
Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes



Abstract
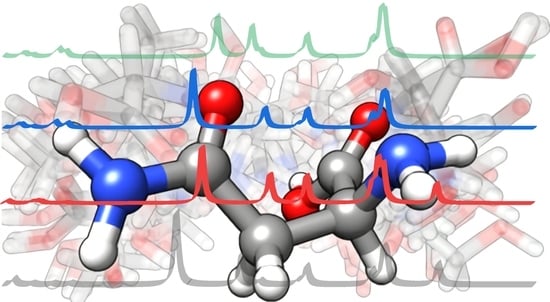
:
1. Introduction
2. Results and Discussion
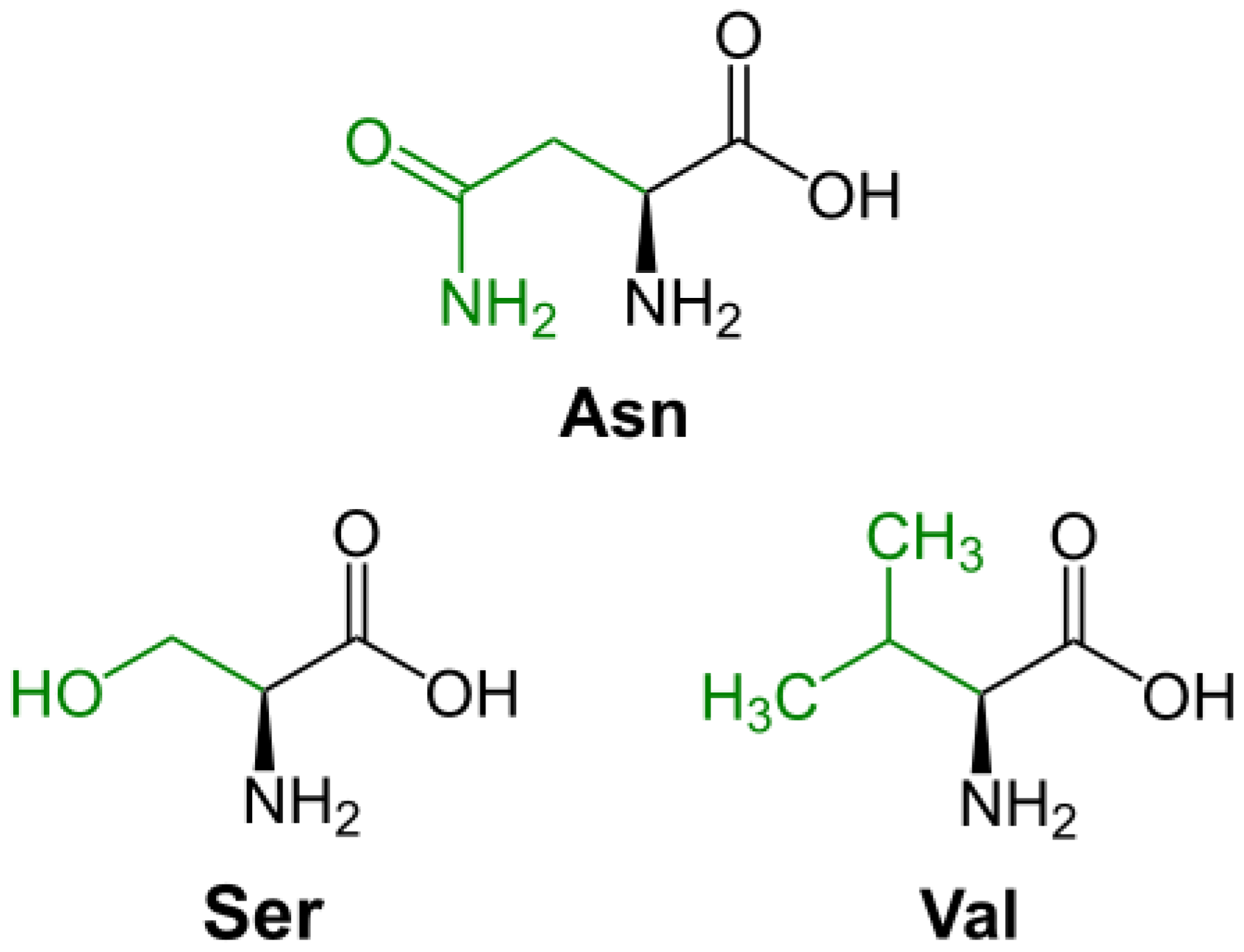
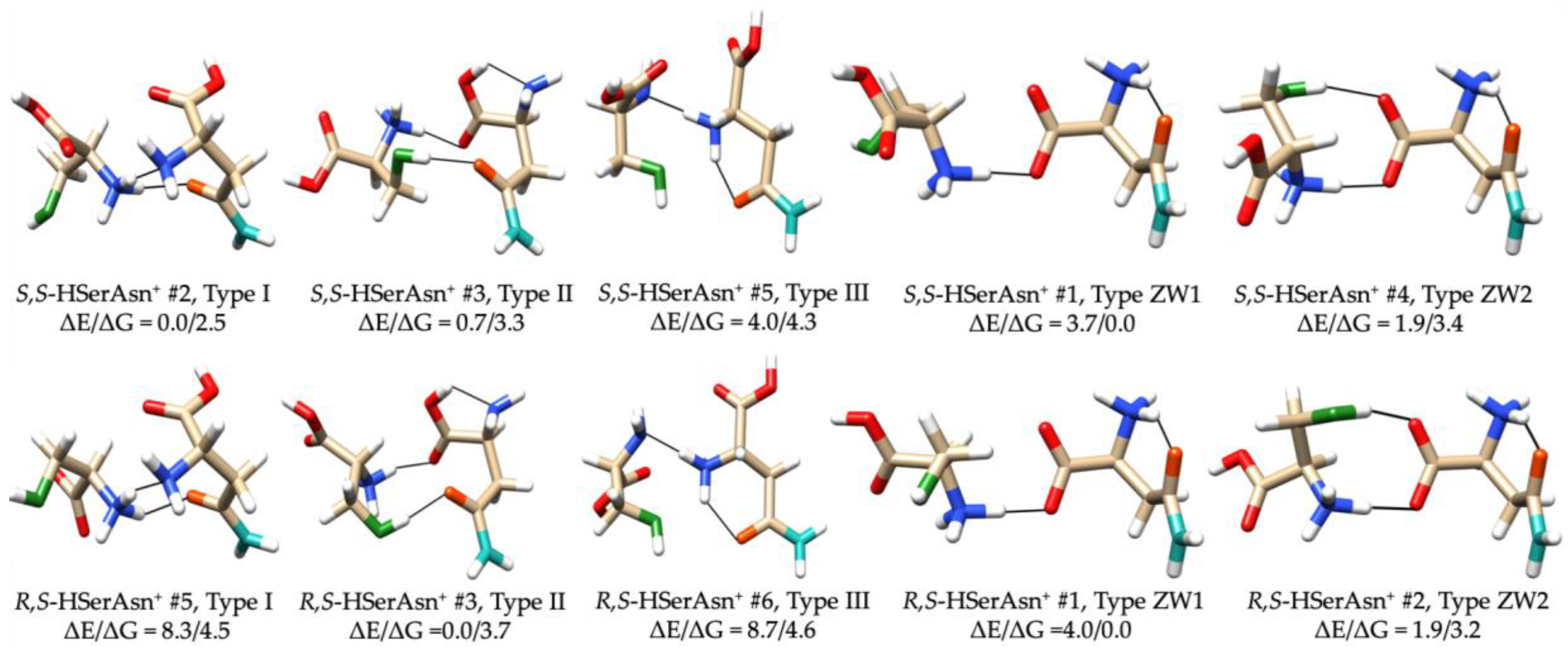
2.1. Possible Low Energy Structures of the Four Diastereomers
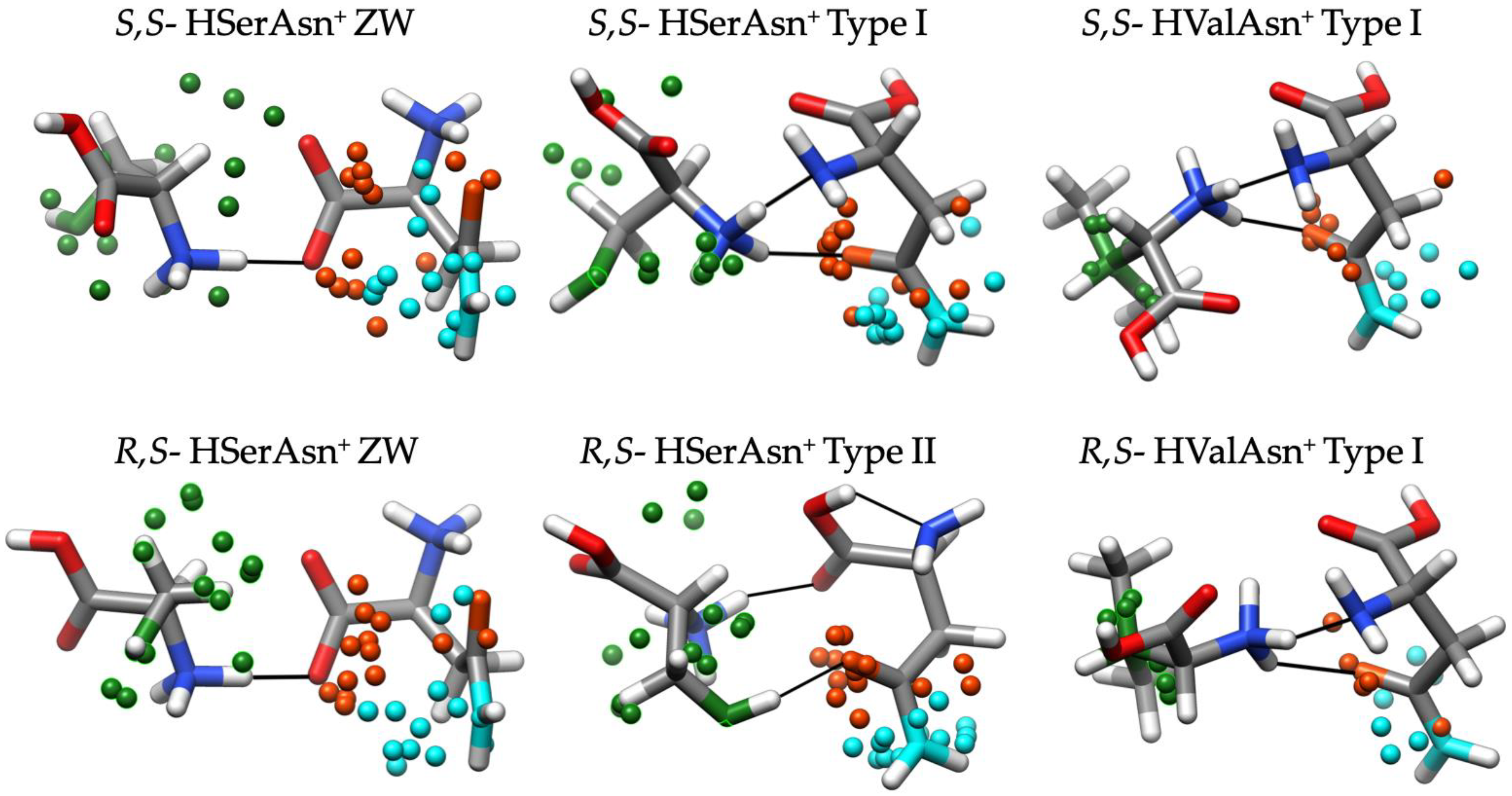
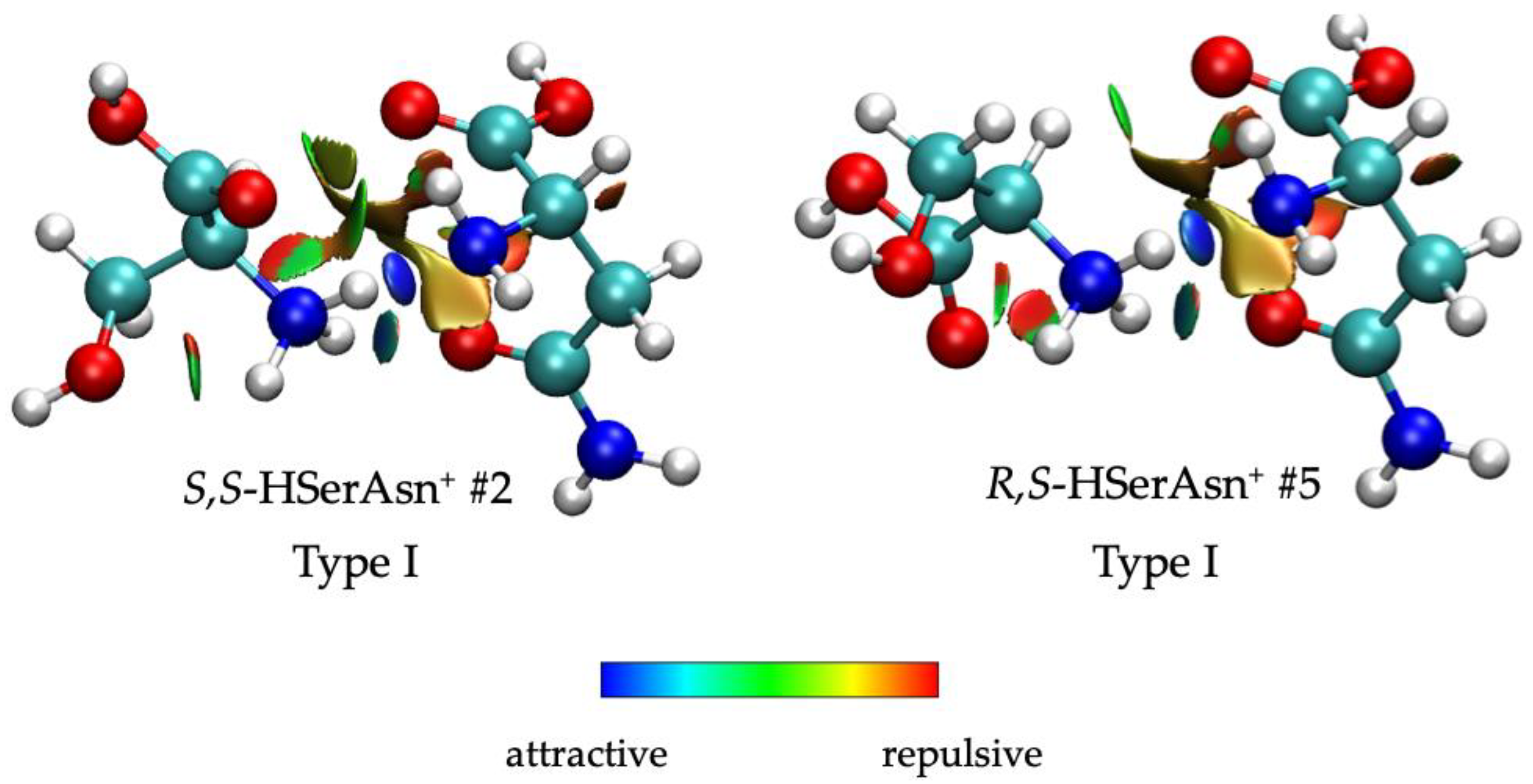
2.2. Different Noncovalent Binding Topologies
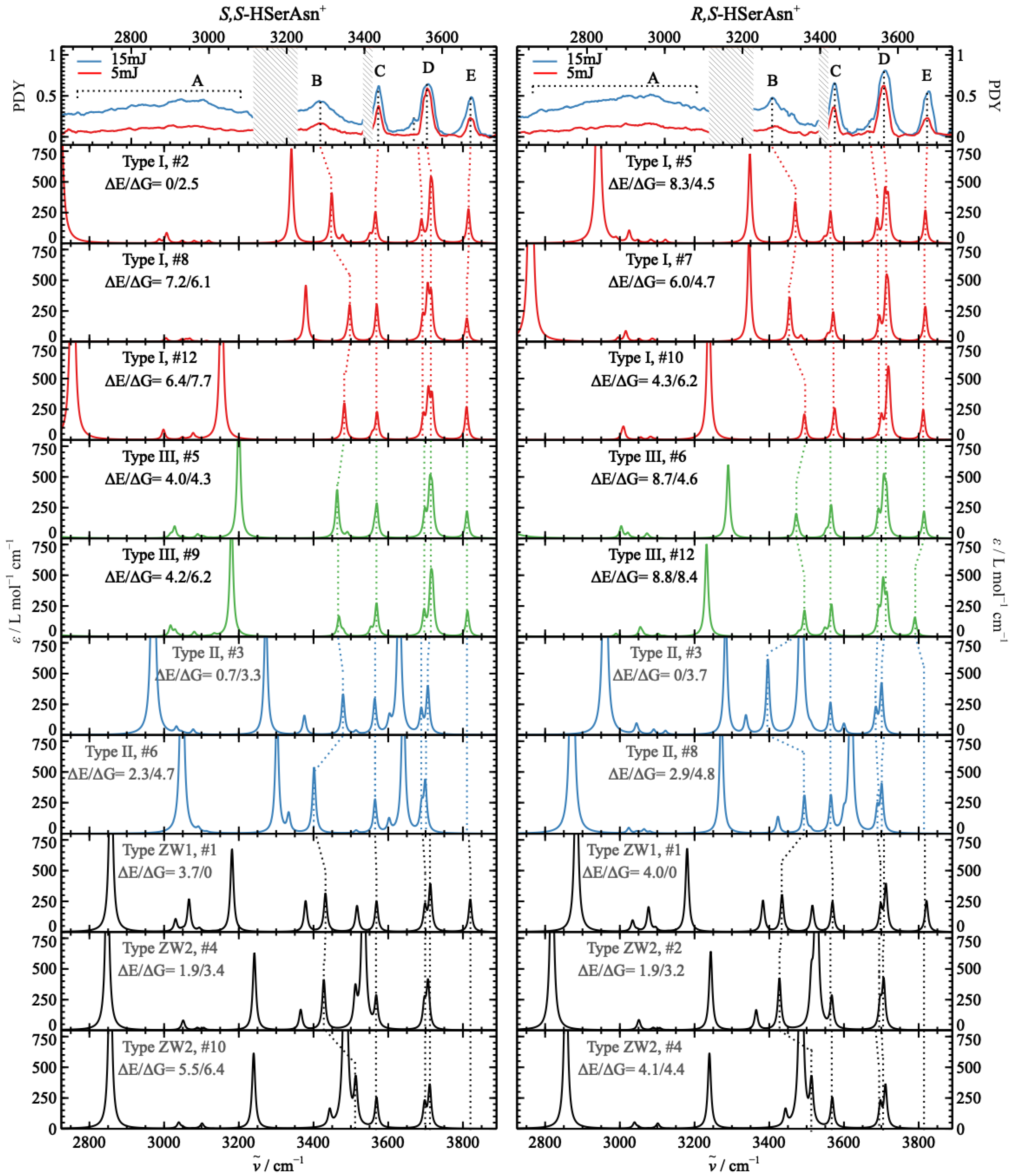
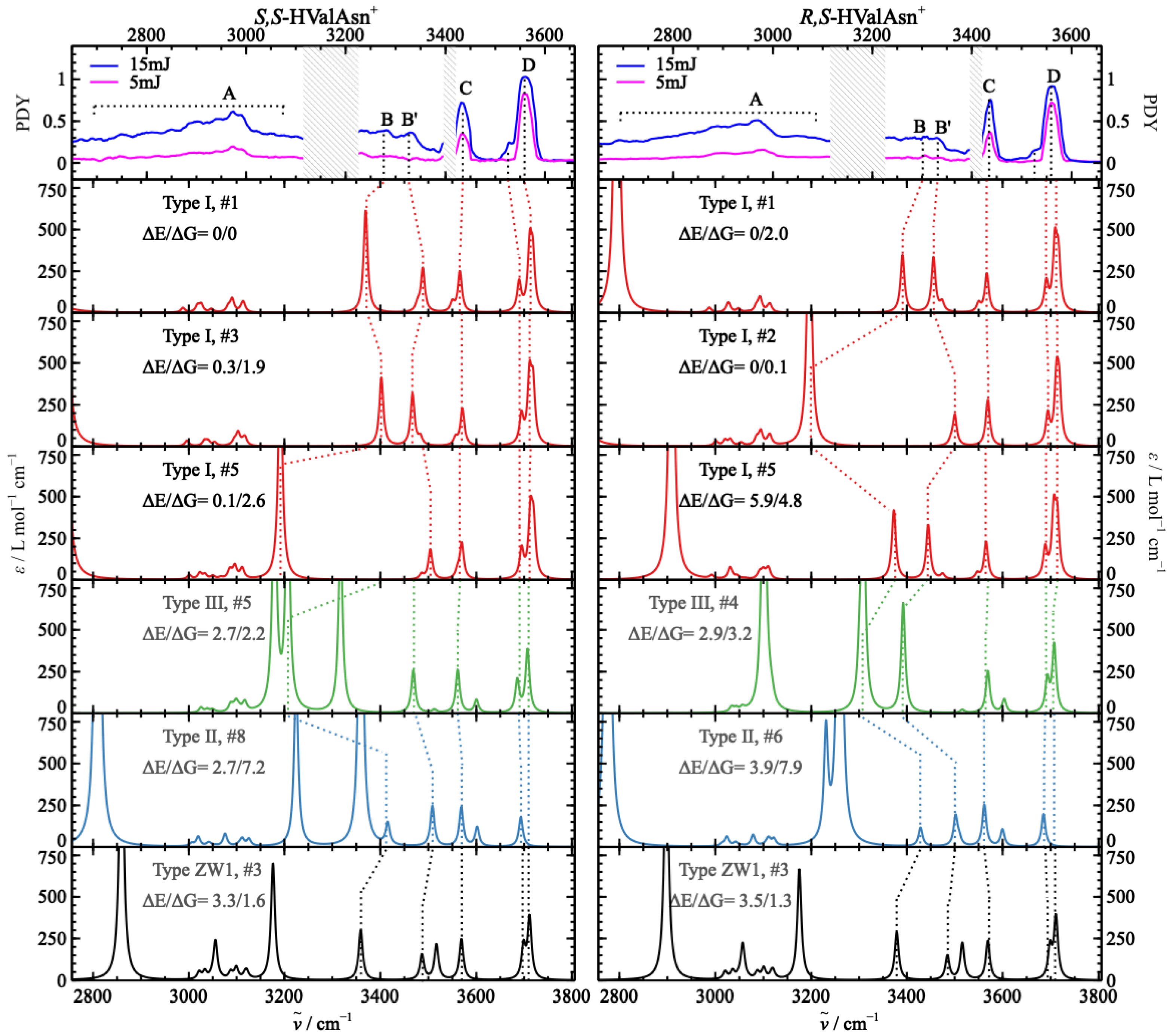
2.3. Comparison of the Experimental and Theoretical IRMPD Spectra
2.4. Chirality Recognition and Kinetic Effects in the IRMPD Spectra
2.5. Fragmentation Channel
3. Materials and Methods
3.1. Sample Preparation
3.2. IRMPD Experimental Details
3.3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Iijima, Y.; Akuzawa, N.; Hatoria, T.; Imai, K.; Kitahara, Y.; Kurabayashi, M. A Lead Intoxication Treated With D-Penicillamine. J. Med. Cases 2014, 5, 187–193. [Google Scholar] [CrossRef]
- Rumsby, P.C.; Shepherd, D.M. The Effect of Penicillamine on Vitamin B6 Function in Man. Biochem. Pharmacol. 1981, 30, 3051–3053. [Google Scholar] [CrossRef]
- Lenz, W. A Short History of Thalidomide Embryopathy. Teratology 1988, 38, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, E.; Yamamoto, T.; Ito, E.; Shibata, N. Understanding the Thalidomide Chirality in Biological Processes by the Self-disproportionation of Enantiomers. Sci. Rep. 2018, 8, 17131. [Google Scholar] [CrossRef] [PubMed]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Zehnacker, A.; Suhm, M.A. Chirality Recognition between Neutral Molecules in the Gas Phase. Angew. Chem. Int. Ed. 2008, 47, 6970–6992. [Google Scholar] [CrossRef] [PubMed]
- Le Barbu, K.; Brenner, V.; Millié, P.; Lahmani, F.; Zehnacker-Rentien, A. An Experimental and Theoretical Study of Jet-Cooled Complexes of Chiral Molecules: The Role of Dispersive Forces in Chiral Discrimination. J. Phys. Chem. A 1998, 102, 128–137. [Google Scholar] [CrossRef]
- Hirata, K.; Mori, Y.; Ishiuchi, S.; Fujii, M.; Zehnacker, A. Chiral Discrimination between Tyrosine and β-Cyclodextrin Revealed by Cryogenic Ion Trap Infrared Spectroscopy. Phys. Chem. Chem. Phys. 2020, 22, 24887–24894. [Google Scholar] [CrossRef]
- Lee, J.J.; Albrecht, M.; Rice, C.A.; Suhm, M.A.; Stamm, A.; Zimmer, M.; Gerhards, M. Adaptive Aggregation of Peptide Model Systems. J. Phys. Chem. A 2013, 117, 7050–7063. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Borho, N.; Xu, Y. Chiral Self-Recognition: Direct Spectroscopic Detection of the Homochiral and Heterochiral Dimers of Propylene Oxide in the Gas Phase. J. Am. Chem. Soc. 2006, 128, 17126–17131. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Seifert, N.A.; Jäger, W.; Xu, Y. Conformational Panorama and Chirality Controlled Structure-Energy Relationship in a Chiral Carboxylic Acid Dimer. Angew. Chem. Int. Ed. 2020, 59, 15703–15710. [Google Scholar] [CrossRef]
- Maris, A.; Giuliano, B.M.; Bonazzi, D.; Caminati, W. Molecular Recognition of Chiral Conformers: A Rotational Study of the Dimers of Glycidol. J. Am. Chem. Soc. 2008, 130, 13860–13861. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.G.; Dian, B.C.; Douglass, K.O.; Geyer, S.M.; Shipman, S.T.; Pate, B.H. A Broadband Fourier transform Microwave Spectrometer Based on Chirped Pulse Excitation. Rev. Sci. Instrum. 2008, 79, 053103. [Google Scholar] [CrossRef]
- Pate, B.H.; Evangelisti, L.; Caminati, W.; Xu, Y.; Thomas, J.; Patterson, D.; Perez, C.; Schnell, M. Quantitative Chiral Analysis by Molecular Rotational Spectroscopy. In Chiral Analysis, Advances in Spectroscopy, Chromatography and Emerging Methods, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 679–729. [Google Scholar] [CrossRef]
- Maitre, P.; Scuderi, D.; Corinti, D.; Chiavarino, B.; Crestoni, M.E.; Fornarini, S. Applications of Infrared Multiple Photon Dissociation (IRMPD) to the Detection of Posttranslational Modifications. Chem. Rev. 2020, 120, 3261–3295. [Google Scholar] [CrossRef] [PubMed]
- Polfer, N.C.; Oomens, J.; Dunbar, R.C. IRMPD Spectroscopy of Metal-Ion/Tryptophan Complexes. Phys. Chem. Chem. Phys. 2006, 8, 2744–2751. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Xu, Y.; Kong, X. Structural Diversity of Di-Metalized Arginine Evidenced by Infrared Multiple Photon Dissociation (IRMPD) Spectroscopy in the Gas Phase. Molecules 2021, 26, 6546. [Google Scholar] [CrossRef]
- Wu, R.; McMahon, T.B. An Investigation of Protonation Sites and Conformations of Protonated Amino Acids by IRMPD Spectroscopy. ChemPhysChem 2008, 9, 2826–2835. [Google Scholar] [CrossRef]
- Fales, B.S.; Fujamade, N.O.; Oomens, J.; Rodgers, M.T. Infrared Multiple Photon Dissociation Action Spectroscopy and Theoreical Studies of Triethyl Phosphate Complexes: Effect of Protonation and Sodium Cationization on Structure. J. Am. Soc. Mass Spectrom. 2011, 22, 1862–1871. [Google Scholar] [CrossRef] [Green Version]
- Andersson, Å.; Poline, M.; Kodambattil, M.; Rebrov, O.; Loire, E.; Maître, P.; Zhaunerchyk, V. Structure of Proton-Bound Methionine and Tryptophan Dimers in the Gas Phase Investigated with IRMPD Spectroscopy and Quantum Chemical Calculations. J. Phys. Chem. A 2020, 124, 2408–2415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooks, R.G.; Zhang, D.; Koch, K.J.; Gozzo, F.C.; Eberlin, M.N. Chiroselective Self-Directed Octamerization of Serine: Implications for Homochirogenesis. Anal. Chem. 2001, 73, 3646–3655. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Tsai, I.-A.; Sabu, S.; Han, C.-C.; Lee, Y.T.; Chang, H.-C.; Tu, S.-Y.; Kung, A.H.; Wu, C.-C. Progressive Stabilization of Zwitterionic Structure in [S(Ser)2–8]+ Studied by Infrared Photodissociation Spectroscopy. Angew. Chem. Int. Ed. 2006, 45, 4130–4134. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Lin, C.; Infusini, G.; Oh, H.-B.; Jiang, H.; Breuker, K.; Wu, C.-C.; Charkin, O.P.; Chang, H.-C.; McLafferty, F.W. Numerous Isomers of Serine Octamer Ions Characterized by Infrared Photodissociation Spectroscopy. ChemPhysChem 2009, 10, 2603–2606. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, F.X.; Yang, G.; Kitova, E.N.; Klassen, J.S.; Xu, Y. Chirality Recognition of the Protonated Serine Dimer and Octamer by Infrared Multiphoton Dissociation Spectroscopy. Phys. Chem. Chem. Phys. 2013, 15, 1873–1886. [Google Scholar] [CrossRef]
- Ren, J.; Wang, Y.-Y.; Feng, R.-X.; Kong, X.-L. Investigation of L/D- Threonine Substituted L-Serine Octamer Ions by Mass Spectrometry and Infrared Photodissociation Spectroscopy. Chin. Chem. Lett. 2017, 28, 537–540. [Google Scholar] [CrossRef]
- Zehnacker, A. Chirality Effects in Gas-Phase Spectroscopy and Photophysics of Molecular and Ionic Complexes: Contribution of Low and Room Temperature Studies. Int. Rev. Phys. Chem. 2014, 33, 151–207. [Google Scholar] [CrossRef]
- Shi, Y.; Du, M.; Ren, J.; Zhang, K.; Xu, Y.; Kong, X. Application of Infrared Multiple Photon Dissociation (IRMPD) Spectroscopy in Chiral Analysis. Molecules 2020, 25, 5152. [Google Scholar] [CrossRef] [PubMed]
- Nugrahani, I.; Jessica, M.A. Amino Acids as the Potential Co-Former for Co-Crystal Development: A Review. Molecules 2021, 26, 3279. [Google Scholar] [CrossRef] [PubMed]
- Kojo, S.; Uchino, H.; Yoshimura, M.; Tanaka, K. Racemic D,L-Asparagine Causes Enantiomeric Excess of Other Coexisting Racemic D,L-Amino Acids During Recrystallization: A Hypothesis Accounting for the Origin of L-Amino Acids in the Biosphere. Chem. Commun. 2004, 19, 2146–2147. [Google Scholar] [CrossRef]
- Blanco, S.; Sanz, M.E.; López, J.C.; Alonso, J.L. Revealing the Multiple Structures of Serine. Proc. Natl. Acad. Sci. USA 2007, 104, 20183–20188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesarri, A.; Cocinero, E.J.; López, J.C.; Alonso, J.L. The Shape of Neutral Valine. Angew. Chem. Int. Ed. 2004, 43, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, C.; Varela, M.; Peña, I.; Mata, S.; Lopez, J.C.; Alonso, J.L. The Conformational Locking of Asparagine. Chem. Commun. 2012, 48, 5934–5936. [Google Scholar] [CrossRef]
- Seo, J.; Warnke, S.; Pagel, K.; Bowers, M.T.; von Helden, G. Infrared Spectrum and Structure of the Homochiral Serine Octamer-Dichloride Complex. Nat. Chem. 2017, 9, 1263–1268. [Google Scholar] [CrossRef]
- Schrödinger, Inc. MacroModel; Version 9.8; Schrödinger, LLC: New York, NY, USA, 2010; Available online: https://www.schrodinger.com/products/macromodel (accessed on 25 November 2021).
- Aradi, B.; Hourahine, B.; Frauenheim, T. DFTB+, A Sparse Matrix-Based Implementation of the DFTB Method. J. Phys. Chem. A 2007, 111, 5678–5684. [Google Scholar] [CrossRef] [PubMed]
- Hourahine, B.; Aradi, B.; Blum, V.; Bonafé, F.; Buccheri, A.; Camacho, C.; Cevallos, C.; Deshaye, M.Y.; Dumitrică, T.; Dominguez, A.; et al. DFTB+, A Software Package for Efficient Approximate Density Functional Theory Based Atomistic Simulations. J. Chem. Phys. 2020, 152, 124101. [Google Scholar] [CrossRef] [PubMed]
- Poline, M.; Rebrov, O.; Larsson, M.; Zhaunerchyk, V. Theoretical Studies of Infrared Signatures ofProton-Bound Amino Acid Dimers with Homochiral and Heterochiral Moieties. Chirality 2020, 32, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Seifert, N.A.; Heger, M.; Thomas, J.; Jäger, W.; Xu, Y. The Rich Conformational Landscape of Perillyl Alcohol Revealed by Broadband Rotational Spectroscopy and Theoretical Modelling. Phys. Chem. Chem. Phys. 2019, 21, 15408–15416. [Google Scholar] [CrossRef]
- Xie, F.; Fusè, M.; Hazrah, A.S.; Jäger, W.; Barone, V.; Xu, Y. Discovering the Elusive Global Minimum in a Ternary Chiral Cluster: Rotational Spectra of Propylene Oxide Trimer. Angew. Chem. Int. Ed. 2020, 59, 22427–22430. [Google Scholar] [CrossRef]
- Pracht, P.; Bauer, C.A.; Grimme, S. Automated and Efficient Quantum Chemical Determination and Energetic Ranking of Molecular Protonation Sites. J. Comput. Chem. 2017, 38, 2618–2631. [Google Scholar] [CrossRef]
- Oswald, S.; Seifert, N.A.; Bohle, F.; Gawrilow, M.; Grimme, S.; Jäger, W.; Xu, Y.; Suhm, M.A. The Chiral Trimer and A Metastable Chiral Dimer of Achiral Hexafluoroisopropanol: A Multi-Messenger Study. Angew. Chem. Int. Ed. 2019, 58, 5080–5084. [Google Scholar] [CrossRef]
- Seifert, N.A.; Thomas, J.; Jäger, W.; Xu, Y. Rotational Spectra and Theoretical Study of Tetramers and Trimers of 2-Fluoroethanol: Dramatic Intermolecular Compensation for Intramolecular Instability. Phys. Chem. Chem. Phys. 2018, 20, 27630–27637. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ng, X.; Seifert, N.A.; Thomas, J.; Jäger, W.; Xu, Y. Rotational Spectroscopy of Chiral Tetreahydro-2-furoic Acid: Conformational Landscape, Conversion and Abundances. J. Chem. Phys. 2018, 149, 224306. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, D.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Györffy, W.; Kats, D.; Korona, T.; Lindh, R.; et al. MOLPRO, Version 2015.1, a Package of ab initio Programs. Available online: https://www.molpro.net/ (accessed on 25 November 2021).
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A General-Purpose Quantum Chemistry Program Package. Wires Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.03; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Alonso, J.L.; López, J.C. Microwave Spectroscopy of Biomolecular Building Blocks. In Gas-Phase IR Spectroscopy and Structure of Biological Molecules; Rijs, A.M., Oomens, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; Volume 364, pp. 335–402. [Google Scholar]
- Miller, R.E. The Vibrational Spectroscopy and Dynamics of Weakly Bound Neutral Complexes. Science 1998, 240, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Saragi, R.T.; Caminati, W.; Lesarri, A. The Hydrogen Bond and Beyond: Perspectives for Rotational Investigations of Non-Covalent Interactions. Chem. Eur. J. 2019, 25, 11402–11411. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Uriarte, I.; Insausti, A.; Vallejo-López, M.; Basterretxea, F.J.; Cochrane, S.A.; Davis, B.G.; Corzana, F.; Cocinero, E.J. Observation of the Unbiased Conformers of Putative DNA-Scaffold Ribosugars. ACS Cent. Sci. 2020, 6, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voronina, L.; Rizzo, T.R. Spectroscopic Studies of Kinetically Trapped Conformations in the Gas Phase: The Case of Triply Protonated Bradykinin. Phys. Chem. Chem. Phys. 2015, 17, 25828–25836. [Google Scholar] [CrossRef]
- Zhu, P.; Yang, G.; Poopari, M.R.; Bie, Z.; Xu, Y. Conformations of Serine in Aqueous Solutions as Revealed by Vibrational Circular Dichroism. ChemPhysChem 2012, 13, 1272–1281. [Google Scholar] [CrossRef]
- Rak, J.; Skurski, P.; Simons, J.; Gutowski, M. Low-Energy Tautomers and Conformers of Neutral and Protonated Arginine. J. Am. Chem. Soc. 2001, 123, 11695–11707. [Google Scholar] [CrossRef]
- Simon, A.; MacAleese, L.; Maître, P.; Lemaire, J.; McMahon, T.B. Fingerprint Vibrational Spectra of Protonated Methyl Esters of Amino Acids in the Gas Phase. J. Am. Chem. Soc. 2007, 129, 2829–2840. [Google Scholar] [CrossRef] [PubMed]
- Heaton, A.L.; Bowman, V.N.; Oomens, J.; Steill, J.D.; Armentrout, P.B. Infrared Multiple Photon Dissociation Spectroscopy of Cationized Asparagine: Effects of Metal Cation Size on Gas-Phase Conformation. J. Phys. Chem. 2009, 113, 5519–5530. [Google Scholar] [CrossRef] [PubMed]
- Heger, H.; Cheramy, J.; Xie, F.; Chen, Z.; Xu, Y. Sturcural and Energetic Properties of Protonated and Sodiated Asparagine Probed by A New Laboratory IRMPD Spectrometer. J. Mol. Spectrosc. 2018, 352, 36–44. [Google Scholar] [CrossRef]
- Andersson, Å.; Poline, M.; Houthuijs, K.J.; van Outersterp, R.E.; Berden, G.; Oomens, J.; Zhaunerchyk, V. IRMPD Spectroscopy of Homo- and Heterochiral Asparagine Proton-Bound Dimers in the Gas Phase. J. Phys. Chem. A 2021, 125, 7449–7456. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras- Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Mara, I.; Alkorta, I.; Espinosa, E.; Molins, E. Relationships Between Interaction Energy, Intermolecular Distance and Electron Density Properties in Hydrogen Bonded Complexes Under External Electric Fields. Chem. Phys. Lett. 2011, 507, 185–189. [Google Scholar]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Borho, N.; Xu, Y. Tailoring the Key in a Molecular Lock-and-Key Model System: The Propylene Oxide⋯ 2-Fluoroethanol Complex. J. Am. Chem. Soc. 2008, 130, 5916–5921. [Google Scholar] [CrossRef]
- Thomas, J.; Jäger, W.; Xu, Y. Chirality Induction and Amplification in the 2, 2, 2-Trifluoroethanol⋅⋅⋅ Propylene Oxide Adduct. Angew. Chem. Int. Ed. 2014, 53, 7277–7280. [Google Scholar] [CrossRef]
- Klyne, J.; Bouchet, A.; Ishiuchi, S.; Fujii, M.; Schneider, M.; Baldauf, C.; Dopfer, O. Probing Chirality Recognition of Protonated Glutamic Acid Dimers by Gas-Phase Vibrational Spectroscopy and First Principles Simulations. Phys. Chem. Chem. Phys. 2018, 20, 28452–28464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, A.L.; Armentrout, P.B. Thermodynamics and Mechanism of Protonated Asparagine Decomposition. J. Am. Mass Spectrom. 2009, 20, 852–866. [Google Scholar] [CrossRef] [Green Version]
- Uddin, K.M.; Warburton, P.L.; Poirier, R.A. Comparisons of Computational and Experimental Thermochemical Properties of α-Amino Acids. J. Phys. Chem. B 2012, 116, 3220–3234. [Google Scholar] [CrossRef] [PubMed]
- Bleiholder, C.; Suhar, S.; Paizs, B. Revising the Proton Affinity Scale of the Naturally Occurring Alpha-Amino Acids. J. Am. Soc. Mass Spectrom. 2006, 17, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Shishido, R.; Huang, Q.; Fujii, A.; Kuo, J. Vibrational Spectroscopy of Protonated Amine-Water Clusters: Tuning Fermi Resonance and Lighting up Dark States. Phys. Chem. Chem. Phys. 2020, 22, 22035–22046. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, W.S.; Marta, R.A.; McMahon, T.B. Proton-Bound 3-Cyanophenylalanine Trimethylamine Clusters: Isomer-Specific Fragmentation Pathways and Evidence of Gas-Phase Zwitterions. J. Phys. Chem. A 2013, 117, 10714–10718. [Google Scholar] [CrossRef]
- Lehnig, R.; Jäger, W. Infrared Spectroscopy of the Antisymmetric Stretching Mode of 16OC18O in Helium Nanodroplets. Chem. Phys. Lett. 2006, 424, 146–150. [Google Scholar] [CrossRef]
- Raston, P.L.; Knapp, C.J.; Jäger, W. Ro-vibrational Spectroscopy of Hydrogen Peroxide Embedded in Superfluid Helium Nanodroplets. Phys. Chem. Chem. Phys. 2011, 13, 18789–18798. [Google Scholar] [CrossRef]
- Prell, J.S.; O’Brien, J.T.; Williams, E.R. IRPD Spectroscopy and Ensemble Measurements: Effects of Different Data Acquisition and Analysis Methods. J. Am. Soc. Mass Spectrom 2010, 21, 800–809. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isomer Ranking a | S,S-HSerAsn+ | R,S-HSerAsn+ | S,S-HValAsn+ | R,S-HValAsn+ |
|---|---|---|---|---|
| Type, Ser/Asn Comp b | Type, Ser/Asn Comp b | Type, Val/Asn Comp b | Type, Val/Asn Comp b | |
| 1 | Type ZW1, HS1+/ZW | Type ZW1, HS1+/ZW | Type I, HV1+/A1 c | Type I, HV1+/A1 c |
| 2 | Type I, HS1+/A1 c | Type II, HS3+/A2 | Type ZW1, HV1+/ZW | Type ZW1, HV1+/ZW |
| 3 | Type II, HS1+/A2 | Type I, HS1+/A1c | Type III, HV1+/A2 | Type III, HV1+/A2 |
| 4 | Type III, S1/HA+ c | Type III, S1/HA+c | Type II, HV3+/A2 | Type II, HV3+/A2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Heger, M.; Al-Jabiri, M.H.; Xu, Y. Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes. Molecules 2022, 27, 38. https://doi.org/10.3390/molecules27010038
Wang H, Heger M, Al-Jabiri MH, Xu Y. Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes. Molecules. 2022; 27(1):38. https://doi.org/10.3390/molecules27010038
Chicago/Turabian StyleWang, Haolu, Matthias Heger, Mohamad H. Al-Jabiri, and Yunjie Xu. 2022. "Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes" Molecules 27, no. 1: 38. https://doi.org/10.3390/molecules27010038
APA StyleWang, H., Heger, M., Al-Jabiri, M. H., & Xu, Y. (2022). Vibrational Spectroscopy of Homo- and Heterochiral Amino Acid Dimers: Conformational Landscapes. Molecules, 27(1), 38. https://doi.org/10.3390/molecules27010038