
Evaluation of Substituted Pyrazole-Based Kinase Inhibitors in One Decade (2011–2020): Current Status and Future Prospects

 ,
,
Abstract
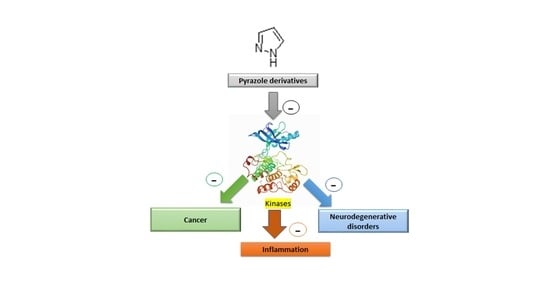
1. Introduction
2. Pyrazole-Based Akt Kinase Inhibitors
2.1. Compound 1
2.2. Compound 2
3. Pyrazole-Based ALK Kinase Inhibitors
Compound 3
4. Pyrazole-Based Apoptosis Signal-Regulating Kinase Inhibitors
Compound 4
5. Pyrazole-Based Aurora Kinase Inhibitors
5.1. Compound 5
5.2. Compound 6
5.3. Compound 7
5.4. Compound 8
5.5. Compound 9
6. Pyrazole-Based BCR-ABL Kinase Inhibitors
6.1. Compound 10
6.2. Compound 11
6.3. Compound 12
7. Pyrazole-Based Calcium-Dependent Kinase Inhibitors
7.1. Compound 13
7.2. Compounds 14 and 15
8. Pyrazole-Based Checkpoint Kinase Inhibitors
8.1. Compounds 16 and 17
8.2. Compound 18
9. Pyrazole-Based Cyclin-Dependent Kinase Inhibitors
9.1. Compound 19
9.2. Compounds 20 and 21
9.3. Compounds 22 and 23
9.4. Compounds 24 and 25
9.5. Compound 26
9.6. Compound 27
9.7. Compound 28
10. Pyrazole-Based EGFR Kinase Inhibitors
Compound 29
11. Pyrazole-Based FGFR Inhibitors
11.1. Compound 30
11.2. Compound 31
11.3. Compound 32
12. Pyrazole-Based IKK Kinase Inhibitors
Compound 33
13. Pyrazole-Based IRAK Inhibitors
13.1. Compound 34
13.2. Compound 35
13.3. Compound 36
14. Pyrazole-Based ITK Inhibitors
14.1. Compounds 37 and 38
14.2. Compound 39
15. Pyrazole-Based JAK Inhibitors
15.1. Compounds 40 and 41
15.2. Compound 42
15.3. Compounds 43 and 44
15.4. Compound 45
16. Pyrazole-Based JNK Inhibitors
16.1. Compound 46
16.2. Compound 47
17. Pyrazole-Based LRRK Inhibitors
17.1. Compounds 48–50
17.2. Compounds 51 and 52
18. Pyrazole-Based Lsrk Inhibitor
Compound 53
19. Pyrazole-Based MEK/ERK Kinase Inhibitors
19.1. Compound 54
19.2. Compound 55
19.3. Compound 56
20. Pyrazole-Based p38α/MAPK14 Kinase Inhibitors
20.1. Compound 57
20.2. Compound 58
20.3. Compound 59
20.4. Compound 60
21. Pyrazole-Based PDK Inhibitors
Compound 61
22. Pyrazole-Based Pim Kinase Inhibitors
22.1. Compound 62
22.2. Compound 63
23. Pyrazole-Based RAF Kinase Inhibitors
23.1. Compound 64
23.2. Compound 65
23.3. Compound 66
23.4. Compound 67
23.5. Compound 68
23.6. Compound 69
23.7. Compound 70
23.8. Compounds 71 and 72
24. Pyrazole-Based ROS Kinase Inhibitors
Compound 73
25. Pyrazole-Based Src Kinase Inhibitor
Compound 74
26. Pyrazole-Based TGFβ/ALK Kinase Inhibitors
26.1. Compounds 75 and 76
26.2. Compounds 75 and 77
26.3. Compound 78
26.4. Compound 79
26.5. Compound 80
26.6. Compound 81
26.7. Compound 82
27. Pyrazole-Based Trk Inhibitors
27.1. Compounds 83–85
27.2. Compound 86
27.3. Compound 87
27.4. Compound 88
28. Pyrazole-Based VEGFR Kinase Inhibitors
28.1. Compound 89
28.2. Compound 90
28.3. Compound 91
29. Pyrazole-Based Multikinase Inhibitors
29.1. Compound 92
29.2. Compounds 93 and 94
29.3. Compound 95
29.4. Compound 96
29.5. Compound 97
29.6. Compound 98
29.7. Compound 99
29.8. Compound 100
30. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.; Al-Aizari, F.; Ansar, M.H. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef]
- Yerragunta, V.; Suman, D.; Anusha, V.; Patil, P.; Naresh, M. Pyrazole and its biological activity. PharmaTutor 2014, 2, 40–48. [Google Scholar]
- Faria, J.V.; Vegi, P.F.; Miguita, A.G.C.; Dos Santos, M.S.; Boechat, N.; Bernardino, A.M.R. Recently reported biological activities of pyrazole compounds. Bioorg. Med. Chem. 2017, 25, 5891–5903. [Google Scholar] [CrossRef]
- Rauch, J.; Volinsky, N.; Romano, D.; Kolch, W. The secret life of kinases: Functions beyond catalysis. Cell Commun. Signal. 2011, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Kannaiyan, R.; Mahadevan, D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Shaw, A.T.; Yasothan, U.; Kirkpatrick, P. Crizotinib. Nat. Rev. Drug Discov. 2011, 10, 897–898. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Erdafitinib: First Global Approval. Drugs 2019, 79, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Yasothan, U.; Kirkpatrick, P. Ruxolitinib. Nat. Rev. Drug Discov. 2012, 11, 103–104. [Google Scholar] [CrossRef]
- Zhan, W.; Xu, L.; Dong, X.; Dong, J.; Yi, X.; Ma, X.; Qiu, N.; Li, J.; Yang, B.; Zhou, Y.; et al. Design, synthesis and biological evaluation of pyrazol-furan carboxamide analogues as novel Akt kinase inhibitors. Eur. J. Med. Chem. 2016, 117, 47–58. [Google Scholar] [CrossRef]
- Zhan, W.; Che, J.; Xu, L.; Wu, Y.; Hu, X.; Zhou, Y.; Cheng, G.; Hu, Y.; Dong, X.; Li, J. Discovery of pyrazole-thiophene derivatives as highly Potent, orally active Akt inhibitors. Eur. J. Med. Chem. 2019, 180, 72–85. [Google Scholar] [CrossRef]
- Fushimi, M.; Fujimori, I.; Wakabayashi, T.; Hasui, T.; Kawakita, Y.; Imamura, K.; Kato, T.; Murakami, M.; Ishii, T.; Kikko, Y.; et al. Discovery of Potent, Selective, and Brain-Penetrant 1H-Pyrazol-5-yl-1H-pyrrolo[2,3-b]pyridines as Anaplastic Lymphoma Kinase (ALK) Inhibitors. J. Med. Chem. 2019, 62, 4915–4935. [Google Scholar] [CrossRef]
- Xin, Z.; Himmelbauer, M.K.; Jones, J.H.; Enyedy, I.; Gilfillan, R.; Hesson, T.; King, K.; Marcotte, D.J.; Murugan, P.; Santoro, J.C.; et al. Discovery of CNS-Penetrant Apoptosis Signal-Regulating Kinase 1 (ASK1) Inhibitors. ACS Med. Chem. Lett. 2020, 11, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Yokozawa, T.; Sakura, T.; Watanabe, T.; Fujisawa, S.; Yamauchi, T.; Uike, N.; Ando, K.; Kihara, R.; Tobinai, K.; et al. A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leuk. Res. 2011, 35, 1384–1389. [Google Scholar] [CrossRef]
- Sessa, F.; Villa, F. Structure of Aurora B-INCENP in complex with barasertib reveals a potential transinhibitory mechanism. Acta Cryst. F Struct. Biol. Commun. 2014, 70, 294–298. [Google Scholar] [CrossRef]
- Li, X.; Lu, X.; Xing, M.; Yang, X.-H.; Zhao, T.-T.; Gong, H.-B.; Zhu, H.-L. Synthesis, biological evaluation, and molecular docking studies of N,1,3-triphenyl-1H-pyrazole-4-carboxamide derivatives as anticancer agents. Bioorg. Med. Chem. Lett. 2012, 22, 3589–3593. [Google Scholar] [CrossRef]
- Sharma, M.C.; Sharma, S.; Bhadoriya, K.S. QSAR studies on pyrazole-4-carboxamide derivatives as Aurora A kinase inhibitors. J. Taibah Univ. Sci. 2016, 10, 107–114. [Google Scholar] [CrossRef][Green Version]
- Zheng, Y.; Zheng, M.; Ling, X.; Liu, Y.; Xue, Y.; An, L.; Gu, N.; Jin, M. Design, synthesis, quantum chemical studies and biological activity evaluation of pyrazole–benzimidazole derivatives as potent Aurora A/B kinase inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3523–3530. [Google Scholar] [CrossRef]
- Bavetsias, V.; Pérez-Fuertes, Y.; McIntyre, P.J.; Atrash, B.; Kosmopoulou, M.; O’Fee, L.; Burke, R.; Sun, C.; Faisal, A.; Bush, K.; et al. 7-(Pyrazol-4-yl)-3H-imidazo[4,5-b]pyridine-based derivatives for kinase inhibition: Co-crystallisation studies with Aurora-A reveal distinct differences in the orientation of the pyrazole N1-substituent. Bioorg. Med. Chem. Lett. 2015, 25, 4203–4209. [Google Scholar] [CrossRef]
- Lakkaniga, N.R.; Zhang, L.; Belachew, B.; Gunaganti, N.; Frett, B.; Li, H.-y. Discovery of SP-96, the first non-ATP-competitive Aurora Kinase B inhibitor, for reduced myelosuppression. Eur. J. Med. Chem. 2020, 203, 112589. [Google Scholar] [CrossRef]
- Hu, L.; Zheng, Y.; Li, Z.; Wang, Y.; Lv, Y.; Qin, X.; Zeng, C. Design, synthesis, and biological activity of phenyl-pyrazole derivatives as BCR–ABL kinase inhibitors. Bioorg. Med. Chem. 2015, 23, 3147–3152. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Cao, T.; Lv, Y.; Ding, Y.; Yang, L.; Zhang, Q.; Guo, M. Design, synthesis, and biological activity of 4-(imidazo[1,2-b]pyridazin-3-yl)-1H-pyrazol-1-yl-phenylbenzamide derivatives as BCR–ABL kinase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5830–5835. [Google Scholar] [CrossRef] [PubMed]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed]
- Study of Efficacy and Safety of Asciminib in Combination With Imatinib in Patients with Chronic Myeloid Leukemia in Chronic Phase (CML-CP). Available online: https://clinicaltrials.gov/ct2/show/NCT03578367?id=NCT03578367&draw=2&rank=1&load=cart (accessed on 24 November 2021).
- Zhan, J.-Y.; Ma, J.; Zheng, Q.-C. Molecular dynamics investigation on the Asciminib resistance mechanism of I502L and V468F mutations in BCR-ABL. J. Mol. Graph. Model. 2019, 89, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Large, J.M.; Osborne, S.A.; Smiljanic-Hurley, E.; Ansell, K.H.; Jones, H.M.; Taylor, D.L.; Clough, B.; Green, J.L.; Holder, A.A. Imidazopyridazines as potent inhibitors of Plasmodium falciparum calcium-dependent protein kinase 1 (PfCDPK1): Preparation and evaluation of pyrazole linked analogues. Bioorg. Med. Chem. Lett. 2013, 23, 6019–6024. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Hulverson, M.A.; Choi, R.; Arnold, S.L.M.; Zhang, Z.; McCloskey, M.C.; Whitman, G.R.; Hackman, R.C.; Rivas, K.L.; Barrett, L.K.; et al. Development of 5-Aminopyrazole-4-carboxamide-based Bumped-Kinase Inhibitors for Cryptosporidiosis Therapy. J. Med. Chem. 2019, 62, 3135–3146. [Google Scholar] [CrossRef] [PubMed]
- Galal, S.A.; Abdelsamie, A.S.; Shouman, S.A.; Attia, Y.M.; Ali, H.I.; Tabll, A.; El-Shenawy, R.; El Abd, Y.S.; Ali, M.M.; Mahmoud, A.E.; et al. Part I: Design, synthesis and biological evaluation of novel pyrazole-benzimidazole conjugates as checkpoint kinase 2 (Chk2) inhibitors with studying their activities alone and in combination with genotoxic drugs. Eur. J. Med. Chem. 2017, 134, 392–405. [Google Scholar] [CrossRef]
- Galal, S.A.; Khairat, S.H.M.; Ali, H.I.; Shouman, S.A.; Attia, Y.M.; Ali, M.M.; Mahmoud, A.E.; Abdel-Halim, A.H.; Fyiad, A.A.; Tabll, A.; et al. Part II: New candidates of pyrazole-benzimidazole conjugates as checkpoint kinase 2 (Chk2) inhibitors. Eur. J. Med. Chem. 2018, 144, 859–873. [Google Scholar] [CrossRef]
- Galal, S.A.; Khattab, M.; Shouman, S.A.; Ramadan, R.; Kandil, O.M.; Kandil, O.M.; Tabll, A.; El Abd, Y.S.; El-Shenawy, R.; Attia, Y.M.; et al. Part III: Novel checkpoint kinase 2 (Chk2) inhibitors; design, synthesis and biological evaluation of pyrimidine-benzimidazole conjugates. Eur. J. Med. Chem. 2018, 146, 687–708. [Google Scholar] [CrossRef]
- Jorda, R.; Schütznerová, E.; Cankař, P.; Brychtová, V.; Navrátilová, J.; Kryštof, V. Novel arylazopyrazole inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. 2015, 23, 1975–1981. [Google Scholar] [CrossRef]
- Ganga Reddy, V.; Srinivasa Reddy, T.; Lakshma Nayak, V.; Prasad, B.; Reddy, A.P.; Ravikumar, A.; Taj, S.; Kamal, A. Design, synthesis and biological evaluation of N-((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)-1,3-diphenyl-1H-pyrazole-4-carboxamides as CDK1/Cdc2 inhibitors. Eur. J. Med. Chem. 2016, 122, 164–177. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Sonawane, Y.A.; Taylor, M.A.; Kizhake, S.; Zahid, M.; Natarajan, A. Synthesis of aminopyrazole analogs and their evaluation as CDK inhibitors for cancer therapy. Bioorg. Med. Chem. Lett. 2018, 28, 3736–3740. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Mol. Cancer Ther. 2009, 8, 324. [Google Scholar] [CrossRef]
- Santo, L.; Vallet, S.; Hideshima, T.; Cirstea, D.; Ikeda, H.; Pozzi, S.; Patel, K.; Okawa, Y.; Gorgun, G.; Perrone, G.; et al. AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene 2010, 29, 2325–2336. [Google Scholar] [CrossRef]
- Harras, M.F.; Sabour, R. Design, synthesis and biological evaluation of novel 1,3,4-trisubstituted pyrazole derivatives as potential chemotherapeutic agents for hepatocellular carcinoma. Bioorg. Chem. 2018, 78, 149–157. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Doctor, Z.M.; Ficarro, S.B.; Marto, J.A.; Kim, N.D.; Sim, T.; Gray, N.S. Synthesis and structure activity relationships of a series of 4-amino-1H-pyrazoles as covalent inhibitors of CDK14. Bioorg. Med. Chem. Lett. 2019, 29, 1985–1993. [Google Scholar] [CrossRef]
- Cheng, C.; Yun, F.; Ullah, S.; Yuan, Q. Discovery of novel cyclin-dependent kinase (CDK) and histone deacetylase (HDAC) dual inhibitors with potent in vitro and in vivo anticancer activity. Eur. J. Med. Chem. 2020, 189, 112073. [Google Scholar] [CrossRef] [PubMed]
- Poddutoori, R.; Samajdar, S.; Mukherjee, S. Substituted pyrazole derivatives as selective cdk12/13 inhibitors. U.S. Patent No. 10,894,786, 10 October 2019. [Google Scholar]
- Zhang, W.-M.; Xing, M.; Zhao, T.-T.; Ren, Y.-J.; Yang, X.-H.; Yang, Y.-S.; Lv, P.-C.; Zhu, H.-L. Synthesis, molecular modeling and biological evaluation of cinnamic acid derivatives with pyrazole moieties as novel anticancer agents. RSC Adv. 2014, 4, 37197–37207. [Google Scholar] [CrossRef]
- Gavine, P.R.; Mooney, L.; Kilgour, E.; Thomas, A.P.; Al-Kadhimi, K.; Beck, S.; Rooney, C.; Coleman, T.; Baker, D.; Mellor, M.J.; et al. AZD4547: An Orally Bioavailable, Potent, and Selective Inhibitor of the Fibroblast Growth Factor Receptor Tyrosine Kinase Family. Cancer Res. 2012, 72, 2045. [Google Scholar] [CrossRef]
- Search Results of the Term AZD4547. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=AZD4547&cntry=&state=&city=&dist= (accessed on 24 November 2021).
- Tucker, J.A.; Klein, T.; Breed, J.; Breeze, A.L.; Overman, R.; Phillips, C.; Norman, R.A. Structural Insights into FGFR Kinase Isoform Selectivity: Diverse Binding Modes of AZD4547 and Ponatinib in Complex with FGFR1 and FGFR4. Structure 2014, 22, 1764–1774. [Google Scholar] [CrossRef]
- Search Results of the Term CH5183284. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=CH5183284&cntry=&state=&city=&dist= (accessed on 24 November 2021).
- Ebiike, H.; Taka, N.; Matsushita, M.; Ohmori, M.; Takami, K.; Hyohdoh, I.; Kohchi, M.; Hayase, T.; Nishii, H.; Morikami, K.; et al. Discovery of [5-Amino-1-(2-methyl-3H-benzimidazol-5-yl)pyrazol-4-yl]-(1H-indol-2-yl)methanone (CH5183284/Debio 1347), An Orally Available and Selective Fibroblast Growth Factor Receptor (FGFR) Inhibitor. J. Med. Chem. 2016, 59, 10586–10600. [Google Scholar] [CrossRef]
- Yamani, A.; Zdżalik-Bielecka, D.; Lipner, J.; Stańczak, A.; Piórkowska, N.; Stańczak, P.S.; Olejkowska, P.; Hucz-Kalitowska, J.; Magdycz, M.; Dzwonek, K.; et al. Discovery and optimization of novel pyrazole-benzimidazole CPL304110, as a potent and selective inhibitor of fibroblast growth factor receptors FGFR (1–3). Eur. J. Med. Chem. 2020, 210, 112990. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, M.; Kumar, N.; Baldi, A.; Chandra, R.; Arockia Babu, M.; Madan, J. Chloro and bromo-pyrazole curcumin Knoevenagel condensates augmented anticancer activity against human cervical cancer cells: Design, synthesis, in silico docking and in vitro cytotoxicity analysis. J. Biomol. Struct. Dyn. 2020, 38, 200–218. [Google Scholar] [CrossRef] [PubMed]
- McElroy, W.T.; Tan, Z.; Ho, G.; Paliwal, S.; Li, G.; Seganish, W.M.; Tulshian, D.; Tata, J.; Fischmann, T.O.; Sondey, C.; et al. Potent and Selective Amidopyrazole Inhibitors of IRAK4 That Are Efficacious in a Rodent Model of Inflammation. ACS Med. Chem. Lett. 2015, 6, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Altman, M.D.; Baker, J.; Brubaker, J.D.; Chen, H.; Chen, Y.; Kleinschek, M.A.; Li, C.; Liu, D.; Maclean, J.K.F.; et al. Identification of N-(1H-pyrazol-4-yl)carboxamide inhibitors of interleukin-1 receptor associated kinase 4: Bicyclic core modifications. Bioorg. Med. Chem. Lett. 2015, 25, 5384–5388. [Google Scholar] [CrossRef]
- Xue, C.; Huang, L.; Li, H.; Li, T.; Huabin, L.; Huabin, L. Pyrazole Compounds, Pharmaceutical Compositions Thereof And Use Thereof. 2020-03-05, 2020. US Patent No. 20210340124, 4 November 2021. [Google Scholar]
- Pastor, R.M.; Burch, J.D.; Magnuson, S.; Ortwine, D.F.; Chen, Y.; De La Torre, K.; Ding, X.; Eigenbrot, C.; Johnson, A.; Liimatta, M.; et al. Discovery and optimization of indazoles as potent and selective interleukin-2 inducible T cell kinase (ITK) inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 2448–2452. [Google Scholar] [CrossRef] [PubMed]
- Burch, J.D.; Lau, K.; Barker, J.J.; Brookfield, F.; Chen, Y.; Chen, Y.; Eigenbrot, C.; Ellebrandt, C.; Ismaili, M.H.A.; Johnson, A.; et al. Property- and Structure-Guided Discovery of a Tetrahydroindazole Series of Interleukin-2 Inducible T-Cell Kinase Inhibitors. J. Med. Chem. 2014, 57, 5714–5727. [Google Scholar] [CrossRef]
- Burch, J.D.; Barrett, K.; Chen, Y.; DeVoss, J.; Eigenbrot, C.; Goldsmith, R.; Ismaili, M.H.A.; Lau, K.; Lin, Z.; Ortwine, D.F.; et al. Tetrahydroindazoles as Interleukin-2 Inducible T-Cell Kinase Inhibitors. Part II. Second-Generation Analogues with Enhanced Potency, Selectivity, and Pharmacodynamic Modulation In Vivo. J. Med. Chem. 2015, 58, 3806–3816. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Zang, J.; Zhu, M.; Gao, Q.; Wang, B.; Xu, W.; Zhang, Y. Design, Synthesis, and Antitumor Evaluation of 4-Amino-(1H)-pyrazole Derivatives as JAKs Inhibitors. ACS Med. Chem. Lett. 2016, 7, 950–955. [Google Scholar] [CrossRef]
- Hoffman, R.; Prchal, J.T.; Samuelson, S.; Ciurea, S.O.; Rondelli, D. Philadelphia Chromosome–Negative Myeloproliferative Disorders: Biology and Treatment. Biol. Blood Marrow Transplant. 2007, 13, 64–72. [Google Scholar] [CrossRef]
- Hanan, E.J.; van Abbema, A.; Barrett, K.; Blair, W.S.; Blaney, J.; Chang, C.; Eigenbrot, C.; Flynn, S.; Gibbons, P.; Hurley, C.A.; et al. Discovery of Potent and Selective Pyrazolopyrimidine Janus Kinase 2 Inhibitors. J. Med. Chem. 2012, 55, 10090–10107. [Google Scholar] [CrossRef] [PubMed]
- Zak, M.; Hanan, E.J.; Lupardus, P.; Brown, D.G.; Robinson, C.; Siu, M.; Lyssikatos, J.P.; Romero, F.A.; Zhao, G.; Kellar, T.; et al. Discovery of a class of highly potent Janus Kinase 1/2 (JAK1/2) inhibitors demonstrating effective cell-based blockade of IL-13 signaling. Bioorg. Med. Chem. Lett. 2019, 29, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.; Schroeder, G.M.; Hart, A.C.; Inghrim, J.; Grebinski, J.; Tokarski, J.S.; Lorenzi, M.V.; You, D.; McDevitt, T.; Penhallow, B.; et al. Discovery of a Highly Selective JAK2 Inhibitor, BMS-911543, for the Treatment of Myeloproliferative Neoplasms. ACS Med. Chem. Lett. 2015, 6, 850–855. [Google Scholar] [CrossRef]
- Doma, A.; Kulkarni, R.; Palakodety, R.; Sastry, G.N.; Sridhara, J.; Garlapati, A. Pyrazole derivatives as potent inhibitors of c-Jun N-terminal kinase: Synthesis and SAR studies. Bioorg. Med. Chem. 2014, 22, 6209–6219. [Google Scholar] [CrossRef]
- Oh, Y.; Jang, M.; Cho, H.; Yang, S.; Im, D.; Moon, H.; Hah, J.-M. Discovery of 3-alkyl-5-aryl-1-pyrimidyl-1H-pyrazole derivatives as a novel selective inhibitor scaffold of JNK3. J. Enzym. Inhib. Med. Chem. 2020, 35, 372–376. [Google Scholar] [CrossRef]
- Estrada, A.A.; Chan, B.K.; Baker-Glenn, C.; Beresford, A.; Burdick, D.J.; Chambers, M.; Chen, H.; Dominguez, S.L.; Dotson, J.; Drummond, J.; et al. Discovery of Highly Potent, Selective, and Brain-Penetrant Aminopyrazole Leucine-Rich Repeat Kinase 2 (LRRK2) Small Molecule Inhibitors. J. Med. Chem. 2014, 57, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Greshock, T.J.; Sanders, J.M.; Drolet, R.E.; Rajapakse, H.A.; Chang, R.K.; Kim, B.; Rada, V.L.; Tiscia, H.E.; Su, H.; Lai, M.-T.; et al. Potent, selective and orally bioavailable leucine-rich repeat kinase 2 (LRRK2) inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2631–2635. [Google Scholar] [CrossRef]
- Stotani, S.; Gatta, V.; Medarametla, P.; Padmanaban, M.; Karawajczyk, A.; Giordanetto, F.; Tammela, P.; Laitinen, T.; Poso, A.; Tzalis, D.; et al. DPD-Inspired Discovery of Novel LsrK Kinase Inhibitors: An Opportunity To Fight Antimicrobial Resistance. J. Med. Chem. 2019, 62, 2720–2737. [Google Scholar] [CrossRef]
- Majik, M.S.; Gawas, U.B.; Mandrekar, V.K. Next generation quorum sensing inhibitors: Accounts on structure activity relationship studies and biological activities. Bioorg. Med. Chem. 2020, 28, 115728. [Google Scholar] [CrossRef]
- Choi, W.-K.; El-Gamal, M.I.; Choi, H.S.; Baek, D.; Oh, C.-H. New diarylureas and diarylamides containing 1,3,4-triarylpyrazole scaffold: Synthesis, antiproliferative evaluation against melanoma cell lines, ERK kinase inhibition, and molecular docking studies. Eur. J. Med. Chem. 2011, 46, 5754–5762. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, M.I.; Choi, H.S.; Yoo, K.H.; Baek, D.; Oh, C.-H. Antiproliferative Diarylpyrazole Derivatives as Dual Inhibitors of the ERK Pathway and COX-2. Chem. Biol. Drug Des. 2013, 82, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.-H.; Ren, Z.-L.; Zhou, B.-G.; Li, Q.-S.; Chu, M.-J.; Liu, D.-H.; Mo, K.; Zhang, L.-S.; Yao, X.-K.; Cao, H.-Q. Discovery of N-(benzyloxy)-1,3-diphenyl-1H-pyrazole-4-carboxamide derivatives as potential antiproliferative agents by inhibiting MEK. Bioorg. Med. Chem. 2016, 24, 4652–4659. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Anbar, H.S.; Tarazi, H.; Oh, C.-H. Discovery of a potent p38α/MAPK14 kinase inhibitor: Synthesis, in vitro/in vivo biological evaluation, and docking studies. Eur. J. Med. Chem. 2019, 183, 111684. [Google Scholar] [CrossRef] [PubMed]
- Getlik, M.; Grütter, C.; Simard, J.R.; Nguyen, H.D.; Robubi, A.; Aust, B.; van Otterlo, W.A.L.; Rauh, D. Structure-based design, synthesis and biological evaluation of N-pyrazole, N′-thiazole urea inhibitors of MAP kinase p38α. Eur. J. Med. Chem. 2012, 48, 1–15. [Google Scholar] [CrossRef]
- Röhm, S.; Schröder, M.; Dwyer, J.E.; Widdowson, C.S.; Chaikuad, A.; Berger, B.-T.; Joerger, A.C.; Krämer, A.; Harbig, J.; Dauch, D.; et al. Selective targeting of the αC and DFG-out pocket in p38 MAPK. Eur. J. Med. Chem. 2020, 208, 112721. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.K.; Blakskjær, P.; Chaikuad, A.; Christensen, A.B.; Dietvorst, J.; Holmkvist, J.; Knapp, S.; Kořínek, M.; Larsen, L.K.; Pedersen, A.E.; et al. Novel p38α MAP kinase inhibitors identified from yoctoReactor DNA-encoded small molecule library. MedChemComm 2016, 7, 1332–1339. [Google Scholar] [CrossRef]
- Mohammed, I.E.-G.; Mohammed, S.A.-M.; Mahmoud, M.G.E.-D.; Ji-Sun, S.; Kyung-Tae, L.; Kyung Ho, Y.; Chang-Hyun, O. Synthesis, in vitro Antiproliferative and Antiinflammatory Activities, and Kinase Inhibitory effects of New 1,3,4-triarylpyrazole Derivatives. Anti-Cancer Agents Med. Chem. 2017, 17, 75–84. [Google Scholar] [CrossRef]
- Lee, D.; Pagire, H.S.; Pagire, S.H.; Bae, E.J.; Dighe, M.; Kim, M.; Lee, K.M.; Jang, Y.K.; Jaladi, A.K.; Jung, K.-Y.; et al. Discovery of Novel Pyruvate Dehydrogenase Kinase 4 Inhibitors for Potential Oral Treatment of Metabolic Diseases. J. Med. Chem. 2019, 62, 575–588. [Google Scholar] [CrossRef]
- Hu, H.; Wang, X.; Chan, G.K.Y.; Chang, J.H.; Do, S.; Drummond, J.; Ebens, A.; Lee, W.; Ly, J.; Lyssikatos, J.P.; et al. Discovery of 3,5-substituted 6-azaindazoles as potent pan-Pim inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5258–5264. [Google Scholar] [CrossRef]
- Wang, X.; Blackaby, W.; Allen, V.; Chan, G.K.Y.; Chang, J.H.; Chiang, P.-C.; Diène, C.; Drummond, J.; Do, S.; Fan, E.; et al. Optimization of Pan-Pim Kinase Activity and Oral Bioavailability Leading to Diaminopyrazole (GDC-0339) for the Treatment of Multiple Myeloma. J. Med. Chem. 2019, 62, 2140–2153. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Choi, H.S.; Cho, H.-G.; Hong, J.H.; Yoo, K.H.; Oh, C.-H. Design, Synthesis, and Antiproliferative Activity of 3,4-Diarylpyrazole-1-carboxamide Derivatives against Melanoma Cell Line. Arch. Der Pharm. 2011, 344, 745–754. [Google Scholar] [CrossRef]
- Caballero, J.; Alzate-Morales, J.H.; Vergara-Jaque, A. Investigation of the Differences in Activity between Hydroxycycloalkyl N1 Substituted Pyrazole Derivatives as Inhibitors of B-Raf Kinase by Using Docking, Molecular Dynamics, QM/MM, and Fragment-Based De Novo Design: Study of Binding Mode of Diastereomer Compounds. J. Chem. Inf. Modeling 2011, 51, 2920–2931. [Google Scholar] [CrossRef]
- Wang, S.-F.; Zhu, Y.-L.; Zhu, P.-T.; Makawana, J.A.; Zhang, Y.-L.; Zhao, M.-Y.; Lv, P.-C.; Zhu, H.-L. Design, synthesis and biological evaluation of novel 5-phenyl-1H-pyrazole derivatives as potential BRAFV600E inhibitors. Bioorg. Med. Chem. 2014, 22, 6201–6208. [Google Scholar] [CrossRef]
- Gamal El-Din, M.M.; El-Gamal, M.I.; Abdel-Maksoud, M.S.; Yoo, K.H.; Oh, C.-H. Design, synthesis, broad-spectrum antiproliferative activity, and kinase inhibitory effect of triarylpyrazole derivatives possessing arylamides or arylureas moieties. Eur. J. Med. Chem. 2016, 119, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; El-Gamal, M.I.; Tarazi, H.; Choi, H.S.; Oh, C.-H. Design and synthesis of a new series of highly potent RAF kinase-inhibiting triarylpyrazole derivatives possessing antiproliferative activity against melanoma cells. Future Med. Chem. 2016, 8, 2197–2211. [Google Scholar] [CrossRef] [PubMed]
- Gamal El-Din, M.M.; El-Gamal, M.I.; Abdel-Maksoud, M.S.; Yoo, K.H.; Oh, C.H. Design, synthesis, in vitro potent antiproliferative activity, and kinase inhibitory effects of new triarylpyrazole derivatives possessing different heterocycle terminal moieties. J. Enzym. Inhib. Med. Chem. 2019, 34, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Gamal El-Din, M.M.; El-Gamal, M.I.; Abdel-Maksoud, M.S.; Yoo, K.H.; Baek, D.; Choi, J.; Lee, H.; Oh, C.-H. Design, synthesis, and in vitro antiproliferative and kinase inhibitory effects of pyrimidinylpyrazole derivatives terminating with arylsulfonamido or cyclic sulfamide substituents. J. Enzym. Inhib. Med. Chem. 2016, 31, 111–122. [Google Scholar] [CrossRef]
- Tarazi, H.; El-Gamal, M.I.; Oh, C.-H. Discovery of highly potent V600E-B-RAF kinase inhibitors: Molecular modeling study. Bioorg. Med. Chem. 2019, 27, 655–663. [Google Scholar] [CrossRef]
- El-Gamal, M.I.; Park, B.-J.; Oh, C.-H. Synthesis, in vitro antiproliferative activity, and kinase inhibitory effects of pyrazole-containing diarylureas and diarylamides. Eur. J. Med. Chem. 2018, 156, 230–239. [Google Scholar] [CrossRef]
- Park, B.S.; Al-Sanea, M.M.; Abdelazem, A.Z.; Park, H.M.; Roh, E.J.; Park, H.-M.; Yoo, K.H.; Sim, T.; Tae, J.S.; Lee, S.H. Structure-based optimization and biological evaluation of trisubstituted pyrazole as a core structure of potent ROS1 kinase inhibitors. Bioorg. Med. Chem. 2014, 22, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Karim, S.S.; Anwar, M.M.; Mohamed, N.A.; Nasr, T.; Elseginy, S.A. Design, synthesis, biological evaluation and molecular docking studies of novel benzofuran–pyrazole derivatives as anticancer agents. Bioorganic Chem. 2015, 63, 1–12. [Google Scholar] [CrossRef]
- Sekimata, K.; Sato, T.; Sakai, N.; Watanabe, H.; Mishima-Tsumagari, C.; Taguri, T.; Matsumoto, T.; Fujii, Y.; Handa, N.; Honma, T.; et al. Bis-Heteroaryl Pyrazoles: Identification of Orally Bioavailable Inhibitors of Activin Receptor-Like Kinase-2 (R206H). Chem. Pharm. Bull. 2019, 67, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Sekimata, K.; Sakai, N.; Watanabe, H.; Mishima-Tsumagari, C.; Taguri, T.; Matsumoto, T.; Fujii, Y.; Handa, N.; Tanaka, A.; et al. Structural Basis of Activin Receptor-Like Kinase 2 (R206H) Inhibition by Bis-heteroaryl Pyrazole-Based Inhibitors for the Treatment of Fibrodysplasia Ossificans Progressiva Identified by the Integration of Ligand-Based and Structure-Based Drug Design Approaches. ACS Omega 2020, 5, 11411–11423. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.H.; Sreenu, D.; Krishnaiah, M.; Subrahmanyam, V.B.; Rao, K.S.; Nagendra Mohan, A.V.; Park, C.-Y.; Son, J.-Y.; Son, D.-H.; Park, H.-J.; et al. Synthesis and biological evaluation of 1-substituted-3(5)-(6-methylpyridin-2-yl)-4-(quinoxalin-6-yl)pyrazoles as transforming growth factor-β type 1 receptor kinase inhibitors. Eur. J. Med. Chem. 2011, 46, 3917–3925. [Google Scholar] [CrossRef]
- Zhao, L.-M.; Guo, Z.; Xue, Y.-J.; Min, J.Z.; Zhu, W.-J.; Li, X.-Y.; Piao, H.-R.; Jin, C.H. Synthesis and Evaluation of 3-Substituted-4-(quinoxalin-6-yl) Pyrazoles as TGF-β Type I Receptor Kinase Inhibitors. Molecules 2018, 23, 3369. [Google Scholar] [CrossRef]
- Jin, C.H.; Krishnaiah, M.; Sreenu, D.; Subrahmanyam, V.B.; Park, H.-J.; Park, S.-J.; Sheen, Y.Y.; Kim, D.-K. 4-([1,2,4]Triazolo[1,5-a]pyridin-6-yl)-5(3)-(6-methylpyridin-2-yl)imidazole and -pyrazole derivatives as potent and selective inhibitors of transforming growth factor-β type I receptor kinase. Bioorg. Med. Chem. 2014, 22, 2724–2732. [Google Scholar] [CrossRef]
- Zhu, W.-J.; Cui, B.-W.; Wang, H.M.; Nan, J.-X.; Piao, H.-R.; Lian, L.-H.; Jin, C.H. Design, synthesis, and antifibrosis evaluation of 4-(benzo-[c][1,2,5]thiadiazol-5-yl)-3(5)-(6-methyl- pyridin-2-yl)pyrazole and 3(5)-(6-methylpyridin- 2-yl)-4-(thieno-[3,2,-c]pyridin-2-yl)pyrazole derivatives. Eur. J. Med. Chem. 2019, 180, 15–27. [Google Scholar] [CrossRef]
- Thress, K.; MacIntyre, T.; Wang, H.; Whitston, D.; Liu, Z.-Y.; Hoffmann, E.; Wang, T.; Brown, J.L.; Webster, K.; Omer, C.; et al. Identification and preclinical characterization of AZ-23, a novel, selective, and orally bioavailable inhibitor of the Trk kinase pathway. Mol. Cancer Ther. 2009, 8, 1818. [Google Scholar] [CrossRef]
- Wang, T.; Lamb, M.L.; Block, M.H.; Davies, A.M.; Han, Y.; Hoffmann, E.; Ioannidis, S.; Josey, J.A.; Liu, Z.-Y.; Lyne, P.D.; et al. Discovery of Disubstituted Imidazo[4,5-b]pyridines and Purines as Potent TrkA Inhibitors. ACS Med. Chem. Lett. 2012, 3, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Momose, T.; Katsuno, K.; Fushimi, N.; Muranaka, H.; Handa, C.; Ozawa, T.; Kinoshita, T. The juxtamembrane region of TrkA kinase is critical for inhibitor selectivity. Bioorg. Med. Chem. Lett. 2017, 27, 1233–1236. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Johnson, P.D.; Zachary, T.; Roush, N.; Zhu, Y.; Bowen, S.J.; Janssen, A.; Duclos, B.A.; Williams, T.; Javens, C.; et al. Deciphering the Allosteric Binding Mechanism of the Human Tropomyosin Receptor Kinase A (hTrkA) Inhibitors. ACS Chem. Biol. 2019, 14, 1205–1216. [Google Scholar] [CrossRef] [PubMed]
- Shirahashi, H.; Toriihara, E.; Suenaga, Y.; Yoshida, H.; Akaogi, K.; Endou, Y.; Wakabayashi, M.; Takashima, M. The discovery of novel 3-aryl-indazole derivatives as peripherally restricted pan-Trk inhibitors for the treatment of pain. Bioorg. Med. Chem. Lett. 2019, 29, 2320–2326. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef]
- Miyamoto, N.; Sakai, N.; Hirayama, T.; Miwa, K.; Oguro, Y.; Oki, H.; Okada, K.; Takagi, T.; Iwata, H.; Awazu, Y.; et al. Discovery of N-[5-({2-[(cyclopropylcarbonyl)amino]imidazo[1,2-b]pyridazin-6-yl}oxy)-2-methylph enyl]-1,3-dimethyl-1H-pyrazole-5-carboxamide (TAK-593), a highly potent VEGFR2 kinase inhibitor. Bioorg. Med. Chem. 2013, 21, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.G.; Reddy, T.S.; Jadala, C.; Reddy, M.S.; Sultana, F.; Akunuri, R.; Bhargava, S.K.; Wlodkowic, D.; Srihari, P.; Kamal, A. Pyrazolo-benzothiazole hybrids: Synthesis, anticancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model. Eur. J. Med. Chem. 2019, 182, 111609. [Google Scholar] [CrossRef]
- Dawood, D.H.; Nossier, E.S.; Ali, M.M.; Mahmoud, A.E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. [Google Scholar] [CrossRef]
- Thaher, B.A.; Arnsmann, M.; Totzke, F.; Ehlert, J.E.; Kubbutat, M.H.G.; Schächtele, C.; Zimmermann, M.O.; Koch, P.; Boeckler, F.M.; Laufer, S.A. Tri- and Tetrasubstituted Pyrazole Derivates: Regioisomerism Switches Activity from p38MAP Kinase to Important Cancer Kinases. J. Med. Chem. 2012, 55, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Strocchi, E.; Fornari, F.; Minguzzi, M.; Gramantieri, L.; Milazzo, M.; Rebuttini, V.; Breviglieri, S.; Camaggi, C.M.; Locatelli, E.; Bolondi, L.; et al. Design, synthesis and biological evaluation of pyrazole derivatives as potential multi-kinase inhibitors in hepatocellular carcinoma. Eur. J. Med. Chem. 2012, 48, 391–401. [Google Scholar] [CrossRef]
- Curtin, M.L.; Robin Heyman, H.; Frey, R.R.; Marcotte, P.A.; Glaser, K.B.; Jankowski, J.R.; Magoc, T.J.; Albert, D.H.; Olson, A.M.; Reuter, D.R.; et al. Pyrazole diaminopyrimidines as dual inhibitors of KDR and Aurora B kinases. Bioorg. Med. Chem. Lett. 2012, 22, 4750–4755. [Google Scholar] [CrossRef]
- Abdel-Maksoud, M.S.; El-Gamal, M.I.; Gamal El-Din, M.M.; Oh, C.H. Design, synthesis, in vitro anticancer evaluation, kinase inhibitory effects, and pharmacokinetic profile of new 1,3,4-triarylpyrazole derivatives possessing terminal sulfonamide moiety. J. Enzym. Inhib. Med. Chem. 2019, 34, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Nossier, E.S.; Abd El-Karim, S.S.; Khalifa, N.M.; El-Sayed, A.S.; Hassan, E.S.I.; El-Hallouty, S.M. Kinase Inhibitory Activities and Molecular Docking of a Novel Series of Anticancer Pyrazole Derivatives. Molecules 2018, 23, 3074. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Manhi, F.M.; Mahmoud, M.A.A.; Abdelwahab, A. Uses of dimedone to synthesis pyrazole, isoxazole and thiophene derivatives with antiproliferative, tyrosine kinase and Pim-1 kinase inhibitions. Med. Chem. Res. 2020, 29, 1536–1551. [Google Scholar] [CrossRef]
- Zheng, Y.-G.; Wang, J.-A.; Meng, L.; Pei, X.; Zhang, L.; An, L.; Li, C.-L.; Miao, Y.-L. Design, synthesis, biological activity evaluation of 3-(4-phenyl-1H-imidazol-2-yl)-1H-pyrazole derivatives as potent JAK 2/3 and aurora A/B kinases multi-targeted inhibitors. Eur. J. Med. Chem. 2021, 209, 112934. [Google Scholar] [CrossRef] [PubMed]
- A Phase I, First in Man Study to Evaluate the Safety and Tolerability of a panRAF Inhibitor (CCT3833/BAL3833)in Patients with Solid Tumours (PanRAF). Available online: https://clinicaltrials.gov/ct2/show/NCT02437227?id=NCT02437227&draw=2&rank=1&load=cart (accessed on 24 November 2021).
- Saturno, G.; Lopes, F.; Niculescu-Duvaz, I.; Niculescu-Duvaz, D.; Zambon, A.; Davies, L.; Johnson, L.; Preece, N.; Lee, R.; Viros, A.; et al. The paradox-breaking panRAF plus SRC family kinase inhibitor, CCT3833, is effective in mutant KRAS-driven cancers. Ann. Oncol. 2020, 32, 269–278. [Google Scholar] [CrossRef]
- Dean, E.J.; Banerji, U.; Girotti, R.; Niculescu-Duvaz, I.; Lopes, F.; Davies, L.; Niculescu-Duvaz, D.; Dhomen, N.; Ellis, S.; Ali, Z.; et al. A Phase 1 first-in-human trial to evaluate the safety and tolerability of CCT3833, an oral panRAF inhibitor, in patients with advanced solid tumours, including metastatic melanoma. J. Clin. Oncol. 2016, 34, TPS9597. [Google Scholar] [CrossRef]







































































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | Inhibitor | IC50 Value in Cell-Free Assay (nM) | Other Biological Activities |
|---|---|---|---|
| AKT1 |  | 61 | Antiproliferative activity against HCT116 and OVCAR-8 cell lines (IC50 = 7.76 and 9.76 µM, respectively) |
 | 1.3 | Antiproliferative activity against HCT116 colon cancer cell line (IC50 = 0.95 µM). Reduction of tumor size by 42% in the MM1S model. | |
| ALK |  | 2.9 | Reduction of phosphorylation of ALK in hippocampus in a dose-dependent manner at 30 mg/kg and higher. Inhibited phosphorylation in prefrontal cortex at 100 mg/kg. |
| ASK1 |  | - | Good CNS penetration. Weak potency against hERG, CYP3A4, and CYP2C9. |
| Aurora |  (Barasertib, AZD1152) | 0.37 (Aurora B) | Passed phase I clinical trials in Japanese and Western volunteers suffering from advanced acute myeloid leukemia. |
 | 160 (Aurora A) | IC50 values against HCT116 colon cancer and MCF7 breast cancer cell lines are 0.39 and 0.46 µM, respectively. | |
 | 28.9 (Aurora A) 2.2 (Aurora B) | IC50 values against U937 (leukemia), K562 (leukemia), A549 (lung), LoVo (colon), and HT29 (colon) cancer cell lines are 5.106, 5.003, 0.487, 0.789, and 0.381 µM, respectively. | |
 | 35 (Aurora A) 75 (Aurora B) | Antiproliferative activity against SW620 and HCT116 colon cancer cell lines (IC50 = 0.35 and 0.34 µM, respectively). | |
 | 0.316 (Aurora B) | Antiproliferative activity against MDA-MB-468 with IC50 value equal to 107 nM. | |
| BCR-ABL |  | 14.2 | Antiproliferative activity against the K562 leukemia cell line with an IC50 value equal to 0.27 µM. |
 | 8.5 | Antiproliferative activity against the K562 leukemia cell line with an IC50 value less than 2 nM. | |
 | 0.5 | IC50 = 25 nM against ABL (T315I). Clinical candidate for CML. | |
| Calcium-dependent kinase |  | 56 | Anti-parasitic activity against Plasmodium falciparum with an IC50 value of 0.262 µM. |
 | 0.7 | Anti-parasitic activity against Cryptosporidium parvum with an EC50 value of 0.41 µM. | |
 | 2.5 | Anti-parasitic activity against Cryptosporidium parvum with an EC50 value of 0.51 µM. | |
| Checkpoint kinase 2 |  | 48.4 | Antiproliferative activity against HepG2 (hepatocellular carcinoma), HeLa (cervical), and MCF7 (breast) cancer cell lines. |
 | 17.9 | Antiproliferative activity against HepG2 (hepatocellular carcinoma), HeLa (cervical), and MCF7 (breast) cancer cell lines (IC50 = 10.8, 11.8, and 10.4 µM, respectively). | |
 | 41.64 | Modest potency against HepG2, HeLa, and MCF7 cell lines with 2-digit micromolar IC50 values. | |
| Cyclin-dependent kinases |  | 420 (CDK4) | Modest antiproliferative activity against K562, MCF7, and RPMI-8226 cancer cell lines. Induced apoptosis in RPMI-8226 cells. |
 | - | IC50 values against MCF7 cells (IC50 = 0.13 µM), MIAPaCa pancreatic cancer cell line (IC50 = 0.28 µM), and HeLa cervical cancer cell line (IC50 = 0.21 µM). | |
 | - | IC50 values against MCF7 cells (IC50 = 0.15 µM), MIAPaCa pancreatic cancer cell line (IC50 = 0.34 µM), and HeLa cervical cancer cell line (IC50 = 0 0.73 µM). | |
 | 24 (CDK2) 23 (CDK5) | Induction of apoptosis in the MiaPaCa2 pancreatic cancer cell line. | |
 | 10–210 (multiple CDK inhibitor) | Induction of apoptosis in colon cancer and multiple myeloma cells. | |
 | 2380 (CDK1) | Antiproliferative activity against hepatocellular carcinoma (HepG2, Huh7, and SNU-475), colon cancer (HCT116), and renal cancer (UO-31) cell lines (IC50 = 0.05, 0.065, 1.93, 1.68, and 1.85 µM, respectively). | |
 | 1520 (CDK1) | Antiproliferative activity against hepatocellular carcinoma (HepG2, Huh7, and SNU-475), colon cancer (HCT116), and renal cancer (UO-31) cell lines (IC50 = 0.028, 1.83, 1.70, 0.035, and 2.24 µM, respectively). | |
 | 88 (CDK14) | Antiproliferative activity against HCT116 colorectal cancer cell line (IC50 = 1.14 µM). | |
 | 8.63 (CDK1) 0.30 (CDK2) | Inhibitory effect against HDAC1, HDAC2, and HDAC3 (IC50 = 6.40, 0.25, and 45.0 nM, respectively). Antiproliferative activity against HCT116 colorectal cancer cell line (IC50 = 0.71 µM). | |
 | 9 (CDK12) 5.8 (CDK13) | - | |
| EGFR |  | 210 | Antiproliferative effect against MCF-7 breast cancer cell line (IC50 = 0.30 μM). |
| FGFR |  | 0.2 (FGFR1) 2.5 (FGFR2) 1.8 (FGFR3) 165 (FGFR4) | Orally bioavailable, clinical candidate for lymphoma, glioma, lung, breast, gastric, and esophageal types of cancer. |
 | 9.3 (FGFR1) 7.6 (FGFR2) 22 (FGFR3) 290 (FGFR4) | Antiproliferative IC50 values against gastric SNU-16 and colon HCT116 cancer cell lines are 17 nM and 5.9 µM, respectively. | |
 | 0.75 (FGFR1) 0.5 (FGFR2) 3.05 (FGFR3) 87.9 (FGFR4) | Antiproliferative activity against FGFR-2-amplified SNU-16 gastric cancer cell line (IC50 = 85.64 nM). | |
| IKK |  | - | Antiproliferative activity against HeLa cervical cancer cell line (IC50 = 14.2 μg/mL). |
| IRAK4 |  | 5 | Strong potency (IC50 = 83 nM) against lipopolysaccharide-induced THP1-XBlue cells. |
 | 0.4 | Good permeability (25 × 10−6 cm/s) in MDCK cells. | |
 | 0.51 | - | |
| ITK |  | Ki = 0.1 nM | Multi-kinase inhibitor. |
 | Ki = 0.5 nM | Higher kinase selectivity, permeability, and oral bioavailability than compound 37. | |
 | Ki = 0.09 nM | Improved potency, selectivity, and less toxicity. | |
| JAK |  | 3.4 (JAK1) 2.2 (JAK2) 3.5 (JAK3) | Antiproliferative activity: IC50 against PC-3 IC50 = 1.08 μM, MCF-7 IC50 = 1.33 μM, HEL IC50 = 1.08 μM, K562 IC50 = 0.77 μM, MOLT4 IC50 = 1.61 μM. |
 | - | Antiproliferative activity against HEL (IC50 = 0.35 μM) and K562 (IC50 = 0.37 μM). | |
 | Ki = 2.5 nM (JAK2) | Potent inhibitory activity in a JAK2-driven SET2 cell-based assay (IC50 = 131 nM). Low potential for reversible inhibition of five major human CYP450 isozymes, and good in vitro permeability profile. | |
 | Ki = 0.21 nM (JAK1) Ki = 0.088 nM (JAK2) | IC50 of 4.7 nM in IL-13 stimulated BEAS-2B cells. | |
 | Ki = 0.31 nM (JAK1) Ki = 0.14 nM (JAK2) | IC50 of 6.4 nM in the IL-13-pSTAT6 cell-based assay. | |
 | 75(JAK1) 1.1 (JAK2) 360 (JAK3) | In vivo reduction of reticulocytes and subsequent reductions in red blood cell mass as well as a decrease in platelets. | |
| JNK |  | 2800 (JNK1) | In vivo anti-inflammatory activity against carrageenan-induced paw edema model in rats. |
 | 227 (JNK3) | - | |
| LRRK |  | 9 (LRRK2) | - |
 (GNE-0877) | 0.7 (LRRK2) | Improved human hepatocyte stability, brain exposure, and lower ability to inhibit or induce CYP compared to compound 48. | |
 (GNE-9605) | 2 (LRRK2) | Improved human hepatocyte stability, brain exposure, and lower ability to inhibit or induce CYP compared to compound 48. | |
 | - | - | |
 | Ki = 84 nM (wild-type LRRK2) and 39 nM (G2019S mutant type LRRK2) | 98% oral bioavailability. | |
| LsrK |  | 119,000 | - |
| MEK/ERK |  | - | Antiproliferative activity against the A375P melanoma cell line (IC50 = 6.7 µM). |
 | - | Antiproliferative activity against the MDA-MB-435 melanoma cell line (IC50 = 2.7 µM). Kinase inhibition was confirmed by Western blotting. IC50 = 0.30 µM against COX-2. | |
 | - | IC50 = 91 nM against recombinant proteins of the RAF-MEK-ERK cascade. GI50 of 1.18, 2.11, and 0.26 μM against HeLa, MCF-7, and A549 cell lines, respectively. | |
| P38α/MAPK14 |  | 22 | Inhibition of TNF-α production in lipopolysaccharide-stimulated THP-1 human cells. In vivo anti-inflammatory activity. |
 | 135 | Poor cellular permeability due to its highly charged carboxylate group. Its ethyl ester analogue could inhibit phosphorylation of MK2 in HeLa cells (IC50 value = 6 µM) but its IC50 value against p38α is 639 nM. | |
 (VPC00628) | 7 | High selectivity against p38α and p38β. | |
 | 515 | Antiproliferative activity against RPMI-8226 and K-562 leukemia cell lines in addition to the MDA-MB-468 breast cancer cell line (IC50 values are 1.71, 3.42, and 6.70 µM, respectively). | |
| PDK4 |  | 84 | Enhanced glucose tolerance in a diet-induced obesity model in mice. Alleviated the allergic reactions in a passive cutaneous anaphylaxis model in mice. |
| Pim |  | Ki = 0.073 nM (Pim1), 0.473 (Pim2), and 0.041 (Pim3) | Antiproliferative activity against MM1.s myeloma cell line (IC50 = 0.64 µM). |
 | Ki = 0.03 nM (Pim1), 0.1 (Pim2), and 0.02 (Pim3) | Promising in vivo activity against MM1.s and RPMI 8226 mice models of multiple myeloma. | |
| RAF |  | - | Antiproliferative activity against the A375P melanoma cell line (IC50 = 4.5 µM). |
 | 0.04 (wild-type B-RAF) | Its trans isomer (with the hydroxyl group behind the plane) is less potent against the kinase (IC50 = 0.09 nM). | |
 | 330 (V600E-B-RAF) | Antiproliferative activity against WM266.4 and A375 melanoma cell lines with IC50 values of 2.63 and 3.16 µM, respectively | |
 | 770 (V600E-B-RAF) and 1500 (RAF1) | One-digit micromolar IC50 values against different cancer cell lines. | |
 | 2.98 (V600E-B-RAF) | Antiproliferative activity against the A375 melanoma cells (IC50 = 1.82 µM). | |
 | 99.17% inhibition at 10 µM (V600E-B-RAF) | IC50 values within sub-micromolar range (0.27–0.92 µM) against nine cancer cell lines of nine cancer types. Against the A375 melanoma cell line, its IC50 value is 0.82 µM. | |
 | - | 78.04%, 74.47%, and 72.46% inhibition at 10 µM concentration against RAF1, V600E-B-RAF, and V600K-B-RAF kinases, respectively. | |
 | 7 (V600E-B-RAF) | Mean IC50 value against the NCI nine subpanels was within the range of 1.98–3.26 µM. | |
 | 390 (V600E-B-RAF) | IC50 values are within the submicromolar range against most of the tested cell lines (NCI-60 panel). Induced apoptosis in the RPMI-8226 leukemia cell line with an EC50 of 1.52 µM. | |
| ROS |  | 13.6 | - |
| Src |  | 59% inhibition at 10 µM concentration | Antiproliferative activity against CCRF-CEM and MOLT-4 leukemia cell lines (IC50 = 1.00 µM against both of them). |
| TGFβ/ALK |  | 684 (R206H mutated ALK2) | Promising lead compound for treatment of fibrodysplasia ossificans progressive. |
 | 25.6 (R206H mutated ALK2) | Good permeability and in vivo pharmacokinetic properties. | |
 | 9.4 | Promising lead compound for treatment of fibrodysplasia ossificans progressive. Improved aqueous solubility compared to compound 75. | |
 | 13 (TGFβ type 1/ALK5) | Inhibited luciferase activity by 80% at 0.1 μM. In-cell kinase inhibition. | |
 | 280 (TGFβ type 1/ALK5) | >35-fold more selective against ALK5 compared to p38α MAPK. | |
 | 18 (TGFβ type 1/ALK5) | In-cell kinase inhibition. | |
 | 69 (TGFβ type 1/ALK5) | Inhibitory effects against the p38α kinase (IC50 = 104 nM). | |
 | 30 (TGFβ type 1/ALK5) | Potential inhibitor of collagen I and α-SMA protein and mRNA expressions in TGFβ-induced LX-2 human hepatic stellate cells. | |
| Trk |  | 2 (TrkA) 8 (TrkB) | - |
 | - | Potent inhibitor of TrkA with an IC50 value of 0.5 nM in a cellular assay. 29% oral bioavailability. High aqueous solubility and safety against hERG. | |
 | - | Potent inhibitor of TrkA with an IC50 value of 0.5 nM in a cellular assay.54% oral bioavailability. High aqueous solubility and safety against hERG. | |
 | 2.7 (TrkA) | Higher selectivity against TrkA than TrkB and TrkC. | |
 | - | Rapid association rate with the TrkA crystal structure, thus binds to the inactive conformation of the kinase (i.e., type II TrkA inhibitor). | |
 | 0.2 (TrkA) | In vivo activity in CFA-induced thermal hypersensitivity model. | |
| VEGFR |  | 0.95 (VEGFR2) | Decreased the proliferation of VEGF stimulated HUVEC with an IC50 of 0.30 nM. In vivo anticancer activity in a mouse xenograft model of human lung adenocarcinoma A549 cells. |
 | 97 (VEGFR2) | Antiproliferative activity against HT-29 colon cell line (IC50 = 3.32 μM), PC-3 prostate cells (IC50 = 3.17 μM), A549 lung cells (IC50 = 3.87 μM), and U87MG glioblastoma cells (IC50 = 6.77 μM). | |
 | - | In-cell kinase inhibition. Antiproliferative activity against the MCF-7 cell line (IC50 = 18.35 µM). | |
| Multikinase inhibitors |  | Inhibitor of VEGFR2, Src, B-RAF (wild-type), V600E-B-RAF, EGFR (wild-type), and L858R-EGFR with IC50 values of 34, 399, 270, 592, 113, and 31 nM, respectively. | |
 | Multi-kinase inhibitory effects against AKT2, GSK-3β, PI3K, EGFR, IGFR, CDK2, Aurora A, and MAPK. | Antiproliferative activity against SNU449 hepatocellular carcinoma cell line. | |
 | Multikinase inhibitory effects against AKT2, GSK-3β, PI3K, EGFR, IGFR, CDK2, Aurora A, and MAPK. | Antiproliferative activity against SNU449 hepatocellular carcinoma cell line. | |
 | Dual KDR/Aurora B activity | Narrow therapeutic index. | |
 | Inhibitory effect against B-RAF (wild-type), V600E-B-RAF, p38α, JNK1, and JNK2 kinases (inhibition % values at 10 µM concentration are 72.56%, 93.67%, 86.54%, 99.05%, and 98.49%, respectively). | Antiproliferative activity against the A498 renal carcinoma cell line (IC50 = 0.33 µM). JNK1 and JNK2 are the most sensitive among them (IC50 = 350 and 360 nM, respectively). | |
 | >94% inhibition of AKT1, AKT2, V600E-B-RAF, EGFR, p38α, and PDGFRβ at 100 µM. | Antiproliferative activity against MCF7 breast cancer cell line (IC50 = 6.53 µM). | |
 | c-Kit, FLT-3, VEGFR-2, EGFR, PDGFR, and Pim-1 kinases (IC50 260–610 nM). | Antiproliferative activity against A549 (lung), H460 (lung), HT29 (colon), MKN-45 (gastric), U87MG (glioma), and SMMC-77217721 (hepatic) cancer cell lines (IC50 values from 0.29–0.42 µM). | |
 | JAK2, JAK3, Aurora A, and Aurora B (IC50 = 166, 57, 939, 583 nM, respectively). | Antiproliferative activity against K562 leukemia cell line and HCT116 colon cancer cell line (IC50 = 6.726 and 15.054 µM, respectively). | |
 | Inhibited B-RAF, C-RAF, and Src kinases both in vitro and in vivo. | In vivo activity and phase I clinical trials in volunteers with solid tumors. Increased the progression-free survival in a patient suffering from KRAS (G12V) spindle cell sarcoma. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Gamal, M.I.; Zaraei, S.-O.; Madkour, M.M.; Anbar, H.S. Evaluation of Substituted Pyrazole-Based Kinase Inhibitors in One Decade (2011–2020): Current Status and Future Prospects. Molecules 2022, 27, 330. https://doi.org/10.3390/molecules27010330
El-Gamal MI, Zaraei S-O, Madkour MM, Anbar HS. Evaluation of Substituted Pyrazole-Based Kinase Inhibitors in One Decade (2011–2020): Current Status and Future Prospects. Molecules. 2022; 27(1):330. https://doi.org/10.3390/molecules27010330
Chicago/Turabian StyleEl-Gamal, Mohammed I., Seyed-Omar Zaraei, Moustafa M. Madkour, and Hanan S. Anbar. 2022. "Evaluation of Substituted Pyrazole-Based Kinase Inhibitors in One Decade (2011–2020): Current Status and Future Prospects" Molecules 27, no. 1: 330. https://doi.org/10.3390/molecules27010330
APA StyleEl-Gamal, M. I., Zaraei, S.-O., Madkour, M. M., & Anbar, H. S. (2022). Evaluation of Substituted Pyrazole-Based Kinase Inhibitors in One Decade (2011–2020): Current Status and Future Prospects. Molecules, 27(1), 330. https://doi.org/10.3390/molecules27010330