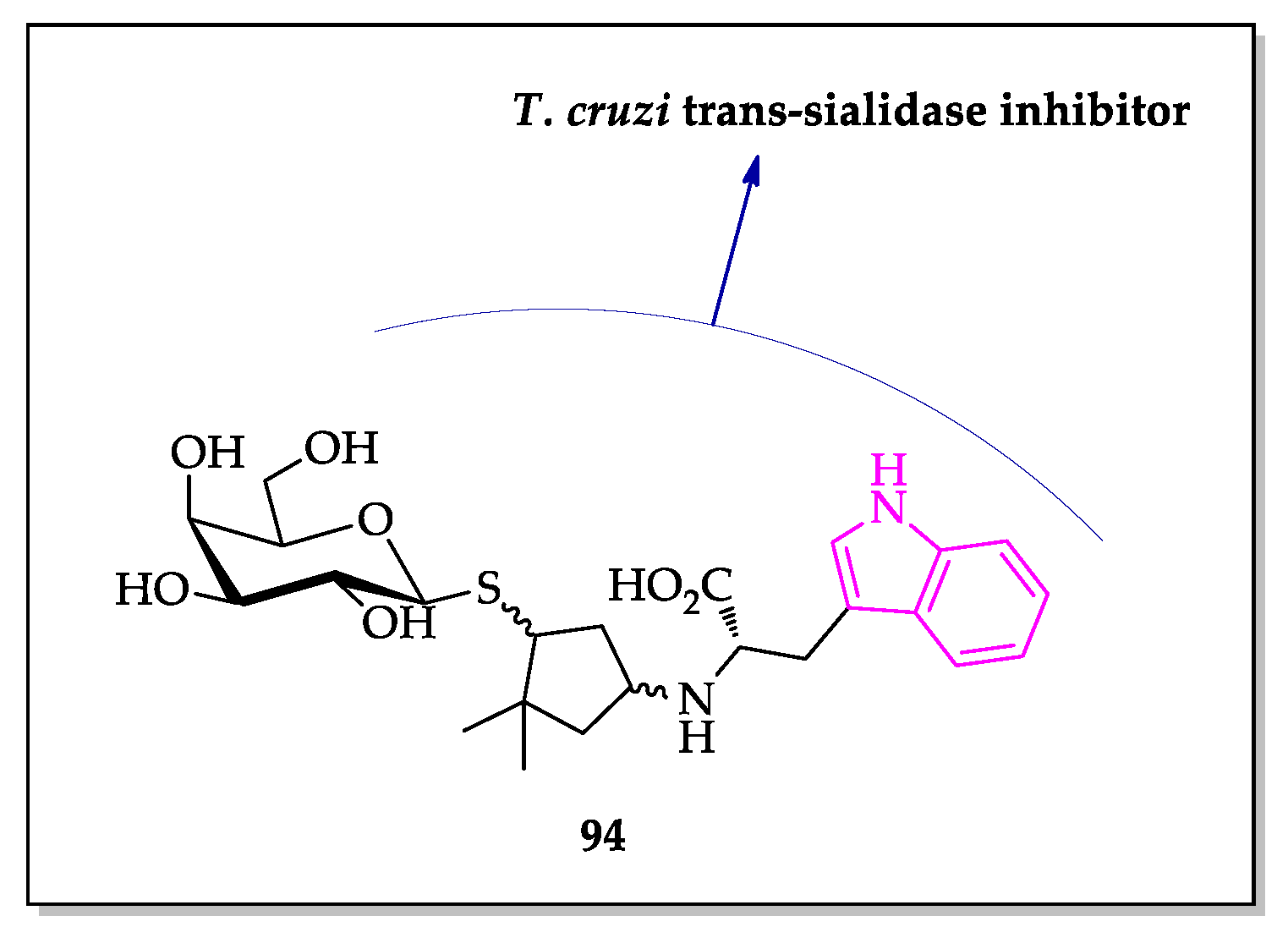
- Harrison, J.A.; Kartha, K.P.R.; Fournier, E.J.L.; Lowary, T.L.; Malet, C.; Nilsson, U.J.; Hindsgaul, O.; Schenkman, S.; Naismith, J.H.; Field, R.A. Probing the acceptor substrate binding site of Trypanosoma cruzi trans-sialidase with systematically modified substrates and glycoside libraries. Org. Biomol. Chem. 2011, 9, 1653–1660. [Google Scholar] [CrossRef] [Green Version]
- Doyle, P.S.; Chen, C.K.; Johnston, J.B.; Hopkins, S.D.; Leung, S.S.F.; Jacobson, M.P.; Engel, J.C.; McKerrow, J.H.; Podust, L.M. A nonazole CYP51 inhibitor cures Chagas’ disease in a mouse model of acute infection. Antimicrob. Agents Chemother. 2010, 54, 2480–2488. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.K.; Doyle, P.S.; Yermalitskaya, L.V.; Mackey, Z.B.; Ang, K.K.H.; McKerrow, J.H.; Podust, L.M. Trypanosoma cruzi CYP51 inhibitor derived from a Mycobacterium tuberculosis screen hit. PLoS Negl. Trop. Dis. 2009, 3, e372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
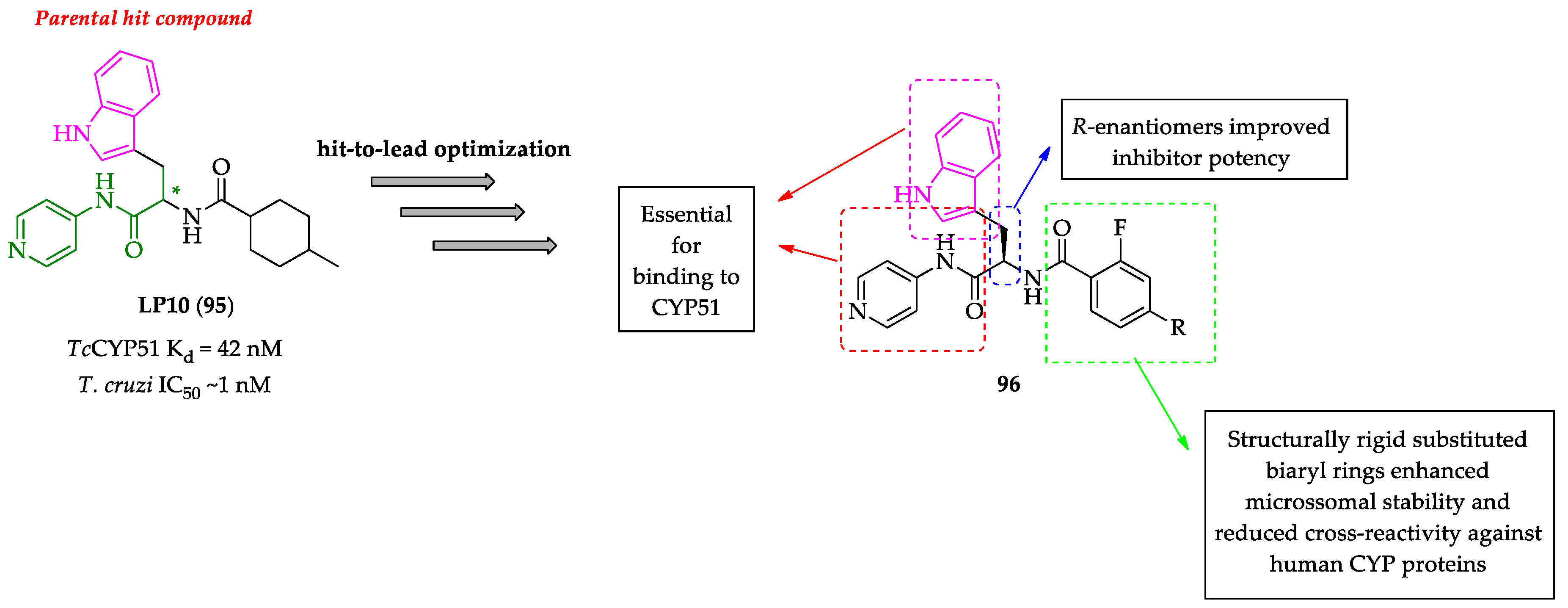
- Choi, J.Y.; Calvet, C.M.; Gunatilleke, S.S.; Ruiz, C.; Cameron, M.D.; McKerrow, J.H.; Podust, L.M.; Roush, W.R. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-chagas agents. J. Med. Chem. 2013, 56, 7651–7668. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.Y.; Calvet, C.M.; Vieira, D.F.; Gunatilleke, S.S.; Cameron, M.D.; McKerrow, J.H.; Podust, L.M.; Roush, W.R. R-configuration of 4-aminopyridyl-based inhibitors of CYP51 confers superior efficacy against Trypanosoma cruzi. ACS Med. Chem. Lett. 2014, 5, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.M.; Vieira, D.F.; Choi, J.Y.; Kellar, D.; Cameron, M.D.; Siqueira-Neto, J.L.; Gut, J.; Johnston, J.B.; Lin, L.; Khan, S.; et al. 4-Aminopyridyl-based CYP51 inhibitors as anti-Trypanosoma cruzi drug leads with improved pharmacokinetic profile and in vivo potency. J. Med. Chem. 2014, 57, 6989–7005. [Google Scholar] [CrossRef] [Green Version]
- Vieira, D.F.; Choi, J.Y.; Calvet, C.M.; Siqueira-Neto, J.L.; Johnston, J.B.; Kellar, D.; Gut, J.; Cameron, M.D.; McKerrow, J.H.; Roush, W.R.; et al. Binding mode and potency of N-indolyloxopyridinyl-4-aminopropanyl-based inhibitors targeting Trypanosoma cruzi CYP51. J. Med. Chem. 2014, 57, 10162–10175. [Google Scholar] [CrossRef] [PubMed]
- Ryczak, J.; Papini, M.; Lader, A.; Nasereddin, A.; Kopelyanskiy, D.; Preu, L.; Jaffe, C.L.; Kunick, C. 2-Arylpaullones are selective antitrypanosomal agents. Eur. J. Med. Chem. 2013, 64, 396–400. [Google Scholar] [CrossRef]
- Orban, O.C.F.; Korn, R.S.; Benítez, D.; Medeiros, A.; Preu, L.; Loaëc, N.; Meijer, L.; Koch, O.; Comini, M.A.; Kunick, C. 5-Substituted 3-chlorokenpaullone derivatives are potent inhibitors of Trypanosoma brucei bloodstream forms. Bioorg. Med. Chem. 2016, 24, 3790–3800. [Google Scholar] [CrossRef]
- Maiwald, F.; Benítez, D.; Charquero, D.; Dar, M.A.; Erdmann, H.; Preu, L.; Koch, O.; Hölscher, C.; Loaëc, N.; Meijer, L.; et al. 9- and 11-substituted 4-azapaullones are potent and selective inhibitors of African trypanosoma. Eur. J. Med. Chem. 2014, 83, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Ferrigno, F.; Biancofiore, I.; Malancona, S.; Ponzi, S.; Paonessa, G.; Graziani, R.; Bresciani, A.; Gennari, N.; Di Marco, A.; Kaiser, M.; et al. Discovery of 2-(1H-imidazo-2-yl)piperazines as a new class of potent and non-cytotoxic inhibitors of Trypanosoma brucei growth in vitro. Bioorg. Med. Chem. Lett. 2018, 28, 3689–3692. [Google Scholar] [CrossRef]
- Kryshchyshyn, A.; Kaminskyy, D.; Karpenko, O.; Gzella, A.; Grelier, P.; Lesyk, R. Thiazolidinone/thiazole based hybrids – New class of antitrypanosomal agents. Eur. J. Med. Chem. 2019, 174, 292–308. [Google Scholar] [CrossRef]
- Cacchi, S.; Fabrizi, G.; Parisi, L.M. Preparation of indoles from o-alkynyltrifluoroacetanilides through the aminopalladation-reductive elimination process. Synthesis 2004, 2004, 1889–1894. [Google Scholar] [CrossRef]
- Guillon, J.; Boudot, C.; Cohen, A.; Savrimoutou, S.; Rubio, S.; Milano, V.; Marchivie, M.; Azas, N.; Mullié, C.; Sonne, P.; et al. Synthesis of 1H-3-{4-[(3-dimethylaminopropyl)aminomethyl]phenyl}-2-phenylindole and evaluation of its antiprotozoal activity. Molbank 2019, 2019, M1060. [Google Scholar] [CrossRef] [Green Version]
- Cockram, P.E.; Dickie, E.A.; Barrett, M.P.; Smith, T.K. Halogenated tryptophan derivatives disrupt essential transamination mechanisms in bloodstream form Trypanosoma brucei. PLoS Negl. Trop. Dis. 2020, 14, e0008928. [Google Scholar] [CrossRef] [PubMed]
- Lepovitz, L.T.; Meis, A.R.; Thomas, S.M.; Wiedeman, J.; Pham, A.; Mensa-Wilmot, K.; Martin, S.F. Design, synthesis, and evaluation of novel anti-trypanosomal compounds. Tetrahedron 2020, 76, 131086. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, F.S.; Santos, H.; Lima, S.R.; Conti, C.; Rodrigues, M.T.; Zeoly, L.A.; Ferreira, L.L.G.; Krogh, R.; Andricopulo, A.D.; Coelho, F. Discovery of highly potent and selective antiparasitic new oxadiazole and hydroxy-oxindole small molecule hybrids. Eur. J. Med. Chem. 2020, 201, 112418. [Google Scholar] [CrossRef]
- Klug, D.M.; Mavrogiannaki, E.M.; Forbes, K.C.; Silva, L.; Diaz-Gonzalez, R.; Pérez-Moreno, G.; Ceballos-Pérez, G.; Garcia-Hernández, R.; Bosch-Navarrete, C.; Cordón-Obras, C.; et al. Lead Optimization of 3,5-Disubstituted-7-Azaindoles for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 2021, 64, 9404–9430. [Google Scholar] [CrossRef] [PubMed]
- Zulfiqar, B.; Shelper, T.B.; Avery, V.M. Leishmaniasis drug discovery: Recent progress and challenges in assay development. Drug Discov. Today 2017, 22, 1516–1531. [Google Scholar] [CrossRef]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R.; et al. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 106–117. [Google Scholar] [CrossRef]
- Ponte-Sucre, A.; Gamarro, F.; Dujardin, J.C.; Barrett, M.P.; López-Vélez, R.; García-Hernández, R.; Pountain, A.W.; Mwenechanya, R.; Papadopoulou, B. Drug resistance and treatment failure in leishmaniasis: A 21st century challenge. PLoS Negl. Trop. Dis. 2017, 11, e0006052. [Google Scholar] [CrossRef]
- Zhang, Z.; Ray, S.; Imlay, L.; Callaghan, L.T.; Niederstrasser, H.; Mallipeddi, P.L.; Posner, B.A.; Wetzel, D.M.; Phillips, M.A.; Smith, M.W. Total synthesis of (+)-spiroindimicin A and congeners unveils their antiparasitic activity. Chem. Sci. 2021, 12, 10388–10394. [Google Scholar] [CrossRef]
- Tonin, L.T.D.; Panice, M.R.; Nakamura, C.V.; Rocha, K.J.P.; dos Santos, A.O.; Ueda-Nakamura, T.; da Costa, W.F.; Sarragiotto, M.H. Antitrypanosomal and antileishmanial activities of novel N-alkyl-(1-phenylsubstituted-β-carboline)-3-carboxamides. Biomed. Pharmacother. 2010, 64, 386–389. [Google Scholar] [CrossRef]
- Pedroso, R.B.; Tonin, L.T.D.; Ueda-Nakamura, T.; Dias Filho, B.P.; Sarragiotto, M.H.; Nakamura, C.V. Beta-carboline-3-carboxamide derivatives as promising antileishmanial agents. Ann. Trop. Med. Parasitol. 2011, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Ashok, P.; Chander, S.; Chow, L.M.C.; Wong, I.L.K.; Singh, R.P.; Jha, P.N.; Sankaranarayanan, M. Synthesis and in vitro anti-leishmanial activity of (4-arylpiperazin-1-yl)(1-(thiophen-2-yl)-9H-pyrido[3,4-b]indol-3-yl)methanone derivatives. Bioorg. Chem. 2017, 70, 100–106. [Google Scholar] [CrossRef]
- Ashok, P.; Chander, S.; Smith, T.K.; Prakash Singh, R.; Jha, P.N.; Sankaranarayanan, M. Biological evaluation and structure activity relationship of 9-methyl-1-phenyl-9H-pyrido[3,4-b]indole derivatives as anti-leishmanial agents. Bioorg. Chem. 2019, 84, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, I.; Mohn, T.; Sedlacek, N.; Hamburger, M. Composition of Indigo naturalis. Planta Med. 2009, 75, 860–863. [Google Scholar] [CrossRef] [PubMed]
- Vougogiannopoulou, K.; Skaltsounis, A.L. From tyrian purple to kinase modulators: Naturally halogenated indirubins and synthetic analogues. Planta Med. 2012, 78, 1515–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Z.; Wei, C.; Wang, J.; Xu, Y.; Bai, G.; Yao, Q.; Zhang, L.; Chen, Y. Anticancer potential of indirubins in medicinal chemistry: Biological activity, structural modification, and structure-activity relationship. Eur. J. Med. Chem. 2021, 223, 113652. [Google Scholar] [CrossRef]
- Xingi, E.; Smirlis, D.; Myrianthopoulos, V.; Magiatis, P.; Grant, K.M.; Meijer, L.; Mikros, E.; Skaltsounis, A.L.; Soteriadou, K. 6-Br-5methylindirubin-3′oxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: Exploitation of GSK-3 for treating leishmaniasis. Int. J. Parasitol. 2009, 39, 1289–1303. [Google Scholar] [CrossRef] [Green Version]
- Efstathiou, A.; Gaboriaud-Kolar, N.; Smirlis, D.; Myrianthopoulos, V.; Vougogiannopoulou, K.; Alexandratos, A.; Kritsanida, M.; Mikros, E.; Soteriadou, K.; Skaltsounis, A.L. An inhibitor-driven study for enhancing the selectivity of indirubin derivatives towards leishmanial Glycogen Synthase Kinase-3 over leishmanial cdc2-related protein kinase 3. Parasites Vectors 2014, 7, 234. [Google Scholar] [CrossRef] [Green Version]
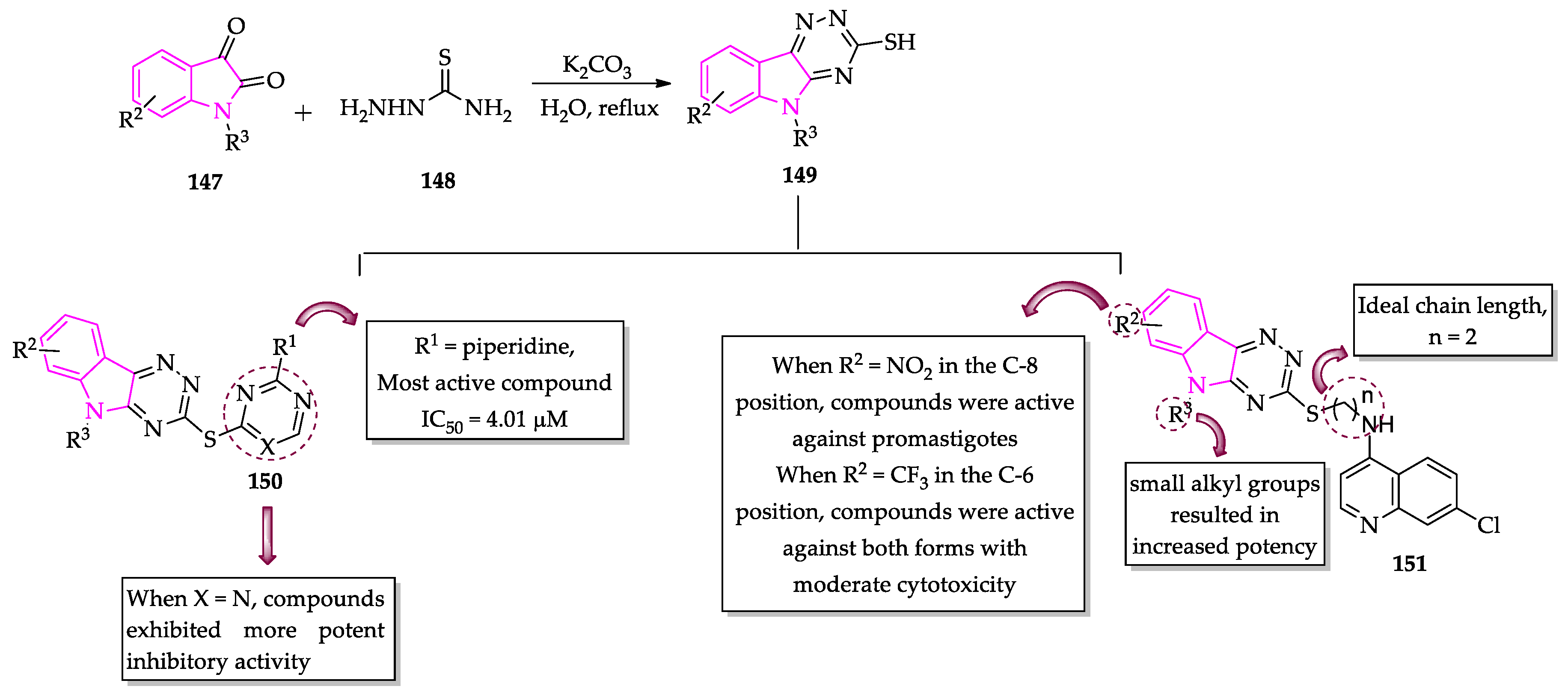
- Gupta, L.; Sunduru, N.; Verma, A.; Srivastava, S.; Gupta, S.; Goyal, N.; Chauhan, P.M.S. Synthesis and biological evaluation of new [1,2,4]triazino[5,6-b]indol-3-ylthio-1,3,5-triazines and [1,2,4]triazino[5,6-b]indol-3-ylthio-pyrimidines against Leishmania donovani. Eur. J. Med. Chem. 2010, 45, 2359–2365. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Pandey, A.K.; Shivahare, R.; Srivastava, K.; Gupta, S.; Chauhan, P.M.S. Triazino indole-quinoline hybrid: A novel approach to antileishmanial agents. Bioorg. Med. Chem. Lett. 2014, 24, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.S.; Al-Kahraman, Y.M.S.A.; Mpadi, D.; Yasinzai, M. Synthesis of N-(1-methyl-1H-indol-3-yl)methyleneamines and 3,3-diaryl-4-(1-methyl-1H-indol-3-yl)azetidin-2-ones as potential antileishmanial agents. Bioorg. Med. Chem. Lett. 2012, 22, 5704–5706. [Google Scholar] [CrossRef]
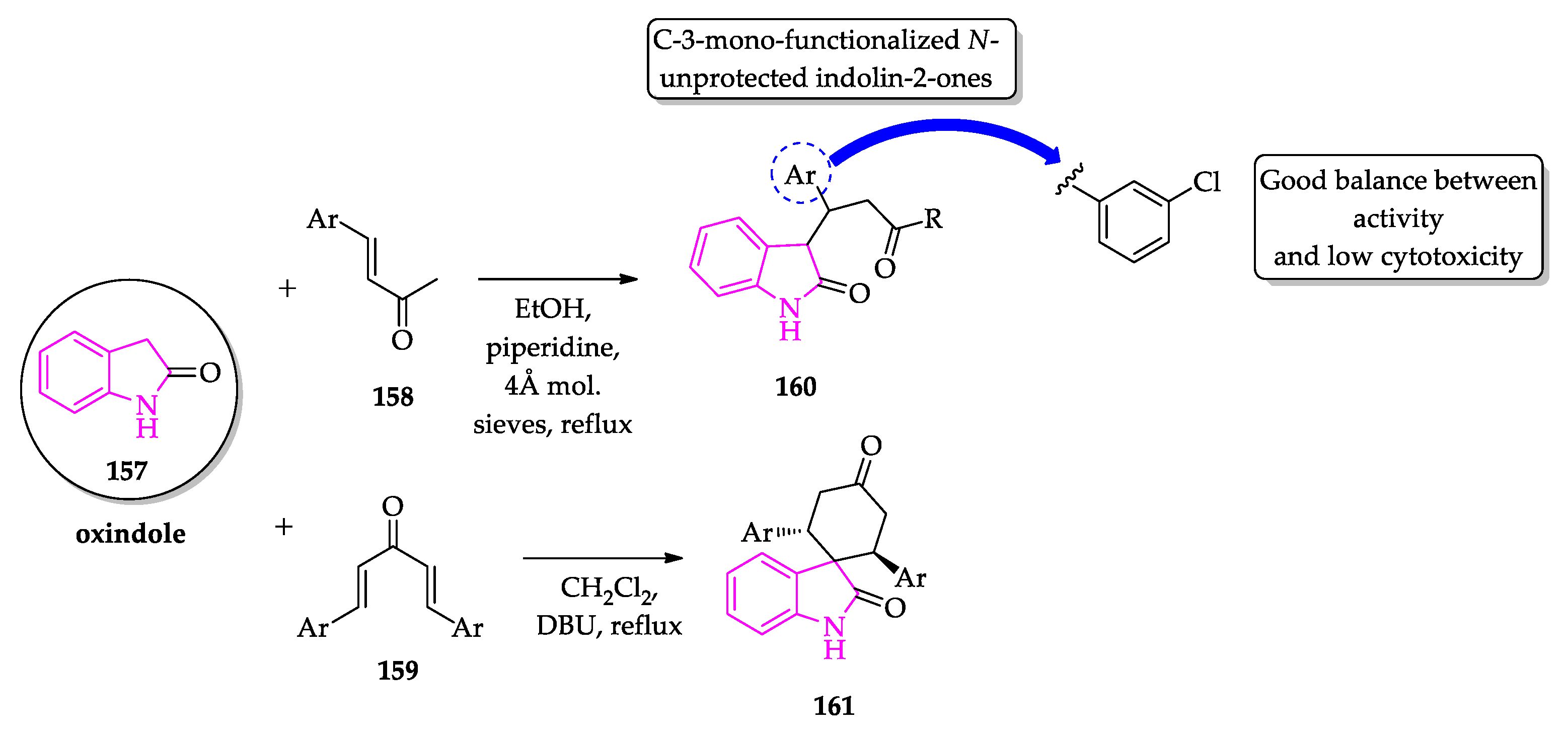
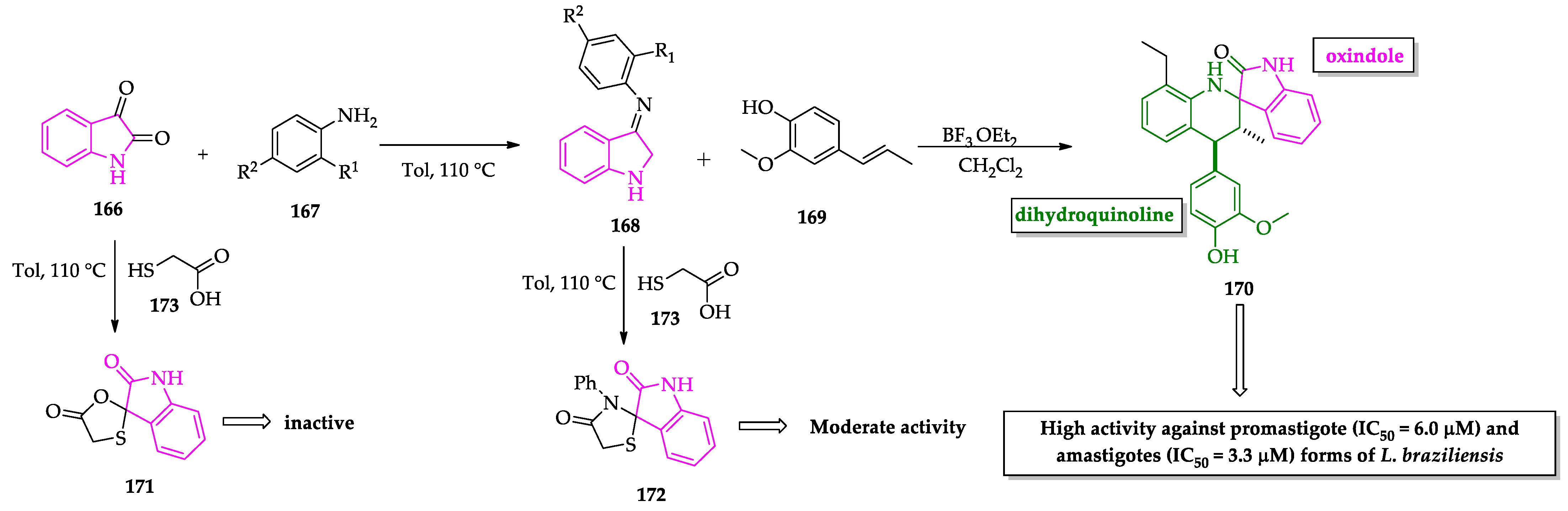
- Scala, A.; Cordaro, M.; Grassi, G.; Piperno, A.; Barberi, G.; Cascio, A.; Risitano, F. Direct synthesis of C3-mono-functionalized oxindoles from N-unprotected 2-oxindole and their antileishmanial activity. Bioorg. Med. Chem. 2014, 22, 1063–1069. [Google Scholar] [CrossRef] [PubMed]
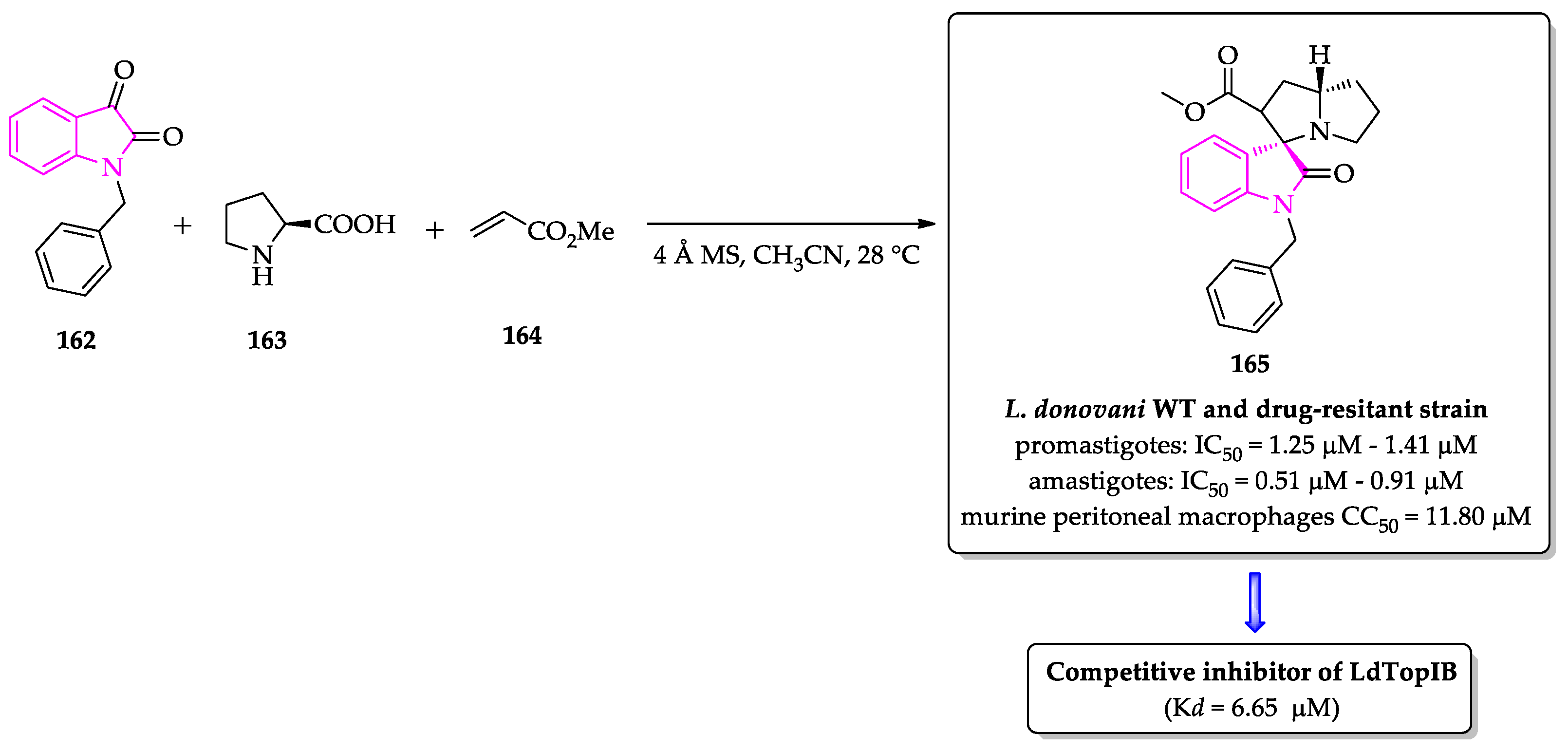
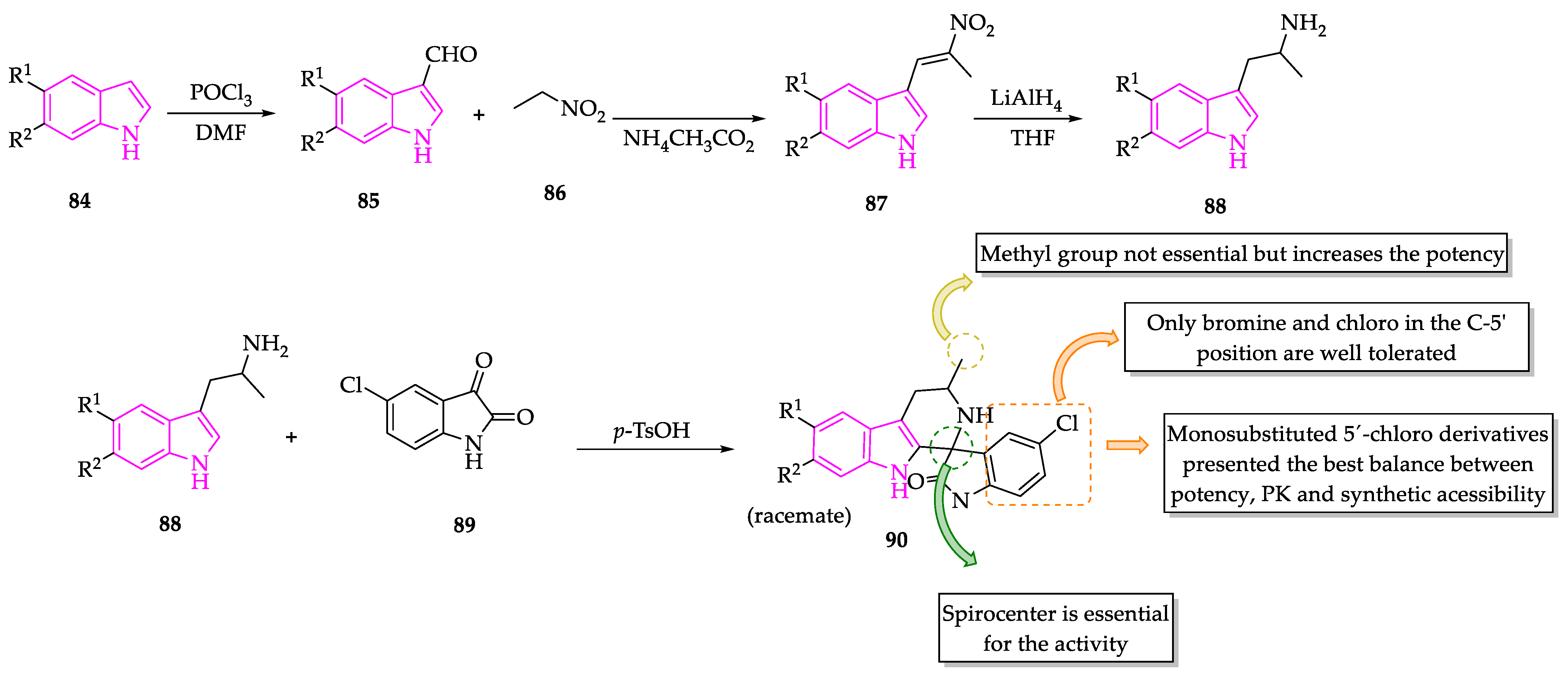
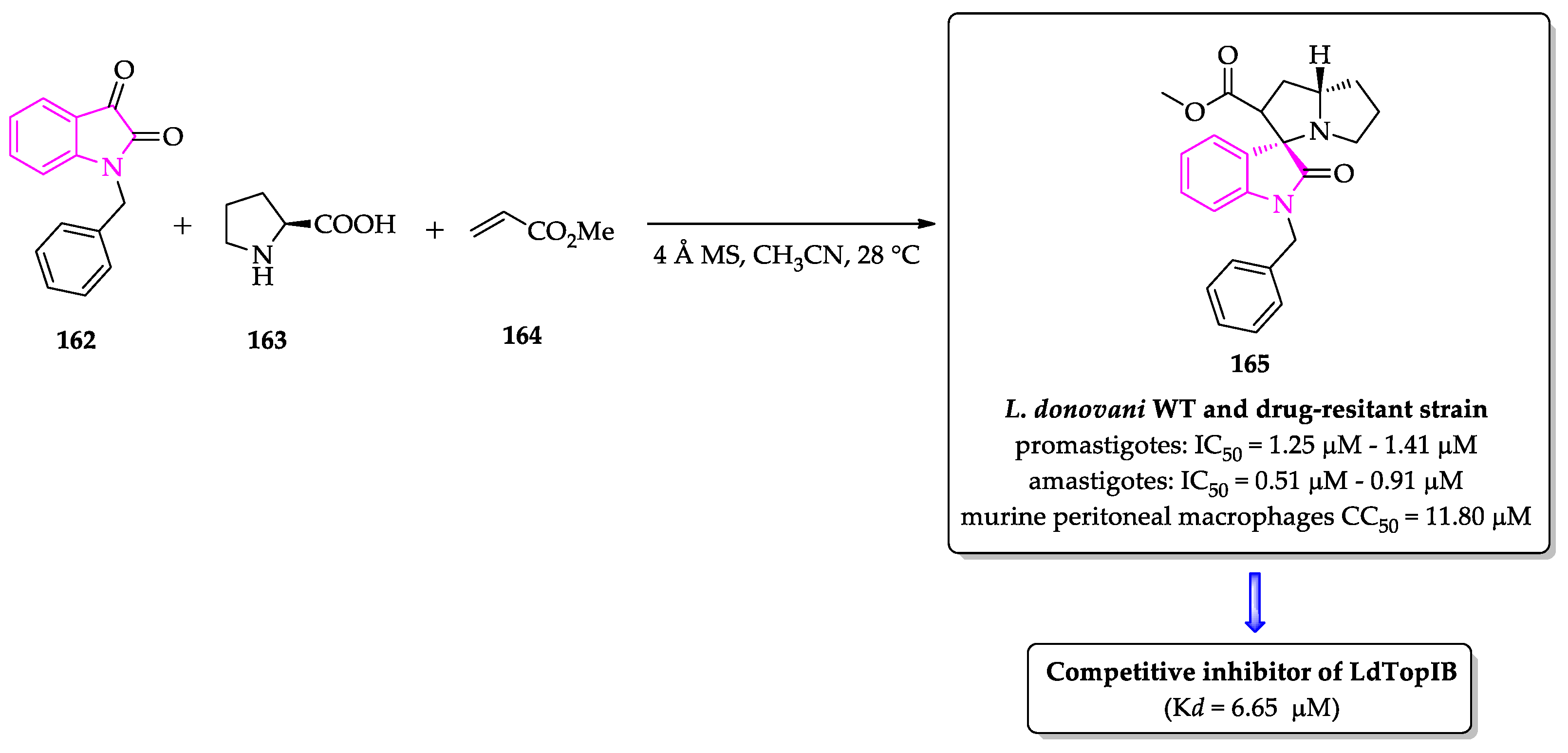
- Saha, S.; Acharya, C.; Pal, U.; Chowdhury, S.R.; Sarkar, K.; Maiti, N.C.; Jaisankar, P.; Majumder, H.K. A novel spirooxindole derivative inhibits the growth of Leishmania donovani parasites both in vitro and in vivo by targeting type IB topoisomerase. Antimicrob. Agents Chemother. 2016, 60, 6281–6293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
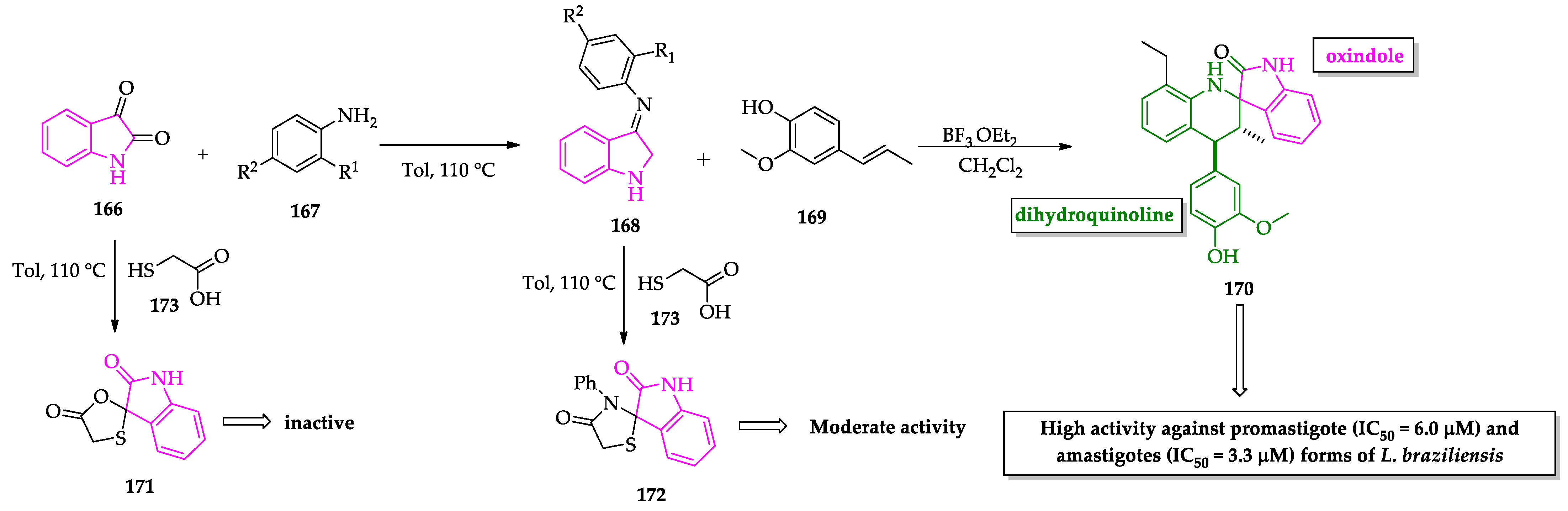
- Leañez, J.; Nuñez, J.; García-Marchan, Y.; Sojo, F.; Arvelo, F.; Rodriguez, D.; Buscema, I.; Alvarez-Aular, A.; Serrano-Martín, X. Anti-leishmanial effect of spiro dihydroquinoline-oxindoles on volume regulation decrease and sterol biosynthesis of Leishmania braziliensis. Exp. Parasitol. 2019, 198, 31–38. [Google Scholar] [CrossRef]
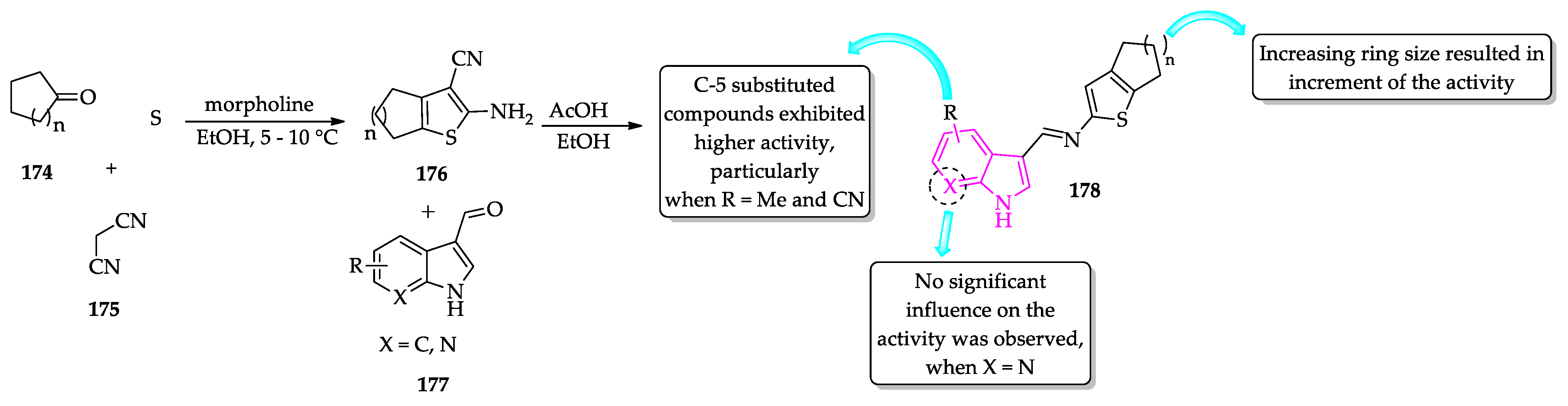
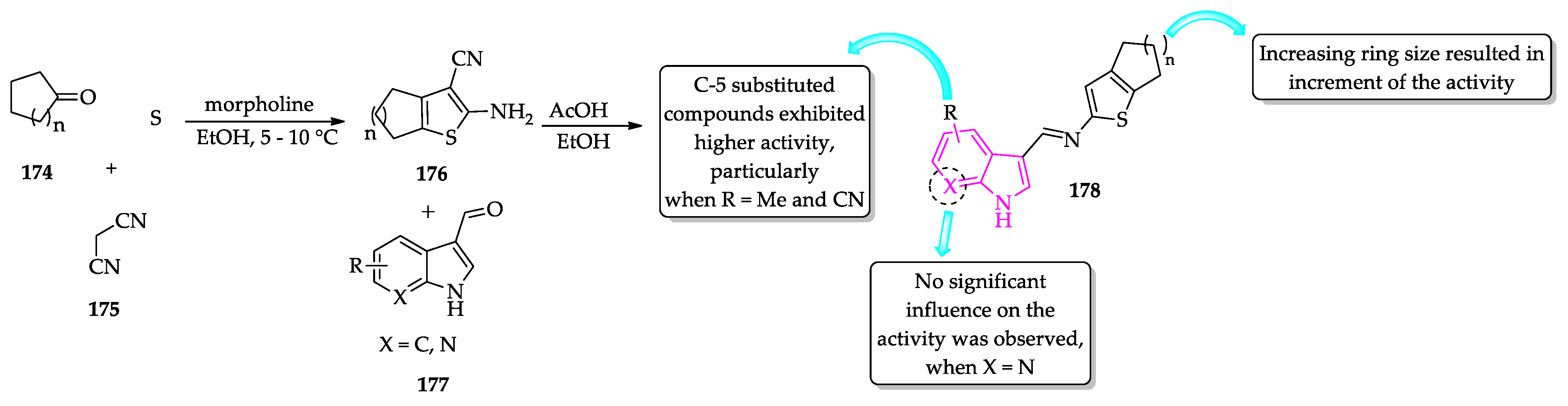
- Rodrigues, K.A.D.F.; Dias, C.N.D.S.; Neris, P.L.D.N.; Rocha, J.D.C.; Scotti, M.T.; Scotti, L.; Mascarenhas, S.R.; Veras, R.C.; De Medeiros, I.A.; Keesen, T.D.S.L.; et al. 2-Amino-thiophene derivatives present antileishmanial activity mediated by apoptosis and immunomodulation in vitro. Eur. J. Med. Chem. 2015, 106, 1–14. [Google Scholar] [CrossRef]
- Félix, M.B.; de Souza, E.R.; de Lima, M.d.C.A.; Frade, D.K.G.; Serafim, V.d.L.; Rodrigues, K.A.d.F.; Néris, P.L.d.N.; Ribeiro, F.F.; Scotti, L.; Scotti, M.T.; et al. Antileishmanial activity of new thiophene–indole hybrids: Design, synthesis, biological and cytotoxic evaluation, and chemometric studies. Bioorg. Med. Chem. 2016, 24, 3972–3977. [Google Scholar] [CrossRef] [PubMed]
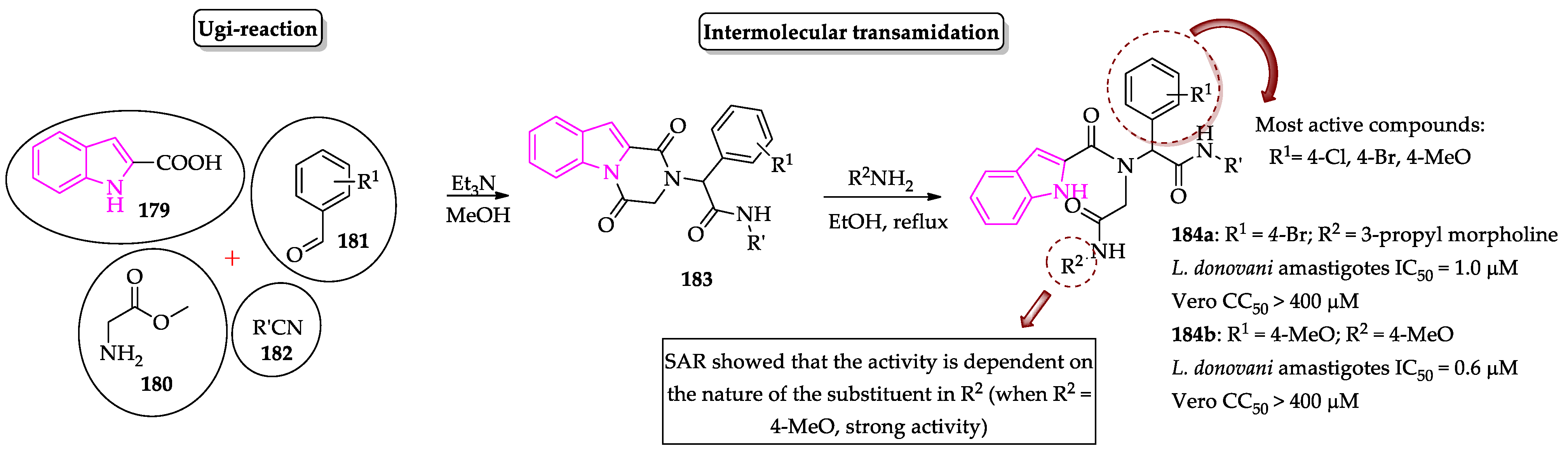
- Pandey, S.; Khan, S.; Singh, A.; Gauniyal, H.M.; Kumar, B.; Chauhan, P.M.S. Access to indole- and pyrrole-fused diketopiperazines via tandem Ugi-4CR/intramolecular cyclization and its regioselective ring-opening by intermolecular transamidation. J. Org. Chem. 2012, 77, 10211–10227. [Google Scholar] [CrossRef]
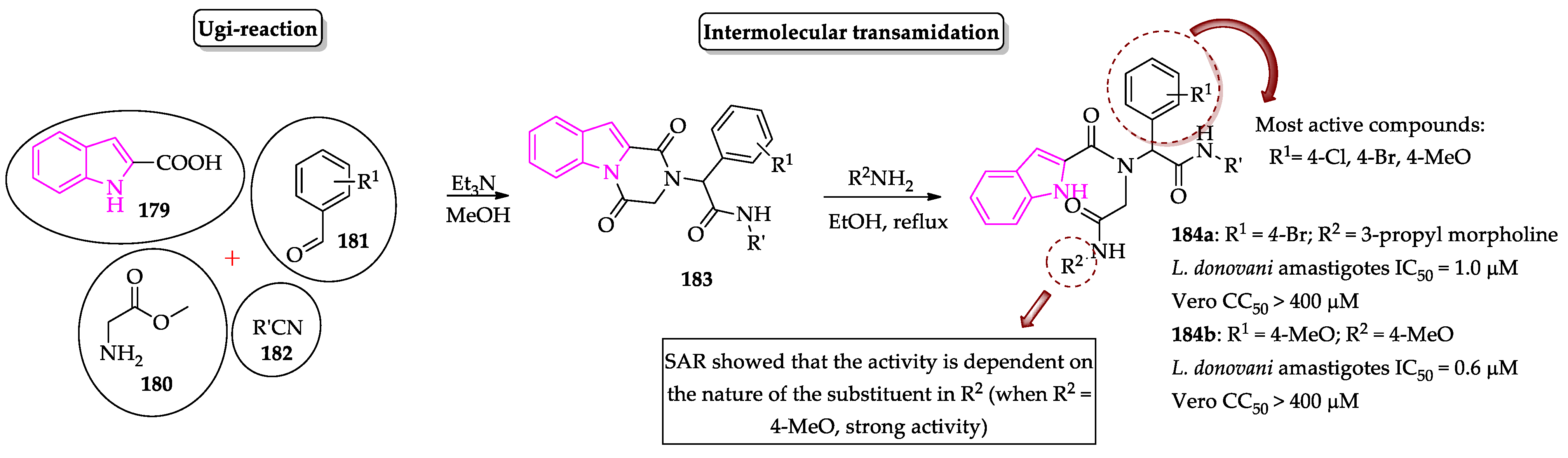
- Pandey, S.; Chauhan, S.S.; Shivahare, R.; Sharma, A.; Jaiswal, S.; Gupta, S.; Lal, J.; Chauhan, P.M.S. Identification of a diverse indole-2-carboxamides as a potent antileishmanial chemotypes. Eur. J. Med. Chem. 2016, 110, 237–245. [Google Scholar] [CrossRef]
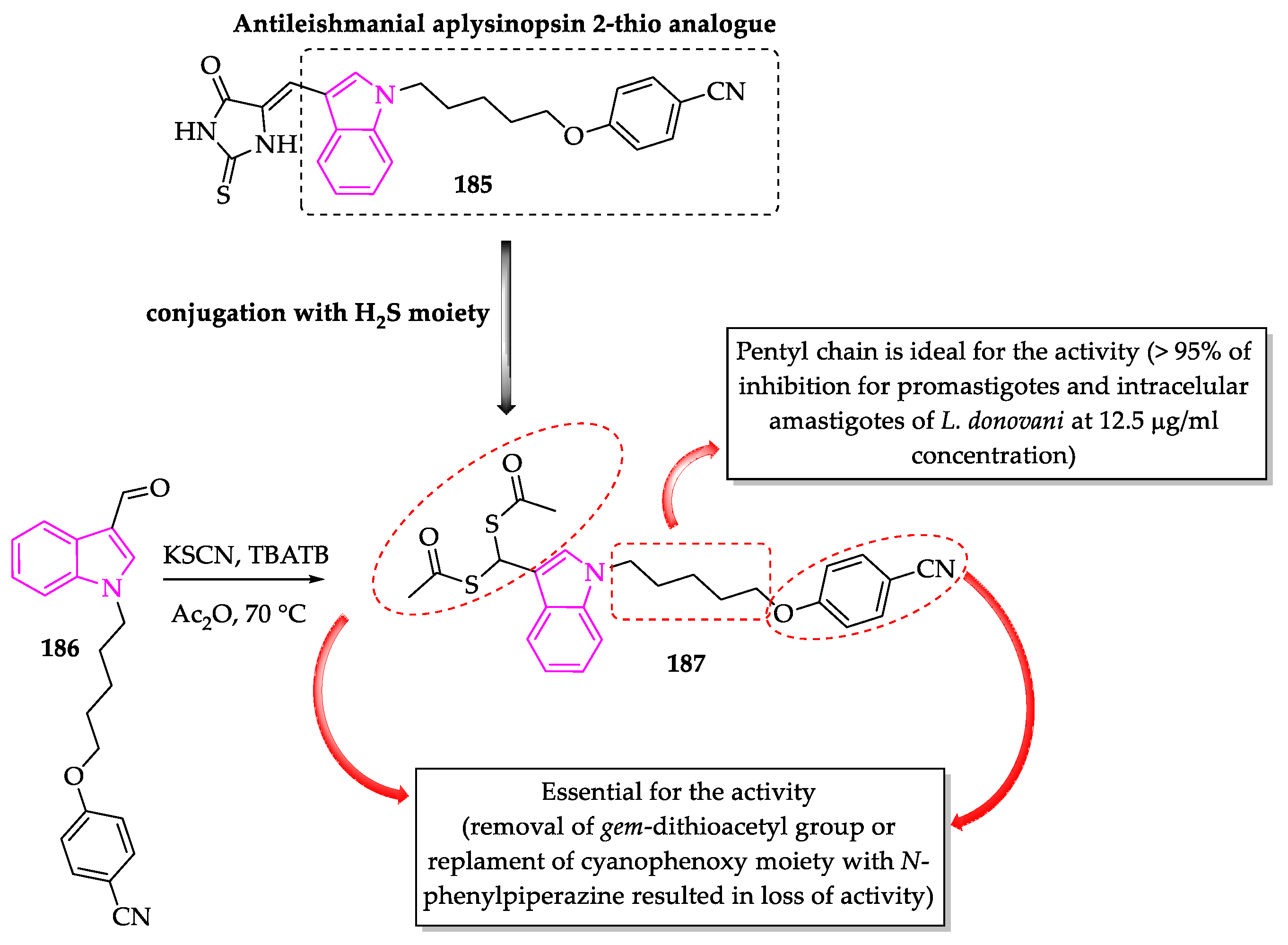
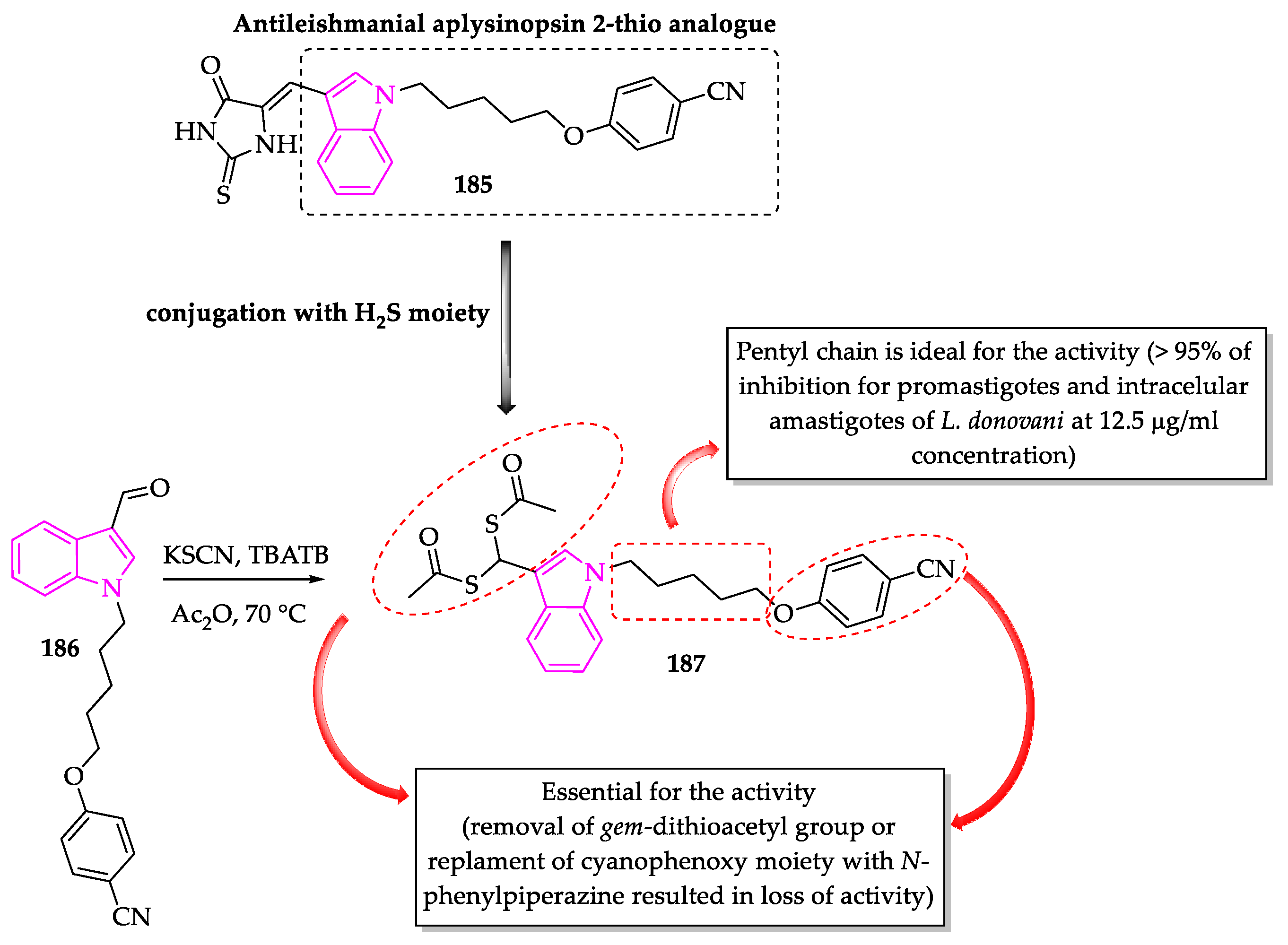
- Porwal, S.; Gupta, S.; Chauhan, P.M.S. gem-Dithioacetylated indole derivatives as novel antileishmanial agents. Bioorg. Med. Chem. Lett. 2017, 27, 4643–4646. [Google Scholar] [CrossRef]
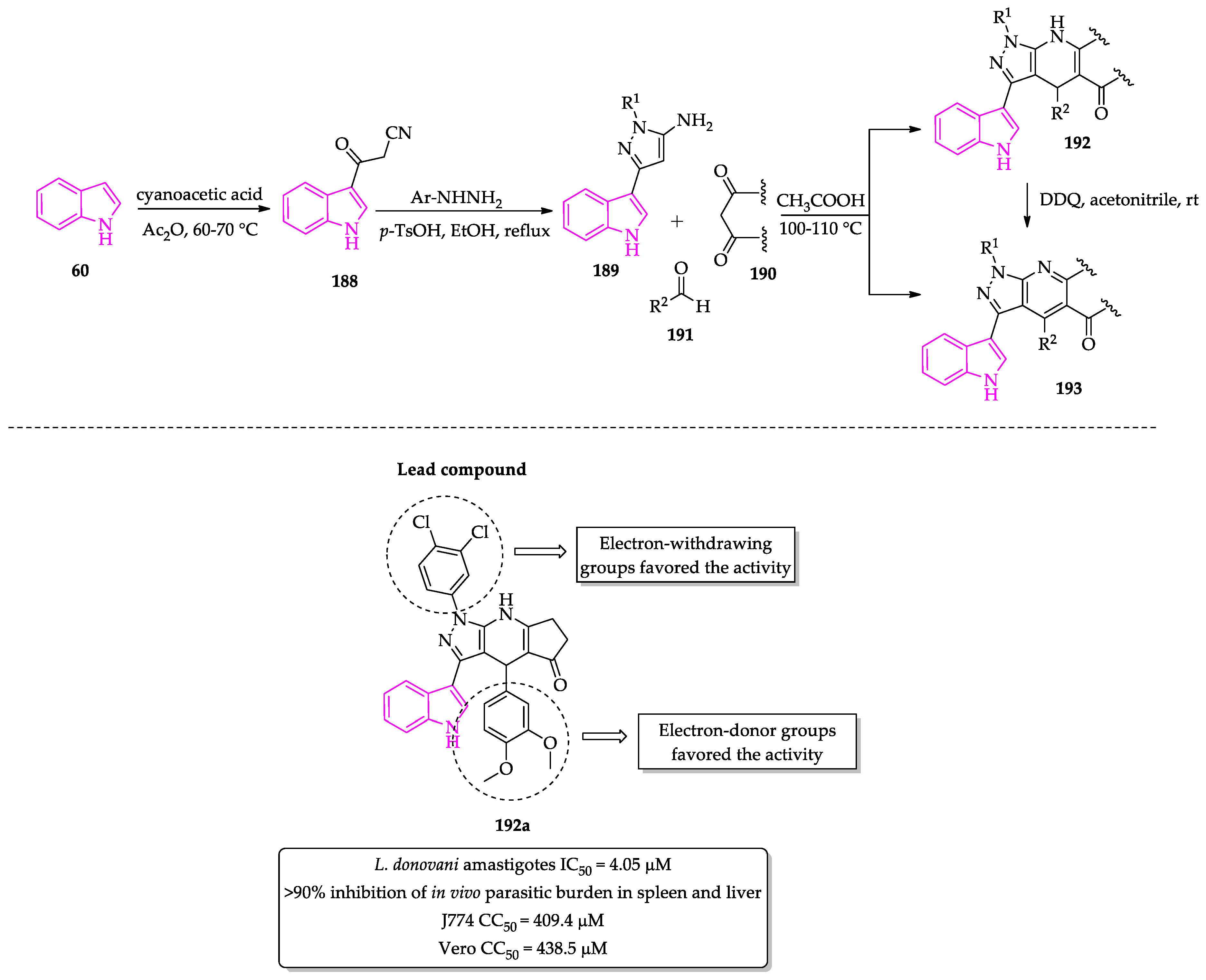
- Anand, D.; Yadav, P.K.; Patel, O.P.S.; Parmar, N.; Maurya, R.K.; Vishwakarma, P.; Raju, K.S.R.; Taneja, I.; Wahajuddin, M.; Kar, S.; et al. Antileishmanial Activity of Pyrazolopyridine Derivatives and Their Potential as an Adjunct Therapy with Miltefosine. J. Med. Chem. 2017, 60, 1041–1059. [Google Scholar] [CrossRef]
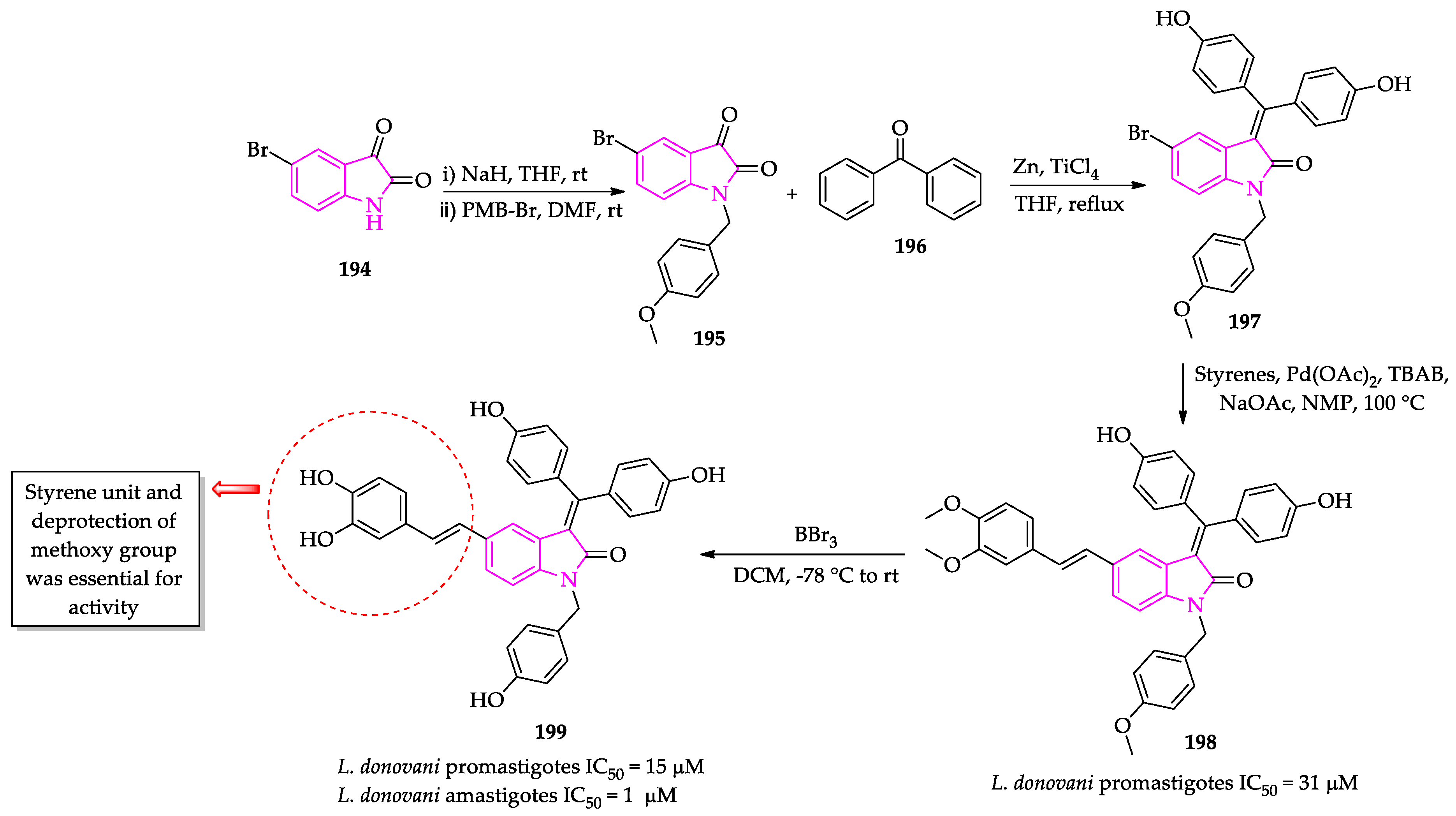
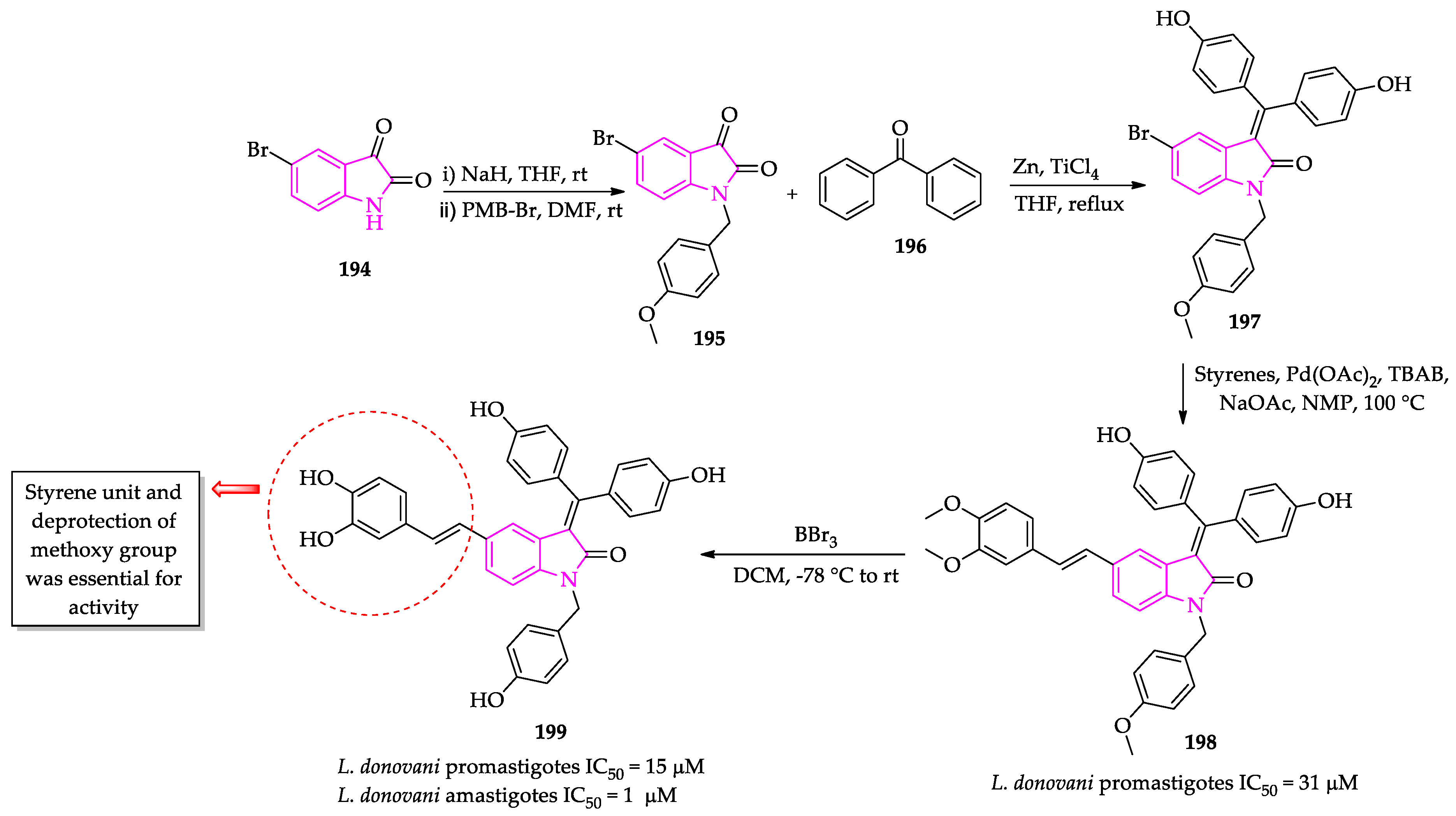
- Pal, A.; Ganguly, A.; Chowdhuri, S.; Yousuf, M.; Ghosh, A.; Barui, A.K.; Kotcherlakota, R.; Adhikari, S.; Banerjee, R. Bis-arylidene oxindole-betulinic acid conjugate: A fluorescent cancer cell detector with potent anticancer activity. ACS Med. Chem. Lett. 2015, 6, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Yousuf, M.; Mukherjee, D.; Dey, S.; Chatterjee, S.; Pal, A.; Sarkar, B.; Pal, C.; Adhikari, S. Synthesis and biological evaluation of polyhydroxylated oxindole derivatives as potential antileishmanial agent. Bioorg. Med. Chem. Lett. 2018, 28, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
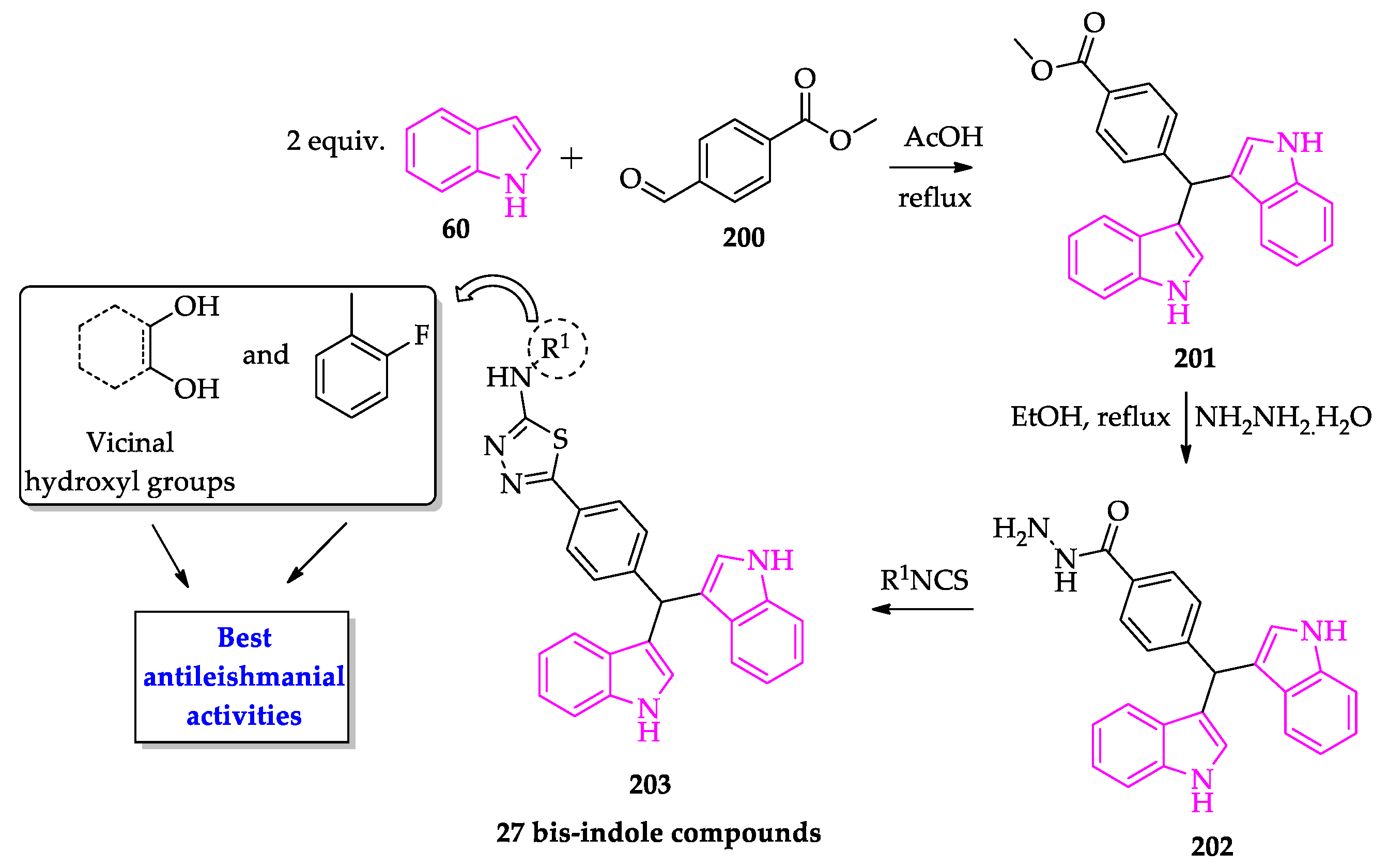
- Taha, M.; Uddin, I.; Gollapalli, M.; Almandil, N.B.; Rahim, F.; Farooq, R.K.; Nawaz, M.; Ibrahim, M.; Alqahtani, M.A.; Bamarouf, Y.A.; et al. Synthesis, anti-leishmanial and molecular docking study of bis-indole derivatives. BMC Chem. 2019, 13, 102. [Google Scholar] [CrossRef] [PubMed]
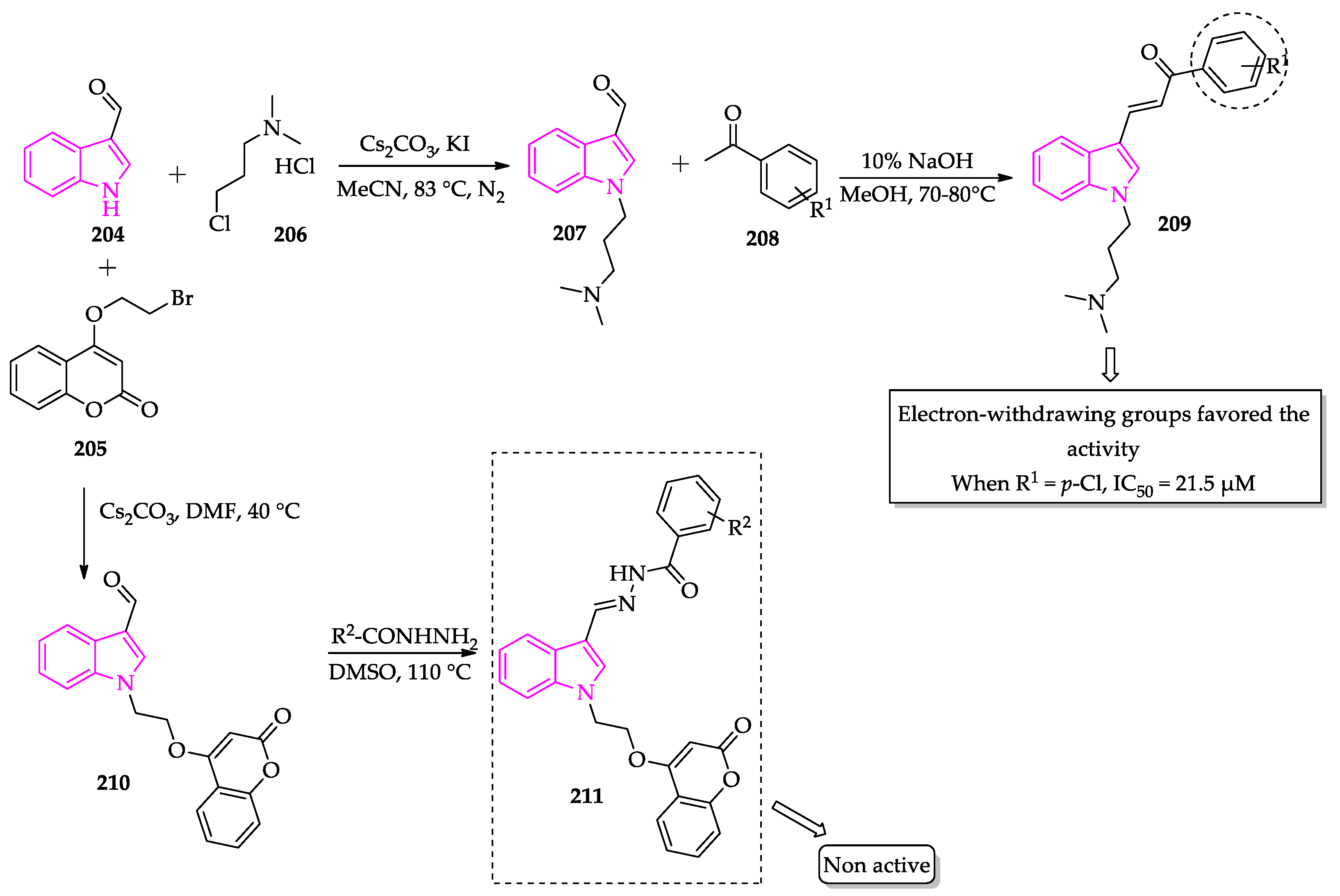
- Tiwari, S.; Kirar, S.; Banerjee, U.C.; Neerupudi, K.B.; Singh, S.; Wani, A.A.; Bharatam, P.V.; Singh, I.P. Synthesis of N-substituted indole derivatives as potential antimicrobial and antileishmanial agents. Bioorg. Chem. 2020, 99, 103787. [Google Scholar] [CrossRef]
- Da Silva, P.R.; de Oliveira, J.F.; da Silva, A.L.; Queiroz, C.M.; Feitosa, A.P.S.; Duarte, D.M.F.A.; da Silva, A.C.; de Castro, M.C.A.B.; Pereira, V.R.A.; da Silva, R.M.F.; et al. Novel indol-3-yl-thiosemicarbazone derivatives: Obtaining, evaluation of in vitro leishmanicidal activity and ultrastructural studies. Chem. Biol. Interact. 2020, 315, 108899. [Google Scholar] [CrossRef] [PubMed]
- Schadich, E.; Kryshchyshyn-Dylevych, A.; Holota, S.; Polishchuk, P.; Džubak, P.; Gurska, S.; Hajduch, M.; Lesyk, R. Assessing different thiazolidine and thiazole based compounds as antileishmanial scaffolds. Bioorg. Med. Chem.Lett. 2020, 30, 127616. [Google Scholar] [CrossRef]




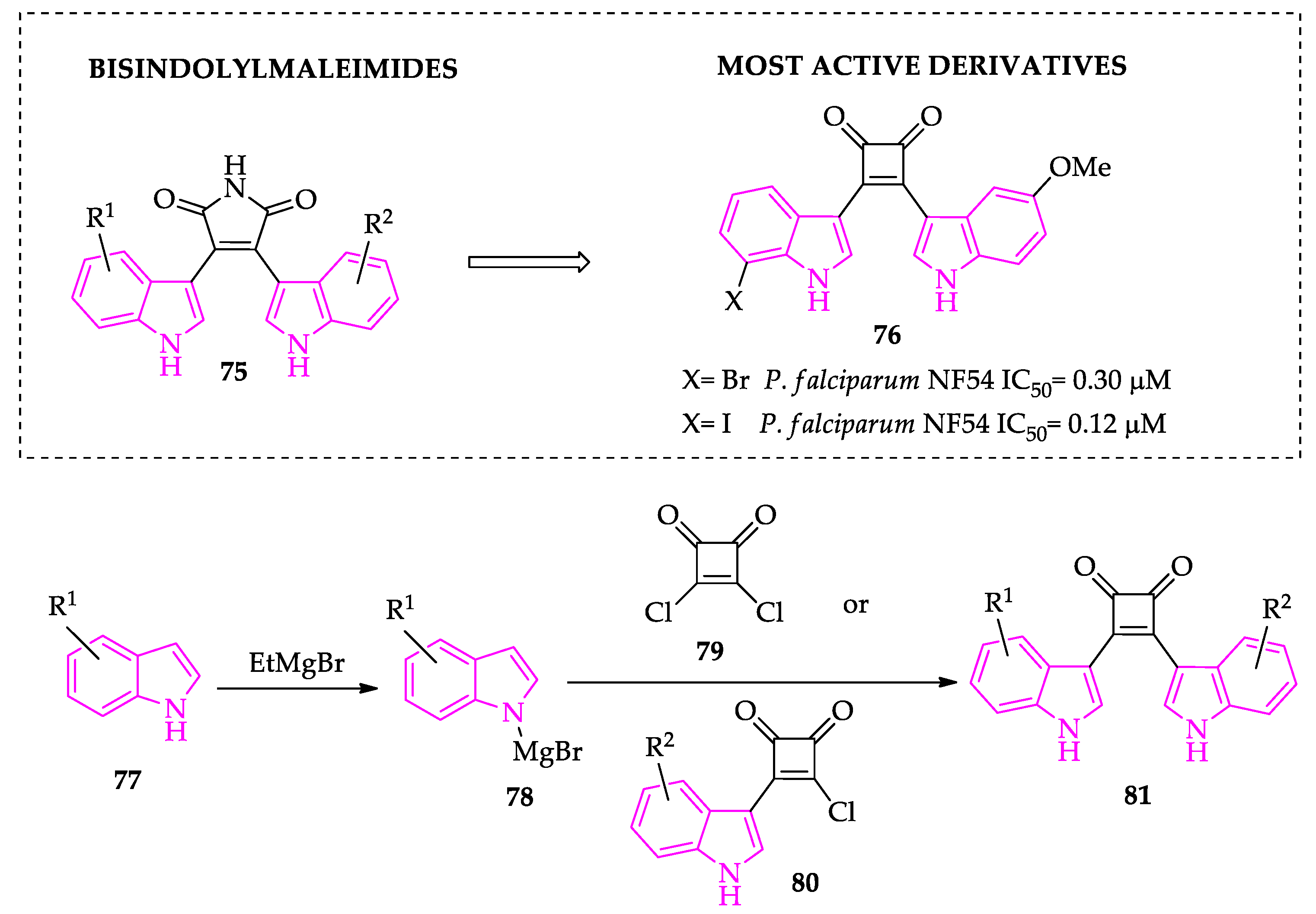
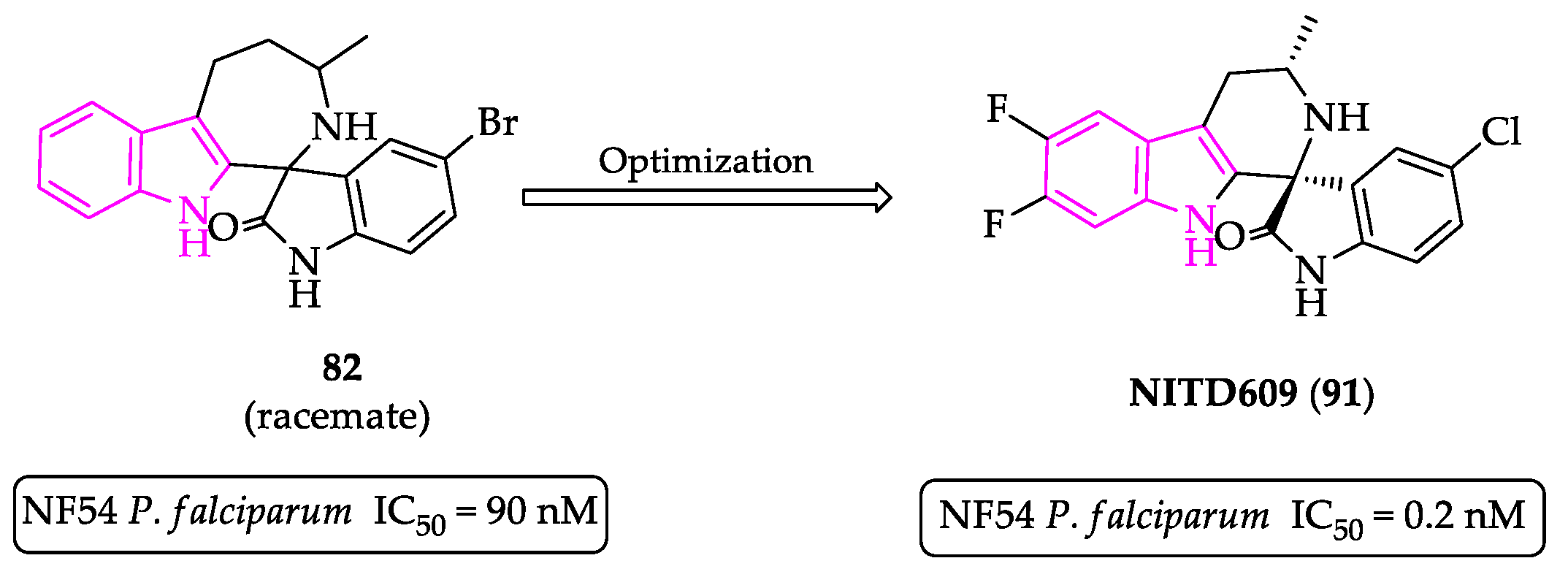
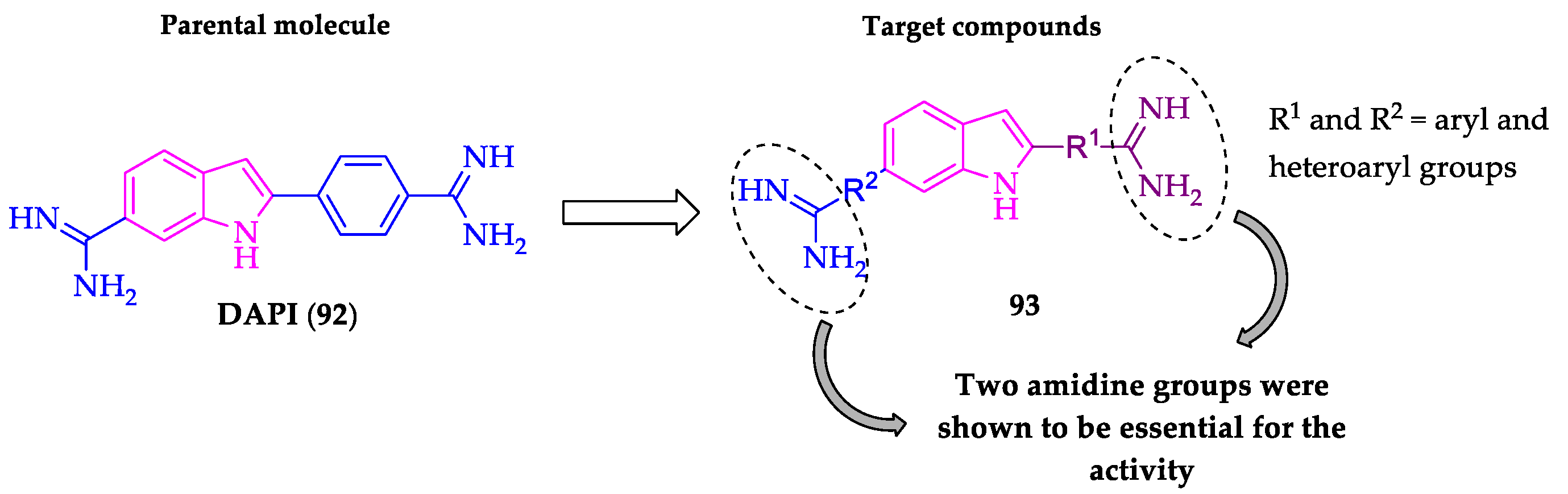
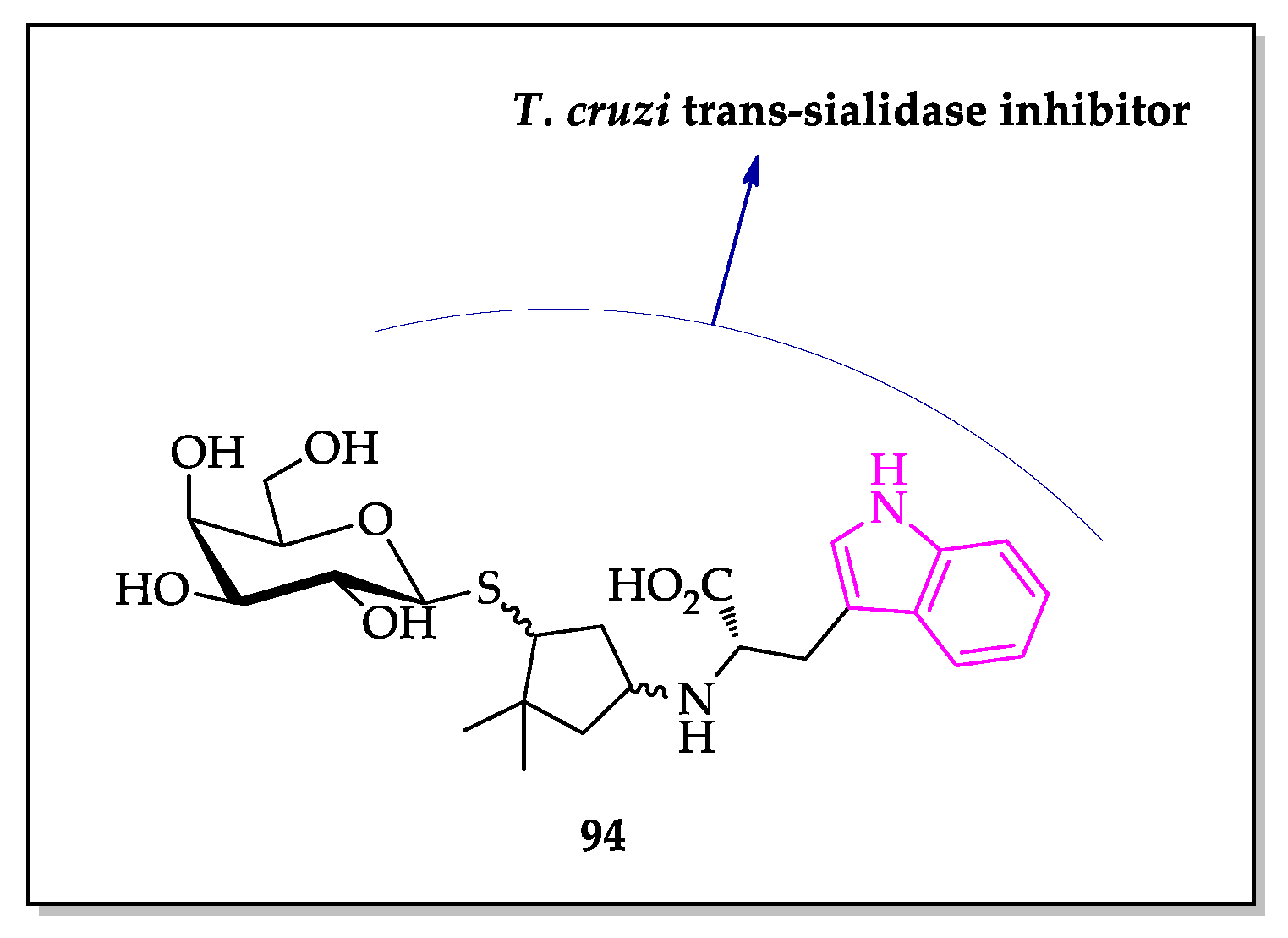
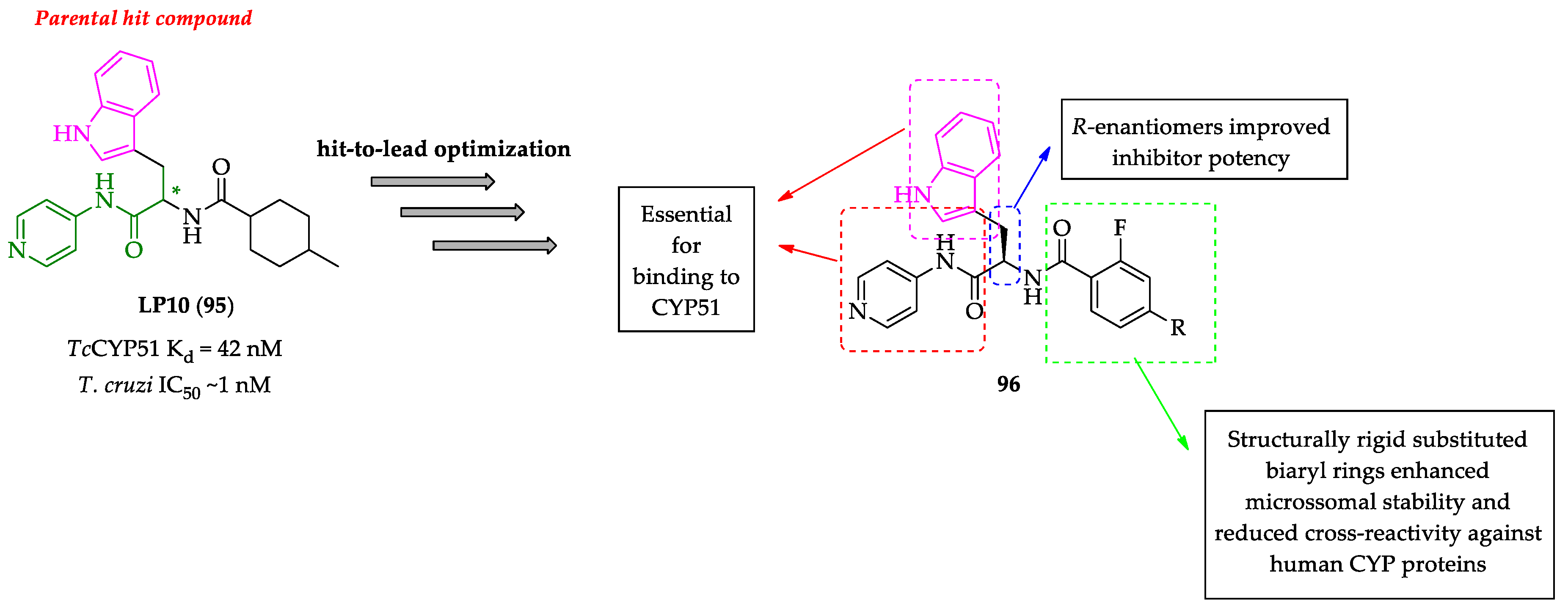
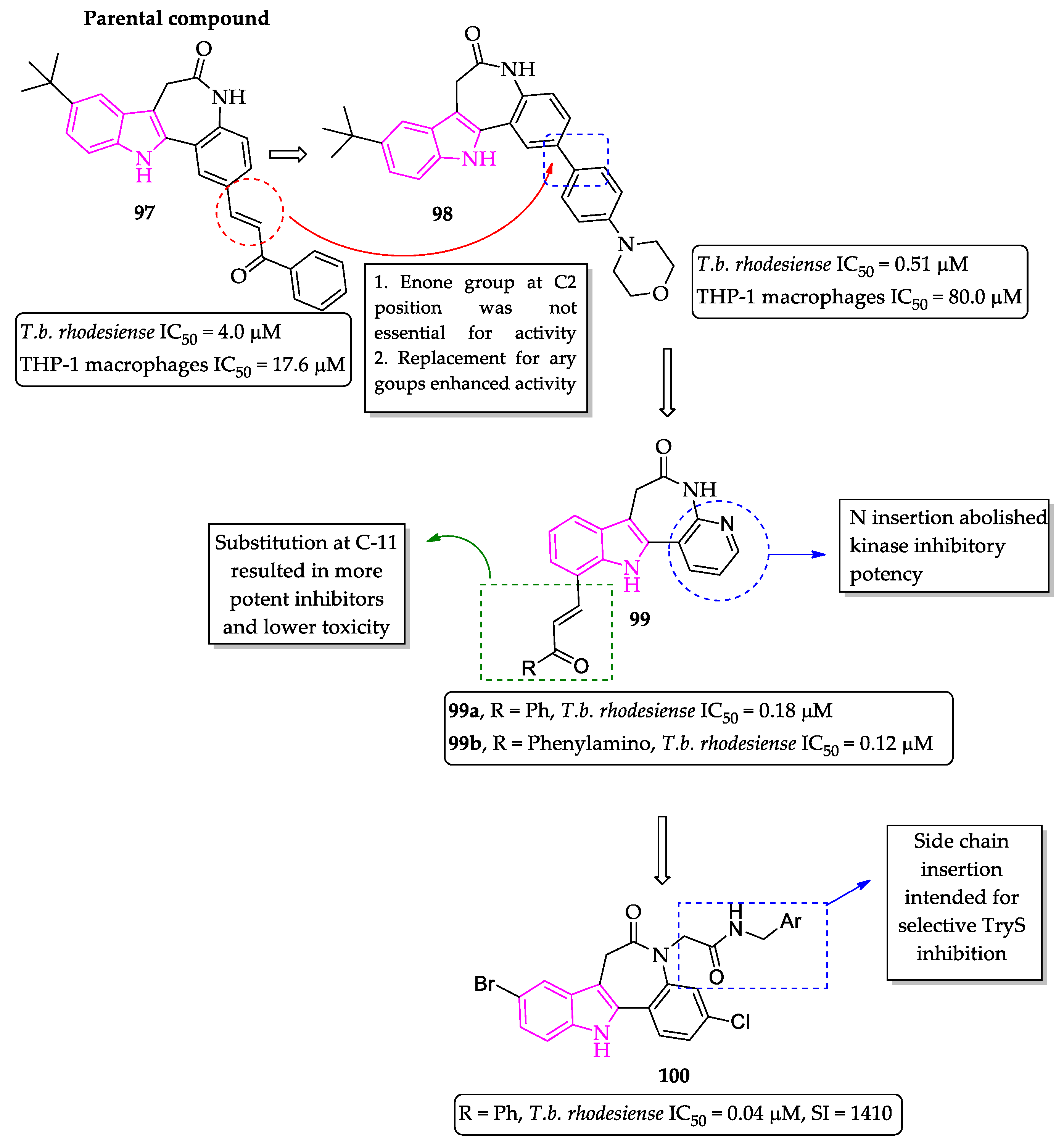
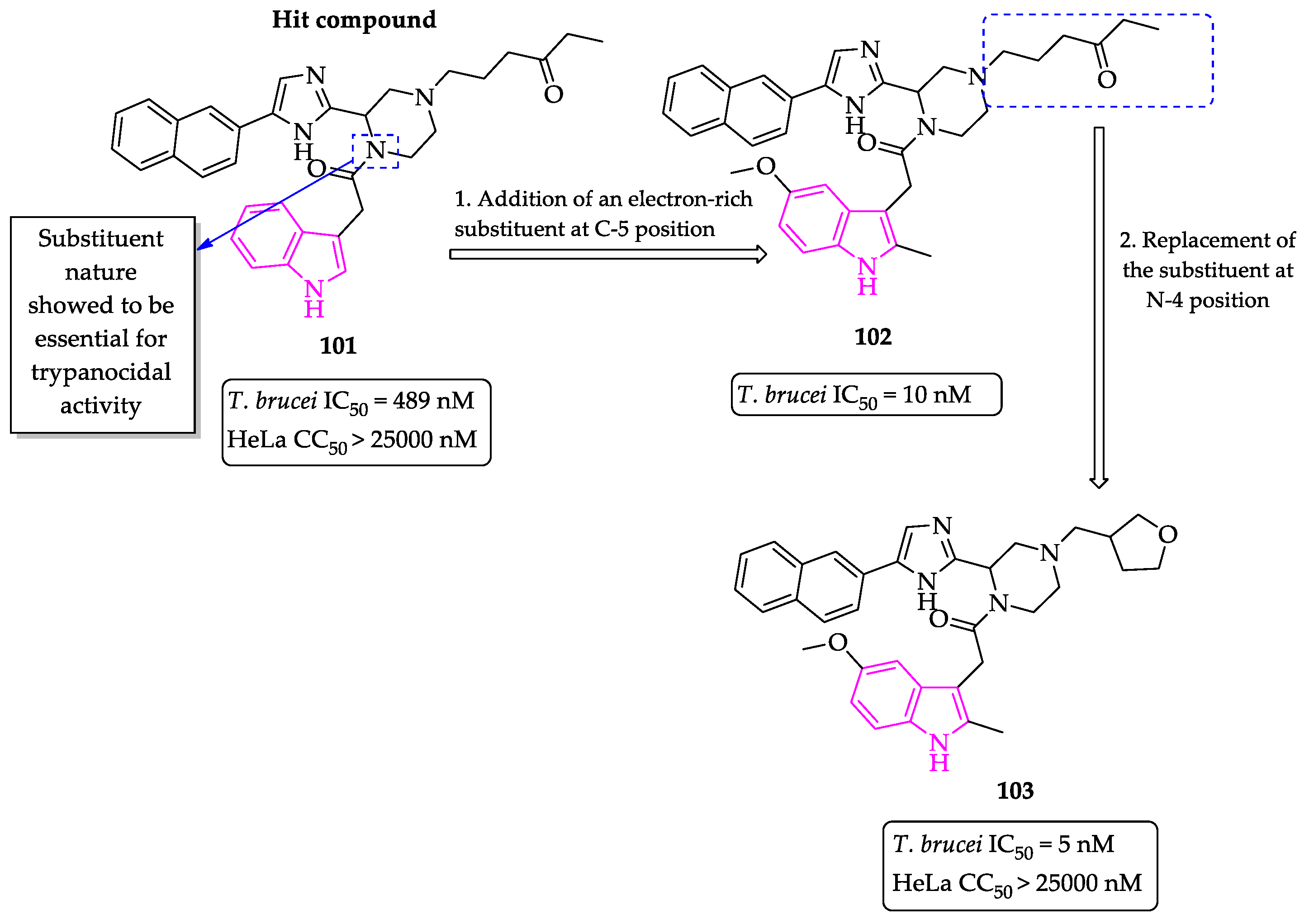
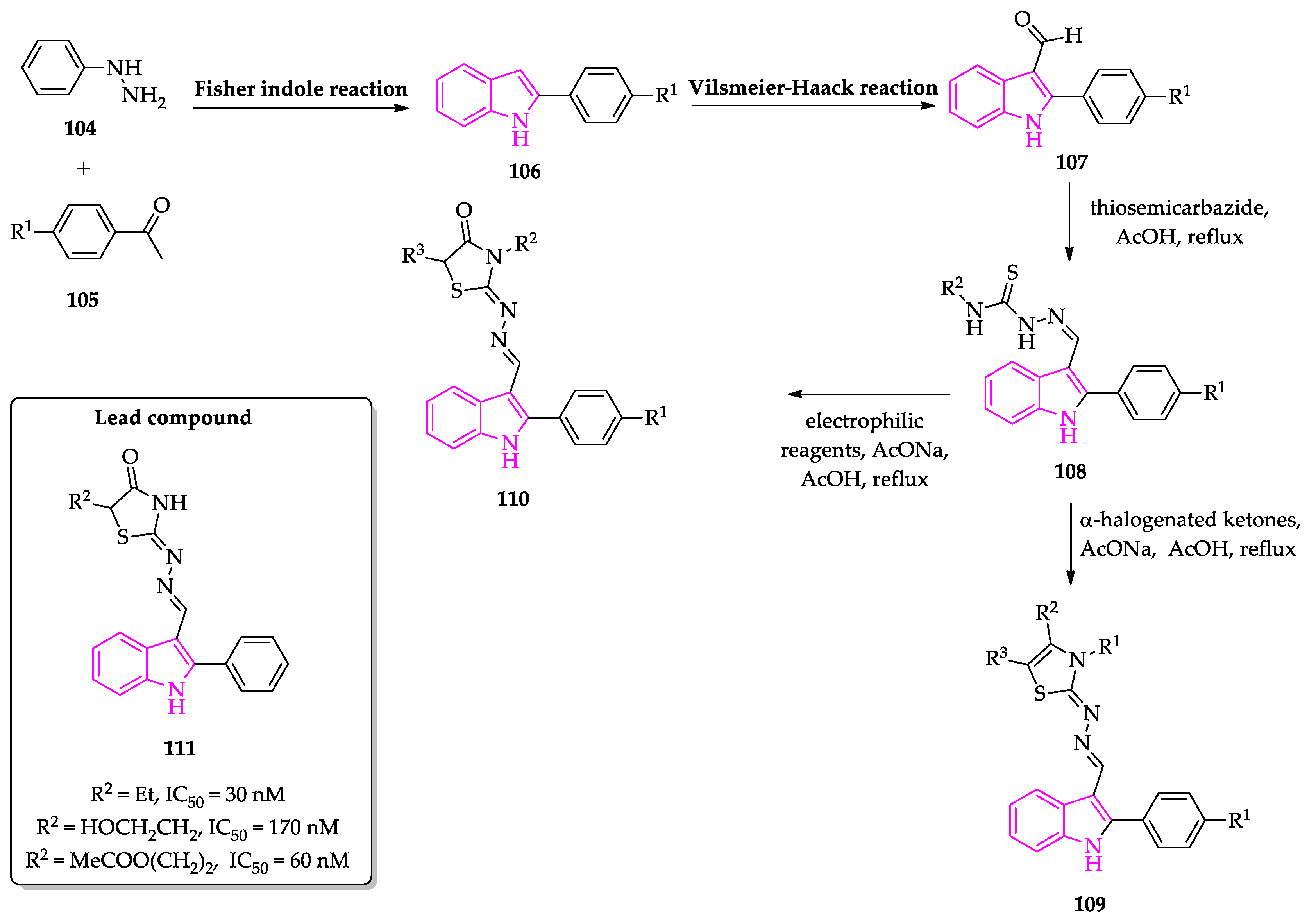

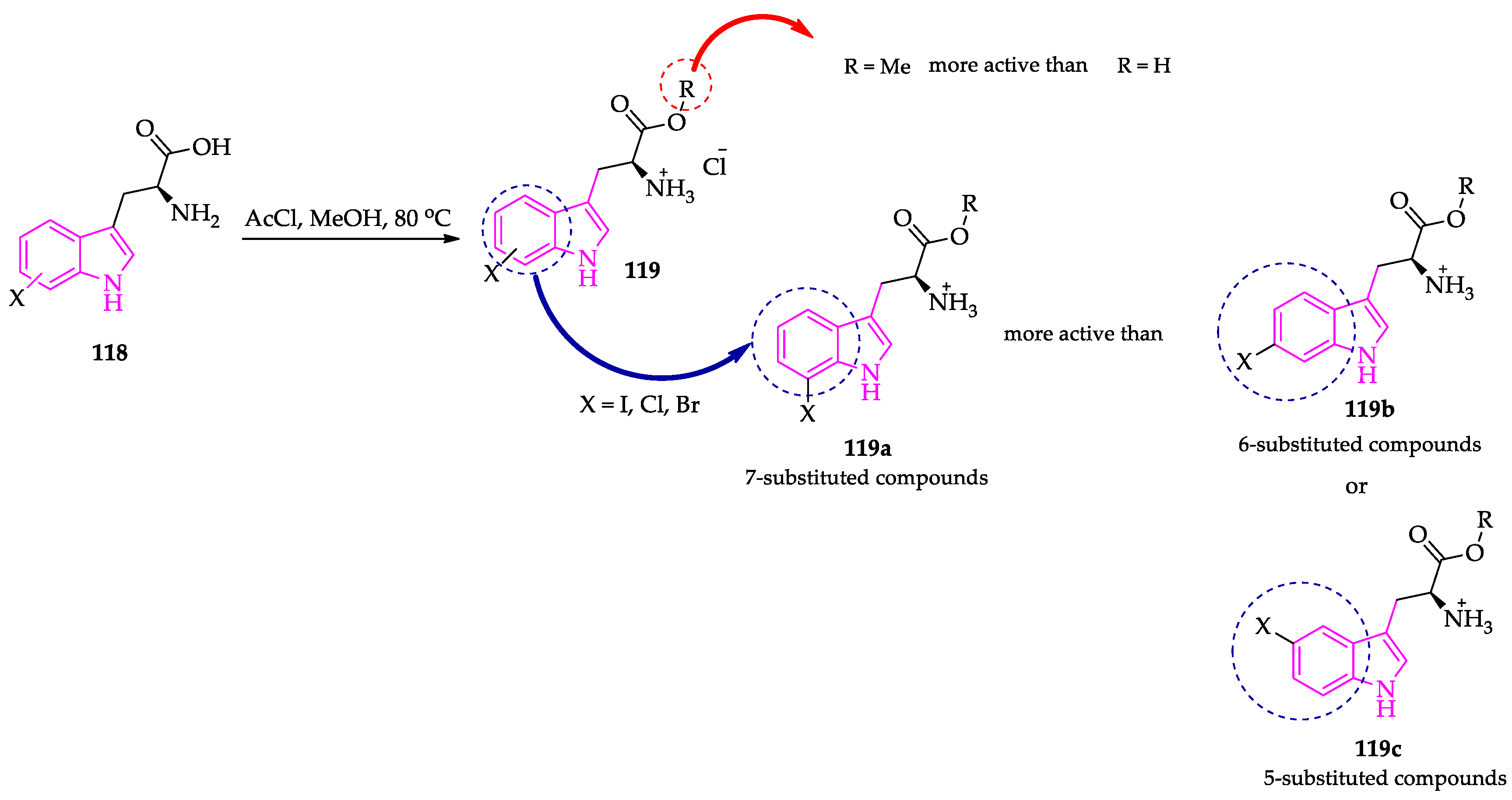
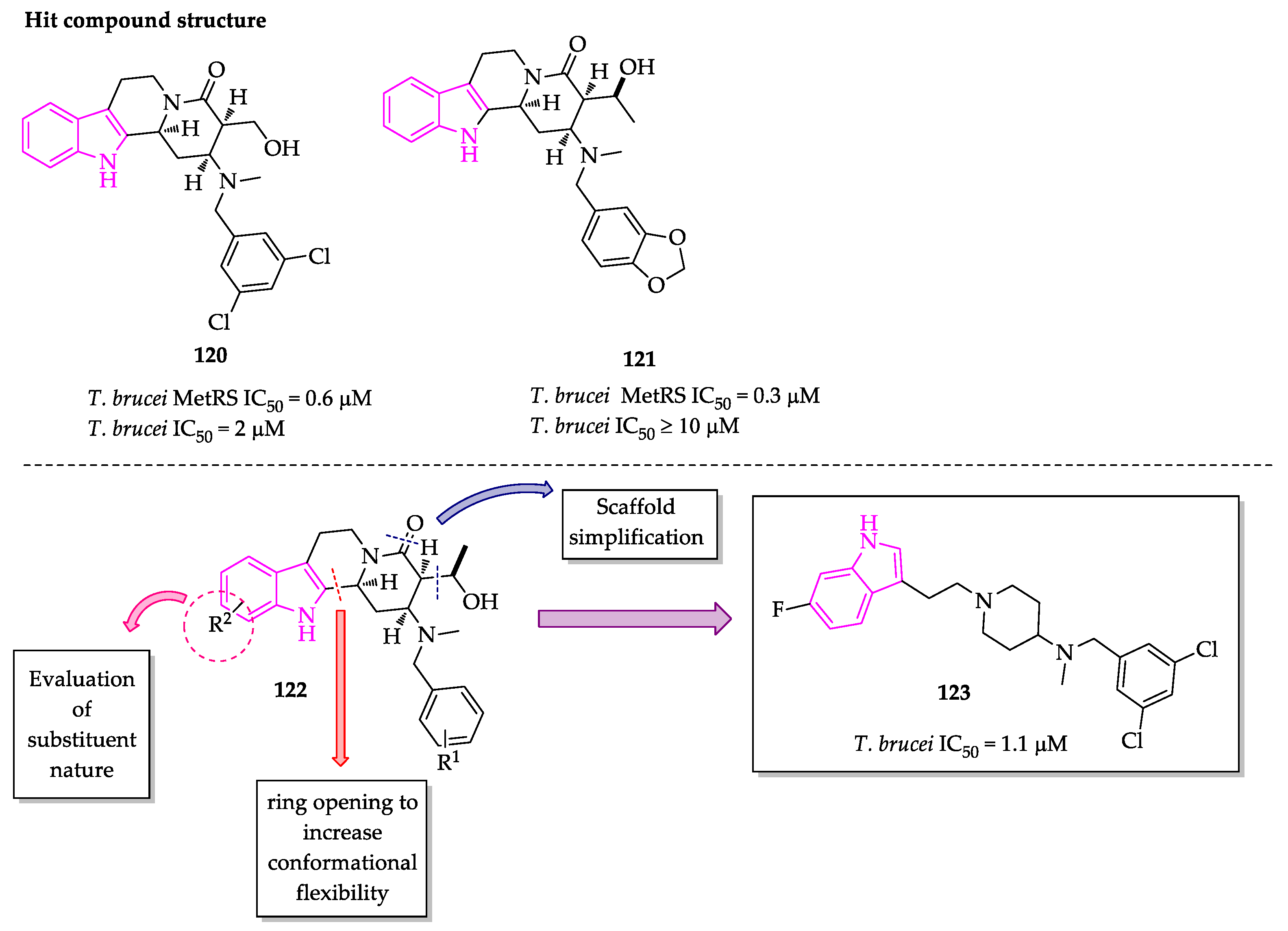
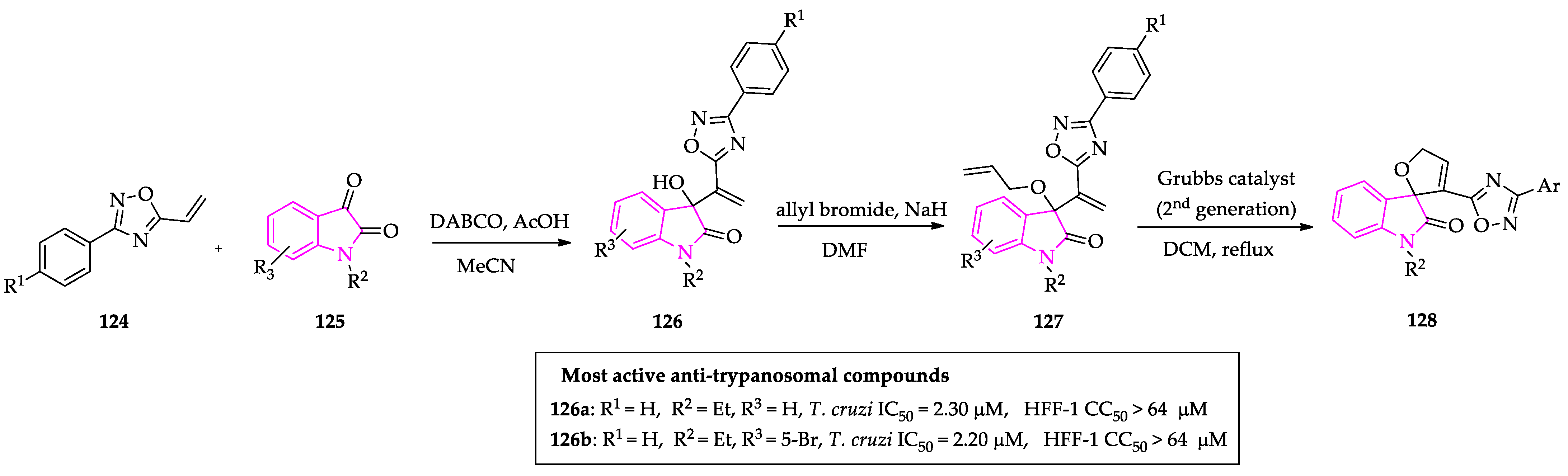


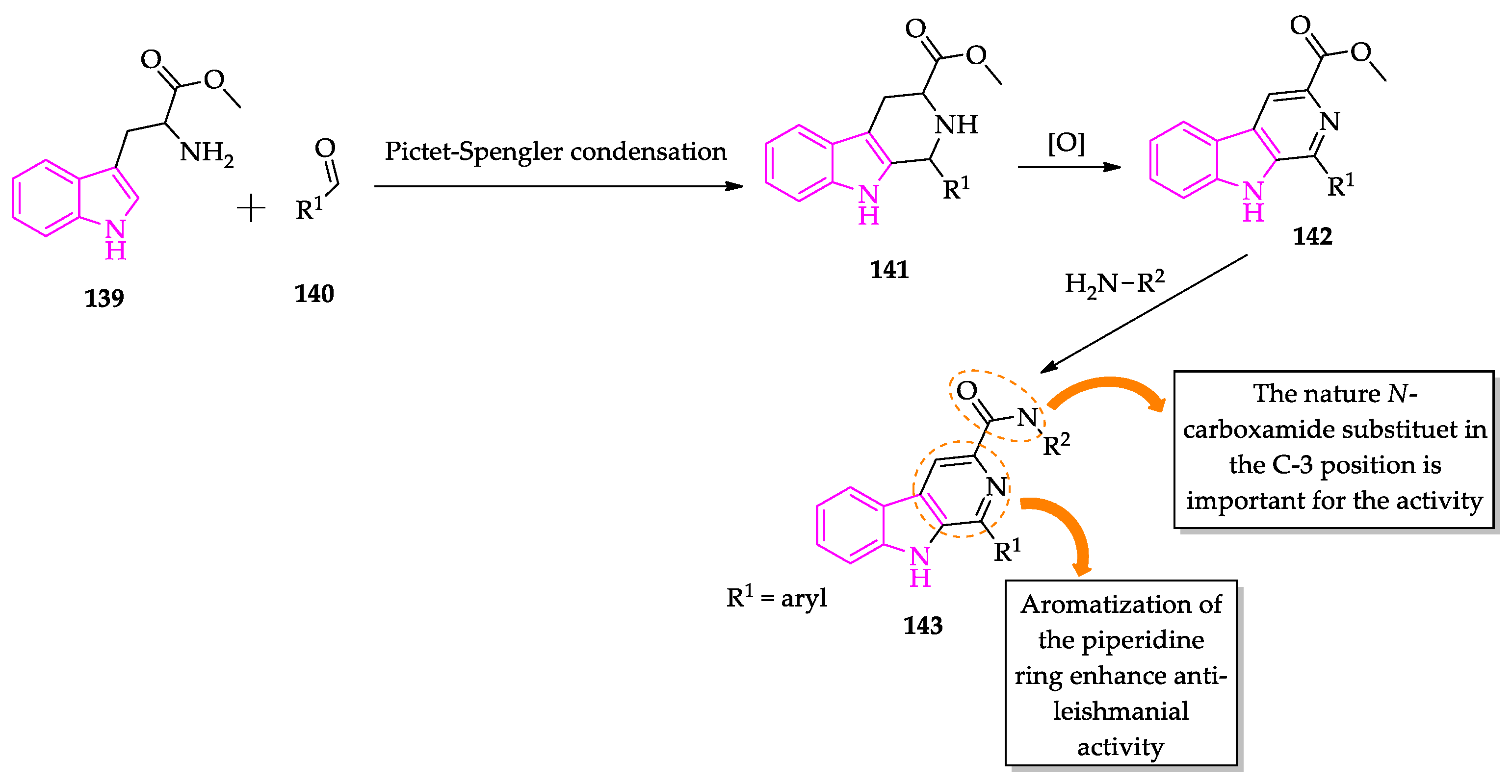

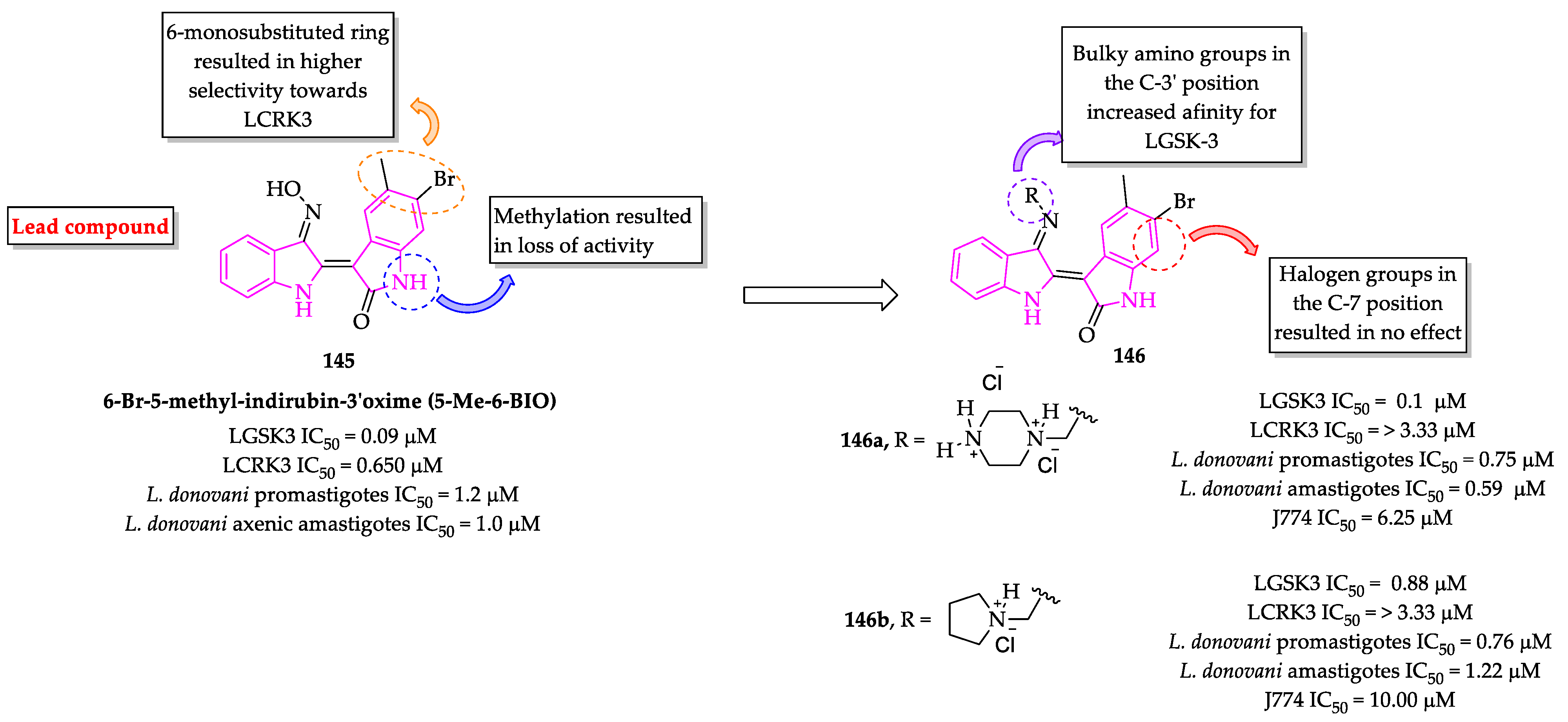


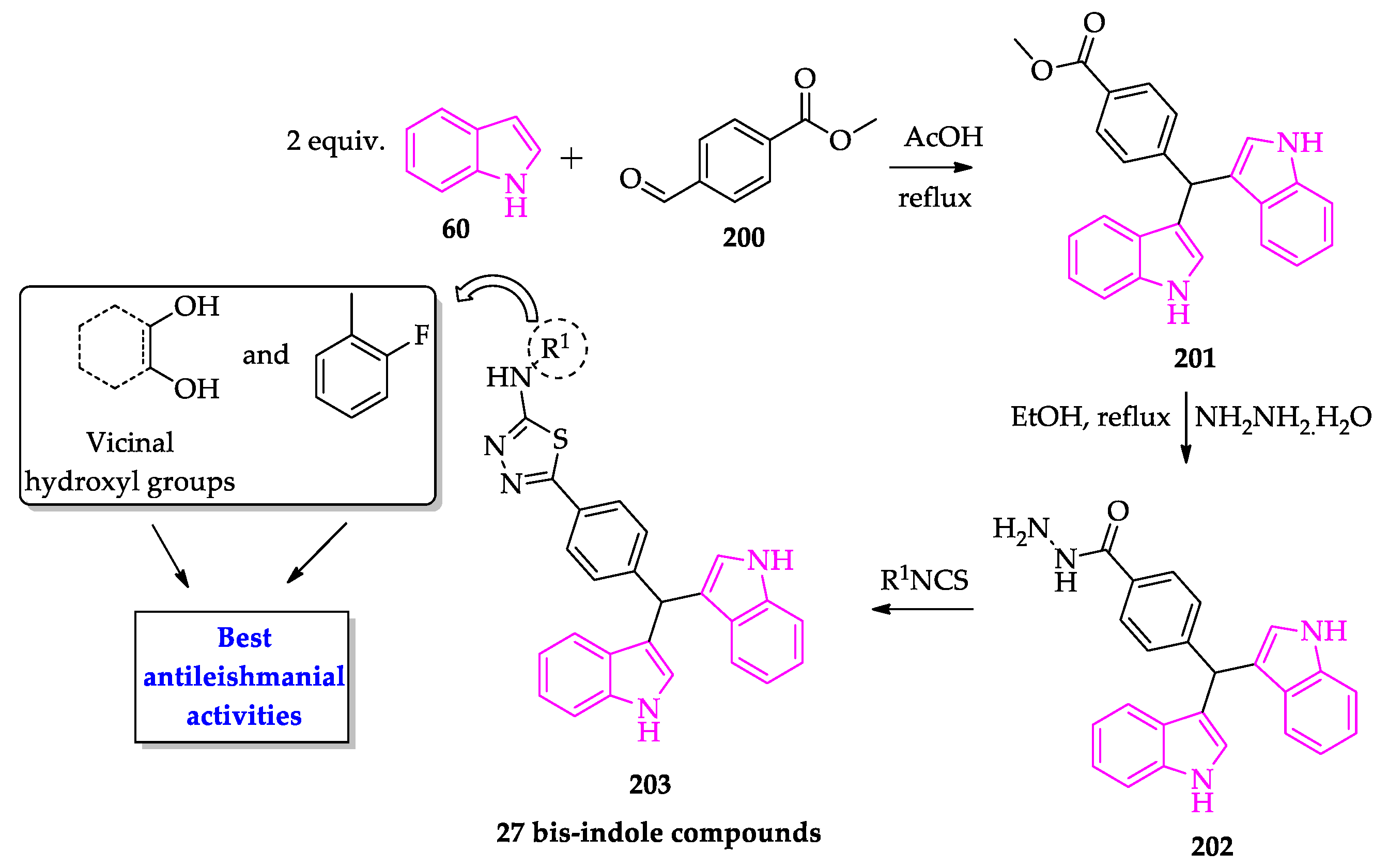
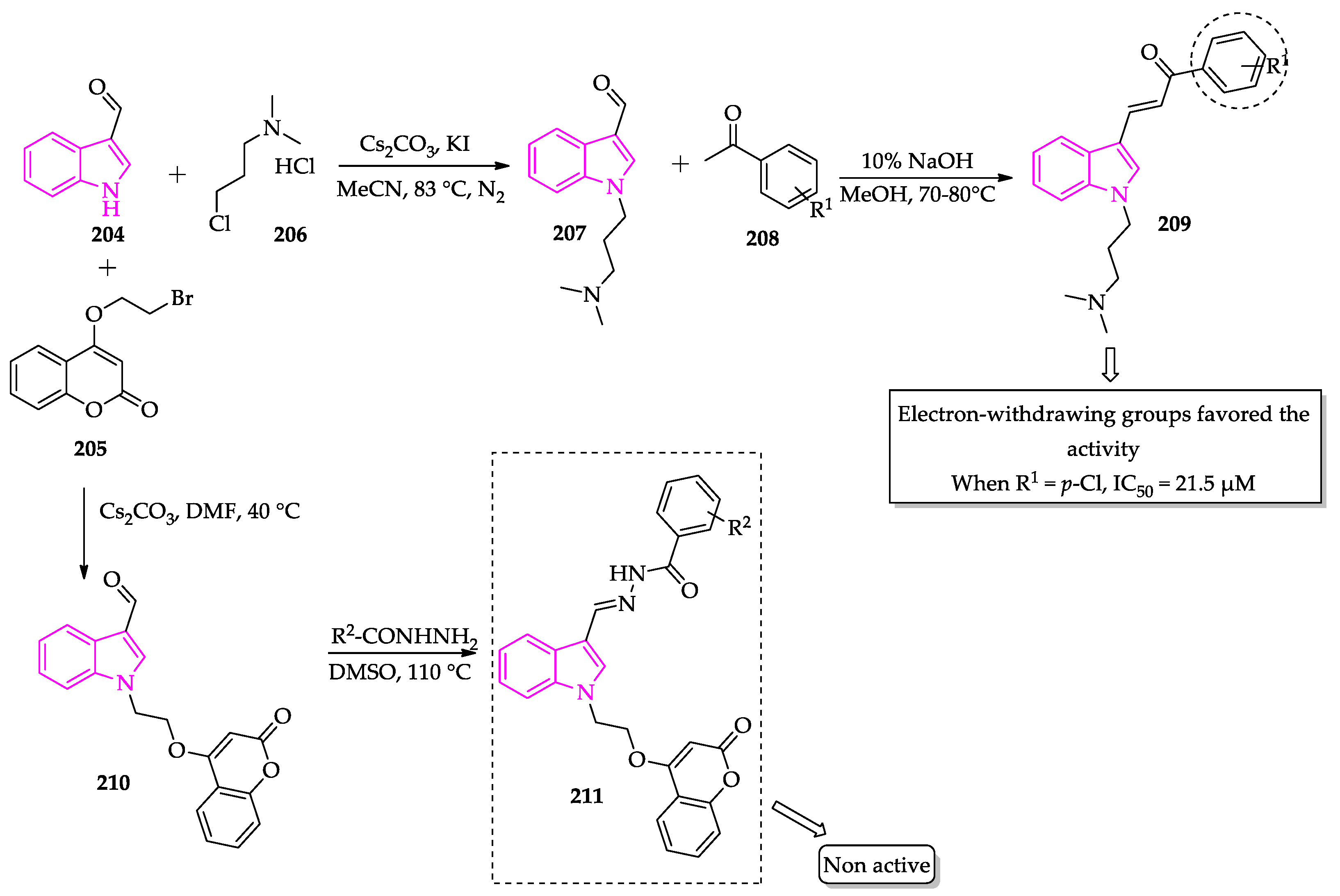

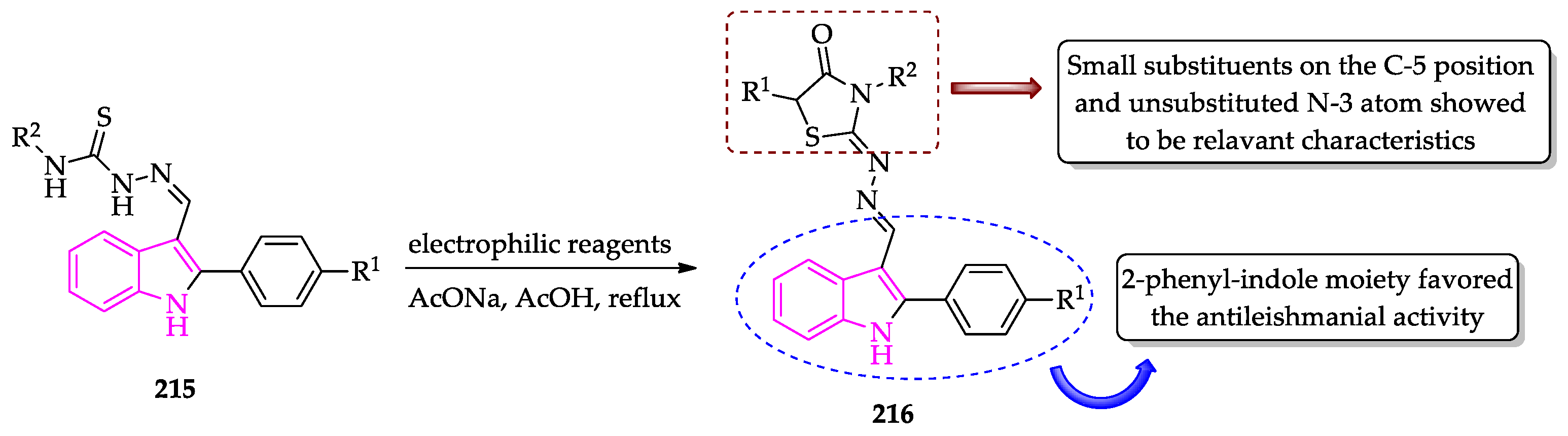
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacheco, P.A.F.; Santos, M.M.M. Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis. Molecules 2022, 27, 319. https://doi.org/10.3390/molecules27010319
Pacheco PAF, Santos MMM. Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis. Molecules. 2022; 27(1):319. https://doi.org/10.3390/molecules27010319
Chicago/Turabian StylePacheco, Paulo A. F., and Maria M. M. Santos. 2022. "Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis" Molecules 27, no. 1: 319. https://doi.org/10.3390/molecules27010319
APA StylePacheco, P. A. F., & Santos, M. M. M. (2022). Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis. Molecules, 27(1), 319. https://doi.org/10.3390/molecules27010319